
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sydnonimines: synthesis, properties and applications in chemical biology
Alfonso
Fumanal Idocin
 a,
Simon
Specklin
*a and
Frédéric
Taran
*b
a,
Simon
Specklin
*a and
Frédéric
Taran
*b
aUniversité Paris-Saclay, Inserm, CNRS, CEA, Laboratoire d’Imagerie Biomédicale Multimodale Paris-Saclay (BioMaps), Orsay 91401, France. E-mail: simon.specklin@cea.fr
bDépartement Médicaments et Technologies pour la Santé, CEA-DMTS-SCBM, Université Paris-Saclay, 91191 Gif-sur-Yvette, France. E-mail: frederic.taran@cea.fr
First published on 6th March 2025
Abstract
Sydnonimines are intriguing compounds belonging to the mesoionic family. To date, only a limited number of research groups have studied their chemistry and use in organic synthesis, medicinal chemistry and chemical biology. This review aims at providing an overview of the synthesis and the properties of sydnonimines and the most recent developments in their use as tools for chemical biology. The recent discovery that sydnonimines can act as a dipole to undergo bioorthogonal click-and-release reactions with cycloalkynes has stimulated a renewed interest from the scientific community. Given the high potential of these mesoionics, we believe that major developments are to be expected in the field of bioorthogonal chemistry.
 From left to right Frédéric Taran, Alfonso Fumanal Idocin, Simon Specklin | Alfonso Fumanal Idocin (middle) is a second-year PhD student at the French Alternative Energies and Atomic Energy Commission (CEA), working under the supervision of Bertrand Kuhnast, Simon Specklin, and Frédéric Taran. He secured a Marie Skłodowska-Curie fellowship as part of the ISOBIOTICS European project, which focuses on the isotopic labeling of biological drugs. His research project involves labeling these biologics with 18F using a click-and-release strategy that incorporates sydnonimines. Simon Specklin (right) is a researcher in a biomedical imaging laboratory at the French Alternative Energies and Atomic Energy Commission (CEA). After completing a PhD in chemistry at Strasbourg University under the supervision of Pr. Patrick Pale in 2010, he spent two years as a postdoctoral fellow at the organic chemistry laboratory led by Pr. Janine Cossy at ESPCI Paris. He then joined Dr Frédéric Taran group at the CEA working on bioorthogonal reactions. He later secured a permanent position at the CEA/SHFJ in 2015. His research focuses on the development of new methodologies for fluorine-18 radiolabeling of various targets, ranging from low-molecular-weight compounds to biologics. Frédéric Taran (left) is group Leader in the department of chemistry at the French Alternative Energies and Atomic Energy Commission (CEA) located at Saclay. Dr Taran secured a PhD in chemistry at the Paris XI University under the supervision of Dr Charles Mioskowski. In 1996, he moved to a post-doctoral position with Prof. Sir Derek Barton at Texas A&M University (USA) and then came back to CEA in 1998 as permanent researcher. His research aims at developing new reagents for bioorthogonal chemistry to address important problems in the fields of bioconjugation, labelling, imaging and drug delivery. |
1 Introduction
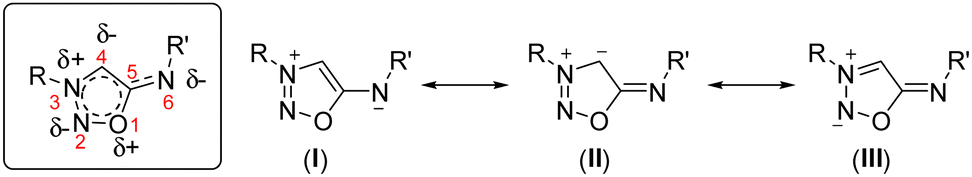
Sydnonimines (SI), also called iminosydnones, are intriguing members of the mesoionic family, a term coined in 1949 by Baker and Ollis, amalgamating “mesomeric” and “ionic”.1 This group encompasses 5-membered dipolar heterocycles that cannot be adequately represented by a single charged structure. Well-known examples in literature include sydnones and münchnones while SI remain comparatively underexplored.2 Structurally close to sydnones, SI differ by the presence of an exocyclic nitrogen instead of oxygen. Single-crystal X-ray analysis reveals that SI exhibit a longer exocyclic C–N bond (1.30–1.32 Å) compared to the exocyclic C–O bond in sydnones (1.19–1.22 Å), though still retaining partial C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N double-bond character, consistent with the resonance structures shown in Scheme 1.3
N double-bond character, consistent with the resonance structures shown in Scheme 1.3
 | ||
| Scheme 1 Resonance structures of sydnonimines. | ||
First reported in 1957,4SI initially gained attention in the 1970s for their pharmacological properties as nitric oxide (NO) donors,5 leading to the development of several drugs, two of which remain on the market nowadays.
Beyond their biological activities, SI also function as ligands for metals; their deprotonation produces anionic and abnormal N-heterocyclic carbenes (NHCs),6 allowing the formation of novel metal complexes.
There has been a resurgence of interest for SI in recent years related to their potential applications in chemical biology. Their role as dipoles in bioorthogonal cycloadditions has been particularly noteworthy, driving innovations in areas such as cleavable linkers for target fishing, immunoprofiling and drug delivery.
This review aims to provide a comprehensive overview of the latest advancements in SI research, focusing on their chemistry, biological activities, applications as metal ligands, and emerging roles in chemical biology.
2 Synthesis and functionalization of SI
2.1 General synthesis
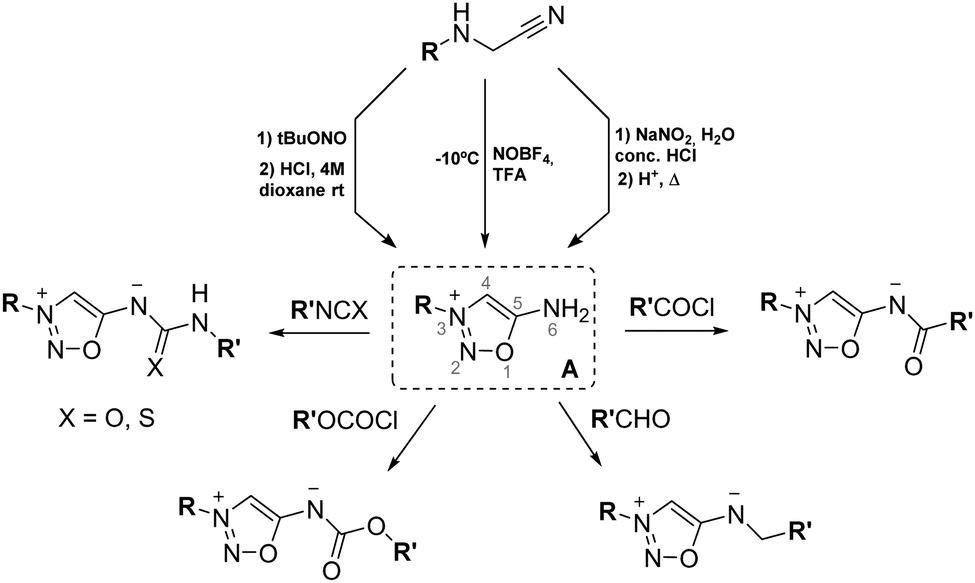
The main synthetic route to SI is still the one initially published in 1957 by Brookes and Walker4 and the team of Ohta.7 Both teams pioneered the same synthetic pathway based on the nitrosylation of α-aminonitriles followed by cyclisation in strong acidic conditions to afford SI. The nitrosylation step was initially described using sodium nitrite in acidic conditions but is also alternatively carried out with alkyl nitrites. SI are then generated upon cyclisation with various acids, HCl being the most common, leading to SI as hydrochlorides. A new addition to this widely used strategy has been disclosed recently for the construction of SI cycles.8 Instead of generating NO+in situ from nitrites, the work of Shuvaev et al. leverages the use of NOBF4 salt. This reagent has a good solubility in organic media, enabling a one-step procedure for nitrosylation and subsequent cyclisation leading to SI. This synthetic pathway tolerates substituted α-aminonitriles and exhibits enhanced yields compared to the classical method.All of these synthetic conditions lead to synthon A bearing a poorly nucleophilic amine in position 6. Functionalization of this amine, leading to SI mesoionic derivatives, therefore requires strong electrophiles such as acylchlorides, isocyanates, chloroformates or aldehydes (Scheme 2). These functionalizations are described in detail in the following paragraph.
 | ||
| Scheme 2 General synthetic routes to sydnonimines. | ||
Recently, the team of Lamaty reported the synthesis of SI using mechanochemistry.9 Starting from a primary amine, an efficient one-pot procedure leading to SI cyclisation was developed by vibratory milling ball grinding. The synthesis of linsidomine and feprosidnine (two bioactive SI, see Section 3) analogues was notably achieved in solvent free conditions with very high yields after a straightforward extraction. This method lead to SI as hexafluorophosphates salts, which can be interesting for applications requiring non-nucleophilic anion.
Due to the growing interest in the biological properties of SI, numerous derivatives have been synthesized since their discovery, expanding the chemical space of these compounds. One of the structural key points of SI is the exocyclic nitrogen, whose functionalization plays a crucial role in stabilizing their structure toward high pH levels. Many carbonyl derivatives were particularly synthesized leading to amides, carbamates, ureas, and thiourea functions in position 6.10 The synthesis of these derivatives is mostly based on the reaction of SI salts with strong electrophiles. Sulfonyl derivatives were also described very early in the development of SI,11 whereas 6-N-phosphorylated SI were only scarcely studied.12
An interesting approach from a retrosynthetic perspective was developed with the use of p-nitrophenyl carbonates as activated intermediates (Scheme 3). Reaction of these compounds with an alcohol or an amine provided a fast access to a variety of SI carbamates or ureas upon heating. Particularly useful for drug design, this strategy was first used to synthesize derivatives of the drug molsidomine.13 The team of O’Hagan also used this activated intermediate for the synthesis of amino acid SI derivatives,14 and for the attachment of SI to peptidic drugs.15
 | ||
| Scheme 3 Functionalization of position 6 through activated intermediates. | ||
Based on these findings, our group reported the synthesis of another activated intermediate and generalized this retrosynthetic approach for the synthesis of diverse SI.16SI salts were readily converted to carbonylimidazole derivatives upon reaction with CDI. Further activation of imidazole by alkylation with MeI provided highly reactive carbonyl- and thiocarbonylimidazolium intermediates which reacted smoothly with amines (primary and secondary), alcohols (aliphatic or aromatic), thiols or other nucleophiles under mild conditions. Functionalized biologically relevant compounds have also been readily substituted with a SI using this three steps procedure starting from SI hydrochlorides.17
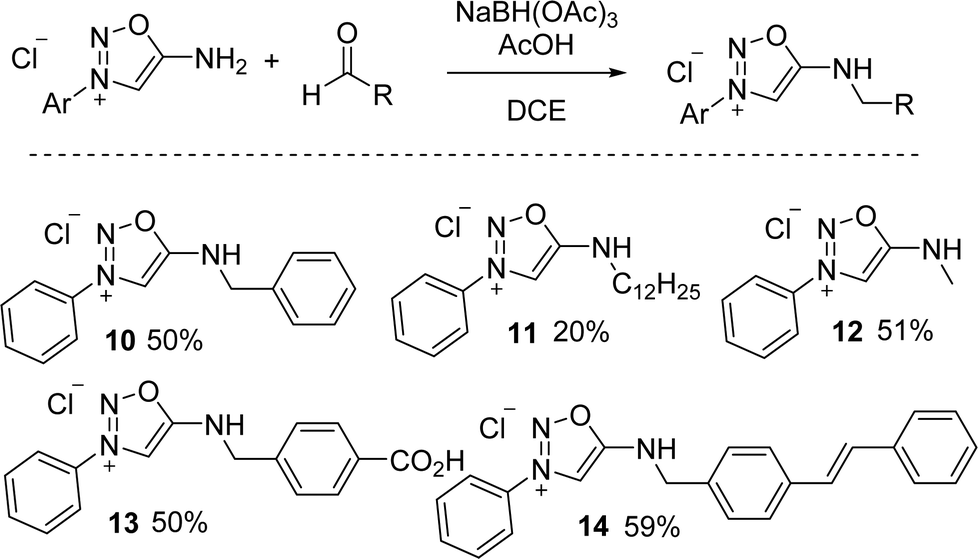
6-N-alkyl SI can also be prepared using reductive amination from synthon A.18 After extensive screening, sodium triacetoxyborohydride was found to be the best reducing agent for this reaction. The use of apolar and aprotic solvents was also found critical to minimize the competing ring opening of the SI cycle. A variety of 6-N-alkyl SI were thus reported with moderate yields from synthon A and aliphatic or aromatic aldehydes (Scheme 4).
