DOI:
10.1039/D5CC00599J
(Feature Article)
Chem. Commun., 2025,
61, 6055-6068
Transition-metal-catalyzed auxiliary-assisted C–H functionalization using vinylcyclopropanes and cyclopropanols
Received
3rd February 2025
, Accepted 25th March 2025
First published on 25th March 2025
Abstract
Transition-metal-catalyzed chelation-assisted C–H functionalization exploiting small strained rings has surfaced as an appealing strategy, offering a robust platform for the construction of complex molecules in a step- and atom-economic fashion. In this vein, three-membered rings, viz. vinylcyclopropanes (VCPs) and cyclopropanols, have emerged as staple coupling partners due to their inherent ring strain. Moreover, their strain release serves as a potent driving force, unlocking new possibilities in molecular engineering via sequential C–H functionalization and ring scission. Recently, significant progress has been made in this emerging domain employing the aforementioned rings. This review article focuses on directing group (DG)-assisted C–H functionalization adopting VCPs and cyclopropanols as potential coupling partners until November 2024. The advancements are organized based on the type of functionalizations achieved.

Shubhajit Basak
| Shubhajit Basak was born and brought up in West Bengal, India. He received his BSc and MSc in Chemistry from the University of North Bengal, India. Currently, he has been pursuing a PhD under the supervision of Professor T. Punniyamurthy at the Indian Institute of Technology Guwahati. His research interest is focused on the transition-metal-catalyzed directed C–H functionalization of heterocycles. |
Tripti Paul
| Tripti Paul was born and brought up in West Bengal, India. She received her BSc and MSc in Chemistry from the University of North Bengal, India. Presently, she has been pursuing her PhD under the supervision of Professor T. Punniyamurthy at the Indian Institute of Technology Guwahati with a Prime Minister's Research Fellowship. Her research work is based on the catalytic C–H functionalization of heterocycles. |

Santu Mandal
| Santu Mandal was born and brought up in West Bengal, India. He received his BSc from Vidyasagar University, India and MSc from NIT Rourkela, India. Currently, he has been pursuing a PhD under the supervision of Professor T. Punniyamurthy at the Indian Institute of Technology Guwahati, India. His research interest is focused on C–H bond functionalization under transition-metal-catalysis. |

Madhab Barman
| Madhab Barman was born and brought up in West Bengal, India. He received his BSc from Vidyasagar University, India, and MSc from University of Hyderabad, India. Currently, he has been pursuing a PhD under the supervision of Professor T. Punniyamurthy at the Indian Institute of Technology Guwahati. His research interest is focused on the directed C–H functionalization of heterocycles. |

Maniya V Nanjegowda
| Maniya V N was born in Karnataka, India. He completed his BSc and MSc in Chemistry from University of Mysore, India. Presently, he is pursuing his PhD with a Prime Minister's Research Fellowship under the supervision of Professor T. Punniyamurthy at the Indian Institute of Technology Guwahati. His research interest is transition-metal-catalyzed regioselective C–H functionalization towards sustainable organic synthesis. |

Tharmalingam Punniyamurthy
| Tharmalingam Punniyamurthy is Professor of Organic Chemistry at the Indian Institute of Technology Guwahati. His research interests include the site-selective C–H functionalization and expansion of strained ring systems for the construction of heterocyclic scaffolds. He is a fellow of the Indian Academy of Sciences, the National Academy of Sciences, India, the Indian National Science Academy and the Royal Society of Chemistry, and a recipient of a CRSI Silver Medal and J C Bose National Fellowship. |
1. Introduction
The emergence of DG-assisted C–H functionalization utilizing transition-metal (TM) catalysis has garnered significant attention due to the rapid construction of C–C/C–heteroatom bonds in a predictable way.1 Moreover, the omnipresence of C–C bonds as pivotal structural constituents in organic molecules underscores the importance of their formation and cleavage in synthetic chemistry.2 In this paradigm, selective C–C cleavage has led to a leading-edge synthetic method for the construction of structurally complex organic molecules. However, the lack of bond polarization and high bond strength make C–C activation a formidable challenge. Along this line, tremendous efforts have thus been devoted to enabling TM-catalyzed selective C–C bond cleavage.3 A useful approach involves the employment of strained small-ring systems as versatile synthons.4 The strain-release energy associated with their cleavage serves as a substantial driving force, rendering them ideal scaffolds for TM-catalyzed C–C activation. In particular, three or four-membered rings manifest as a suitable candidate as the thermodynamic barrier for C–C bond scission is compensated by their inherent strain (Fig. 1A). Belonging to the class of three-membered rings, VCPs and cyclopropanols can function as versatile building blocks for the synthetic elaboration of organic scaffolds.5 In addition, the facile ring opening of VCP may involve β-carbon elimination to execute C–C bond cleavage, whereas ring-opening of cyclopropanol forms an organometallic γ-oxo alkyl-metal intermediate, leading to C–C bond formation (Fig. 1B).
 |
| | Fig. 1 Reactivity profile of VCP and cyclopropanol with C–H substrates under TM-catalysis. | |
Furthermore, due to the enhanced reactivity of small rings, they are susceptible to rapid decomposition, homocoupling or attack by available nucleophiles. Consequently, the prudent choice of TM-catalysts and appropriate C–H substrates is crucial for an impactful transformation. In this context, an assortment of TM-catalysts and substrates tailored with DGs are adopted for harnessing the reactivity of these rings. In addition, the installation of DGs or pre-existing directing functionality in a substrate is paramount to define the reaction pathway, offering advanced regiocontrol. Frequently implemented DGs include N-containing functionalities due to their strong chelating ability and the formation of thermodynamically stable cyclometalated intermediates. However, recent years have witnessed tremendous attention to the utilization of weak-chelating DGs,6 resulting in a less stable metallacycle, thus increasing the propensity to react with the strained rings. From this viewpoint, the compatibility among DG-tethered substrates, ring systems and metal-catalysts requires a comprehensive assessment.
In pursuit of addressing the persistent synthetic obstacle, the integration of C–H functionalization and C–C cleavage in a unified tandem process employing strained rings furnishes a potential approach to access complex molecular architectures.7 From this perspective, the catalytic ring scission of VCPs and cyclopropanols for DG-assisted C–H functionalization has gained considerable interest. Alongside, the seminal studies by the research groups of Ackermann, Glorius, Gooßen, Li, Shi and many others have occupied a prominent position in the archives of synthetic chemistry. Recently, our group has made a significant contribution to this emerging area utilizing the aforementioned moieties. Given the importance and surge in demand, a current update on directed C–H functionalization exploiting the ring cleavage of VCPs and cyclopropanols would thus be valuable. This article attempts to collate the chronological developments in TM-catalyzed chelation-assisted C–H functionalization with VCPs and cyclopropanols until November 2024. The advances presented are categorized based on the types of transformations, such as allylation, alkenylation, alkylation and annulation (Fig. 2). The reactivity pattern of the associated subunits, key mechanistic underpinnings and important synthetic diversifications in a few instances are addressed. However, the Lewis acid-catalyzed ring opening or cycloaddition reactions of these are well documented,5 and hence not covered here.
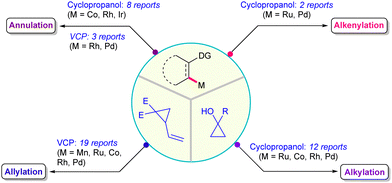 |
| | Fig. 2 General outline of C–H functionalizations with VCPs and cyclopropanols. | |
2. Reactions using vinylcyclopropanes
Readily accessible VCP, due to its tailor-made nature with a strained cyclopropane ring and a conjugated vinyl group, has garnered tremendous attention in organic synthesis.5c–e The inherent ring strain (∼28 kcal mol−1) makes it highly susceptible to ring opening via C–C bond cleavage mediated by Lewis acids, radical pathways and TM-catalysis.5g From the perspective of step- and atom-economy, TM-catalyzed tandem C–H/C–C functionalization offers a highly efficient strategy to access diverse scaffolds with significant applications in medicinal and material science.7 In this context, VCP serves as a promising coupling partner facilitating the synthesis of allyl surrogates or carbocyclic scaffolds that are challenging to produce through conventional methods. This section explores the advancements achieved through C–H functionalization utilizing VCPs.
