
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Direct organocatalytic esterification of carboxylic acids and alcohols by redox neutral sulfur(IV) catalysis via intramolecularly interrupted Pummerrer intermediates†
Ashish
Biswas
,
Priyanka
Pradhan
,
Sumit Ashok
Wakpanjar
and
Pavan K.
Kancharla
 *
*
CHEL-301, Department of Chemistry, Indian Institute of Technology Guwahati, Guwahati, Assam 781039, India. E-mail: pavankancharla@iitg.ac.in
First published on 17th March 2025
Abstract
Design, synthesis, and catalytic activity of new sulfur(IV) based organocatalysts for the direct esterification of carboxylic acids and alcohols is unveiled. The polar nature of the sulfoxide in the phenol-tethered catalyst accelerates the formation of an intramolecularly interrupted Pummerrer intermediate that further facilitates the catalytic esterification reaction via the activation of carboxylic acids.
Esterification is one of the fundamental reactions in organic chemistry. The ester functionality is widely found in nature, and many critical natural products contain ester groups, such as the antitumor drug quiderone1 and taxol2 for treating breast cancer and ovarian cancer. Esterification is a reaction that is also routinely used in the chemical industry to synthesize biodiesel, paints and varnishes, plastics and coatings, and pharmaceuticals. Hence, environmentally benign methods for commercial esterification are in exceptionally high demand. Some of the standard dehydrative Fischer-type esterification methods involve strong Brønsted acids or metal heterogeneous catalysts, which cannot be applied to sensitive organic compounds (Scheme 1a).3–7 Also, one of the components is usually used in high excess to have decent yields of the ester product. Other methods include an extra step of activating the acid functionality into acid chlorides or anhydrides. However, the most used protocols for esterification methods are the Steglich esterification8 (employing DCC and DMAP), Mitsunobu9,10 reaction (employing DEAD and triphenylphosphine) and the Yamaguchi11–13 esterification (2,4,6-trichlorobenzoyl chloride and DMAP) protocol and all of them involve use of stoichiometric amounts of activating agents and produce stoichiometric amount of waste products (Scheme 1b). Also, the need to remove all of these excess by-products make the purification of the ester products more challenging. Attempts have been made to improve the Mitsunobu protocol by various groups by introducing co-oxidants and co-reductants. However, this results only in replacing one stoichiometric reagent with the other. Nacsa14 and coworkers reported an excellent tri-methoxy N-phenylphenothiazine catalysed electrocatalytic direct esterification of carboxylic acids with alcohols at room temperature.
 | ||
| Scheme 1 (a) and (b) Previous literature on esterification reactions and (c) current work. | ||
Recently, Denton15a–c and coworkers introduced a very intriguing and ground-breaking phosphine oxide catalyst with phenoxy tether that helps in performing the Mitsunobu type esterification under catalytic redox neutral conditions with only water as the sole by-product, however, also resulting in stereoinversion at the alcohol center. This reaction is facilitated by the strong oxophilicity of the cationic P(V) intermediate and the regeneration of the catalyst, resulting in the formation of a strong P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond. On the other hand, the formation of the cationic P(V) intermediate is slow due to the very strong PO, which results in longer reaction times. We were intrigued to understand the mechanistic difference by replacing PO with an SO. We envisaged that a sulfoxide-based catalyst with a phenolic tether would also result in a similar cyclic intermediate via an intramolecular interrupted Pummerrer-type intermediate under mildly acidic conditions. However, it would be intriguing to study the reactivity of such an intermediate in esterification reactions. Also, sulfoxide being more polar, we envisaged a mechanistic switch in the esterification reaction.
O bond. On the other hand, the formation of the cationic P(V) intermediate is slow due to the very strong PO, which results in longer reaction times. We were intrigued to understand the mechanistic difference by replacing PO with an SO. We envisaged that a sulfoxide-based catalyst with a phenolic tether would also result in a similar cyclic intermediate via an intramolecular interrupted Pummerrer-type intermediate under mildly acidic conditions. However, it would be intriguing to study the reactivity of such an intermediate in esterification reactions. Also, sulfoxide being more polar, we envisaged a mechanistic switch in the esterification reaction.