 | ||
| Scheme 4 Preparation of 6-N-alkyl SI by reductive amination. | ||
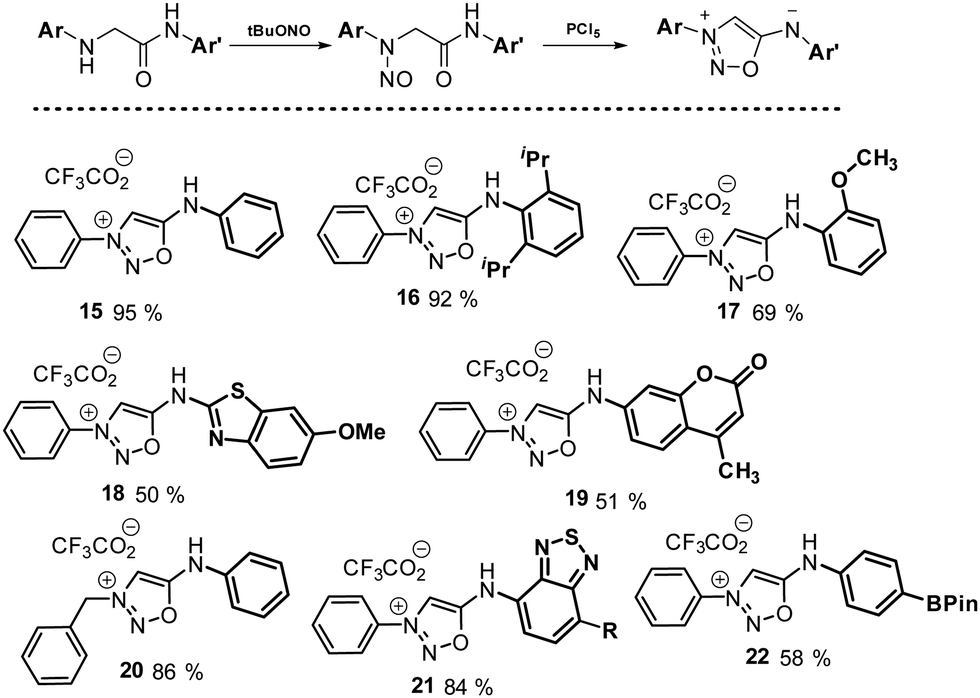
Although leading to many SI derivatives, the synthetic pathway based on derivatization of synthon A cannot provide all possible structures. For instance, all arylation attempts using metal-catalyzed coupling reactions were found unsuccessful. Consequently, 3-N, 6-N-diaryl sydnonimines were never obtained until a new synthetic route was recently developed.18 The latter was based on the cyclisation of N-nitrosamide intermediates obtained from nitrosylation of amino-amides using tert-butyl nitrite. After screening of several reagents, PCl5 was identified as the most effective dehydrating reagent leading to 3,6-diaryl-SI in reasonable yields. This set of conditions allowed to generate a diversity of compounds with an important electronic and steric tolerance for the aryl group (Scheme 5).
 | ||
| Scheme 5 Synthetic route to 3-N, 6-N-diaryl-SI. | ||
However, the use of PCl5 as a reagent limited the scope of the methodology. A complementary approach was thus developed to access diaryl-SI using sydnones as starting material.19 Reaction of sydnones with Tf2O has been described to generate the corresponding triflate in position 6 which can undergo a SNAr reaction with carbon nucleophiles to form sydnonemethide compounds.20 Using the same strategy with amines as nucleophiles, SI can be generated in mild conditions (Scheme 6). Moderate yields are obtained, due to the very high reactivity of the sydnone-triflate intermediate and the competing formation of the corresponding triflated amine. The scope of the reaction is however interesting, as primary, secondary, aliphatic and aromatic amines can be used leading to a variety of SI derivatives.
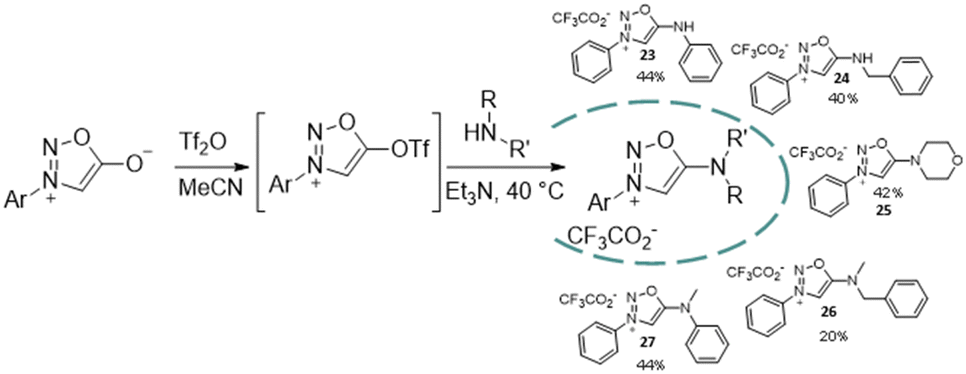 | ||
| Scheme 6 Synthesis of SI from sydnones. | ||
2.2 Functionalization in position 4-C
Functionalization in position 4 of SI is highly common and represents a significant part of the reports involving SI synthesis. This position can be substituted before the SI cyclisation,21 but the most common way is to harness the specific reactivity of this position.SI can be deprotonated with a lithium base at the 4-C position resulting in a lithium adduct that can react with a variety of electrophiles (Scheme 7). Several SI were obtained using non-enolizable aldehydes allowing the introduction of an α-hydroxyalkyl group.22,23 In the case of 6-N-phosphorus SI derivatives, 4-(α-hydroxy) derivatives undergo an intramolecular substitution to form a bicyclic SI.12 Additionally, formylation of 4-lithiosydnnimines has been reported by DMF activation with dimethylsulfate leading to 4-formylsydnone imine. Introduction of other carbon-based substituents has been reported from 4-lithiosydnonimine,24 which have been involved in further condensation and Knoevenagel reactions.25
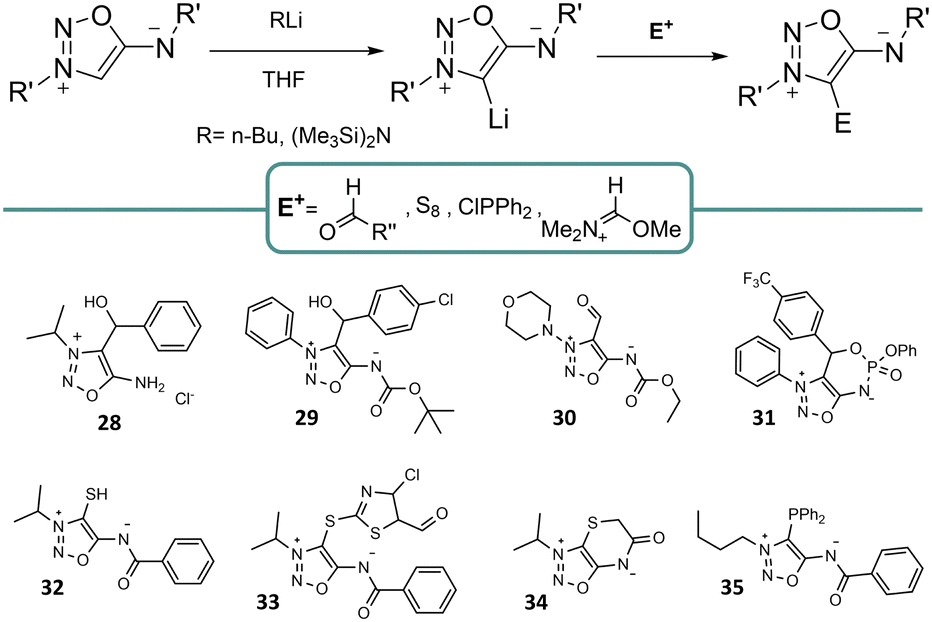 | ||
| Scheme 7 Functionalization in the 4-C position of SI. | ||
Apart from the introduction of carbon based substituents, 4-thio derivatives of SI can be obtained by reaction with S8 to generate the lithium thiolate. This species can be either alkylated to generate different 4-mercapto derivatives, which can then be oxidized to form sulfoxide derivatives,26 or generate the 4-thiol derivative when treated with water.27 Thiolates can also be used to form linear gold complexes or bidentate mercury complexes. However, 4-thiols seem to cleave the SI core where the exocyclic nitrogen atom is not substituted by electron-withdrawing groups. Lastly, 4-thio derivatives can also be intramolecularly alkylated to form bicyclic derivatives.28 On the other hand, 4-phosphorus derivatives are synthesized by reacting the corresponding halophosphine with lithiated SI.29 The corresponding SI-phosphine can coordinate palladium, giving rise to a five-membered metallocycle where the SI derivative acts as a bidentate ligand.
The position 4 of SI can also be easily halogenated, offering the possibility to modify their electronic properties. Overall, 4-bromo derivatives were found to be more stable and easier to synthesize than 4-chloro and 4-iodo derivatives.30 However, our group reported a selective methodology to chlorinate the 4-C position using tBuOCl as chlorinating agent.31 4-bromo and 4-iodo derivatives, easily prepared using N-bromo or N-chlorosuccinimide, are good substrates for Suzuki and Sonogashira coupling reactions (Scheme 8).32 To the best of our knowledge, no 4-fluoro derivative of SI are reported, in contrast to sydnones.33 Halogenation of the 4-C position has proven to enhance the reactivity of SI as dipoles (vide infra), thus making this functionalization very attractive for bioorthogonal chemistry.
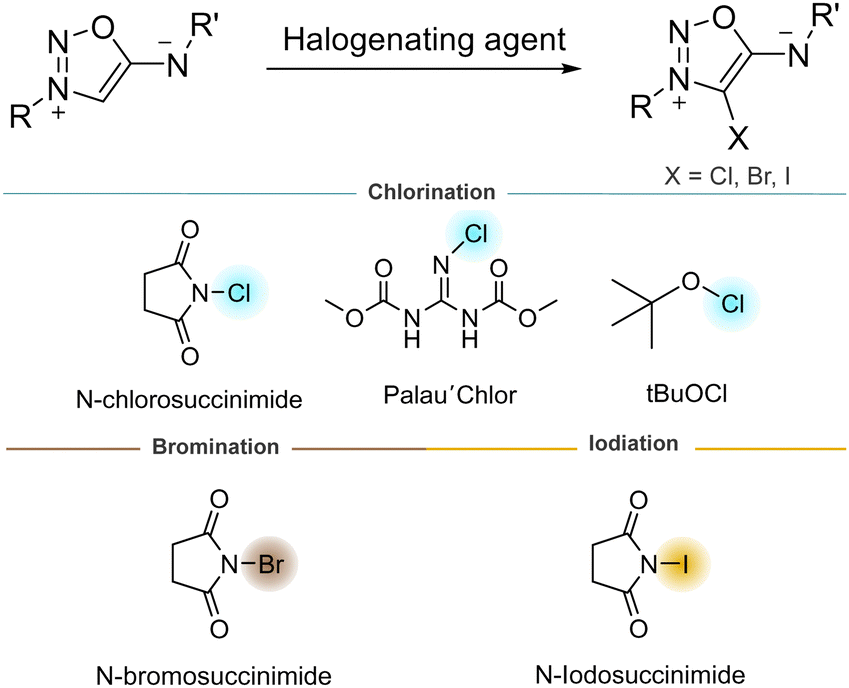 | ||
| Scheme 8 Halogenation of 4-C position of SI. | ||
3 Biological activities of SI
Shortly after the initial synthesis reports of SI, several research groups began investigating the biological potential of this unique class of mesoionic compounds. Early screenings in the 1960s suggested a range of promising biological effects,4 which now include antipyretic and anti-inflammatory effects,34 monoamine oxidase (MAO) inhibition,35 antispasmodic effects, and antitumoral activities.36 These diverse biological applications are largely attributed to the NO-donor capabilities of SI, which are greatly influenced by the nature of the substituents attached to their core.3.1 Applications as vasodilators
The vasodilatory properties of SI rapidly emerged as their most emblematic application. Molsidomine, a vasodilating drug developed in the 1970s, has been widely used to treat stable and unstable angina pectoris, a form of chest pain due to reduced blood flow to the heart. The active metabolite of molsidomine, SIN-1, was first synthesized in 1969. Researchers observed that SIN-1 undergoes alkaline decomposition, yielding the derivative SIN-1C,37 which was associated with a hypotensive effect in animals.38 This effect was also observed in a molsidomine derivative, later identified as an effective prodrug with favorable biodistribution properties.39 A complete mechanism for molsidomine activation was later proposed by Bohn et al.40 In this mechanism, molsidomine is hydrolyzed by liver esterases to produce SIN-1, which is unstable at physiological pH and is converted into an open form, SIN-1A. SIN-1A then reacts with dissolved oxygen to generate nitric oxide (NO) and superoxide anion. Due to their stoichiometric release, these species can form peroxynitrite (ONOO−), reducing the availability of free NO (Scheme 9). | ||
| Scheme 9 Proposed mechanism for molsidomine activation in vivo. | ||
To enhance the NO availability and reduce undesired toxic peroxynitrite formation, a hybrid molecule incorporating a radical scavenger functionality was described by Acharya et al.41 The prodrug SA-4 was developed to release SA-2, which demonstrated notable biological activities, including elevating superoxide dismutase levels and protecting photoreceptor cells from oxidative stress induced by H2O2 (Scheme 10). The same authors then demonstrated the beneficial effect of SA-2 in preventing retinal ganglion cells death in animal models.42
 | ||
| Scheme 10 Structure of a hybrid molecule containing SI. | ||
Recently, molsidomine and acetylsalicylic acid have been combined into a hybrid molecule by Szöke et al. in order to increase the effect of aspirin in ischemic heart conditions and decrease gastrointestinal damage.43 Recent studies report the synthesis of N-nitroso-3-morpholinosydnonimine, which can liberate two molecules of NO while reducing the generation of toxic peroxynitrite.44 Other vasodilating drug candidates include pirsidomine and its active hydrolyzed form (darsidomine),45,46 although they have been discarded in phase 2 trials.