2.1. Allylation
The significance of allylated derivatives in natural products and medicinal compounds has made the allylation strategy a key area of scholarly interest. Alkenes are widely utilized as coupling partners in TM-catalyzed C–H allylation.8 Recently, direct allylation through the synergistic activation of C–H and C–C bonds in a single step has gained significant attention, employing VCPs as a coupling partner. In 2015, Wang and co-workers realized a Rh(III)-catalyzed C–H allylation of N-methoxy benzamides using VCPs (Scheme 1A).9 The reaction exhibits good compatibility with aliphatic alkenyl C–H functionalization and the sterically bulky substituents of the ester group of VCP gave better E/Z-selectivity. Subsequently, an elegant approach was demonstrated by the Ackermann group utilizing cost-effective Co(III)-catalysis for strong chelation-assisted indole and arene C–H allylation with VCPs (Scheme 1B).10 The reaction selectively yielded the thermodynamically less stable Z-alkenes under mild reaction conditions. The origin of the Z-selectivity using Co-catalysis has been supported by the DFT studies, which implied that the key C–C cleavage of VCP is the diastereoselectivity-determining step, whose energetic span is less for the formation of the Z-diastereomer compared to that of the E-diastereomer. This might be attributed to shorter Co–C bonds, leading to more compact organometallic species. Notably, the sterically hindered pivalate additive was essential for the reaction.
 |
| | Scheme 1 Metal-catalyzed C–H allylation of (hetero)arenes with VCPs. | |
Later, the Glorius group reported the economically viable Mn(I)-catalyzed 2-pyridyl-directed C–H allylation of (hetero)arenes using VCPs with excellent E/Z-selectivities (Scheme 1C).11 Interestingly, the methodology showed remarkable efficiency under silver and solvent free conditions, exhibiting excellent functional group tolerance. At the same time, the Ackermann group accomplished 2-pyridyl-assisted E-selective C–H allylation of (hetero)arenes with VCPs utilizing MnBr(CO)5 as the catalyst (Scheme 1D).12 The DFT study revealed that London dispersion interactions play a significant role in stabilizing the (E)-transition-state. Moreover, the allylation strategy was well suited for the late-stage modification of amino-acids. Furthermore, Zhu and Song groups reported a Rh(III)-catalyzed C7–H allylation of indolines with VCPs (Scheme 2).13 A wide range of substrates were amenable to provide the allylated products in high yields with good functional group compatibility.
 |
| | Scheme 2 C7-allylation of indolines under Rh(III)-catalysis. | |
In 2019, Gooßen and co-workers developed the Ru-catalyzed ortho-C–H allylation of aromatic acids with VCPs (Scheme 3A).14 A broad spectrum of allylarenes was synthesized in high yields and stereoselectivities. In addition, the allylation can be combined with follow-up steps to enable the synthesis of functionalized coumarins. In the same year, Yoshino and Matsunaga demonstrated the Co-catalyzed C–H allylation of NH-free benzimidates using VCPs under mild reaction conditions (Scheme 3B).15 The key features include Z-selective C–H allylation with an unaltered imidate group, which enables a second C–H functionalization and cyclization, leading to important heterocycles. Later, the Song and Zhu groups updated the allylation strategy enhancing its synthetic value with a focus on green and sustainable principles. This work featured a solvent-free microwave-assisted direct C7-allylation of indolines with VCPs at room temperature (Scheme 4A).16 A wide range of substrates were tolerated to afford the allylated indolines in high yields and E/Z-selectivities. The scope of the procedure was extended for the allylation of tetrahydroisoquinolines. Meanwhile, the same group reported bisallylation using imidazopyridines as intrinsic DGs with VCPs (Scheme 4B).17 The reaction proceeds with a five-membered ruthenacycle, which furnished a mono-allylated metal complex via C–H/C–C functionalization with one molecule of VCP. The mono-allylated intermediate undergoes a 2nd C–H functionalization through a rollover mechanism, leading to bis-allylation with another molecule of VCP.
 |
| | Scheme 3 Metal-catalyzed ortho-allylation of (hetero)arenes. | |
 |
| | Scheme 4 (A) Microwave-assisted CH-allylation of indolines and tetrahydroisoquinolines. (B) Bis-allylation strategy under Ru(II)-catalysis. | |
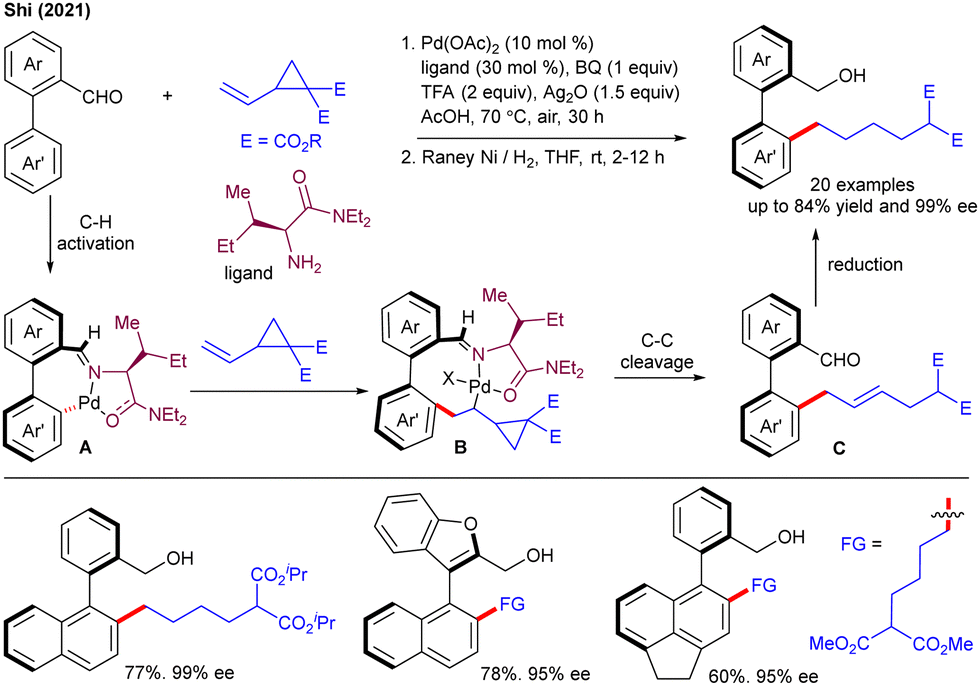
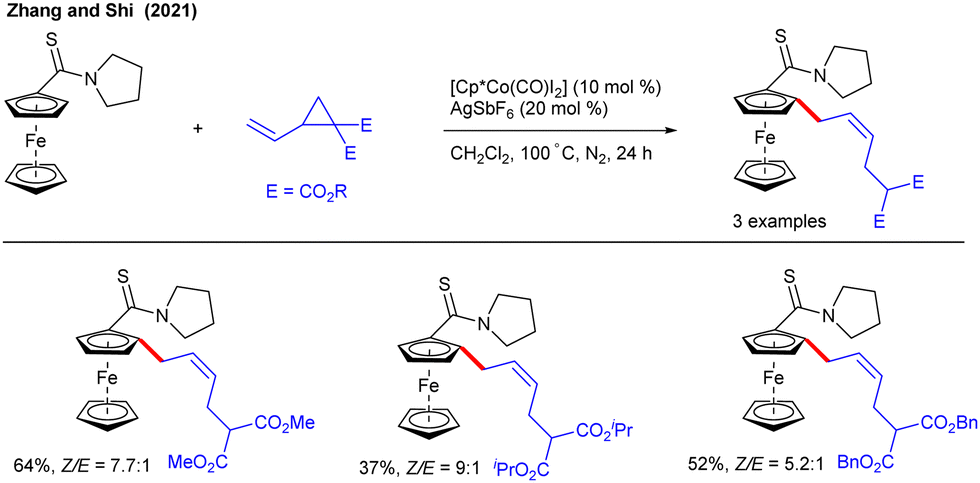
In the ensuing year, the Shi group described an enantioselective synthesis of axially chiral biaryls with VCPs under Pd(II)-catalysis (Scheme 5).18 The ligand acts as a transient DG, governing the stereochemistry of the product by making an enantioenriched palladacycle Avia C–H activation. The coordination and insertion of VCP gives B that undergoes C–C cleavage and protonolysis to produce the allylation product C. The latter was subjected to RANEY®-Ni/H2 reduction to give the desired product in moderate to high yields. The synthetic utility was shown by gram-scale synthesis and further synthetic transformations to access various axially chiral biaryls with up to >99% ee. Soon after, the Zhang and Shi groups documented the Co(III)-catalyzed C–H allylation of ferrocenes with VCPs using thioamide as the DG (Scheme 6).19 The reaction showed good Z-selectivity when stirred with 10 mol % of [Cp*Co(CO)I2] and 20 mol% of AgSbF6 in CH2Cl2 at 100 °C for 24 h. The Z-selectivity might be due to the geometric orientation of the intermediate during the key C–C cleavage of VCP under Co-catalysis.10
 |
| | Scheme 5 Pd(II)-catalyzed enantioselective functionalization of biaryls. | |
 |
| | Scheme 6 Co(III)-catalyzed C−H-allylation of ferrocene. | |
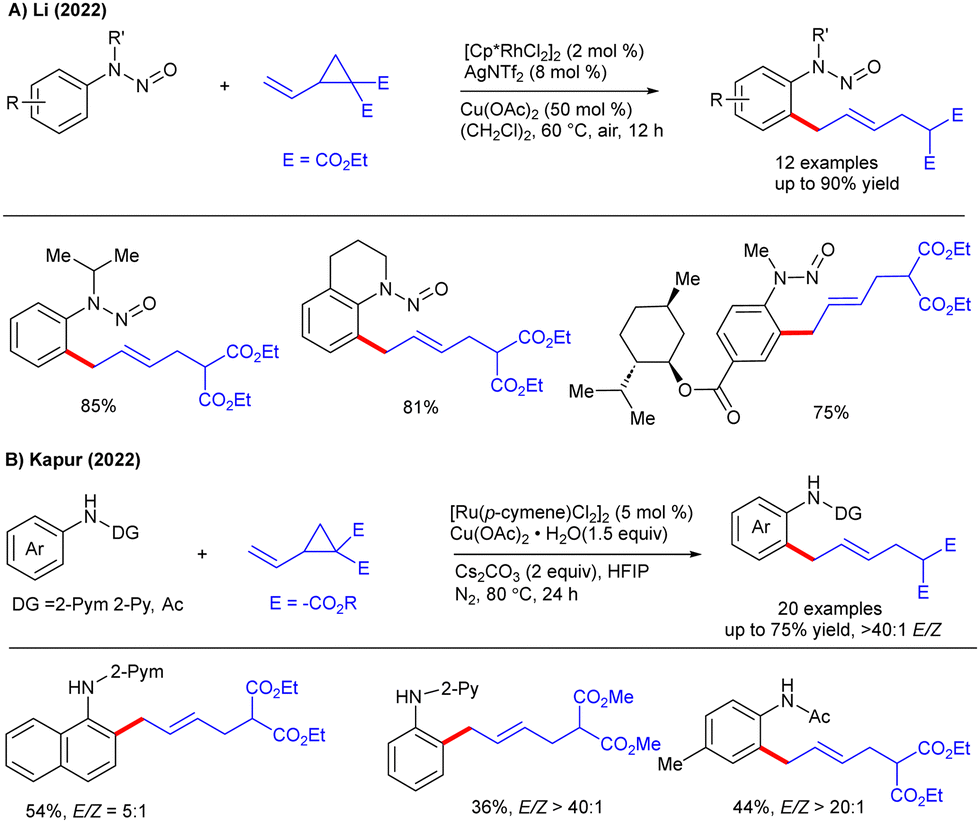
Later, Li and co-workers developed a switchable C- and N-allylation with chemo- and site-selectivities using N-nitroso anilines (Scheme 7A).20 The C–H allylation was compatible with a diverse array of substrates under Rh(III)-catalysis. Furthermore, the Kapur group demonstrated an allylation under Ru-catalysis to afford anilines with excellent E/Z-selectivities (Scheme 7B).21 Most of the anilines and VCPs responded well with good functional group tolerance. The authors noted that Cu(OAc)2·H2O and Cs2CO3 were crucial, as their absence led to significantly lower yields.
