
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient detection of 1H, 15N correlations in hydrogen bonded low molecular catalyst–substrate intermediates without selective 15N-labelling†
Christian L.
Scholtes
 ,
Julian
Ilgen
and
Ruth M.
Gschwind
*
,
Julian
Ilgen
and
Ruth M.
Gschwind
*
Institut für Organische Chemie, Universität Regensburg, Universitätsstraße 31, D-93053 Regensburg, Germany. E-mail: ruth.gschwind@chemie.uni-regensburg.de
First published on 11th April 2025
Abstract
To date, SOFAST approaches have generally been limited to biomolecules. We present the applicability of SOFAST-HMQC techniques to small molecules in the slow-tumbling regime offering a time-efficient characterization of catalyst substrate hydrogen bonds with nitrogen at natural abundance. This extends NMR access to a broader range of catalyst substrate combinations.
Hydrogen bond activation plays a special role in biochemistry, synthesis, and organo-catalysis. In the latter, hydrogen bonds exhibit different potentials to activate the corresponding substrate. Traditionally, they induce a lowering of the activation barrier by decreasing the energy of the LUMO orbital.1 Prime examples are substrates including nitrogen atoms near weak electrophilic centres, which become more reactive through hydrogen bonding. The characterization of hydrogen bonds can generally be performed via NMR spectroscopy often omitted in synthetic studies. This neglect results mainly from the absence of stable hydrogen bonds under reaction conditions. However, if this condition is fulfilled, the chemical shift of the proton can already give some insight into the character of the corresponding hydrogen bond. A more detailed picture can be achieved when the hydrogen bond is established to a nucleus, which provides an NMR active isotope. Nitrogen is a prime example offering the naturally occurring 15N with a nuclear spin of 1/2. However, only every fourth in 1000 nitrogen atoms are naturally 15N isotopes. Additionally, its low gyromagnetic ratio (low γ-nucleus, 0.1 of 1H) makes 15N a quite challenging nucleus for NMR investigations. Nevertheless, since 15N chemical shifts are very sensitive to tiny variations of the chemical environment, if hydrogen bonds are established to a nitrogen atom, NMR properties can give valuable insights helping to understand structural features, molecular assemblies,2 and also the on-going reaction mechanism.3,4 In the case of pyridines and imines, the Steiner–Limbach correlation between 1H and 15N chemical shift characterizes the nature of the investigated hydrogen bond.3–6 However, the low natural abundance of 15N and its low gyromagnetic ratio need some workarounds to monitor the 15N chemical shift efficiently. On the one hand, inverse detection schemes such as HMQC, HSQC and HMBC (and variants)7 are popular whereby the sensitivity gain relies on the excitation and detection of 1H as the high-gamma nucleus. On the other hand, the system can be (selectively) 15N-labelled to deal with its low natural abundance, which is the established procedure in NMR investigations of biomacromolecules. In contrast to biomolecules, small to mid-size molecules come with a much broader structural variety rendering selective 15N-labelling inefficient and expensive, thus requiring expensive 15N precursors and/or extended chemical synthesis. In these cases, fast NMR acquisition techniques promise a practical solution to overcome the extended experimental time of NMR experiments involving nuclei with low natural abundance such as 15N by accumulating a high number of scans. Herein, the accelerated acquisition is generally addressed by two main principles – and combinations of them – either based on changed sampling techniques, namely non-uniform sampling (NUS)8 and single-scan 2D-NMR experiments,9 or by reducing the relaxation time. The latter includes a bundle of experiments such as ASAP-HSQC/HMQC, ALSOFAST-HSQC, IMPACT-H(N)MBC,10,11 Fast12 or SOFAST-HMQC.13 Some of them are developed in the framework of NMR applications for large biomolecules and are more common in these studies, while others are applicable in a broader range from small to large systems. SOFAST-HMQC (band-selective optimized flip-angle short-transient) became a standard in bio-NMR with manifold reported applications such as in proteins, and nucleic acids14,15 both in vitro and in cellulo.16,17 In contrast, there are only a limited number of reported examples in small molecules, herein for 1H, 13C-correlations in metabolomic studies18 and for the detection of NH⋯O