3.2 Applications as plant growth stimulants
Cherepanov et al. reported the properties of SI as plant growth regulators, focusing on 4-C derivatives (Scheme 11).23,47,48 The effect can range from herbicidal to growth stimulant depending on the dose. However, plant species and the SI structure (in particular the 6-N and 3-N substituents) play an important role on modulating this effect.49 In addition, ferrocenyl-containing sydnonimines have proven to act as antidotes to herbicides such as Zinger WP.50,51 | ||
| Scheme 11 Structure of SI with plant growth stimulation activities. | ||
3.3 Applications as psychostimulants
SI containing an amphetamine moiety have psychostimulant properties with reduced toxicity and addictiveness compared to the free amphetamine.52,53 The first psychoactive SI drug was mesocarb (also known as sydnocarb or MLR-1017). First developed in 1971 and commercialized until 2008, mesocarb was prescribed in Russia for psychiatric disorders.54 Mesocarb constitutes a racemic mixture, the active enantiomer was later renamed armesocarb (MLR-1019). Armesocarb functions as a dopamine reuptake inhibitor by obstructing the dopamine transporter.55 Feprosidnine is another psychoactive SI drug prescribed in Russia to treat depression. Feprosidnine differentiates from armesocarb by having the 6-N atom unsubstituted (Scheme 12). | ||
| Scheme 12 Structure of SI with psychostimulant activities. | ||
3.4 Applications for cancer treatment
Nitrosulindac is a well-known nitric oxide-donating non-steroidal anti-inflammatory drug (NO-NSAID), inhibiting proliferation and inducing apoptosis in human prostatic epithelial cells.56 In 2013, O’Hagan et al. synthesized several SI analogues of nitrosulindac displaying similar or better activities than the parent drug (Scheme 13).57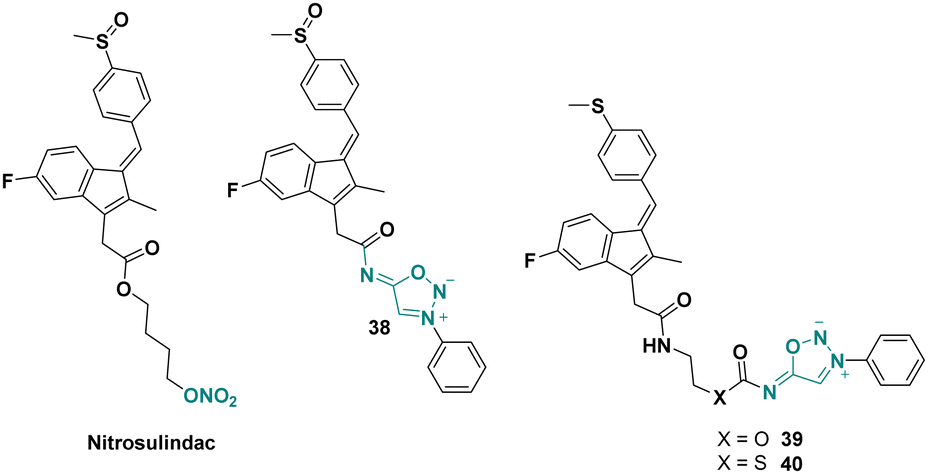 | ||
| Scheme 13 Structures of SI analogues of nitrosulindac. | ||
In 2014, O’Hagan et al. used the NO donor properties of SI to enhance the biological activity of abiraterone,15 a drug used for the treatment of late stage, metastatic prostate cancer. The authors developed a molecule bearing abiraterone, SI and an integrin binding RGD peptide sequence presenting significant cytotoxicity towards cancer cell lines.
3.5 Applications as trypanocidal agent
In 2003, Hoffmann et al. reported the synthesis of several SI derivatives with notable trypanocidal properties.58 The compounds were designed around an SI motif derived from molsidomine and incorporated purine or melaminophenyl groups, which facilitate recognition by a trypanosomal purine transporter. Their trypanocidal effects are hypothesized to result from the release of nitric oxide and superoxide, which may disrupt the parasite's cellular replication (Scheme 14).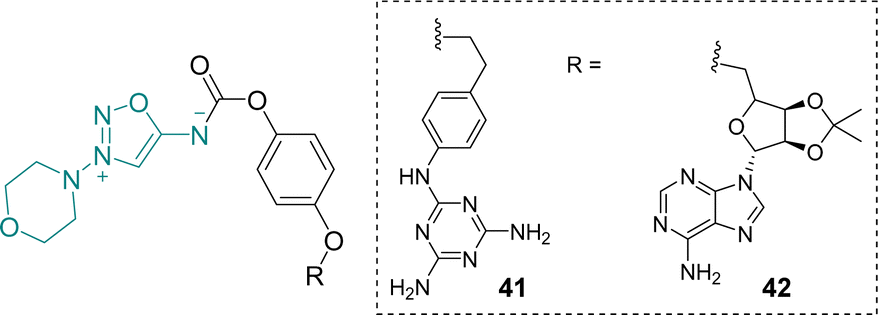 | ||
| Scheme 14 Structures of SI with trypanocidal activities. | ||
3.6 Applications as insecticidal agent
Triflumezopyrim is a mesoionic insecticide recently developed by DuPont acting as an efficient inhibitor for the nicotinic acetylcholine receptor (Scheme 15).59 To evaluate the role of the mesoionic structure on the biological activity, Qian et al. reported in 2021 several SI derivatives whose lethality against Mythimna separate are comparable to the one of the parent drug.60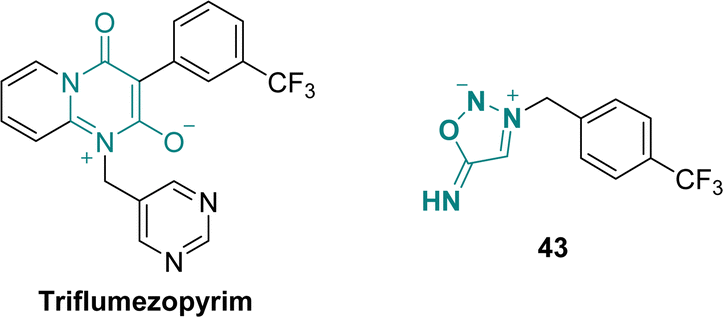 | ||
| Scheme 15 Structures of SI analogues of triflumezopyrim. | ||
This study provided valuable information regarding the relation between the structure of the mesoionic and its insecticidal properties, providing useful background for the design of novel mesoionic insecticides.
4 SI as metal ligands
In addition to their biological activities, sydnonimine derivatives are also studied for their ability to generate anionic N-heterocylic carbenes (NHC) and abnormal NHCs (Scheme 16). It is worth mentioning that carbene formation has been only explored with sydnonimine derivatives substituted in position 6-N, usually 6-N-acyl derivatives. These compounds are conjugated heterocyclic mesomeric betaines (CMB) which can generate both NHCs upon deprotonation of position 4-C. The resonance forms show the possibility of forming the anionic abnormal NHC (A′), the anionic normal NHC (B′), and another resonance form (C′) where the two negative charges are located at the 4-C carbon atom, one deallocated in the π-system and the other in the sp2 hybrid orbital.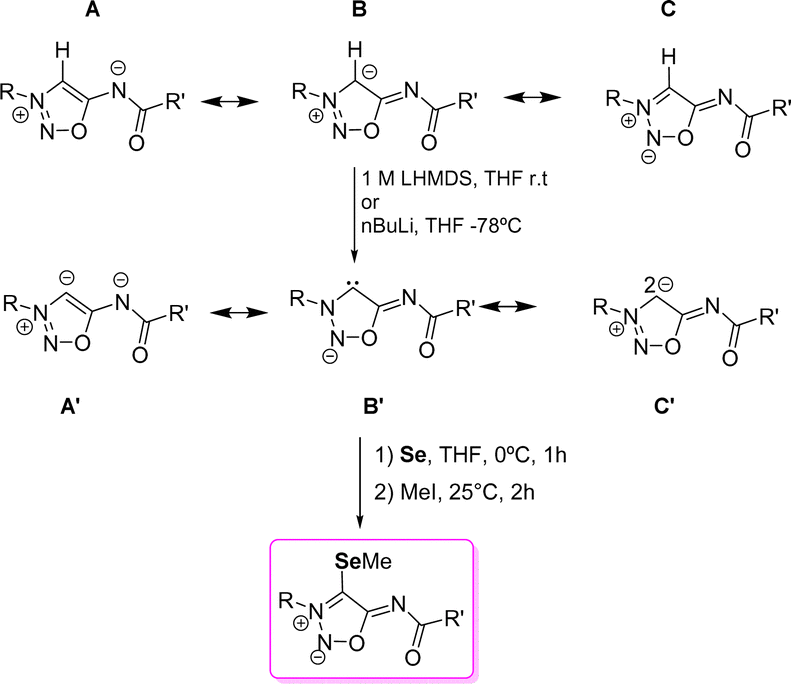 | ||
| Scheme 16 Resonance forms of sydnonimines and their NHC forms. Carbene trapping with Se. | ||
In 2000, Cherepanov et al.61 reported the first 4-lithium derivatives of SI, prepared by deprotonating the 4-C position with nBuLi; however, they noted that this approach led to the formation of unstable carbenes. Schmidt's group similarly found that deprotonation with nBuLi resulted in rapid degradation at room temperature. Nevertheless, treatment of SI with 1 M LiHMDS in THF at room temperature proved to be optimal for 4-C deprotonation, stabilizing the anionic carbene in THF for several weeks. However, more recent data suggest that this stability may be overestimated, with maximum stability at room temperature reported to last only about 10 hours.47 Density functional theory (DFT) calculations indicated that the lithium atom positioned between the 4-C carbon and the carbonyl oxygen is essential for this stabilization, with additional influence from the substituents at the 3-N and 6-N positions. The deprotonated 4-C atom shows a 13C NMR shift ranging from 142.1 ppm to 159.8 ppm, which is considered high-field for NHCs.
DFT calculations by T. Freese et al.62 reveal differences in the HOMO–LUMO orbitals of the molsidomine carbene, used as an SI model, compared to 1,3-dimethylimidazol-2-ylidene (44), which represents standard NHCs. These calculations indicate altered π-accepting properties in molsidomine relative to regular NHCs, with an inversion in the HOMO orbital configuration. Specifically, the HOMO orbital in molsidomine is a π-orbital with a substantial contribution from the non-hybridized p-orbital of the 4-C atom, while the HOMO−1 corresponds to the σ-orbital typical of NHCs (see Fig. 1). Additionally, the LUMO orbitals of molsidomine are higher in energy and contain a significant coefficient from the carbene carbon atom.