 |
| | Scheme 7 Directed ortho-C–H-allylation of arenes. | |
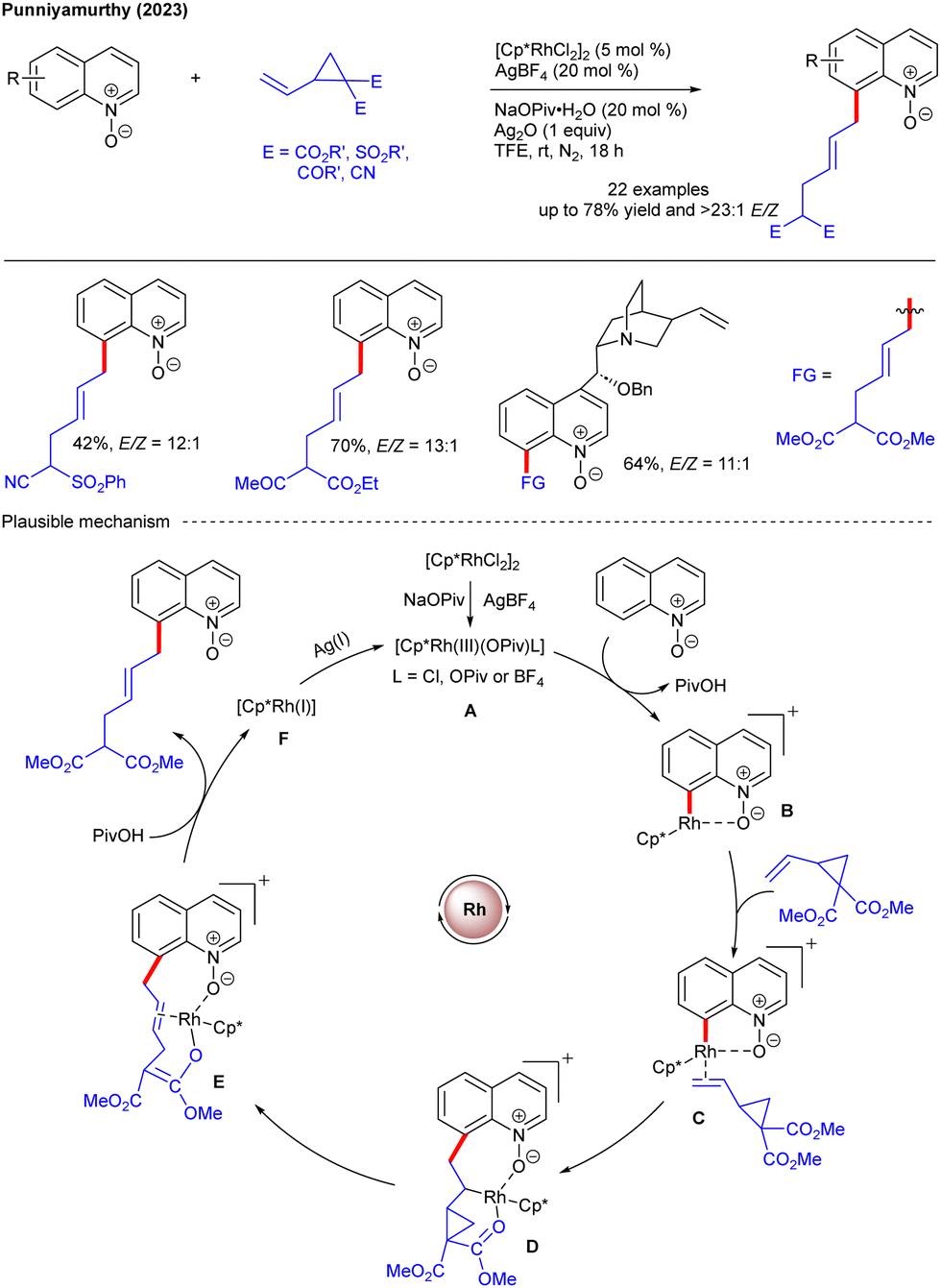
Recently, our group developed distal C8–H allylation of quinoline N-oxides using VCPs under Rh(III)-catalysis at ambient temperature (Scheme 8).22 The reaction features broad substrate scope with excellent E/Z-selectivities. In this reaction, [Cp*RhCl2]2 with AgBF4 and NaOPiv generates active Rh(III)-species A, which activates the C8–H bond of the substrate and produces B. Co-ordination and migratory insertion of VCP resulted in a seven-membered metallacycle D. Energetically favoured β-carbon elimination via C–C bond cleavage and protodemetalation yielded the target product. Ag(I) oxidizes Rh(I) species F to regenerate the active Rh(III)-species and complete the catalytic cycle. In continuation of our previous interest and due to the prevalence of allyl arenes in numerous natural products and pharmaceuticals, we reported sulfoxonium ylide-directed Co(III)-catalyzed C–H allylation of (hetero)arenes (Scheme 9).23 In the post-synthetic modifications, sulfoxonium ylide DG was seamlessly converted into various derivatives as illustrated. Furthermore, the ubiquity of the allylated indole moiety in numerous drug molecules motivated the development of an efficient methodology for weak chelation-assisted distal C4–H allylation of indoles under redox-neutral ruthenium(II) catalysis, using VCPs (Scheme 10).24 The reaction proceeds with active [Ru]-complex A, which eventually activates the C4–H bond of the indole substrate to produce the ruthenacycle B. Co-ordination of VCP followed by 1,2-migratory insertion may lead to the formation of D. Chelation with the ester functionality assists β-carbon elimination to furnish E. Finally, protodemetalation leads to product formation and regenerates the active catalyst A. The regioselectivity, substrate scope and E/Z-selectivities are the key features. Moreover, the procedure was extended to benzothiophene, tetrahydroquinoline, carbazole, α-tetralone and indoline moieties for selective allylation.