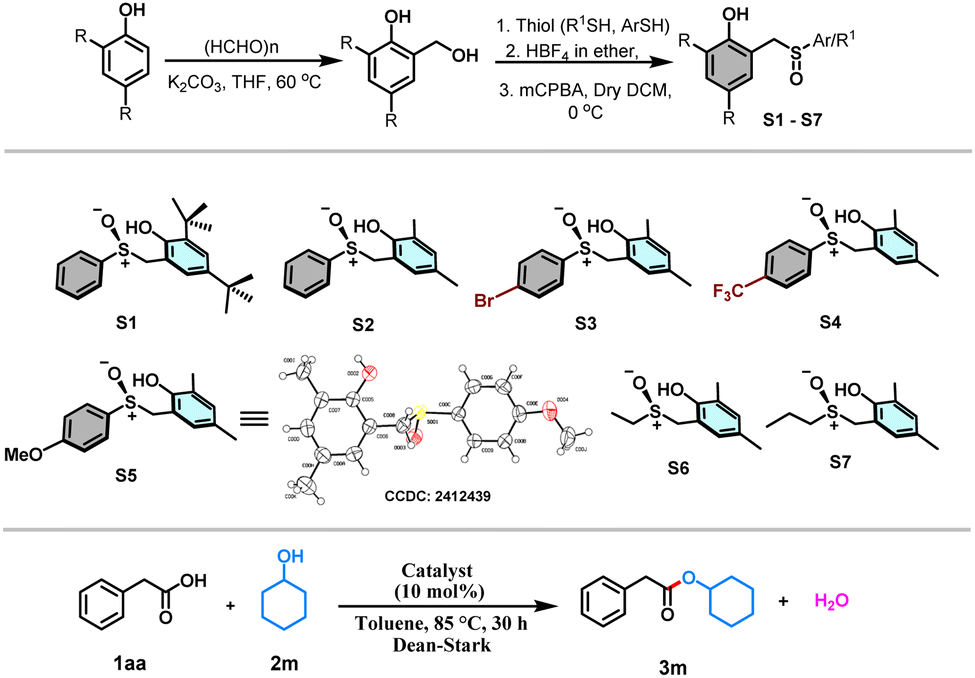
With this view in mind, herein, we designed the synthesis of sulfoxide-based organocatalysts for the direct esterification of carboxylic acids with alcohols via a redox neutral S(IV) catalysis (Scheme 1c). Sulfur(IV) compounds play an extraordinary role in organic chemistry.16–19 Our synthetic strategy started by reacting 2,4-disubstituted phenols with formaldehyde, resulting in the formation of ortho-(hydroxymethyl)phenols (Table 1). Subsequent reactions of these substrates with various thiophenols under acid catalysis followed by oxidation resulted in synthesizing the desired S(IV) catalysts with a phenolic tether. Catalysts S1–S7, with variations from both the phenolic and thiol components, have been synthesized to test the efficacy of these catalysts toward the organocatalytic esterification reaction (Table 1). The crystal structure obtained for one of the catalysts S5 helped the unambiguous characterization of the structures of the catalysts. With the catalysts (S1–S7) in hand, we tested their ability toward esterification between 1.5 equiv. of secondary alcohol, cyclohexanol (2m) and 1 equiv. of phenylacetic acid (1aa) as the model substrates with 10 mol% of the catalyst and toluene as a solvent under Dean–Stark conditions (Table 1). The sterically more hindered S1 provided the ester product in 30% yield, whereas the sterically less hindered S2 yielded only 45% of the product (Table 1). Later, the electron-withdrawing groups like Br and CF3 containing catalysts S3 and S4, respectively, and electron donating OMe group containing catalyst S5 were tested. Surprisingly, the yield significantly dropped with S3 and S4, whereas the OMe containing S5 performed the best providing the expected product in 85% yield (Table 1). Later, the catalysts S6 and S7 derived from alkyl thiols were also tested. However, the yields of the product were only moderate. Hence, the catalyst S5 is taken as the optimized catalyst. Surprisingly, the increase in the catalytic amount to 15 and 20 mol% led to a decrease in the yield of the ester product (Table 1). Hence, 10 mol% of S5 has been taken as the optimized condition for the esterification reaction. A range of alcohols were successfully acylated with phenylacetic acid (Scheme 2). All the primary alcohols tested including functional groups like alkene and alkyne, were successfully coupled to form the ester with phenylacetic acid in moderate to excellent yields (3a–3k, 40–90% yields, Scheme 2). The symmetric alkyne diol provided only the mono-esterified product 3j in a very moderate 40% yield. Secondary alcohols like cyclobutanol, cyclohexanol, cycloheptanol, and cyclododecanol also reacted very well, providing the coupled products in decent yields (3l–3o, 60–85%, Scheme 2). The method's ability has also been tested in the ester protection of galactose-derived 6-OH, which gave the product 3t in a good 75% yield (Scheme 2). Interestingly, the sterically hindered tert-butanol and the weakly nucleophilic phenol failed