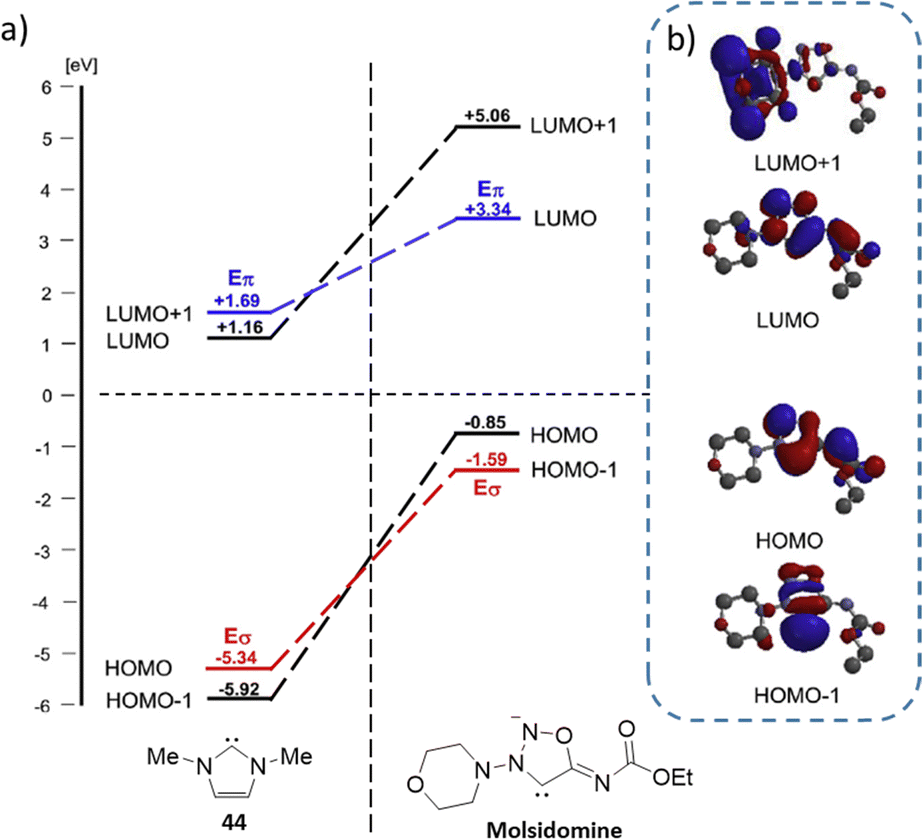 | ||
| Fig. 1 (a) DFT calculation representation of frontier orbital energies of ylidene (55 in comparison to molsidomine carbene). (b) Representation of calculated molecular orbitals of molsidomine. Reproduced from ref. 62 with permission from Elsevier, copyright 2017. | ||
The trapping of a carbene with selenium is a typical procedure to evaluate its π-acceptor strength.63 Higher 77Se NMR shift correlates with higher π-accepting properties of the NHC. In the case of the molsidomine selenium derivative, the NMR shift was 97.2 ppm for the 4-C carbon atom, which could be seen as a proof for the loss of π-accepting properties63 in comparison to the model NHC. The selenium complexes have been prepared by direct reaction with selenium, followed by a methylation with MeI (Scheme 16).
Unlike sydnones, which are widely used as ligands for metals,64,65 only few examples of NHC–metal complexes with SI are found in the literature (Scheme 17).
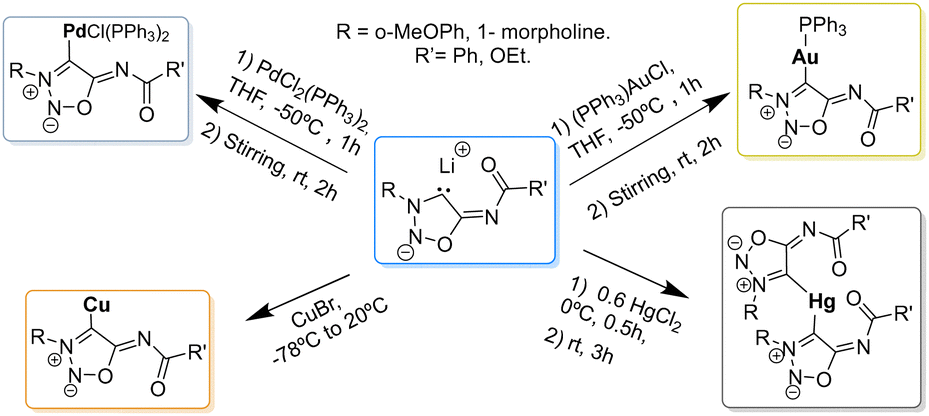 | ||
| Scheme 17 Reactions of SI carbenes with metals. | ||
The first NHC–metal complexes obtained from SI carbenes were described in 2000 by Ilya A. Cherepanov and Valery N. Kalinin.66 Direct reaction of the SI carbenes with CuBr afforded the corresponding Cu-complexes which were found to be good substrates for palladium-catalyzed cross-coupling reactions as seen in Scheme 18.
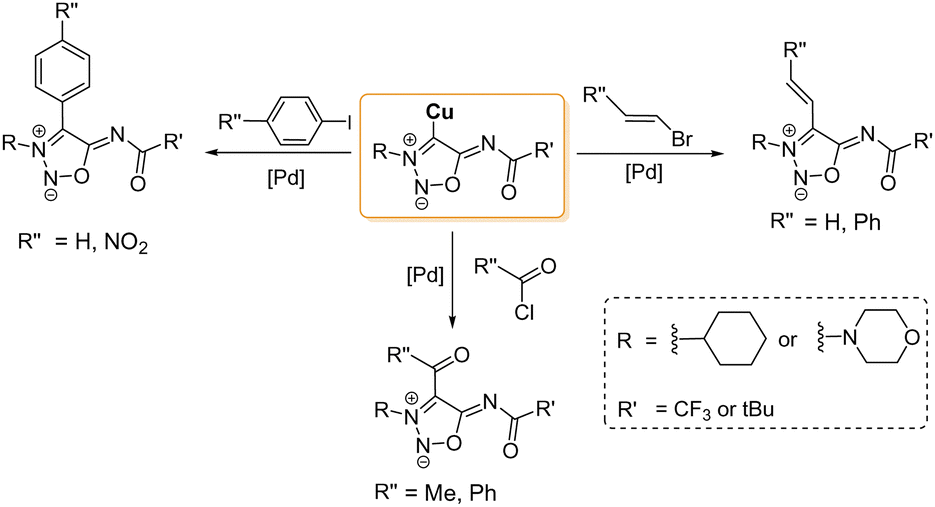 | ||
| Scheme 18 Different cross-coupling reactions with 4-copper SI derivatives. Conditions: Pd(PPh3)4 20 °C, 2–24 h. Yields: 30–65%. | ||
Gold complexes can also be prepared by direct reaction of the deprotonated SI with (PPh3)AuCl. Only one of the two gold complexes described in the literature has crystallographic data available (Fig. 2).62
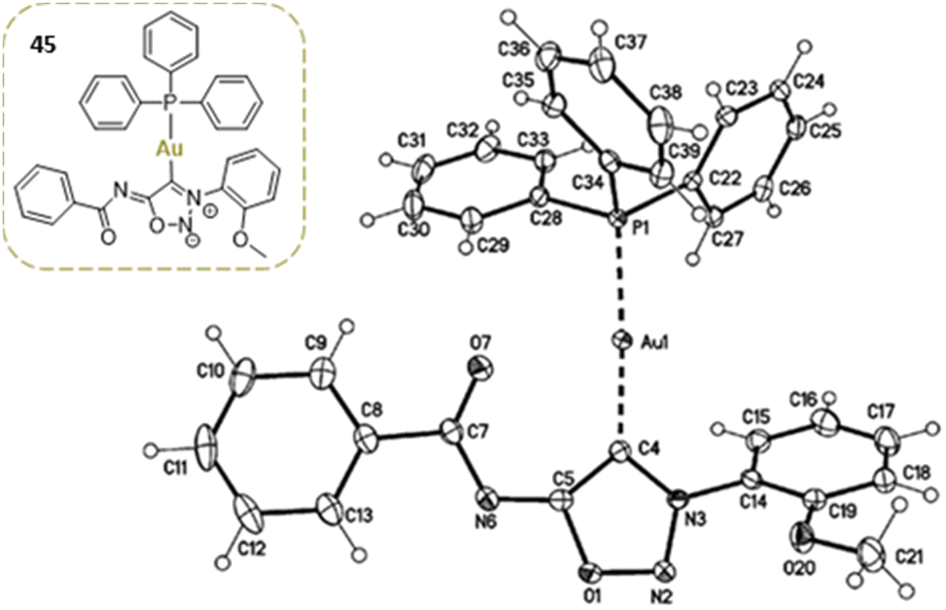 | ||
| Fig. 2 Crystal structure of a sydnonimine NHC–Au complex. Reproduced from ref. 62 with permission from Elsevier, copyright 2017. | ||
Mercury complexes were synthesized by trapping the carbene with HgCl2 at 0 °C. The crystal structure (Fig. 3) showed the coordination of the mercury atom by two SI carbenes, giving rise to bis-NHC complexes.26
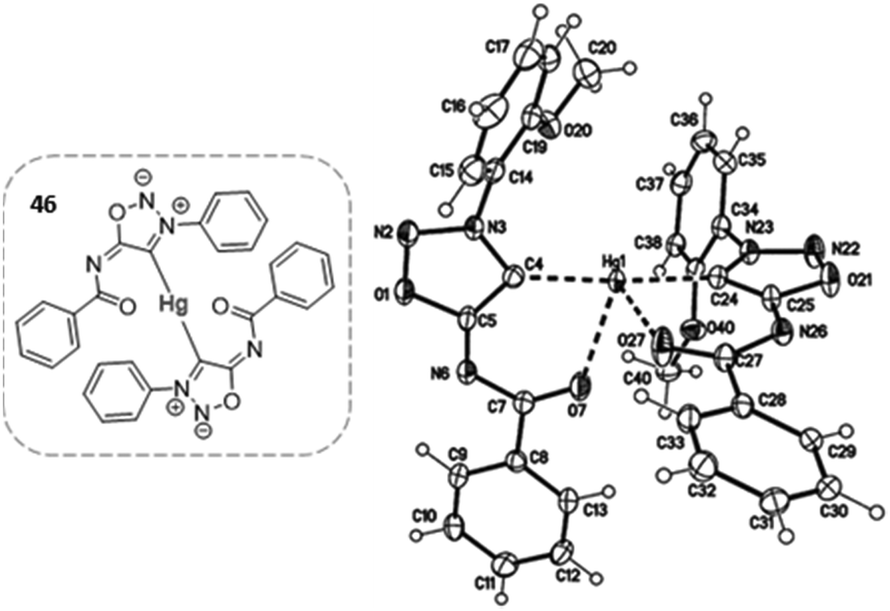 | ||
| Fig. 3 Crystal structure of a sydnonimine bis(NHC)–Hg complex. Reproduced from ref. 26 with permission from Royal Society of Chemistry, copyright 2019. | ||
On the other hand, palladium complexes have been explored to a greater extent.26,67 Freese et al. reported the preparation of palladium complexes under conditions similar to those used for synthesizing gold complexes, involving the direct reaction of SI carbenes at low temperature. These SI-Pd complexes (see Scheme 19) demonstrated notable stability and proved to be effective catalysts for Sonogashira–Hagihara reactions, with some complexes (51 and 52) also showing catalytic activity in Suzuki–Miyaura reactions.