 |
| | Scheme 8 C8–H allylation of quinolines using an N-oxide DG. | |
 |
| | Scheme 9 Sulfoxonium ylide directed ortho-C–H-allylation. | |
 |
| | Scheme 10 Redox-neutral acyl DG-assisted C4–H allylation of indoles. | |
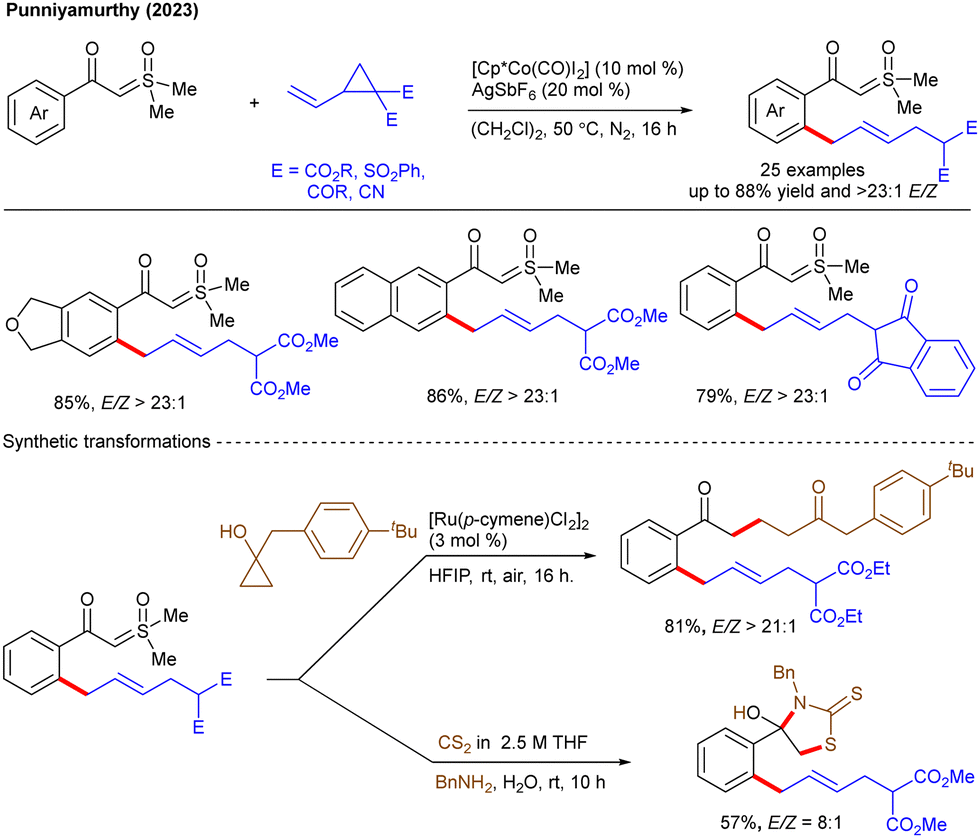
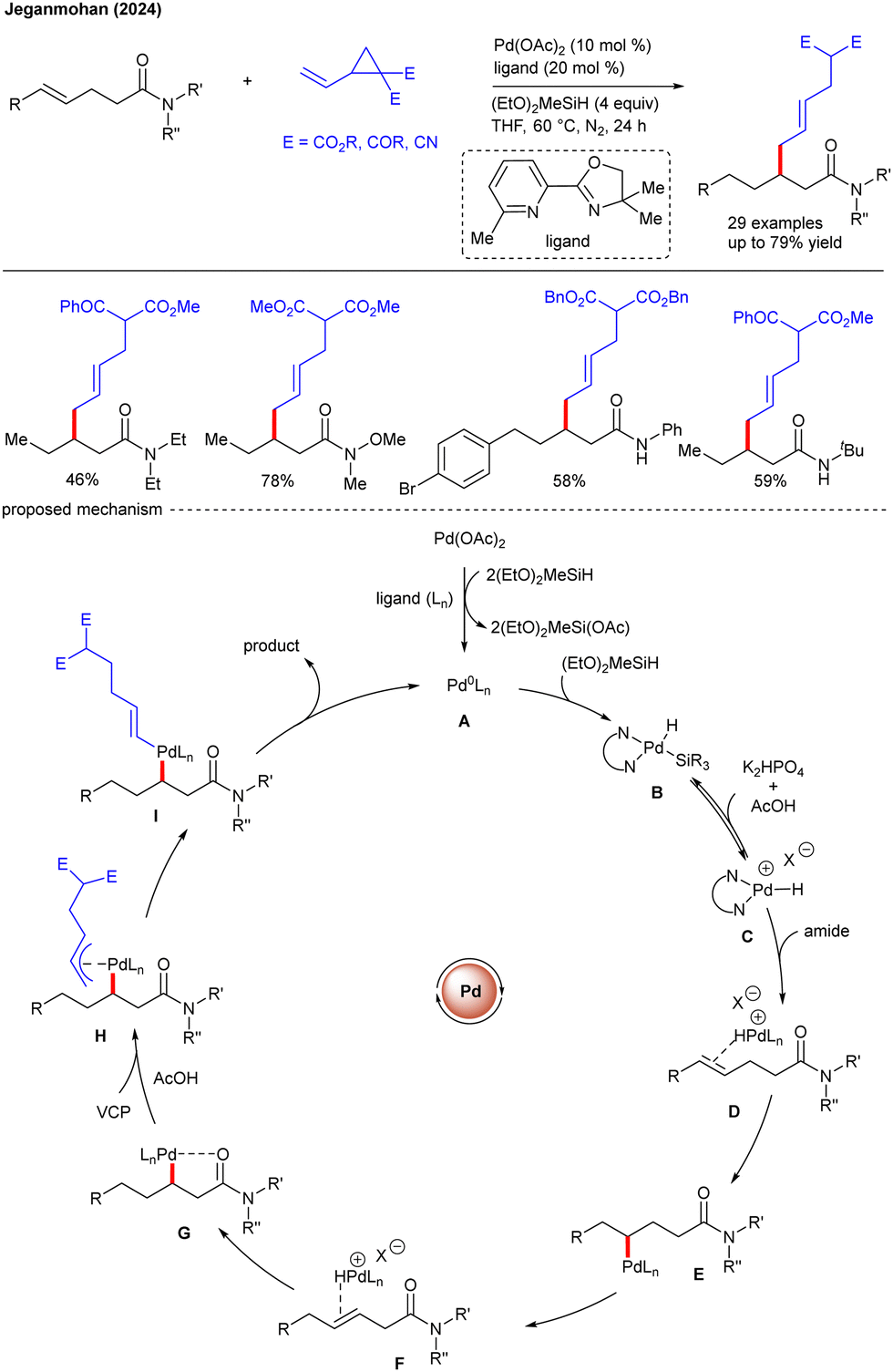
Later, the Baidya group reported an amine directed ortho-C–H functionalization of 2-aminobiaryls with VCPs under high-valent Co-catalysis, enabling regio- and stereo-selective allylation (Scheme 11).25 The innate nucleophilic reactivity of an amine group makes their use as a DG synthetically challenging. The authors have shown the mechanistic experiments and DFT calculations to unravel the reaction pathway. Initially, the Co(III) species A undergoes ortho-C–H activation with aniline via BIES (base-assisted internal electrophilic type substitution) to afford the four-membered cobaltacycle B. Chelation with VCP affords C, which undergoes migratory insertion to provide D. The C–C cleavage of D gives E that undergoes protodemetalation to give the allylation product and the active catalyst A to complete the catalytic cycle. The late-stage modifications of the products lead to valuable benzoisoxazoles and dihydrophenanthridines. Recently, the Jeganmohan group demonstrated the palladium-catalyzed regioselective allylation of unactivated alkenes with VCPs (Scheme 12).26 The reaction aided by weak-coordinating native amide DG was well tolerated over a broad range of substrates with excellent β-selectivity. In addition, secondary amides are proved to be more reactive than primary amides. In contrast to conventional VCP allylation, the mechanism differs significantly as explained by the authors. First, the Pd(0)-complex A with (EtO)2MeSiH generates B that undergoes elimination of (OEt)2MeSi(OAc) using AcOH to yield the cationic (N,N)Pd-H species C. Chelation of the amide gives D that leads to hydride transfer to furnish E, which undergoes β-hydride elimination to afford the isomerized alkene F. Hydride transfer produces the palladacycle G, which leads to chelation with VCP to yield η1-palladium species I. Reductive elimination provides the target product and the Pd(0)-complex to complete the catalytic cycle. Regioselectivity in direct C(alkenyl)–H functionalization remains challenging due to the presence of electronically equivalent alkene hydrogens. Although, the formal Heck and radical reactions have been considerably explored for C(alkenyl)–H functionalization,27 the use of a strained ring to functionalize the C(alkenyl)–H bond is yet to be investigated. More recently, the same group reported 8-aminoquinoline-directed γ-C(alkenyl)–H functionalization with VCPs (Scheme 13).28 The reaction involves the Pd-catalyzed C–H allylation of unactivated alkenes with VCPs to produce the allyl derivatives, which leads to isomerization to give 1,3-conjugated dienes as a major product. Moreover, the authors isolated a six-membered palladacycle and reacted it with VCP to produce the desired product, demonstrating functionalization at the γ-C(alkenyl)–H bond. The phosphine ligand and K2HPO4 base played a crucial role. At elevated temperature, the selectivity shifted, favoring the unmigrated product over the migrated one. The protocol was effective with various substituted amides and VCPs.
 |
| | Scheme 11 Free amine-directed ortho-C–H allylation with VCPs. | |
 |
| | Scheme 12 Regioselective β-C(sp3)–H allylation of amides. | |
 |
| | Scheme 13 Pd-catalyzed γ-C(alkenyl)–H functionalization with VCPs. | |
2.2. Annulation
TM-catalyzed C–H functionalization/annulation is an effective paradigm for synthesizing carbo- and hetero-cycles in step- and atom-economic fashion.29 In this context, Kapur and co-workers developed a catalyst-controlled divergent reactivity of VCPs via tandem C–H/C–C functionalization and annulation (Scheme 14).21 The combination of the Rh-catalyst and mono-N-protected-amino acid (MPAA) ligand is decisive for C–H/C–C functionalization followed by in situ 1,2-aminorhodation-mediated cyclization to produce C2-substituted indoles.