to provide the coupled product. The sterically hindered cholesterol also gave the product in 45% yield (3s, Scheme 2) with a retention in stereochemistry. To understand if there is a neighboring group effect by the alkene functionality, the alkene-reduced cholesterol was also subjected to the reaction conditions, providing the ester product in 40% yield (3r, Scheme 2) again with the retention of the absolute configuration. Later, we tested the scope of various acids under the current protocol with benzyl alcohol as the standard coupling partner (Scheme 2). A variety of acids with a range of functional groups have been successfully coupled to provide the ester products in good to excellent yields (4a–4f, 70–90% yields, Scheme 2). The electron-withdrawing groups NO2 and CF3 containing phenylacetic acids provided greater yields of the ester product relative to electron-donating (OMe) group containing substrate (4g–4i, 75–93% yields, Scheme 2). However, aromatic acids like benzoic acid and heteroaromatic pyridine 2-carboxylic acid failed to react under these conditions. Drug molecules like ibuprofen, naproxen, long-chain carboxylic, and unsaturated fatty acids were esterified in good to excellent yields (4l–4q, 84–91%, Scheme 2). Notably, the acid and base-sensitive Boc, Fmoc groups containing amino acids were also tolerated and afforded the ester products in moderate yields (5a–5e, 51–70% yields, Scheme 2). The gram scale esterification between 1aa and 2c has also provided the product in a good 83% yield (Scheme 3a).
|
|
|||
|---|---|---|---|
| Entry | Catalyst | Catalyst (mol%) | Yieldb (%) |
| a Reaction conditions: 1aa (1.0 equiv. 0.7 mmol), 2m (1.5 equiv. 1.09 mmol), catalyst (10 mol%) in toluene (2 ml) was refluxed in a Dean–Stark apparatus for 30 h. b Isolated yield. | |||
| 1 | S1 | 10 | 30 |
| 2 | S2 | 10 | 45 |
| 3 | S3 | 10 | 15 |
| 4 | S4 | 10 | 10 |
| 5 | S5 | 10 | 85 |
| 6 | S6 | 10 | 20 |
| 7 | S7 | 10 | 58 |
| 8 | S5 | 15 | 55 |
| 9 | S5 | 20 | 65 |
 | ||
Scheme 2 Substrate scope. Reaction conditions: 1aa (1.0 equiv. 0.7 mmol), 2 (1.5 equiv. 1.09 mmol), catalyst S5 (10 mol%) in toluene (2 ml) was refluxed in a Dean–Stark apparatus for 30 h. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Isolated yield. Isolated yield. | ||
 | ||
| Scheme 3 Control experiments. | ||
Control experiments were performed to gain insights into the mechanism of this transformation. As expected, the reaction only gave 10% yield of the product in the absence of any catalyst (see the ESI†). Also, the sulfoxide catalyst with OMe group instead of phenolic group gave only 8% yield of the product confirming the importance of the phenolic substituent in the current transformation (Scheme 3b). We also have performed oxygen labelling experiments in order to understand whether the esterification reaction under the current protocol proceeds via acid activation or alcohol activation. We have synthesized the O18 labelled benzyl alcohol (2ca) and O18 labelled phenylacetic acid (1ab) for the purpose.