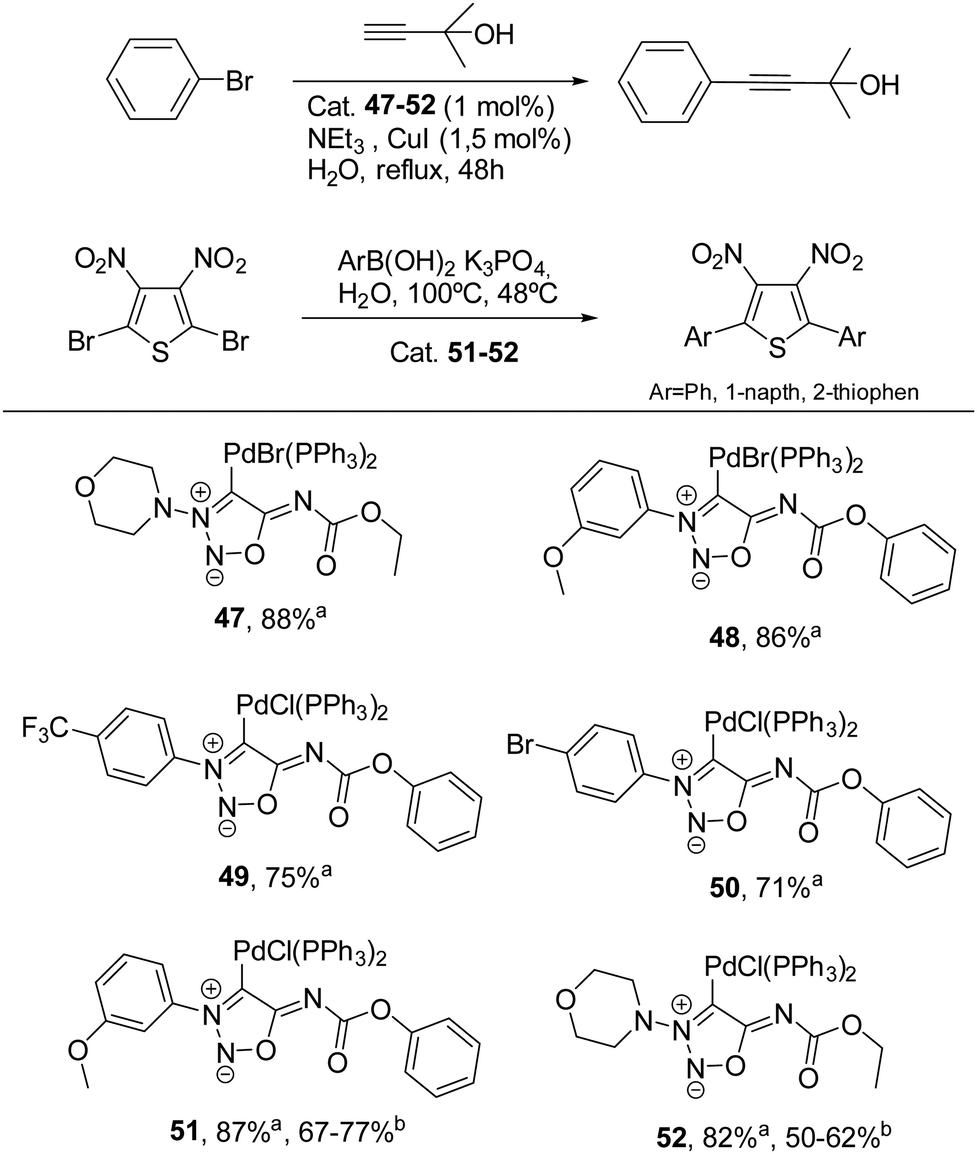 | ||
| Scheme 19 Palladium sydnonimine complexes tested in Sonogashira–Hagihara and Suzuki–Miyaura reactions. aYields for Sonogashira–Hagihara coupling. bYields for Suzuki–Miyaura coupling. | ||
Overall, NHC metal complexes using SI are scarcely reported in the literature and their applications have largely been confined to catalysis. To the best of our knowledge, no biological studies have been carried out or even proposed for these interesting complexes. This gap suggests the potential for discovering interesting applications of a group of NHC metal complexes that have not yet been explored.
5 Sydnonimines as dipoles for cycloaddition reactions
Like sydnones, sydnonimines are masked azomethine imine dipoles able to react in 1,3-dipolar cycloaddition reactions (Scheme 20).68 This property was first reported by B. G. Yashunskii in the 60s using acrylonitrile as dipolarophile and a few years later by K. T. Potts and coworkers who described the formation of a pyrazole product upon reaction with dimethyl acetylenedicarboxylate under refluxing xylene.69,70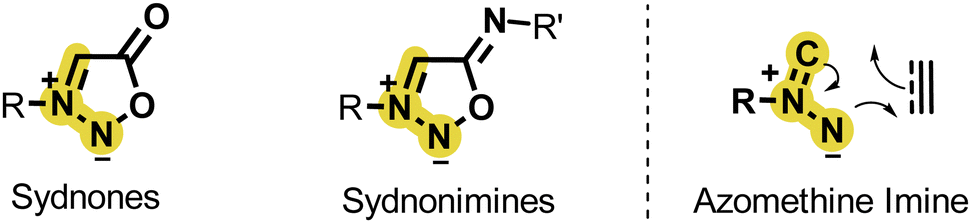 | ||
| Scheme 20 Sydnones and sydnonimines are masked azomethine dipoles. | ||
Surprisingly, following these pioneer two articles, no other paper exploited this reaction until our work published in 2017.71 Among 25 mesoionic compounds screened for their reactivity with cyclooctynes, our group identified sydnonimines (SI) as suitable substrates for strained-promoted cycloaddition reactions. Notably, the reaction occurs at room temperature, yielding a pyrazole product quantitatively through the click reaction between the cyclooctyne and the azomethine imine dipole, accompanied by the release of an isocyanate (Fig. 4). The mechanism involves a (3+2) cycloaddition step, forming a bicyclic intermediate that undergoes a spontaneous retro-Diels–Alder reaction. This reaction, termed strain-promoted sydnonImine cycloaddition (SPSIC), maintains its efficiency in biological fluids such as cell lysate and blood plasma. Consequently, it emerges as a new bioorthogonal click-and-release reaction with potential applications in chemical biology.72 Research efforts aimed at improving the reaction kinetics of SPSIC have underscored the significant influence of substrate structure on the reaction rate constants. Notably, the reaction rate can be enhanced by two orders of magnitude by increasing the strain of the cyclooctyne (Fig. 4). As expected, TMTH (tetramethylthiocycloheptyne) exhibited high reactivity towards SI, although its instability might be an issue. DBCO (dibenzocyclooctyne), offering a balanced compromise between reactivity and stability, is often the preferred cyclooctyne in SPSIC reactions. In addition to cyclooctynes, the structure of SI also significantly influences the SPSIC kinetics. Three positions on the SI scaffold can be modified: positions 3-N, 4-C, and 6-N. Alterations at all three positions have a tremendous impact on the reaction speed, highlighting the importance of optimizing SI structures to enhance the efficiency of the SPSIC reaction (Fig. 4).
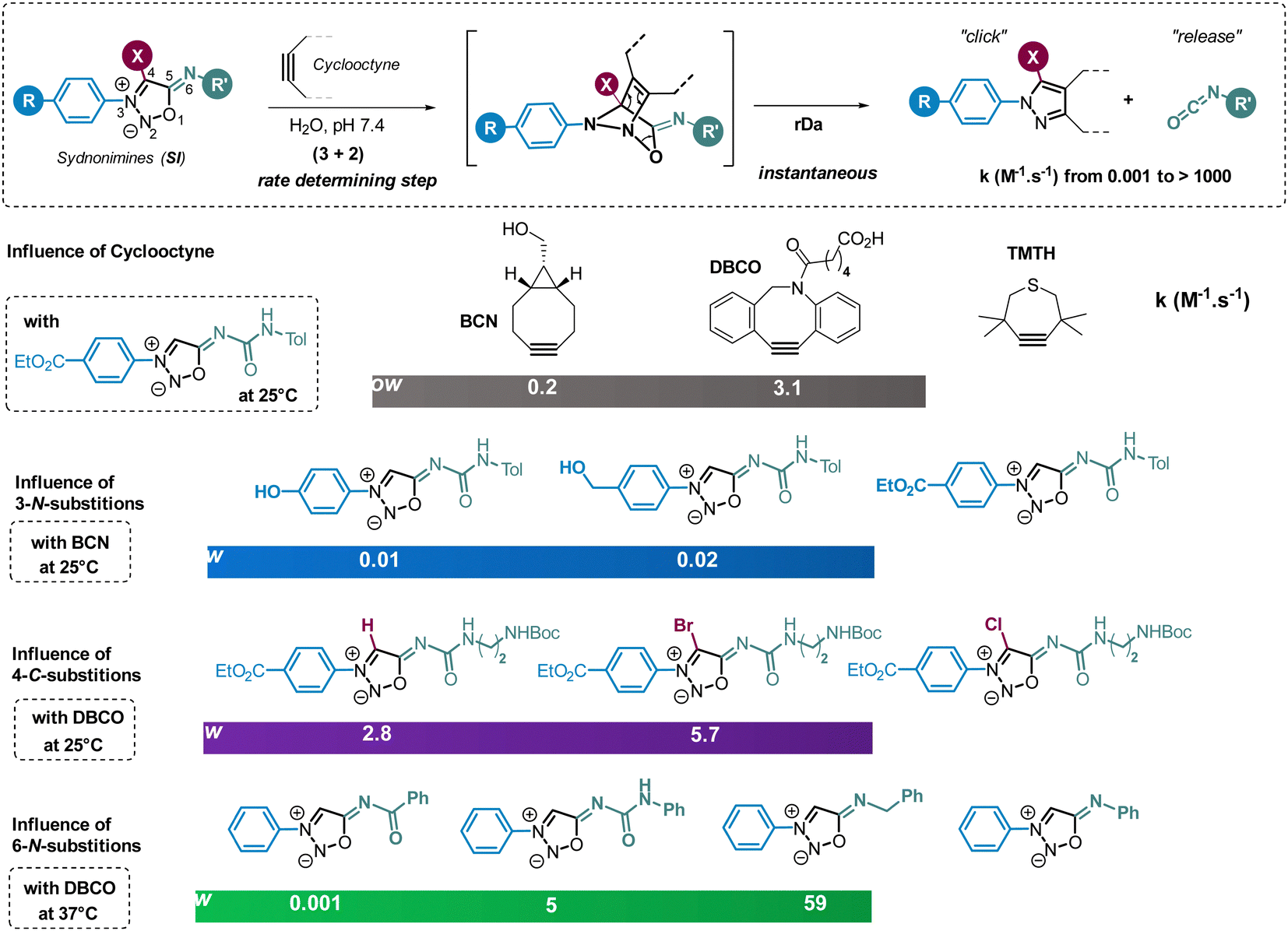 | ||
| Fig. 4 Strained-promoted sydnonimine cycloaddition (SPSIC). Selected examples illustrating the influence of cyclooctyne and SI structures on the SPSIC kinetics. | ||
Electronic effects in position 3-N and 6-N have a particularly strong impact on the reactivity of SI. Electron-withdrawing groups in position 3-N and electron-donating groups in position 6-N favor the active azomethine imine mesomeric form of SI and therefore have a beneficial impact on reaction kinetics.73 Like sydnones,74 the presence of a halogen atom in position 4-C also improves significantly the speed of the SPSIC reaction.75 Among the numerous SI studied, 3-N, 6-N-diaryl sydnonimines appeared the most reactive under physiological conditions affording rate constants from 18 to 458 M−1 s−1 depending on aromatic substitutions.75 6-N-Alkyl sydnonimines react with lower kinetics due to higher pKa of the amine in position 6. For instance only 4% of the reactive mesosionic form of 3-N-phenyl, 6-N-benzyl sydnonimine are present at physiological pH which explains why the rate constant is only of 59 M−1 s−1. On the contrary 85% of this compound is deprotonated at pH 9.5 inducing an enhancement of the SPSIC rate constant up to 1220 M−1 s−1 (Fig. 5).
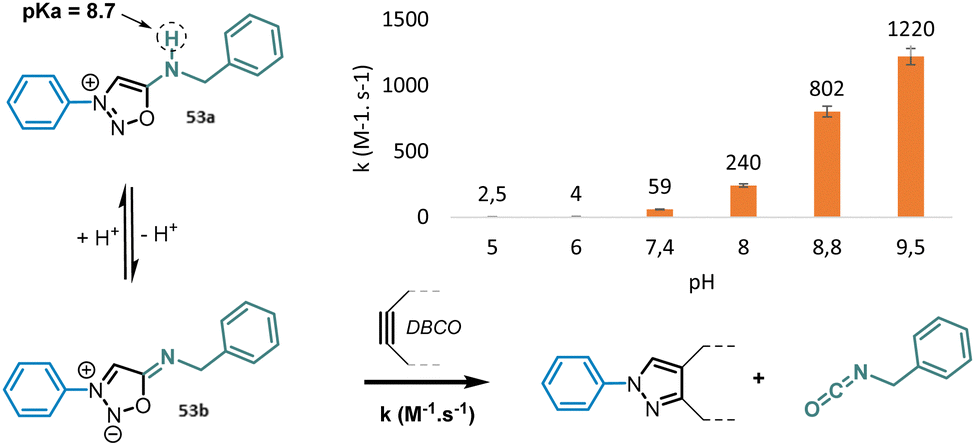 | ||
| Fig. 5 Influence of pH on SPSIC reaction with 6-N-alkyl sydnonimines. | ||
In addition to strained-promoted cycloadditions, 3-N,6-N-diaryl sydnonimines can also react with terminal alkynes under copper-catalysis.76 In this case also the efficiency on this reaction is highly dependent of the SI structure. Although all SI bearing electron-withdrawing groups in position 6 are almost inactive, 6-N-aryl SI display very interesting reactivity toward Cu-catalyzed cycloaddition with alkynes. Only 1% of copper salts are required to catalyze efficiently the reaction which makes sydnonimines even more reactive than sydnones (Scheme 21). The reaction is preferably conducted in tris-buffer in order to quench the released isocyanate. Under these conditions, multi-functionalized substrates can be utilized, resulting in the formation of complex pyrazoles with good yields.