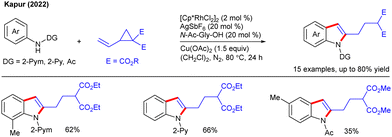 |
| | Scheme 14 Synthesis of C2-substituted indoles via annulative coupling with VCPs. | |
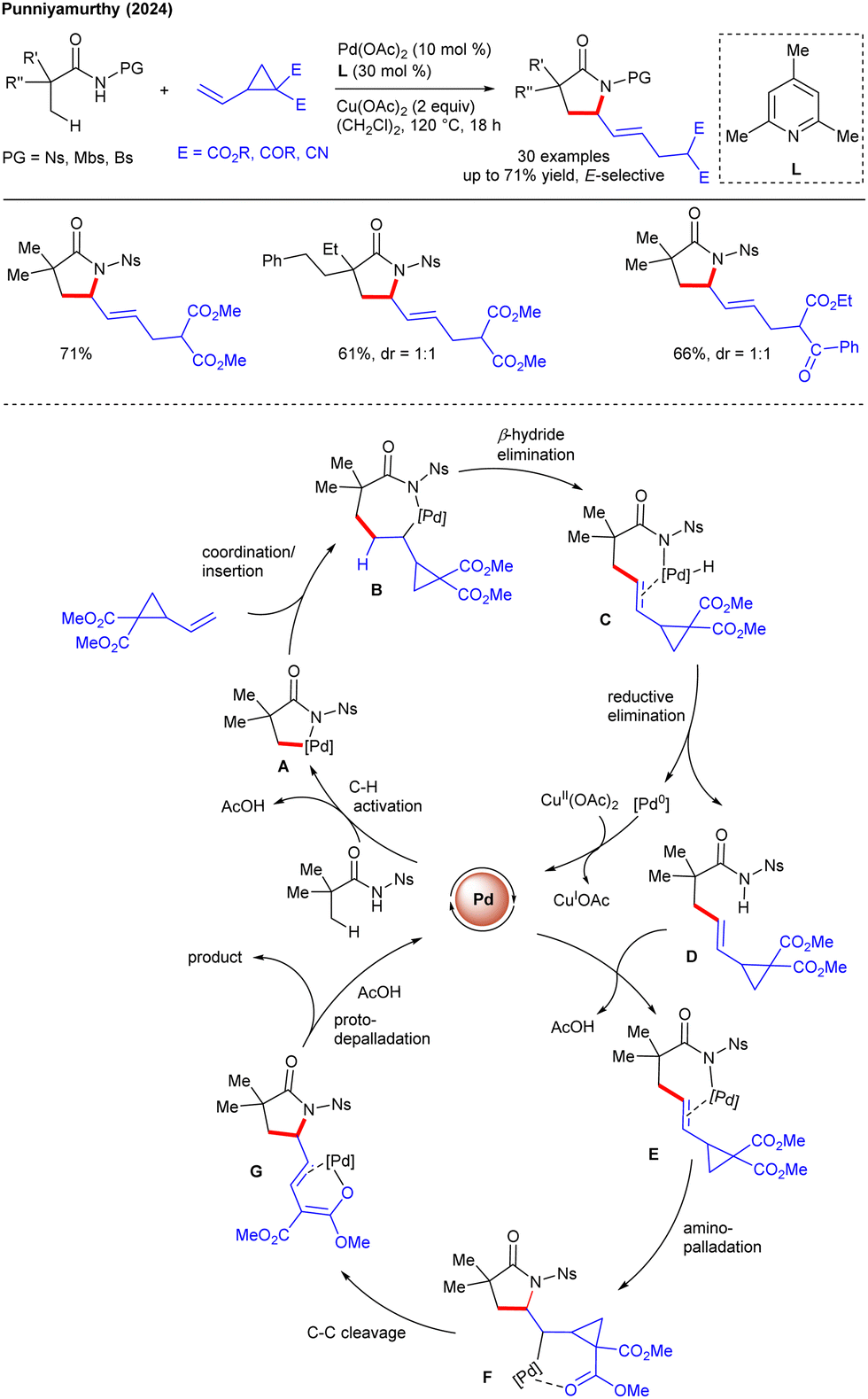
Recently, our group reported the Rh(III)-catalyzed sequential C–H functionalization and annulation of sulfoxonium ylides with VCPs to access cyclopropane-fused α-tetralones (Scheme 15).30 The sulfoxonium ylide served as a traceless DG, eliminating DMSO as a by-product. The protocol shows excellent compatibility with various functional groups and late-stage derivatization of commercial drugs. The proposed mechanism begins with reversible C–H activation to generate rhodacycle A, double bond chelation of VCP followed by migratory insertion into the Rh–C bond generates B. The latter undergoes rearrangement to form C, which evolves into a Rh α-oxo carbene D with the liberation of DMSO. The β-carbon elimination of D leads to E that undergoes intramolecular cyclopropanation to yield fused F (path a), which upon protodemetalation delivers the target product. An alternative pathway (path b) involves reductive migratory insertion forming G, followed by nucleophilic attack of the α-carbon to the proximal carbon, giving the target product. Furthermore, our group reported ligand-promoted Pd(II)-catalyzed C(sp3)–H/C–C functionalization of aliphatic amides with VCPs to produce functionalized γ-lactams (Scheme 16).31 VCP displayed unusual reactivity, favouring alkenylation followed by cyclization instead of the formation of the open chain allyl product. The protocol showcased a wide substrate scope with exclusive (E)-selectivity and post-synthetic utilities. Notably, monodentate sulfonyl-based protecting groups (Ns, Mbs, Bs) proved effective, while bidentate DGs failed to give the desired outcome. Alkyl pyridine (L) promotes the catalytic activity through coordination with palladium, which activates the β-C(sp3)–H bond leading to the formation of five-membered palladium intermediate A. Alkene coordination, followed by migratory insertion generates B, which undergoes β-hydride and reductive elimination to give intermediate D. The latter undergoes aminopalladation and C–C cleavage of cyclopropane, giving rise to G that leads to protodemetalation to afford the target product.
 |
| | Scheme 15 Rh-catalyzed sulfoxonium-ylide directed C–H/C–C functionalization with VCPs. | |
 |
| | Scheme 16 Pd-catalyzed sulfonamide directed C(sp3)–H/C–C functionalization with VCPs. | |
3. Reactions using cyclopropanols
The constrained cyclic framework cyclopropanol, which can be readily synthesized via Kulinkovich or Simmons–Smith cyclopropanation, is an adaptable three-carbon synthon in synthetic chemist's toolbox due to its strain energy and primed reactivity.5a,b Cyclopropanol generates metal-homoenolates or β-keto alkyl radicals under TM-catalysis, enabling it to act as either a nucleophile or an electrophile, depending on the reaction context.5f This remarkable versatility renders cyclopropanol a unique and indispensable resource in modern organic synthesis. Although numerous strategies involving cyclopropanol have been developed, the integration of C–H functionalization with the ring-opening of cyclopropanol remains a burgeoning area of research. Moreover, the single-step incorporation of a three-carbon unit is often essential for the diversification of biologically potent molecules. This section delves into the specific outcomes of C–H functionalization with cyclopropanols.
3.1. Alkylation
TM-catalyzed site-selective C–H alkylation with cyclopropanols set out a way upfront to complex organic molecule synthesis, because it introduces a β-keto alkyl/aryl group that can act as an effective synthetic handle for further useful manipulation. In 2016, Li and co-workers employed cyclopropanols in the Rh-catalyzed oxidative alkylation of arenes via tandem C–H/C–C functionalization (Scheme 17A).32 Both oxime ether and 2-pyrimidyl served as effective DGs for the alkylation providing the products with exclusive site-selectivity. This methodology was further applied for C(sp3)–H alkylation of 8-methyl quinoline affording the γ-aryl ketones. Concurrently, the authors came up with 2-pyrimidyl DG assisted regioselective alkylation of (hetero)arenes using cyclopropanols (Scheme 17B).33 A wide variety of heteroarenes, including indolines, carbazoles, thiophene and pyrroles were effective substrates, providing significant yields of the alkylated products.
 |
| | Scheme 17 Rh-catalyzed coupling of (hetero)arenes with cyclopropanols. | |
Later, a site-selective alkylation of 2-arylquinazolin-4(3H)-one was reported by Peng and co-workers (Scheme 17C).34 This methodology was amenable to a variety of functional groups like alkyl, alkoxy and halogen as well as heteroarenes. However, electron withdrawing substituents on the arylquinazolin-4(3H)-ones provided lower yield. Soon after, an elegant work in this area was disclosed by Shi and co-workers (Scheme 18).18 They used cyclopropanols for the synthesis of axially chiral biaryls via enantioselective tandem C–H/C–C functionalization. The authors suggested that due to the competitive β-hydride elimination and protodemetalation under Pd-catalysis, both alkylation and alkenylation were observed. The key to the success of the procedure was represented by the access of a broad range of axially chiral biaryls in synthetically convenient yields and high enantiopurity. In the same year, the Liu and Liu groups developed the Rh-catalyzed oxidative alkylation of N-aryl-7-azaindoles with regioselectivity and functional group compatibility (Scheme 19).35 In addition, the method proved its potential applicability by transforming the β-aryl ketones into 7-azaindole containing π-extended poly-heterocycles.