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C hydrogen bonds in (beta-)peptides.19 To understand the underrepresentation of small molecules, the two fundaments for speeding up the experiment need to be addressed. First, the SOFAST technique utilizes the potential of Ernst angle pulses from the Fast-HMQC,12 which increase the sensitivity per time unit and scan as a general concept independent of the system and the experimental conditions. Second, it relies on an accelerated relaxation upon selective excitation of one distinct group of protons (e.g., amides). Whether accelerated relaxation upon selective excitation is observed depends on the molecular tumbling regime. Due to different signs of the dipolar cross-relaxation rate at room temperature for fast and slow tumbling molecules, selective excitation is detrimental for small molecules and beneficial for large molecules. Therefore, biomolecules are notorious candidates for the application of SOFAST, which fulfils its full potential in the slow tumbling regime, whereas low molecular compounds are generally assumed to be fast tumbling. The conclusion for small molecules would be to force them into the slow tumbling regime e.g., by lowering the temperature. Given a significant amount of NOE contacts, this should render SOFAST approaches generally applicable to small molecules. Besides reducing cost and labour required for labelling substrates, SOFAST approaches additionally hold the potential to access substrates which cannot be 15N-labelled.
C hydrogen bonds in (beta-)peptides.19 To understand the underrepresentation of small molecules, the two fundaments for speeding up the experiment need to be addressed. First, the SOFAST technique utilizes the potential of Ernst angle pulses from the Fast-HMQC,12 which increase the sensitivity per time unit and scan as a general concept independent of the system and the experimental conditions. Second, it relies on an accelerated relaxation upon selective excitation of one distinct group of protons (e.g., amides). Whether accelerated relaxation upon selective excitation is observed depends on the molecular tumbling regime. Due to different signs of the dipolar cross-relaxation rate at room temperature for fast and slow tumbling molecules, selective excitation is detrimental for small molecules and beneficial for large molecules. Therefore, biomolecules are notorious candidates for the application of SOFAST, which fulfils its full potential in the slow tumbling regime, whereas low molecular compounds are generally assumed to be fast tumbling. The conclusion for small molecules would be to force them into the slow tumbling regime e.g., by lowering the temperature. Given a significant amount of NOE contacts, this should render SOFAST approaches generally applicable to small molecules. Besides reducing cost and labour required for labelling substrates, SOFAST approaches additionally hold the potential to access substrates which cannot be 15N-labelled.
For the CPA (chiral phosphoric acid) catalyst interaction with its imine substrate, we investigated hydrogen bonds using low temperature NMR.3,4 The strength of those H-bonds correlates with the reactivity at synthetic reaction conditions.4 So far, these investigations relied on the 15N-labelling of the substrates to observe intramolecular 1H, 15N-correlations by direct 15N-detection. Under the conditions in which the hydrogen bonds under investigation are observable – below 240 K – the intermediate complexes are in the slow tumbling regime. Moreover, the hydrogen bond protons resonate in a well isolated chemical shift region (12–18 ppm). Both aspects make the SOFAST approach a promising technique for the given systems.