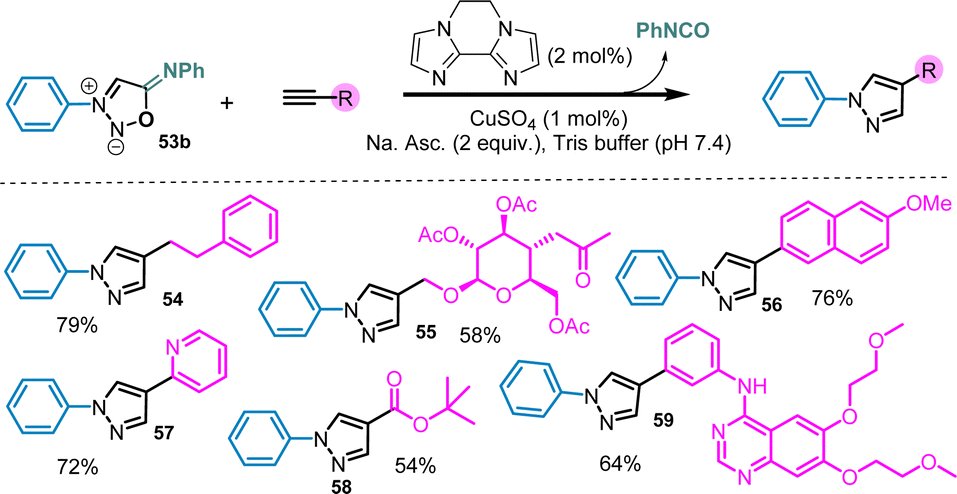 | ||
| Scheme 21 Cu-catalyzed sydnonimine–alkyne cycloaddition. | ||
To take profit of the isocyanate generated from SI substrates during the catalytic process, a two-steps cascade reaction has been developed. Starting from amine-containing alkynes, the formed pyrazoles can then react with the released isocyanate leading to the corresponding urea. This cascade reaction was observed with various amine-containing alkynes affording the expected pyrazole-urea products in moderate to good yields (Scheme 22).
 | ||
| Scheme 22 Cascade reaction. | ||
6 Application of sydnonimines in chemical biology
Because the SPSIC reaction was found to be highly efficient and relatively fast in biological fluids, its use in chemical biology has been the subject of a significant number of research. One potential drawback of the reaction relies on the release of an isocyanate which may react with biological nucleophiles, lowering the bioorthogonality of the process. To address this issue, SI with carbonyl or sulfonyl groups at the 6-position were initially employed for applications in chemical biology. These compounds release acyl- or sulfonyl-isocyanates, which spontaneously hydrolyze in water to form unreactive amide, urea, or sulfonamide products. An added bonus of these specific SI relies on their non-reactivity toward the Cu(I) catalysis.76 This property has been used to develop bifunctional spacers able to fish and purify proteins from complex mixtures. For example sydnonimine 66 was successfully used to carry out an activity-based purification method of β-lactam targets.77 First, a model L,D-transpeptidase was incubated with an alkynylated carbapenem substrate resulting in the formation of an acylenzyme. The alkyne group of the enzyme-bound carbapenem was then exploited for CuAAC attachment with probe 66. After binding to streptavidin magnetic beads and purification, the SPSIC reaction with a DBCO-TAMRA derivative enabled the release and fluorescent labeling of the captured carbapenem target from the streptavidin support (Fig. 6).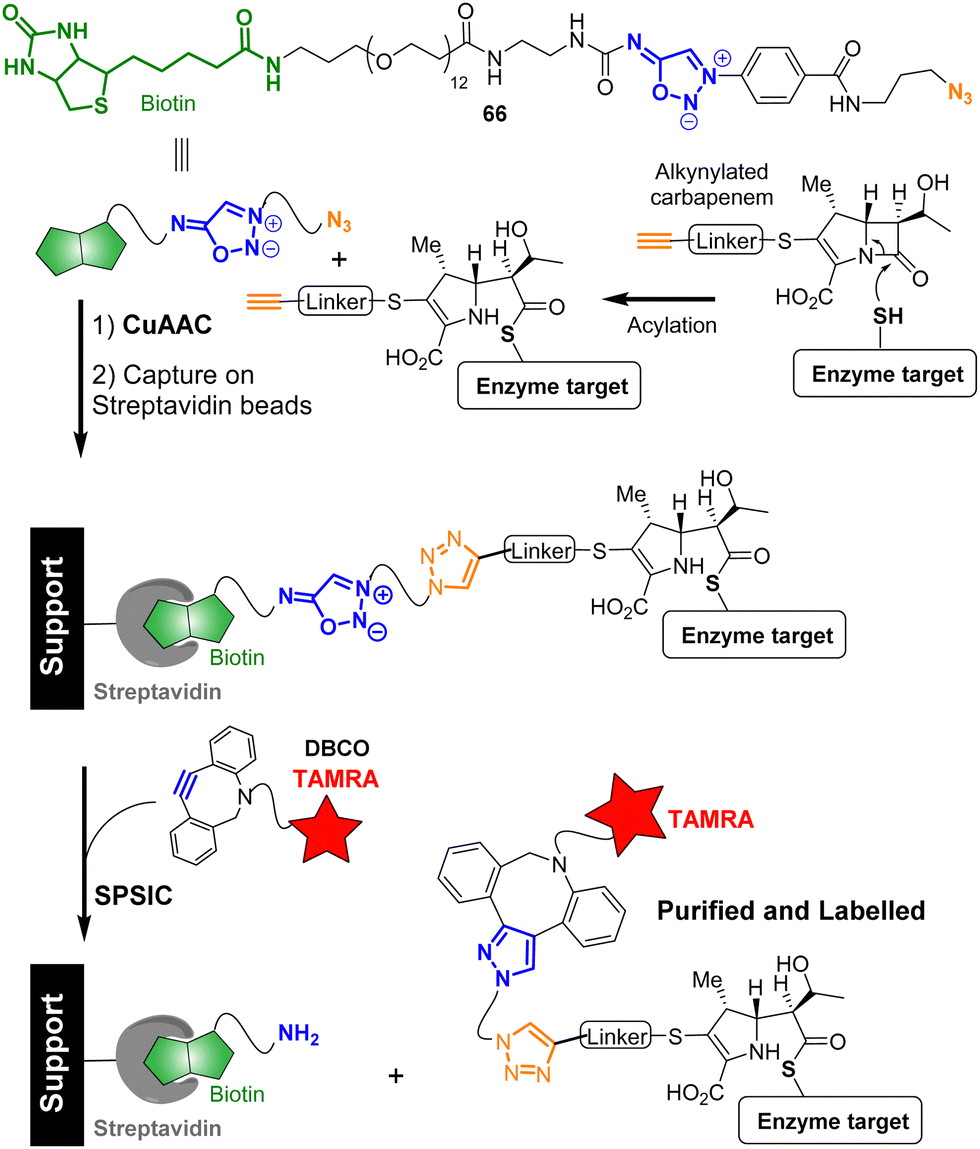 | ||
| Fig. 6 SI as cleavable linkers for capture and release of β-lactam targets. | ||
The use of SI as bioorthogonal cleavable linker has been further explored in the field of bioconjugation. An automated technology has been notably developed to access to antibody conjugates with the precise attachment of one single payload per antibody (degree of conjugation DoC = 1).78 In this process, excess of the native antibody is iteratively engaged into cycles of low-conversion chemical modification with the sydnonimine derivative 67, allowing the monobiotinylation of the antibody, and subsequently immobilized onto a streptavidin column (Fig. 7). Once captured on the solid support, the antibody is detached after addition of a cyclooctyne functionalized by the drug of interest. The desired monoconjugated antibody (i.e., DoC = 1) is then simply collected at the column outlet. First designed to enable mg scale syntheses of stoichiometric antibody–payload conjugates, the experimental setup has been recently adjusted to allow the manipulation of μg quantities of precious antibodies.79
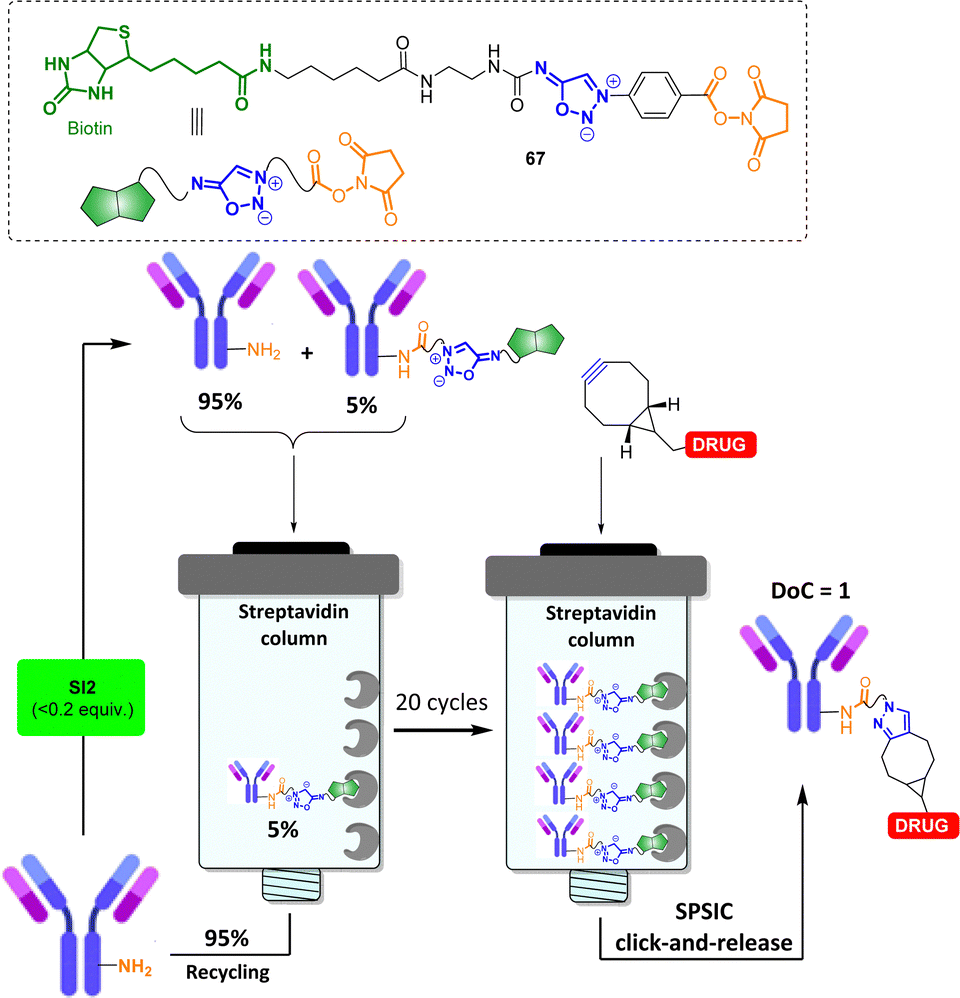 | ||
| Fig. 7 Use of SPSIC to control the degree of conjugation (DoC) of antibody conjugates. | ||
Like sydnones,80SI can efficiently quench several fluorophores, making them valuable tools for developing fluorogenic probes. In 2020, we leveraged this property to design probes capable of generating two distinct fluorescent signals in biological media following the SPSIC reaction (Fig. 8).81 Among the different probes, compound 68 was found the most promising. Upon reaction with DBCO, it simultaneously released two fluorescent species: naphthalimide-urea and pyrazolo-pyridinium. The reaction exhibited sufficiently fast rate constants, allowing successful application in living cells. The probe was designed to be doubly fluorogenic: upon click reaction, the resulting pyrazole exhibits greater fluorescence than 68 due to intramolecular charge transfer (ICT). Additionally, the quenching of the naphthalimide via photoinduced electron transfer (PeT) is suppressed, leading to a significant fluorescence enhancement of the released urea. Confocal microscopy imaging confirmed the efficiency of the dual turn-on SPSIC reaction within cells, further validating the biocompatibility of the approach. Two years later, the group of Liang pursued an alternative strategy to develop even more powerful turn-on probes.82 The design utilized quenchers attached to the 6-N position of the SI core to quench a fluorophore linked at the 3-N position. Among the probes, 69 bearing a Cy5 flurophore and a BHQ quencher was found to display impressive optical properties.