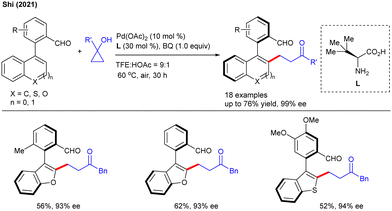 |
| | Scheme 18 Pd-catalyzed atroposelective C–H/C–C functionalization with cyclopropanols. | |
 |
| | Scheme 19 Rh-catalyzed alkylation of N-aryl-7-azaindoles with cyclopropanols. | |
Recently, we accomplished a weak acyl chelation-guided indole C4–H alkylation with cyclopropanols utilizing Rh-catalysis (Scheme 20).36 A wide range of substituted indoles and cyclopropanols were evaluated to check the practicality. The procedure showed potential amenability to other carbonyl DG-tethered (hetero)aryl substrates, e.g. benzo[b]thiophene, indoline and tetrahydroquinoline. To shed light on the reaction pathway, H/D-scrambling experiments were conducted either in the presence or absence of cyclopropanol, which resulted in 12% and 27% deuterium incorporation at the C4-site, respectively. Based on the detailed mechanistic studies, a plausible catalytic cycle was depicted, in which the active Rh-catalyst A activates the C4–H bond to furnish rhodacycle B, which undergoes ligand exchange with cyclopropanol to give C. Subsequent β-carbon and β-hydride elimination leads to the formation of E, which undergoes aryl insertion into the double bond, furnishing eight-membered rhodacycle F. The latter upon protodemetalation produces the alkylated product and Rh(I)-species, which is oxidized to the active catalyst A by Cu(II), completing the catalytic cycle. Furthermore, the Anbarasan group reported the Rh-catalyzed C2–H alkylation of indole with cyclopropanols using an N,N-dialkylcarbamoyl group as the traceless DG (Scheme 21A).37 Moreover, pyrrolo[1,2-a]indole can be obtained from the alkylated product through a reduction followed by cyclization. Concurrently, they utilized Co-catalysis for the C2–H alkylation of N-pyridylindoles via the ring scission of cyclopropanols (Scheme 21A).38 The use of AgSbF6 and Cu(OAc)2·H2O is essential, as they play a key role in synthesizing active catalysts for C–H functionalization. The ketone functionality in the alkyl moiety facilitates the formation of tetrahydrocyclopenta-[b]indole and C2-allylated indole, reflecting the synthetic versatility of the procedure. Later, the authors showed the Rh-catalyzed ortho-alkylation of N-(hetero)aryl pyrazoles with cyclopropanols (Scheme 21B).39 This method enables nonsymmetrical dialkylation, showcasing the practicality of the strategy.
 |
| | Scheme 20 Rh-catalyzed C4-selective alkylation of indoles with cyclopropanols. | |
 |
| | Scheme 21 Metal-catalyzed regioselective alkylation with cyclopropanols. | |
Our group exploited cyclopropanols for the Co-catalyzed C8–H alkylation of quinolines using N-oxide as a weak chelating DG (Scheme 22).40 This protocol was suited for a wide range of quinolines and cyclopropanols to afford the alkylated products.
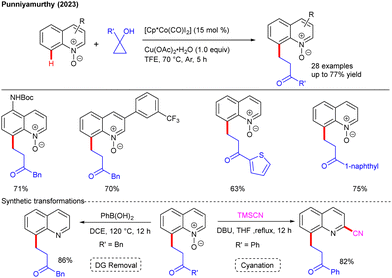 |
| | Scheme 22 Co-catalyzed C8-selective alkylation of quinolines with cyclopropanols. | |
Moreover, the potent synthetic ability of the method was exemplified by late-stage modification of natural products and drug molecules. The directing auxiliary could easily be removed by the treatment of PhB(OH)2. Furthermore, when the target product was treated with TMSCN, a C2-cyanated quinoline derivative was realized. In the same year, the Ru-catalyzed, Cu-mediated C(3)/C(4)-alkylation of N-pyridylisoquinolones was detailed by the Kapur group (Scheme 23A).41 Electron withdrawing groups facilitated the C3-alkylation, while electron donating ones eased C4-alkylated product formation. A detailed mechanistic study along with DFT studies revealed the co-existence of the intermediates, such as homoenolate and β-keto alkyl radical species, which are implicated in the formation of C3- and C4-products, respectively. Furthermore, Volla and co-workers illustrated Rh-catalyzed aldehydic and aryl C–H alkylation with cyclopropanols (Scheme 23B).42 The utility of the method was highlighted by post-synthetic transformation of the products into valuable heterocycles, including pyrrole, pyridazine and indole derivatives.
 |
| | Scheme 23 Ru- and Rh-catalyzed alkylation with cyclopropanols. | |
3.2. Alkenylation
C–H alkenylation utilizing TM-catalysis has attracted immense attention in recent times because of the ability for efficient downstream transformations. In this context, the Hurski group reported Pd-catalyzed removable 2-(neopentylsulfinyl)-aniline assisted arene C–H alkenylation using cyclopropanol as a vinyl ketone surrogate (Scheme 24).43 This methodology was compatible with various functional groups, such as alcohol and nitroaromatics.
 |
| | Scheme 24 Pd-catalyzed C–H alkenylation of arylacetamides with cyclopropanols. | |
More recently, our group accomplished biorelevant intrinsic DG-assisted arene C–H alkenylation with cyclopropanol under Ru-catalysis (Scheme 25).44 A diverse array of α,β-unsaturated ketone functionalities were successfully introduced using DGs like β-, γ- and ε-lactams, isoindolinone, pyridone and morphilinone. Scale-up synthesis and divergent transformations from the alkenylated product demonstrated its profound synthetic potential. A mechanistic blueprint was proposed, where the reaction commenced with the formation of an active Ru-catalyst A, followed by reversible C–H activation leading to the formation of six-membered ruthenacycle B. Ligand exchange with cyclopropanol and β-carbon elimination gives D. Furthermore, β-hydride elimination and aryl insertion into the double bond of E affords F that undergoes β-hydride elimination to furnish the target product and Ru(0)-species, which is oxidized by Cu(II) to regenerate the active catalyst.
 |
| | Scheme 25 Ru-catalyzed arene C–H alkenylation with cyclopropanols. | |
3.3. Annulation
C–H functionalization/annulation under TM-catalysis offers a powerful and modular approach for constructing decorated heterocycles that are omnipresent in natural products and pharmaceuticals. In this regard, cyclopropanol has emerged as an effective and prominent coupling partner. In 2017, the Li group disclosed the Rh(III)-catalyzed annulation of imidamides with cyclopropanols for the construction of 2-substituted quinoline heterocycles (Scheme 26A).45 Interestingly, cyclopropanol served as the C3-synthon in annulation and afforded ample substrate scope via sequential C–H/C–C functionalization and C–C/C–N bond formation. The role of Cu(OAc)2 as an oxidant is crucial for annulation, while Fe(III) salt facilitates electron transfer during the oxidation process. Later, the Liu and Liu groups reported the synthesis of C3-substituted isoindolinones via the Rh(III)-catalyzed cyclization of oxazolines (Scheme 26B).46 The bifunctional nucleophilic oxazoline-directed cyclization entailed the cleavage of three chemical bonds and allowed the formation of C–C, C–N and C–O bonds in a single step. Of note, the ring opening of oxazolines via nucleophilic attack by acetate enabled an efficient route to synthesize substituted isoindolin-1-ones. An array of oxazolines and cyclopropanols reacted smoothly, but the reaction conditions were ineffective for relatively bulky 1,2-disubstituted cyclopropanols as well as less strained cyclobutanol and cyclopentanol. Mechanistically, the catalytic cycle begins with the active catalyst A, which undergoes oxazolinyl-assisted C–H bond activation to afford five membered rhodacycle B. Migratory insertion of the in situ formed enone into an aryl–Rh bond afforded C that undergoes β-hydride elimination to furnish D together with a Rh(I) species. Intramolecular aza-Michael addition of D forms oxazolinium salt E, which upon attack by acetate furnishes the product and the active catalyst is regenerated using a Cu(II) oxidant. In addition, the Yu group reported the fabrication of isoindolin-1-ones via C–H/N–H functionalization and [4+1] annulation of N-(quinolin-8-yl)benzamide with cyclopropanols under Co(II)-catalysis (Scheme 27A).47 The method offers utilization of the earth-abundant Co(OAc)2·H2O as the catalyst along with Ag2CO3 and TEMPO as the oxidants. Among various DGs screened, 8-aminoquinoline was superior to other N,N- and N,O-bidentate chelations. Meanwhile, the Huang and Yang groups showed the Rh-catalyzed C–H/C–C functionalization/cyclization of carboxylic acid with cyclopropanols to afford 3-substituted phthalides and α,β-butenolides (Scheme 27B).48 Remarkably, this catalytic method displays the synthesis of bioactive compounds bearing 3-substituted phthalide scaffolds. Later, the Guan and Wu groups disclosed a coupling of benzamides with cyclopropanols under Ir(III)-catalysis using KOAc as an additive (Scheme 27C).49 Solvent screening revealed that the protic solvents such as MeOH were the optimal choice due to their better solubility for KOAc compared to aprotic solvents. The reaction showcased broad substrate scope via C–H functionalization and annulation, where cyclopropanol served as a one-carbon partner for intermolecular annulation. In 2022, the Liu and Liu groups introduced a three-component synthetic strategy to access structurally diverse benzofuran-3(2H)-ones via sequential C–H/C–C functionalization and annulation of salicylaldehyde, cyclopropanol and alcohols under Rh(III)-catalysis (Scheme 28A).50 The choice of solvent, such as MeOH, was vital. In addition, control experiments suggest that the ortho-hydroxy group plays a pivotal role for initial aldehyde C–H bond functionalization. Recently, the Ling, Chen and Yao groups described the Rh-catalyzed cyclization of substituted pyrazoles with cyclopropanols to form pyrazolo[5,1-a]isoindoles (Scheme 28B).51 A variety of substituted pyrazoles and cyclopropanols were coupled smoothly. Moreover, the protocol enables late-stage functionalization of biologically important compounds. In 2024, the Zhang and Fan groups reported the synthesis of indane fused bicyclic pyrazolidinones through a one-pot reaction of aryl azomethine imines with cyclopropanols under Rh-catalysis (Scheme 28C).52 The reaction was well tolerated with phenyl azomethine imines and cyclopropanols. Mechanistically, the reaction proceeds via C–H/C–C functionalization, where cyclopropanol showed multiple roles by acting as an alkylating agent as well as a masked nucleophile and electrophile for the fabrication of both indanes and bicyclic pyrazolidinones.