As a proof of concept, we show the application and suitability of the SOFAST-HMQC and some extensions (derived experiments) as generally applicable techniques for small molecules in reaction centres on the example of binary complexes formed by a hydrogen bond between a CPA and an imine without prior 15N-labelling. This allows fast access to 15N chemical shifts and scalar couplings, which can be used for the classification of the hydrogen bonds using the theory developed by Limbach et al.5
Furthermore, the acceleration of the longitudinal relaxation time T1 upon selective excitation as the basis of the SOFAST approach as well as the resulting sensitivity enhancement are discussed. As a benchmark system in this study we chose nine different binary complexes already thoroughly investigated by our group.3,4 All combinations of the three imines 1a–c with the three acids (R)-3,3′-bis(3,5-bis(trifluoromethyl)phenyl)-1,1′-binaphthyl-2,2′-diyl hydrogen phosphate (TRIFP, 2a),20 (R)-3,3′-bis(2,4,6-triisopropylphenyl)-1,1′-binaphthyl-2,2′-diyl hydrogen phosphate (TRIP, 2b)21 and tetrafluoroboric acid diethyl ether complex (HBF4·O(CH2CH3)2, 3) have been screened using a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio (Fig. 1).
:1 ratio (Fig. 1).
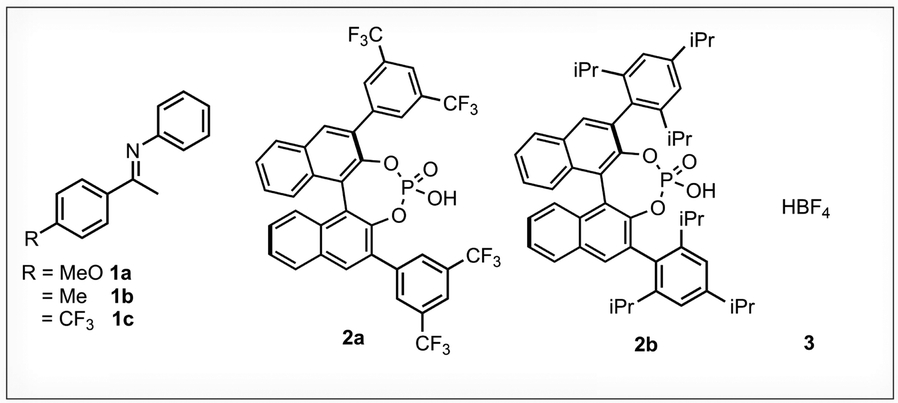 | ||
| Fig. 1 Imine/acid intermediates used as model systems: structures of the investigated imines 1, 3,3′-substituted phosphoric acids 2 and HBF43. All samples have been prepared using a concentration of 50 mM and an imine:acid ratio of 1:1. The imines exist as both E- and Z-isomer. | ||
For all nine binary complexes, the application of 1H, 15N-SOFAST-HMQC has proven successful in an amenable time at natural abundance of 15N and using a standard NMR probe at 180 K. Signals resulting from an effective 15N intermediate concentrations as low as ∼25 μM have been observed using the SOFAST-HMQC pulse sequence offering a signal to noise ratio (S/N) of 18 in about 30 min (see Fig. 2A). Measuring the same sample with a classical HMQC pulse sequence only gave an S/N of 14 in 2 h 40 min.