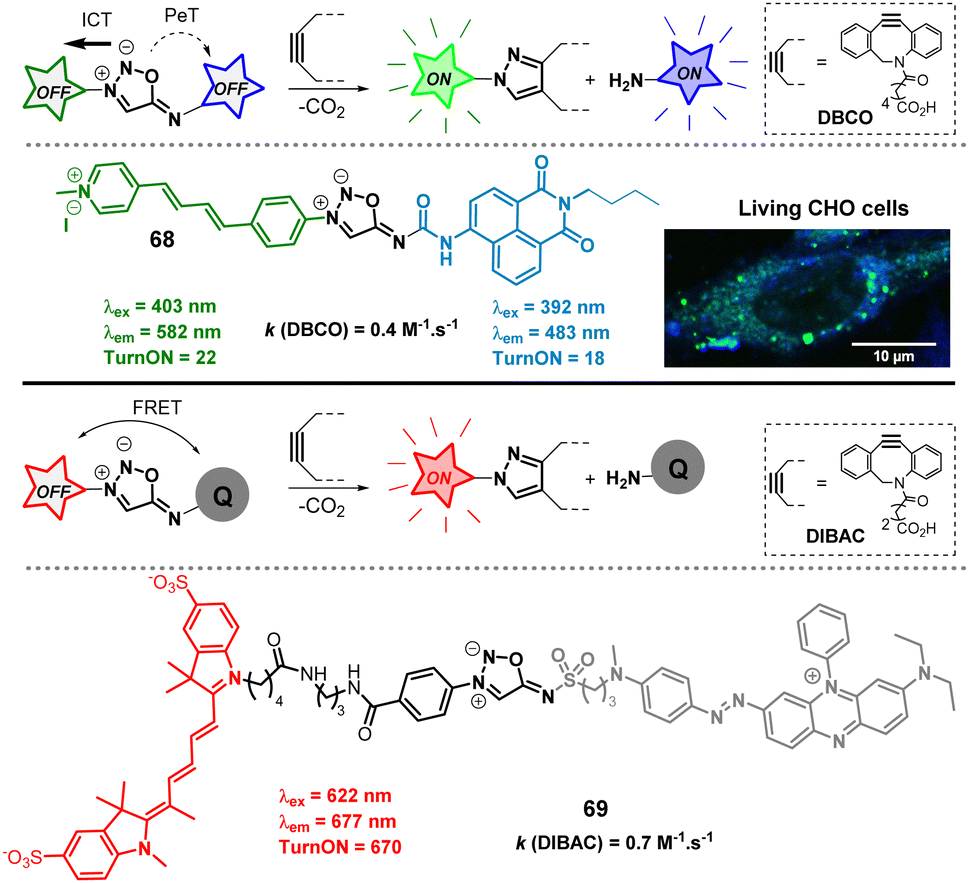 | ||
| Fig. 8 Fluorogenic sydnonimines. ITC = intramolecular charge transfer, PeT = photoinduced electron tranfer; FRET = fluorescence resonance energy transfer. Adapted from ref. 81 with permission from Royal Society of Chemistry, copyright 2020. | ||
In 2023, our group described a new class of SI able to release alkyl or aryl-isocyanates in biological media.75 This type of isocyanates are stable enough to react with bionucleophiles, including proteins. To exploit this property, several probes were developed in order to release fluorescent isocyanates inside cells (Fig. 9). Acting as caged isocyanates, these probes have the potential to label permanently biological materials within the cells which is interesting for cell imaging.83 This has been confirmed when probe 70 was incubated with two cell models HEK293 (human embryonic kidney cells) and A549 (lung cancer cells) and further treated with DBCO. After cell lysis, SDS-PAGE analysis was performed highlighting multiple fluorescent bands on the electrophoresis gel indicating that numerous proteins inside the cells have been efficiently labeled.
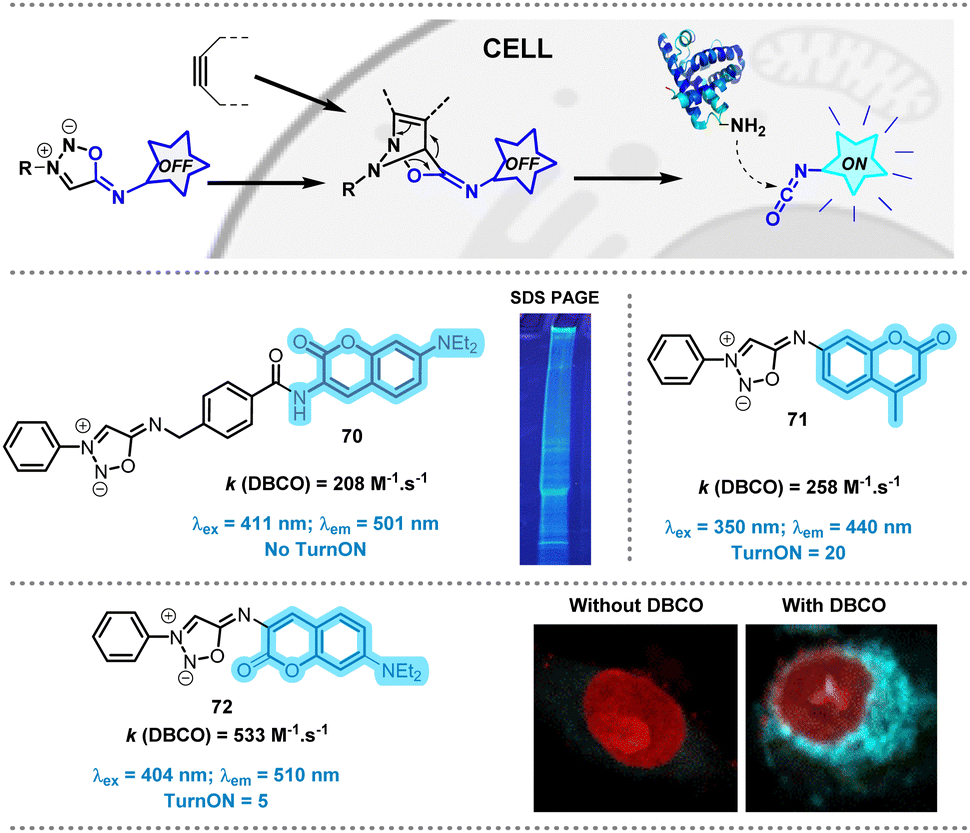 | ||
| Fig. 9 Bioorthogonal release of fluorescent isocyanates in cell. Adapted from ref. 84 with permission from Royal Society of Chemistry, copyright 2024. | ||
Although probe 70 displayed high reactivity toward DBCO, it is not fluorogenic, making the monitoring of the SPSIC reaction in living cells difficult. In order to palliate this drawback, probes 71–72 bearing the coumarine moiety directly branched at position N-6 of the SI core were designed. Both displayed impressive kinetics and exploitable turn-on when mixed with DBCO. 72 was found the most suitable for cell imaging: its high reactivity allowed complete release of coumarin-isocyanate in 30 min inside A549 cells (Fig. 9).84 The fluorescent labelling was resistant to several successive washing steps suggesting irreversible labelling of the cells.
Prodrug strategies utilizing bioorthogonal bond cleavage chemistry have emerged as a promising therapeutic approach, offering significant advantages over traditional prodrug methods, such as a precise spatiotemporal control of drug activation.85 In this context, SI have been used to mask amide, sulfonamide and urea-containing drugs. Liang et al. pioneered this approach by developing a SI capable of releasing the sulfonamide drug Celecoxib through bioorthogonal reaction with DBCO (Fig. 10).86 Following this work, the same group used a similar SI-based prodrug to deliver Celecoxib into the mitochondria of HeLa and HepG2 cancer cells. The study indicated that celecoxib-induced cancer cell death may not involve mitochondria-related pathway.87 One limitation of these SI is their slow kinetics in drug release. To address this, our group developed a method to chlorinate the 4-C position of SI enhancing their reactivity as dipole.88 The resulting chloro-sydnonimines demonstrated much higher reactivity, enabling faster release of drugs containing a terminal sulfonamide, urea and even amide functions. The latter reacted smoothly, with rate constants k > 1 M−1 s−1, whereas their non-chlorinated counterparts were nearly unreactive, highlighting the dramatic beneficial influence of the chlorine atom on SI reactivity.
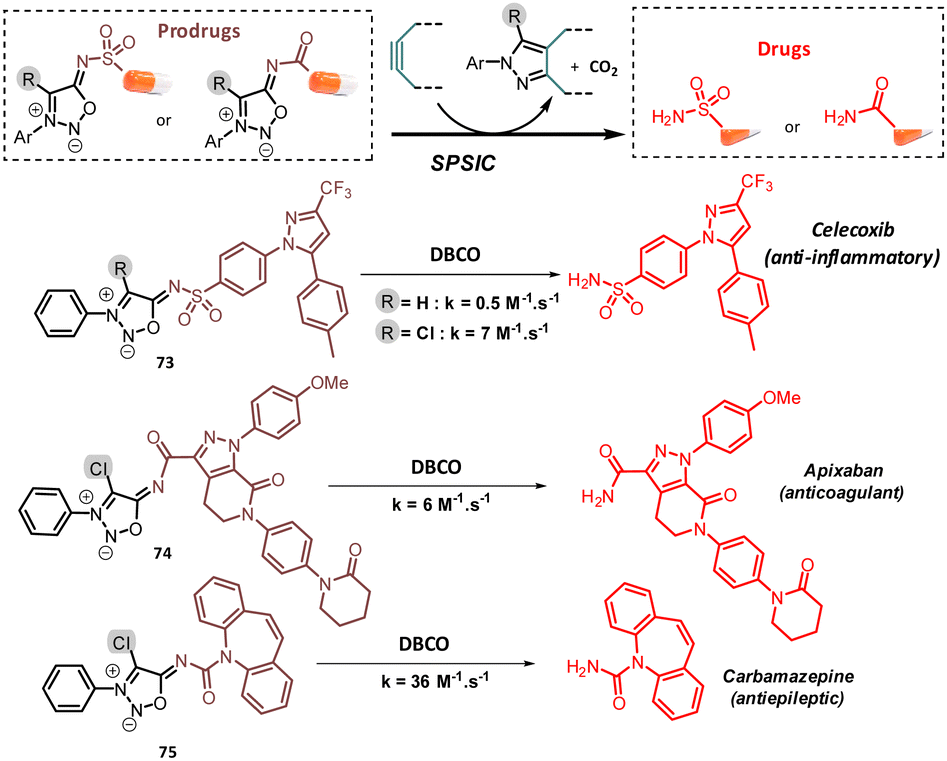 | ||
| Fig. 10 Drug release from SI-prodrugs. | ||
Although several bioorthogonal reactions are efficient and useful when performed in vitro in biological media and even inside cells, only a few were successfully applied in vivo. In addition to bioorthogonality, biostability, non-toxicity, favorable pharmacokinetics and high reaction kinetics are essential prerequisites for in vivo applications. The SPSIC is one of the few reactions that has demonstrated effectiveness in animal models. In 2019, our group provided the first proof-of-concept for the utility of the SPSIC reaction in vivo. We developed a dual tumor-targeting strategy using micelles constructed from the amphiphile 76 containing a SI moiety, which separates the PEG head from the lipophilic chain (Fig. 11).89
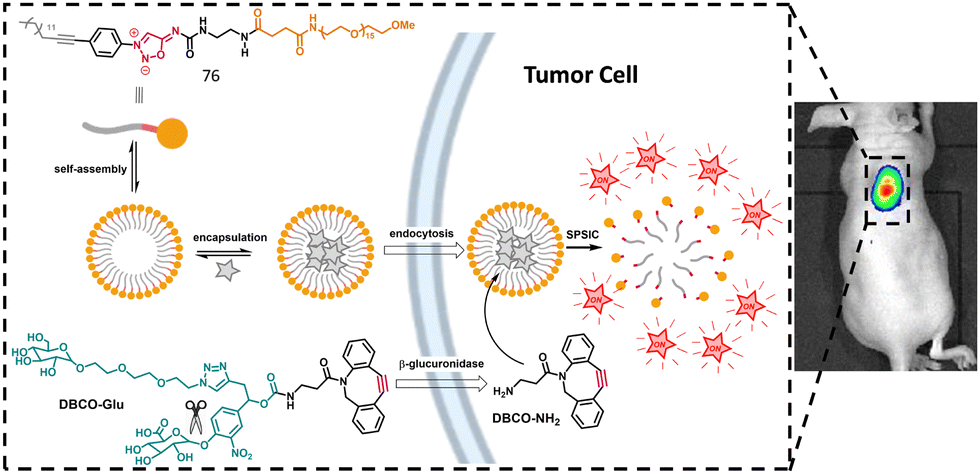 | ||
| Fig. 11 Drug release from cleavable micelles. Adapted from ref. 89 with permission from Wiley, copyright 2019. | ||
Addition of lipophilic cyclooctynes to these micelles induces a high local concentration of both cyclooctyne and 76 inside the micelle core leading to fast cleavage of the amphiphile and micelle disassembly in only a few seconds. To ensure that micelle decomposition and subsequent content release (e.g., fluorophore or drug) occur specifically at the tumor site, we employed a hydrophilic β-glucuronidase-responsive pro-activator (DBCO-Glu). Because of its hydrophilicity, this cyclooctyne is inactive toward the micelle. The activation of DBCO-Glu at the tumor is triggered by β-glucuronidase, an enzyme overexpressed by cancer cells, inducing the detachment of the hydrophilic part of DBCO-Glu to form active DBCO-NH2. Once activated, the bioorthogonal reaction proceeds, resulting in micelle destruction. This strategy was first demonstrated in mice using micelles loaded with a dye. 24 h after micelle injection, tumor accumulation was observed, and the pro-activator DBCO-Glu was subsequently injected, causing the micelles to disassemble and release the dye into malignant tissues. Few years later, this strategy was successfully applied to the release of the anticancer drug Entinostat within cancer cells. An interesting enhancement of the biological activity of the drug was observed using this strategy.90 Very recently, micelles constructed with amphiphile 77 allowed the synthesis of a bioactive urea in cancer cells (Fig. 12).91 Upon SPSIC reaction, 77 releases an isocyanate which reacts rapidly with an encapsulated amine yielding the corresponding urea in the hydrophobic core of the micelles. Application of this strategy was made with the synthesis of the FDA-approved anticancer drug Sorafenib inside living cells. Interestingly, this work proved that bioorthogonal chemistry coupled with confinement could make non-bioorthogonal transformations possible inside living cells.