Interestingly, when the coupling reaction is performed with O18 labelled benzyl alcohol (2ca) with unlabelled acid (1aa), the same amount of O18 labelling is observed in the ester (3ab) product (Scheme 3c). In addition, when the coupling experiment is performed with O18 labelled phenylacetic acid (1ab) unlabelled benzyl alcohol (2c), a significant loss of O18 labelling is observed within the product (3ad) (Scheme 3d). These experiments suggest that the major pathway in the current transformation is via the acid activation, not the alcohol activation. Based on all the above experiments, we propose the mechanism of the current transformation as depicted in (Scheme 4). The sulfoxide catalyst S5 reacts with the acid and forms an initial sulfonium cationic intermediate I1 that can be in equilibrium with intermediates I2 and I3. The intermediate I3 can react with nucleophilic alcohol via an H-bonding with the phenoxide, thus providing the ester product and also regenerating the catalyst S5 (Scheme 4). Also, the pathway involving an SN2 attack on alcohol intermediate I4 is unproductive. Since sulfur is less electronegative, it lacks the driving force for a C–O bond cleavage by a weak nucleophile, unlike the phosphorous analogue.15a
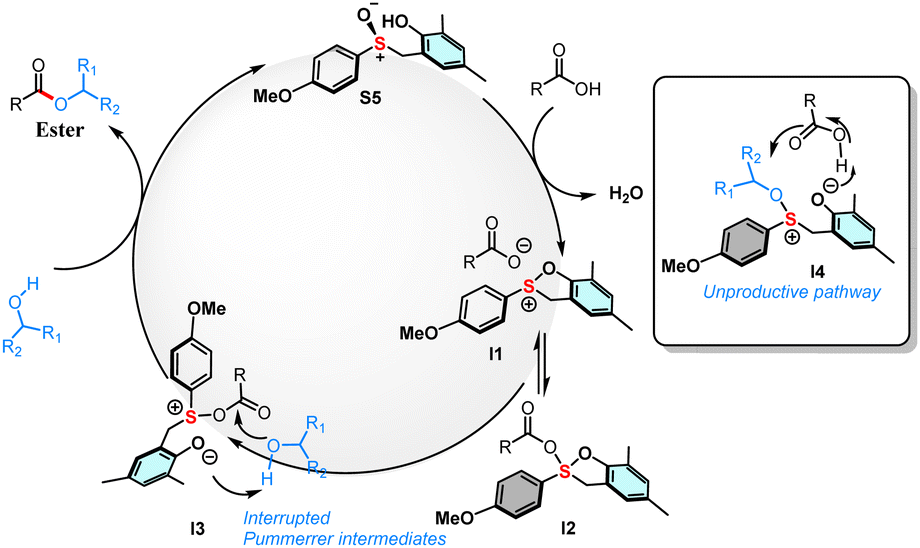 | ||
| Scheme 4 Proposed mechanism. | ||
In conclusion, we have designed and developed a new redox neutral sulfoxide-based S(IV) organocatalysts with a phenolic tether, that can be used for esterification reactions without inversion at the alcohol stereocenter. The substrate scope is broad for the current protocol and we believe that this new class of organocatalysts may find use in catalysing many other organic transformations as well.
PKK conceived the idea and wrote the manuscript. AB has performed all the experiments, PP and SAW performed the control experiments.
We are thankful to the central Instruments facility (CIF), IITG, for NMR, HRMS and other instruments; the Department of Chemistry, IITG for NMR, SC-XRD and other instrumental facilities; PKK is thankful to SERB (DST, New Delhi) for the financial assistance through CRG/2023/002033 and Ministry of Energy India for STARS-2/2023-0671. A. B., P. P. acknowledge IITG and SAW acknowledge UGC for the fellowships.