 | ||
| Fig. 2 SOFAST allows full characterization of H-bonds in catalyst substrate intermediates at natural abundance of 15N: (A) overlay of the 1H, 15N-SOFAST-HMQC spectrum (blue) and the 1H, 15N-SOFAST-CLIP-HMQC spectrum (red) at an effective 15N concentration of 25 μM and 94 μM for Z- and E-1a·2a, respectively. The SOFAST-HMQC spectrum has been acquired with 256 scans and 32 increments within 29 min. The SOFAST-CLIP-HMQC spectrum has been acquired with 1024 scans and 16 increments within 65 min. For clarity the δ(15N) axes of both spectra have been shifted against each other. (B) Constructed Steiner–Limbach curve of the 1H and 15N chemical shifts extracted from the 1H, 15N-SOFAST-HMQC spectra at 15N natural abundance (grey diamonds) in comparison with literature reported data (red open circles) acquired on 15N-labeled samples.4 (C) To date elusive 1H, 31P-coherence obtained via magnetization transfer through the 2hJPH hydrogen bond scalar coupling. 1H, 31P-SOFAST-HMBC spectrum acquired with 256 scans and 32 increments within 30 min. All data have been generated using a standard NMR probe at 180 K and 50 mM 15N-unlabelled complexes in an imine:acid ratio of 1:1. For further details see Sections S4–S7 (ESI†). | ||
This allowed the establishment of the Steiner–Limbach curve in a fraction of the time required in the literature.3,4 The general trend of the spectra is discussed based on the example of 1a·2a (for all other spectra see Sections S4–S6, ESI†). As displayed in Fig. 2A (blue) the 1H, 15N-SOFAST-HMQC was acquired in only 29 min using 256 scans and 32 increments in the indirect dimension. Both 1H, 15N-correlations of the present isomeric complexes are observed with their distinct 15N chemical shifts. The Z-1·2a complexes generally show smaller line widths compared to the corresponding E-configured complexes (e.g. see Section S4.1, ESI†). This is due to the exchange of the latter with the free E-imine (see Section S8, ESI†), which results in a coherence loss over time and generally makes this correlation more tedious to detect. As shown in Fig. 2B all data are in agreement with known literature chemical shifts (open circles) performed using 15N-labelled compounds (∼50 mM) and traditional pulse sequences.4 Based on the mathematical description outlined by Limbach et al., the character of the hydrogen bond can further be corroborated by accessing the 1hJNH coupling constant.5,6
By applying a 90° pulse on the heteronuclear channel prior to acquisition and abstaining from the heteronuclear decoupling, as known from the CLIP-HSQC22 and SOFAST-IPAP-HMQC,23 the SOFAST-HMQC (Fig. 3A(i)) is transformed into the SOFAST-CLIP-HMQC pulse sequence (Fig. 3A(ii)). The extraction of 1hJNH coupling constants from clean in-phase signals becomes accessible, as shown in Fig. 2A (red). The SOFAST-CLIP-HMQC may be acquired both as a 2D and a 1D spectrum, whereby the latter provides a more time-efficient variant when more scans are demanded. This comes of course at the cost of the 15N chemical shift information and is less suitable for overlapping proton signals (see Fig. S15, ESI†).
So far, we focussed only on the 1H, 15N-correlation inside the P–O−⋯H·+N structural moiety. However, the 1H, 31P-correlation corroborates the formation of the hydrogen bond within the binary complex. Therefore, we deployed another member out of the SOFAST family. Previously described by Farjon, the SOFAST-HMBC was used to observe NH⋯OC hydrogen bridges in peptides.19 This pulse sequence (Fig. 3B) allows the long-range correlation of the hydrogen bond proton and the phosphorus of 2. In our experience the detection of these 2hJPH correlations has been tedious or even inaccessible in the case of strong acids. However, using 1H, 31P-SOFAST-HMBC the correlation of both isomeric complexes 1a·2a which have been to date elusive are shown in Fig. 2C.
 | ||
| Fig. 3 SOFAST pulse sequence schemes applied within the study of CPA catalyst imine interactions. (A) (i) SOFAST-HMQC, (ii) SOFAST-CLIP-HMQC (without broadband decoupling), and (B) SOFAST-HMBC. Pulse sequences. Further details are outlined in the ESI† (Section S3). | ||
Finally, revisiting the accelerated relaxation in selective inversion only works effectively for large slow tumbling compounds. Therefore, we evaluated the T1 relaxation using inversion recovery experiments with either broadband or selective pulses on our small molecules in the slow tumbling limit at low temperature. Fig. 4A shows the relaxation profiles performed with selective (open circles) and broadband pulses (circles) on the hydrogen bond proton of the binary complexes E- and Z-1a·2a. An unexpectedly high acceleration of the longitudinal relaxation is observed for the hydrogen bond protons of the binary complexes in contrast to the methyl and methoxy groups (see Section S9, ESI†). Upon selective treatment, a full recovery to the equilibrium state is achieved after about 2 s, whereas recovery takes more than 10 s when broadband pulses are applied. Note that first acquiring a 2D-NOESY spectrum hints at whether relaxation acceleration upon selective treatment might be observed based on the sign and the number of NOE cross-peaks. The observed relaxation acceleration of the H-bond protons becomes obvious from the high number NOE contacts with the same sign as the diagonal peaks (see Section S8, ESI†).