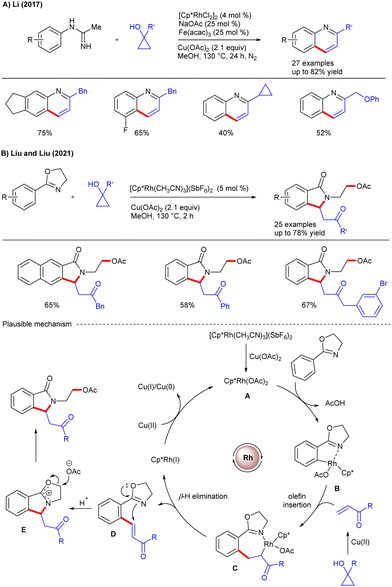 |
| | Scheme 26 Rh-catalyzed annulative coupling with cyclopropanols. | |
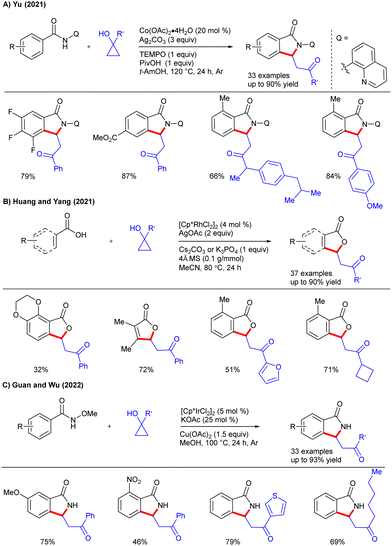 |
| | Scheme 27 Metal-catalyzed C–H functionalization/annulation with cyclopropanols. | |
 |
| | Scheme 28 Rh-catalyzed cascade annulation via C–H/C–C functionalization. | |
4. Conclusion and outlook
Over the preceding decades, directed C–H functionalization under TM-catalysis has evolved into a linchpin strategy for enhancing molecular intricacy. Recently, in view of step-/atom-economy, sequential C–H and C–C functionalization continues to expand the synthetic toolkit for constructing C–C and C–heteroatom bonds. Towards this goal, small ring systems (three- or four-membered) can serve as a cornerstone due to their high reactivity pertaining to strain relief. Being a congener within the three-membered carbocycles, VCPs and cyclopropanols are proven to be promising synthetic precursors that can undergo C–C cleavage en route to important molecular frameworks. A breakthrough has been achieved in the chelation-assisted concomitant C–H and C–C functionalization implementing both the mentioned rings. This article aims to cover the recent findings, including our contributions to DG guided C–H functionalization employing VCPs and cyclopropanols to date. Despite the commendable progress, there is still a synthetic void that requires improvement by introducing either a precisely DG-tailored substrate or efficient and economically viable catalytic system. In this scenario, the traceless/transient DG- or template-assisted strategies would be instrumental in achieving proximal or distal C–H functionalization with the aforementioned rings. Furthermore, the more inert C(sp3)–H functionalization leveraging VCPs and cyclopropanols in the presence of effective chiral ligands could contribute to the exploration of enantioselective transformations. In addition, the utilization of more abundant metal catalysts in combination with electro- and photo-catalysis may exhibit excellent potential for fostering practical and sustainable developments in this direction. We anticipate that this feature article will provide a valuable resource to the synthetic front for future aspiration in this burgeoning field.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
T. P. thanks SERB for the J C Bose National Fellowship (JCB/2022/000037). S. B. and S. M. acknowledge the DST for the INSPIRE Senior Research Fellowship. T. Paul and M. V. N. are thankful to the MoE for a PMR Fellowship. M. B. acknowledges the UGC for the Senior Research Fellowship.
Notes and references
- For selected reviews, see:
(a) J. Wencel-Delord and F. Glorius, Nat. Chem., 2013, 5, 369 CrossRef CAS PubMed;
(b) R.-Y. Zhu, M. E. Farmer, Y.-Q. Chen and J.-Q. Yu, Angew. Chem., Int. Ed., 2016, 55, 10578 CrossRef CAS PubMed;
(c) C. Sambiagio, D. Schönbauer, R. Blieck, T. Dao-Huy, G. Pototschnig, P. Schaaf, T. Wiesinger, M. F. Zia, J. Wencel-Delord, T. Besset, B. U. W. Maes and M. Schnürch, Chem. Soc. Rev., 2018, 47, 6603 RSC;
(d) P. Gandeepan, T. Müller, D. Zell, G. Cera, S. Warratz and L. Ackermann, Chem. Rev., 2019, 119, 2192 CrossRef CAS PubMed;
(e) T. Shah, P. B. De, S. Pradhan and T. Punniyamurthy, Chem. Commun., 2019, 55, 572 RSC;
(f) S. Rej, Y. Ano and N. Chatani, Chem. Rev., 2020, 120, 1788 CrossRef CAS PubMed;
(g) T. Dalton, T. Faber and F. Glorius, ACS Cent. Sci., 2021, 7, 245 CrossRef CAS PubMed;
(h) B. Zhao, B. Prabagar and Z. Shi, Chem, 2021, 7, 2585 CAS;
(i) B. Liu, A. M. Romine, C. Z. Rubel, K. M. Engle and B.-F. Shi, Chem. Rev., 2021, 121, 14957 CrossRef CAS PubMed;
(j) S. Barranco, J. Zhang, S. López-Resano, A. Casnati and M. H. Pérez-Temprano, Nat. Synth., 2022, 1, 841 CAS;
(k) J. H. Docherty, T. M. Lister, G. Mcarthur, M. T. Findlay, P. D. Legarda, J. Kenyon, S. Choudhary and I. Larrosa, Chem. Rev., 2023, 123, 7692 CAS.
- For selected reviews, see:
(a) V. Ritleng, C. Sirlin and M. Pfeffer, Chem. Rev., 2002, 102, 1731 CAS;
(b) N. Rodríguez and L. J. Goossen, Chem. Soc. Rev., 2011, 40, 5030 Search PubMed;
(c) S. Pradhan, P. B. De, T. A. Shah and T. Punniyamurthy, Chem. Asian J., 2020, 15, 4184 CAS;
(d) Y.-F. Liang, M. Bilal, L.-Y. Tang, T.-Z. Wang, Y.-Q. Guan, Z. Cheng, M. Zhu, J. Wei and N. Jiao, Chem. Rev., 2023, 123, 12313 CAS;
(e) S. Basak, T. Paul, S. Mandal, P. Karjee, M. V. Nanjegowda and T. Punniyamurthy, Synthesis, 2023, 3454 CAS.
- For selected reviews, see:
(a) C.-H. Jun, Chem. Soc. Rev., 2004, 33, 610 RSC;
(b) M. Murakami and T. Matsuda, Chem. Commun., 2011, 47, 1100 RSC;
(c) L. Souillart and N. Cramer, Chem. Rev., 2015, 115, 9410 CrossRef CAS PubMed;
(d) M. D. R. Lutz and B. Morandi, Chem. Rev., 2021, 121, 300 CrossRef CAS PubMed.
- For selected reviews, see:
(a) M. Rubin, M. Rubina and V. Gevorgyan, Chem. Rev., 2007, 107, 3117 CrossRef CAS PubMed;
(b) T. Seiser, T. Saget, D. N. Tran and N. Cramer, Angew. Chem., Int. Ed., 2011, 50, 7740 CrossRef CAS PubMed;
(c) D. J. Mack and J. T. Njardarson, ACS Catal., 2013, 3, 272 CrossRef CAS;
(d) C.-Y. D. Huang and A. G. Doyle, Chem. Rev., 2014, 114, 8153 CrossRef CAS PubMed;
(e) G. Fumagalli, S. Stanton and J. F. Bower, Chem. Rev., 2017, 117, 9404 CrossRef CAS PubMed.
- For selected reviews, see:
(a) D. H. Gibson and C. H. DePuy, Chem. Rev., 1974, 74, 605 CrossRef CAS;
(b) O. G. Kulinkovich, Chem. Rev., 2003, 103, 2597 CrossRef CAS PubMed;
(c) H.-U. Reissig and R. Zimmer, Chem. Rev., 2003, 103, 1151 CrossRef CAS PubMed;
(d) T. Hudlicky and J. W. Reed, Angew. Chem., Int. Ed., 2010, 49, 4864 CrossRef CAS PubMed;
(e) L. Jiao and Z.-X. Yu, J. Org. Chem., 2013, 78, 6842 CrossRef CAS PubMed;
(f) T. R. McDonald, L. R. Mills, M. S. West and S. A. L. Rousseaux, Chem. Rev., 2021, 121, 3 CrossRef CAS PubMed;
(g) J. Wang, S. A. Blaszczyk, X. Li and W. Tang, Chem. Rev., 2021, 121, 110 CrossRef CAS PubMed.