Data availability
The data underlying this study are available in the published article and its ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- P. A. Sprengeler, et al. , J. Am. Chem. Soc., 1991, 113, 3533–3542 CrossRef.
- Y. Nakamura and S. Chung-gi, Chem. Lett., 1992, 21, 49–52 CrossRef.
- Comprehensive Organic Synthesis, ed. P. Knochel, G. A. Molander, Elsevier, Amsterdam, 2nd edn, 2014, vol. 6, pp. 1–841 Search PubMed.
- (a) J. W. Bode, et al. , Nature, 2011, 480, 471–479 CrossRef PubMed; (b) J. M. J. Williams, OH Activation for Nucleophilic Substitution, In Sustainable Catalysis, John Wiley & Sons, Hoboken, NJ, 2013, pp. 121–138 Search PubMed.
- R. J. Ouellette and J. D. Rawn, Principles of Organic Chemistry, Elsevier, Amsterdam, 2014, pp. 699–745 Search PubMed.
- J. Otera, Esterification: Methods, Reactions, and Applications, Wiley-VCH, Weinheim, 2003 Search PubMed.
- E. Fischer and A. Speier, Darstellung der Ester, Ber. Dtsch. Chem. Ges., 1895, 28, 3252–3258 CrossRef CAS.
- (a) W. Steglich, et al. , Angew. Chem., Int. Ed. Engl., 1978, 17, 522–524 CrossRef; (b) H. F. Sneddon, et al. , Green Chem., 2021, 23, 6405–6413 RSC.
- G. Boshart, et al. , J. Org. Chem., 1961, 26, 2525–2528 Search PubMed.
- O. Mitsunobu, et al. , Bull. Chem. Soc. Jpn., 1967, 40(50), 2380–2382 CrossRef CAS.
- P. H. Toy, et al. , J. Am. Chem. Soc., 2006, 128, 9636–9637 CrossRef PubMed.
- (a) T. Taniguchi, et al. , Angew. Chem., Int. Ed., 2013, 52, 4613–4617 CrossRef PubMed; (b) T. Taniguchi, et al. , Chem. Sci., 2016, 7, 5148 RSC; (c) D. Hirose, M. Gazvoda, J. Košmrlj and T. Taniguchi, Org. Lett., 2016, 18, 4036–4039 CrossRef CAS PubMed.
- J. A. Buonomo and C. C. Aldrich, Angew. Chem., Int. Ed., 2015, 54, 13041–13044 CrossRef CAS PubMed.
- E. D. Nacsa, J. Am. Chem. Soc., 2023, 145(29), 15680–15687 CrossRef PubMed.
- (a) R. H. Beddoe, K. G. Andrews, V. Magn, J. D. Cuthbertson, J. Saska, A. L. Shannon-Little, S. E. Shanahan, H. F. Sneddon and R. M. Denton, Science, 2019, 365, 910–914 CrossRef CAS PubMed; (b) X. Tang, C. Chapman, M. Whiting and R. M. Denton, Chem. Commun., 2014, 50, 7340–7343 RSC; (c) Y. Zou, J. J. Wong and K. N. Houk, J. Am. Chem. Soc., 2020, 142(38), 16403–16408 CrossRef CAS PubMed.
- D. Kaiser, I. Klose, R. Oost, J. Neuhaus and N. Maulide, Chem. Rev., 2019, 119(14), 8701–8780 CrossRef CAS PubMed.
- T. Wai, H. L. Wang and Y. Tian, et al. , Nat. Chem., 2024, 16, 1301–1311 CrossRef PubMed.
- S. Song, X. Li and J. Wai, et al. , Nat. Catal., 2020, 3, 107–115 CrossRef CAS.
- B. M. Trost and M. Rao, et al. , Angew. Chem., Int. Ed., 2015, 54, 5026–5043 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures for the synthesis of starting materials and products; their spectroscopic data; NMR, HRMS spectra and X-ray crystallographic data of compounds S5. CCDC 2412439. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5cc00556f |
| This journal is © The Royal Society of Chemistry 2025 |