 | ||
| Fig. 4 The principles of SOFAST in action: (A) accelerated relaxation in inversion recovery series using selective (open circles) inversion pulses versus broadband (filled circles). (B) Sensitivity enhancement curves acquired with the 1H-SOFAST experiment (without 15N-filtering) for different flip-angles α (open circles) in comparison with an analogous experiment using broadband 1H excitation (filled circles). At Tscan = 0.25 s enhancement factors up to 3.6 are possible. The shown curves were acquired at 180 K for the hydrogen bond protons of the complex 1a·2a (50 mM, 1:1 ratio) using a standard NMR probe with a base frequency of 600 MHz. For further details see Sections S9 and S10 (ESI†), respectively. | ||
In addition to the relaxation enhancements, SOFAST approaches leverage the Ernst angle to obtain a sensitivity enhancement compared to 90° excitation pulses. The effect on the sensitivity can be studied by monitoring relaxation enhancement curves. These are educative illustrations, since they can be constructed from any pulse sequence, and several effects like the excitation angle can be studied and they show the optimal experimental conditions considering the time Tscan, i.e. the time required for one scan, and sensitivity gain (Fig. 4B). The influence of Tscan and excitation angle α are discussed based on Z-1a·2a. Using a broadband 90° excitation pulse, in the following referred to as a reference, the maximal sensitivity was obtained when Tscan equals around 1 s. Using a 90° selective excitation pulse (black, open circles) the sensitivity maximum shifts to a smaller Tscan value giving a sensitivity gain by a factor of 2.5 together with a time gain factor of 1.6. In agreement with biomolecular literature,13 increasing the selective excitation angles, the maximal sensitivity further shifts to smaller Tscan values linked with a small additional sensitivity gain.
Summing up the results, the successful application of SOFAST based experiments to small molecules inside hydrogen bonded intermediates effectively on a μmolar level was shown. Extending our toolbox for small molecules by these methods renders the 15N-labelling (or any other low abundant nuclei) of our substrates under given conditions obsolete. Given that the system under investigation falls into the slow tumbling regime, and has sufficiently separated protons and NOE contacts, we think that the SOFAST approach can be used as a general concept. This is not only interesting from a time and financial perspective but opens the door to substrates previously not accessible due to a lack of possibilities for 15N labelling.
J. I. conceived and conceptualized the project. C. S. and J. I. planned and performed all experiments. C. S. did analysis and visualization. Interpretation of results, writing and revision was done by all authors. R. G. provided funding and resources.
This work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) through project 426795949, RTG 2620 “Ion Pair Effects in Molecular Reactivity” and project 444632635, TRR 325 “Assembly Controlled Chemical Photocatalysis”.
Data availability
The data underlying this study are available in the published article and its supporting information. The primary NMR data of the figures 2 and 4 as well as those shown in the supporting information, and Bruker pulse sequence codes are available free of charge at: DOI: 10.5281/zenodo.14755142.Conflicts of interest
There are no conflicts to declare.References
- R. Maji, S. C. Mallojjala and S. E. Wheeler, Chem. Soc. Rev., 2018, 47, 1142–1158 RSC.
- M. A. Jinks, M. Howard, F. Rizzi, S. M. Goldup, A. D. Burnett and A. J. Wilson, J. Am. Chem. Soc., 2022, 144, 23127–23133 CrossRef CAS PubMed.
- N. Sorgenfrei, J. Hioe, J. Greindl, K. Rothermel, F. Morana, N. Lokesh and R. M. Gschwind, J. Am. Chem. Soc., 2016, 138, 16345–16354 CrossRef CAS PubMed.