- For selected reviews, see:
(a) K. M. Engle, T.-S. Mei, M. Wasa and J.-Q. Yu, Acc. Chem. Res., 2012, 45, 788 CrossRef CAS PubMed;
(b) S. De Sarkar, W. Liu, S. I. Kozhushkov and L. Ackermann, Adv. Synth. Catal., 2014, 356, 1461 CrossRef CAS;
(c) G. Shi and Y. Zhang, Adv. Synth. Catal., 2014, 356, 1419 CrossRef CAS;
(d) E. N. da Silva Júnior, G. A. M. Jardim, R. S. Gomes, Y.-F. Liang and L. Ackermann, Chem. Commun., 2018, 54, 7398 RSC.
- For selected reviews, see:
(a) Z. Nairoukh, M. Cormier and I. Marek, Nat. Rev. Chem., 2017, 1, 0035 CrossRef;
(b) T. A. Shah, P. B. De, S. Pradhan, S. Banerjee and T. Punniyamurthy, Chem. Asian J., 2019, 14, 4520 CrossRef CAS PubMed;
(c) H. Azizollahi and J.-A. García-López, Molecules, 2020, 25, 5900 CrossRef CAS PubMed;
(d) H.-H. Wang, X.-D. Wang, G.-F. Yin, Y.-F. Zeng, J. Chen and Z. Wang, ACS Catal., 2022, 12, 2330 CrossRef CAS.
-
(a) J. L. Schwarz, F. Schäfers, A. Tlahuext-Aca, L. Lückemeier and F. Glorius, J. Am. Chem. Soc., 2018, 140, 12705 CrossRef CAS PubMed;
(b) S. Tanabe, H. Mitsunuma and M. Kanai, J. Am. Chem. Soc., 2020, 142, 12374 CrossRef CAS PubMed.
- J.-Q. Wu, Z.-P. Qiu, S.-S. Zhang, J.-G. Liu, Y.-X. Lao, L.-Q. Gu, Z.-S. Huang, J. Li and H. Wang, Chem. Commun., 2015, 51, 77 RSC.
- D. Zell, Q. Bu, M. Feldt and L. Ackermann, Angew. Chem., Int. Ed., 2016, 55, 7408 CrossRef CAS PubMed.
- Q. Lu, F. J. R. Klauck and F. Glorius, Chem. Sci., 2017, 8, 3379 RSC.
- T. H. Meyer, W. Liu, M. Feldt, A. Wuttke, R. A. Mata and L. Ackermann, Chem. – Eur. J., 2017, 23, 5443 CrossRef CAS PubMed.
- Q. Wang, C.-L. Zhi, P.-P. Lu, S. Liu, X. Zhu, X. Hao and M.-P. Song, Adv. Synth. Catal., 2019, 361, 1253 CAS.
- Z. Hu, X.-Q. Hu, G. Zhang and L. J. Gooßen, Org. Lett., 2019, 21, 6770 CAS.
- R. Tanaka, I. Tanimoto, M. Kojima, T. Yoshino and S. Matsunaga, J. Org. Chem., 2019, 84, 13203 CrossRef CAS PubMed.
- Q. Wang, L. Shi, S. Liu, C. Zhi, L.-R. Fu, X. Zhu, X.-Q. Hao and M.-P. Song, RSC Adv., 2020, 10, 10883 RSC.
- S. Liu, H. Jiang, W. Liu, X. Zhu, X.-Q. Hao and M.-P. Song, J. Org. Chem., 2020, 85, 15167 CrossRef CAS PubMed.
- H.-M. Chen, G. Liao, C.-K. Xu, Q.-J. Yao, S. Zhang and B.-F. Shi, CCS Chem., 2021, 3, 455 CrossRef CAS.
- Z.-Z. Zhang, G. Liao, H.-M. Chen and B.-F. Shi, Org. Lett., 2021, 23, 2626 CrossRef CAS PubMed.
- W. Ouyang, B. Liu, Y. He, Y. Wen, Y. Gao, Y. Huo, Q. Chen and X. Li, Org. Chem. Front., 2022, 9, 2746 RSC.
- S. K. Keshri, S. Madhavan and M. Kapur, Org. Lett., 2022, 24, 9043 CrossRef CAS PubMed.
- S. Mandal, P. Karjee, S. Saha and T. Punniyamurthy, Chem. Commun., 2023, 59, 2823 RSC.
- S. Saha, H. Bhattacharyya, P. Karjee, B. Debnath, K. Verma and T. Punniyamurthy, Chem. Commun., 2023, 59, 14173 RSC.
- S. Basak, T. Paul and T. Punniyamurthy, Chem. Commun., 2023, 59, 11568 RSC.
- D. Chowdhury, S. Ghosh, K. S. S. V. P. Reddy, S. S. R. K. C. Yamijala and M. Baidya, ACS Catal., 2023, 13, 12543 CrossRef CAS.
- M. S. Keerthana and M. Jeganmohan, Chem. Commun., 2024, 60, 7347 RSC.
-
(a) S. Tang, K. Liu, C. Liu and A. Lei, Chem. Soc. Rev., 2015, 44, 1070 CAS;
(b) K. Zhu, J. Dunne, M. P. Shaver and S. P. Thomas, ACS Catal., 2017, 7, 2353 CAS.
- M. S. Keerthana and M. Jeganmohan, Eur. J. Org. Chem., 2024, e202400749 CrossRef CAS.
- For reviews, see:
(a) D. A. Colby, A. S. Tsai, R. G. Bergman and J. A. Ellman, Acc. Chem. Res., 2012, 45, 814 CAS;
(b) T. Piou and T. Rovis, Acc. Chem. Res., 2018, 51, 170 CAS;
(c) L. Song and E. V. Van der Eycken, Chem. – Eur. J., 2021, 27, 121 CAS.
- S. Saha, B. Debnath, K. Talukdar, P. Karjee, S. Mandal and T. Punniyamurthy, Org. Lett., 2023, 25, 3352 CAS.
- M. Barman, M. Mishra, S. Mandal and T. Punniyamurthy, Org. Lett., 2024, 26, 3722 CrossRef CAS PubMed.
- X. Zhou, S. Yu, L. Kong and X. Li, ACS Catal., 2016, 6, 647 CAS.
- X. Zhou, S. Yu, Z. Qi, L. Kong and X. Li, J. Org. Chem., 2016, 81, 4869 CAS.
- M.-W. Chen, M. Lou, Z. Deng, Q. Yang and Y. Peng, Asian J. Org. Chem., 2021, 10, 192 CAS.
- J. Liu, J. Jiang, Z. Yang, Q. Zeng, J. Zheng, S. Zhang, L. Zheng, S.-S. Zhang and Z.-Q. Liu, Org. Biomol. Chem., 2021, 19, 993 CAS.
- T. Paul, S. Basak and T. Punniyamurthy, Org. Lett., 2022, 24, 6000 CAS.
- K. Ramachandran and P. Anbarasan, Org. Lett., 2022, 24, 6745 CAS.
- K. Ramachandran and P. Anbarasan, Chem. Commun., 2022, 58, 10536 RSC.
- K. Ramachandran and P. Anbarasan, Synlett, 2023, 868 CAS.
- S. Mandal, T. Paul, P. Karjee, M. Barman and T. Punniyamurthy, Org. Lett., 2023, 25, 7805 CrossRef PubMed.
- N. Jha, W. Guo, W.-Y. Kong, D. J. Tantillo and M. Kapur, Chem. – Eur. J., 2023, e202301551 CrossRef CAS PubMed.
- O. P. Dash, D. Garga and C. M. R. Volla, Chem. Commun., 2024, 60, 10576 RSC.
- M. V. Barysevich, M. V. Laktsevich-Iskryk, A. V. Krech, V. N. Zhabinskii, V. A. Khripach and A. L. Hurski, Eur. J. Org. Chem., 2020, 937 CrossRef CAS.
- T. Paul, S. Basak, M. V. Nanjegowda and T. Punniyamyrthy, Org. Lett., 2023, 25, 8975 CrossRef CAS PubMed.
- X. Zhou, Z. Qi, S. Yu, L. Kong, Y. Li, W.-F. Tian and X. Li, Adv. Synth. Catal., 2017, 359, 1620 CAS.
- J. Liu, Z. Yang, J. Jiang, Q. Zeng, L. Zheng and Z.-Q. Liu, Org. Lett., 2021, 23, 5927 CrossRef CAS PubMed.
- J. Chen, C. Yin, J. Zhou and C. Yu, Eur. J. Org. Chem., 2021, 915 CrossRef CAS.
- S. Wang, E. Miao, H. Wang, B. Song, W. Huang and W. Yang, Chem. Commun., 2021, 57, 5929 RSC.
- S. Xu, C. Luo, K. Yan, J. Li, R. Lai, L. Hai, M. Guan and Y. Wu, Synthesis, 2022, 3015 CAS.
- J. Jiang, J. Liu, Z. Yang, L. Zheng and Z.-Q. Liu, Adv. Synth. Catal., 2022, 364, 2540 CrossRef CAS.
- W. Chen, Y. Mao, M. Wang, F. Ling, C. Li, Z. Chen and J. Yao, Org. Biomol. Chem., 2023, 21, 775 RSC.
- X. Yang, X. Cai, X. Zhang and X. Fan, Adv. Synth. Catal., 2024, 366, 141 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Tripti
Paul
,
Tripti
Paul