- K. Rothermel, M. Melikian, J. Hioe, J. Greindl, J. Gramüller, M. Žabka, N. Sorgenfrei, T. Hausler, F. Morana and R. M. Gschwind, Chem. Sci., 2019, 10, 10025–10034 RSC.
- H.-H. Limbach, M. Pietrzak, S. Sharif, P. M. Tolstoy, I. G. Shenderovich, S. N. Smirnov, N. S. Golubev and G. S. Denisov, Chemistry, 2004, 10, 5195–5204 CrossRef CAS PubMed.
- S. Sharif, G. S. Denisov, M. D. Toney and H.-H. Limbach, J. Am. Chem. Soc., 2007, 129, 6313–6327 CrossRef CAS PubMed.
- D. J. Russell, C. E. Hadden, G. E. Martin, A. A. Gibson, A. P. Zens and J. L. Carolan, J. Nat. Prod., 2000, 63, 1047–1049 CrossRef CAS PubMed.
- K. Kazimierczuk, J. Stanek, A. Zawadzka-Kazimierczuk and W. Koźmiński, Prog. Nucl. Magn. Reson. Spectrosc., 2010, 57, 420–434 CrossRef CAS PubMed.
- L. Frydman, T. Scherf and A. Lupulescu, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 15858–15862 CrossRef CAS PubMed.
- J. Furrer, Chem. Commun., 2010, 46, 3396–3398 RSC.
- J. F. K. Limtiaco, D. J. Langeslay, S. Beni and C. K. Larive, J. Magn. Reson., 2011, 209, 323–331 CrossRef CAS PubMed.
- A. Ross, M. Salzmann and H. Senn, J. Biomol. NMR, 1997, 10, 389–396 CrossRef CAS PubMed.
- P. Schanda and B. Brutscher, J. Am. Chem. Soc., 2005, 127, 8014–8015 CrossRef CAS PubMed.
- J. Farjon, J. Boisbouvier, P. Schanda, A. Pardi, J.-P. Simorre and B. Brutscher, J. Am. Chem. Soc., 2009, 131, 8571–8577 CrossRef CAS PubMed.
- B. Sathyamoorthy, J. Lee, I. Kimsey, L. R. Ganser and H. Al-Hashimi, J. Biomol. NMR, 2014, 60, 77–83 CrossRef CAS PubMed.
- E. Luchinat, L. Barbieri, M. Cremonini and L. Banci, J. Biomol. NMR, 2021, 75, 97–107 CrossRef CAS PubMed.
- P. Broft, S. Dzatko, M. Krafcikova, A. Wacker, R. Hänsel-Hertsch, V. Dötsch, L. Trantirek and H. Schwalbe, Angew. Chem., Int. Ed., 2021, 60, 865–872 CrossRef CAS PubMed.
- S. Ghosh, A. Sengupta and K. Chandra, Anal. Bioanal. Chem., 2017, 409, 6731–6738 CrossRef CAS PubMed.
- A. Altmayer-Henzien, V. Declerck, D. J. Aitken, E. Lescop, D. Merlet and J. Farjon, Org. Biomol. Chem., 2013, 11, 7611–7615 RSC.
- M. Rueping, E. Sugiono, C. Azap, T. Theissmann and M. Bolte, Org. Lett., 2005, 7, 3781–3783 CrossRef CAS PubMed.
- S. Hoffmann, A. M. Seayad and B. List, Angew. Chem., 2005, 117, 7590–7593 CrossRef.
- A. Enthart, J. C. Freudenberger, J. Furrer, H. Kessler and B. Luy, J. Magn. Reson., 2008, 192, 314–322 CrossRef CAS PubMed.
- P. Schanda, E. Kupce and B. Brutscher, J. Biomol. NMR, 2005, 33, 199–211 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cc00537j |
| This journal is © The Royal Society of Chemistry 2025 |