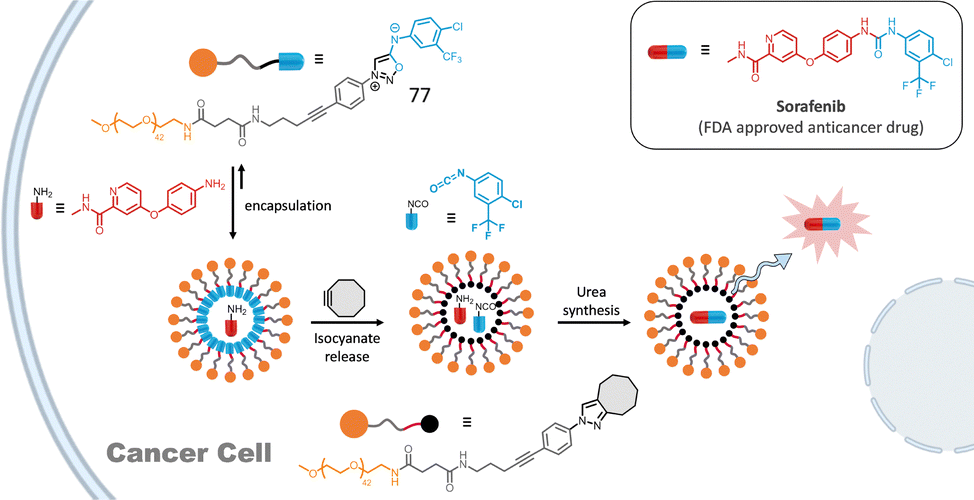 | ||
| Fig. 12 SPSIC-promoted drug construction inside cancer cells. Adapted from ref. 91 with permission from Wiley, copyright 2025. | ||
Recently, Liu and Zhang demonstrated the use of SPSIC for in vivo cancer photothermal therapy (PTT).92 The authors described a strategy based on the injection of two gold nanoparticles: AuNP-1 functionalized by PEGs, cyclooctynes and the tumor-targeting peptide RGD, and AuNP-2 bearing the prodrug sydnonimine-lonidamine 78 (Fig. 13). AuNP-1 were first intravenously injected to mice bearing the breast tumor model 4T1, leading to good tumor-specific enrichment and retention. Then, AuNP-2 were intravenously injected. Once both AuNPs accumulated at the tumor, they react together via SPSIC inducing the formation of Au nanoparticles aggregates and the release of Lonidamine, a drug known to render cancer cells more sensitive to PTT. Mice were then irradiated at 808 nm for PTT treatment. The aggregated AuNPs and the release of Lonidamine showed a synergistic effect leading to significant enhancement of PTT effect and a robust tumor suppression.
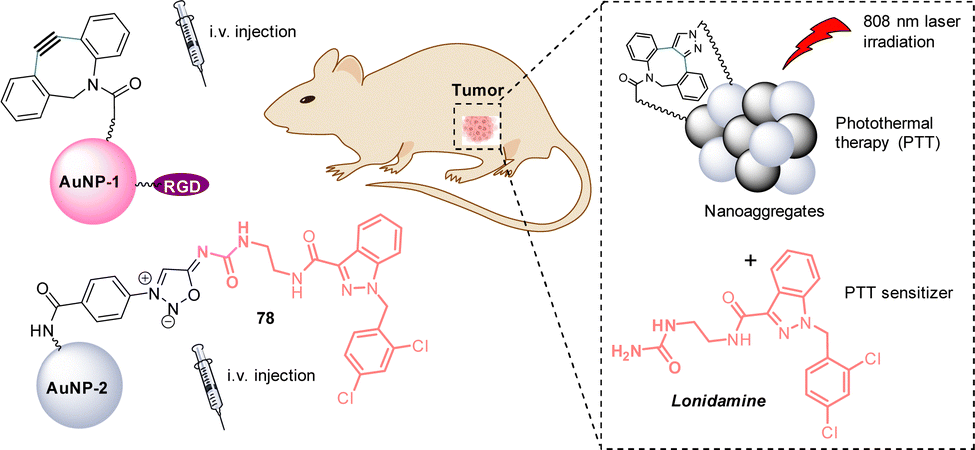 | ||
| Fig. 13 Use of the SPSIC reaction for enhanced photothermal therapy. | ||
Recently, our group described a multiplexed diagnostic approach based on MS-tagged antibodies cleavable by the SPSIC reaction.93 The technology is based on the use MS-tag isotopologues attached to anti-cancer antibodies through a SI linker (Fig. 14). Four FDA-approved therapeutic antibodies raised against biomarkers located at the surface of cancer cells: trastuzumab (TRZ, anti-Her2), Cetuximab (CTX, anti-EGFR), Durvalumab (DUR, anti-PDL1) and Bevacizumab (BVZ, anti-VEGF) were labelled with tris(2,4,6,-trimethoxyphenyl)-phosphonium (TMPP) tags containing a specific number of deuterium atoms. The mixture of four antibodies was then incubated on cancer cells and after washing, a DBCO-TAMRA derivative was added resulting in both fluorescent labeling of the cells and the release of the TMPP tags which can then be quantified by one single mass spectrometry analysis.
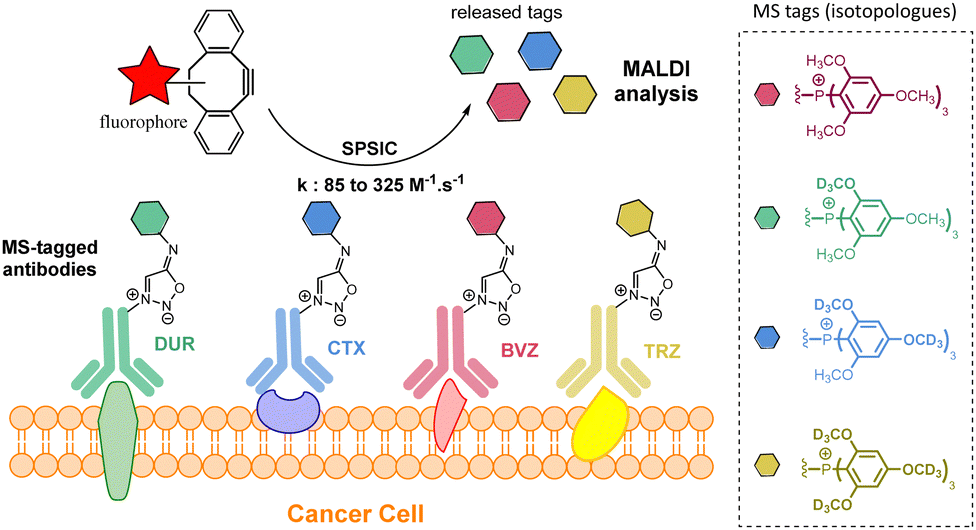 | ||
| Fig. 14 Multiplexed detection of cell membrane receptors using SPSIC. Adapted from ref. 93 with permission from Royal Society of Chemistry, copyright 2024. | ||
In a proof of concept studies, we then demonstrated the potential of this approach for cancer immunoprofiling in tissues and even in vivo. The cocktail of the four antibodies was intravenously injected to mice bearing A431 tumors. Three days later, an excess of DBCO was administered intratumorally. The released TMPP tags were subsequently quantified in the urine of the mice, showing the overexpression of the EGFR, as expected for an A431 tumor model.
Although still at an early of proof-of-concept stage, this work suggests that in vivo tumor characterization may be feasible, opening up promising avenues for diagnostic applications.
Conclusions
After extensive development in the 1970s as NO donors, which led to the commercialization of several drugs for various diseases, sydnonimines (SI) were largely overlooked for nearly 50 years. As a result, only a few synthetic methods exist, and significant synthetic efforts will be necessary to expand the diversity and complexity of SI structures. A key challenge in the coming years will be to develop robust and efficient synthetic routes for preparing libraries of SI, enabling their screening for various biological applications. The primary biological mechanism of action of many SI is associated with their property to release NO. Future research could explore alternative biological activities. Additionally, the polar heterocyclic nature of SI makes it a promising structural analogue of five-membered-ring drugs, offering valuable potential in medicinal chemistry. The recent discovery of the ability of SI to undergo efficient click-and-release reactions in biological media has renewed interest in these compounds. SI now offer promising possibilities for cleaving molecules, releasing compounds from antibodies, and breaking down nanoparticles in vivo, inspiring ambitious future applications in both therapeutic and diagnostic fields. With rate constants reaching up to 103 M−1 sec−1 and no side reactions, leading to 100% release, the SPSIC reaction can be considered as a valuable complementary tool to the widely used inverse electron-demand Diels–Alder (iEDDA) click-and-release reaction involving tetrazines and carbamate-trans-cyclooctenes derivatives.94 While SPSIC has been explored in in vivo applications, it has not yet achieved the same level of success as iEDDA, which have led to the development of a therapeutic treatment for cancer in humans,95 currently in phase 2. The good stability of most sydnonimines in biological media, a weak point of many tetrazines, could be a game changer. In this regard, future in vivo stability studies of these mesoionic compounds will be crucial to further assess their therapeutic potential.Data availability
No primary research results, software or code have been included and no new data were generated or analyzed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
All authors are participant in the EU DN ISOBIOTICS Consortium. The ISOBIOTICS project has received funding from the European Union's Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement no. 101072780.Notes and references
- W. Baker, W. D. Ollis and V. D. Poole, J. Chem. Soc., 1949, 307–314 RSC
.
- K. Porte, M. Riomet, C. Figliola, D. Audisio and F. Taran, Chem. Rev., 2021, 121, 6718–6743 CrossRef CAS PubMed
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., 2016, B72, 171–179 Search PubMed
- P. Brookes and J. Walker, J. Chem. Soc., 1957, 4409–4416 Search PubMed
- Y. Asahi, K. Shinozaki and M. Nagaoka, Chem. Pharm. Bull., 1971, 19, 1079–1088 Search PubMed
- T. Freese, A.-L. Lücke, J. C. Namyslo, M. Nieger and A. Schmidt, Eur. J. Org. Chem., 2018, 1646–1654 Search PubMed
- H. Kato, M. Hashimoto and M. Ohta, Nippon Kagaku Zasshi, 1957, 78, 707 Search PubMed
- A. D. Shuvaev, E. S. Zhilin and L. L. Fershtat, Synthesis, 2023, 1863–1874 Search PubMed
- N. Pétry, F. Luttringer, X. Bantreil and F. Lamaty, Faraday Discuss., 2023, 241, 114–127 Search PubMed
- V. G. Yashunskii and L. E. Kholodov, Russ. Chem. Rev., 1980, 49, 28–45 Search PubMed
- H. U. Daeniker and J. Druey, Helv. Chim. Acta, 1962, 45, 2462–2465 Search PubMed
- A. S. Samarskaya, I. A. Cherepanov, I. A. Godovikov, A. O. Dmitrienko, S. K. Moiseev, V. N. Kalinin and E. Hey-Hawkins, Tetrahedron, 2018, 74, 2693–2702 CrossRef CAS
- L. Soulère, P. Hoffmanna and F. Bringaud, J. Heterocycl. Chem., 2003, 40, 943–947 Search PubMed
- A. Nortcliffe, N. P. Botting and D. O’Hagan, Org. Biomol. Chem., 2013, 11, 4657–4671 RSC
- A. Nortcliffe, I. N. Fleming, N. P. Botting and D. O’Hagan, Tetrahedron, 2014, 70, 8343–8347 CrossRef CAS
- M. Riomet, K. Porte, L. Madegard, P. Thuéry, D. Audisio and F. Taran, Org. Lett., 2020, 22, 2403–2408 Search PubMed
- C. Kitoun, M. Fonvielle, N. Sakkas, M. Lefresne, F. Djago, Q. Blancart Remaury, P. Poinot, M. Arthur, M. Etheve-Quelquejeu and L. Iannazzo, Org. Lett., 2020, 22, 8034–8038 CrossRef CAS PubMed
- M. Ribéraud, K. Porte, A. Chevalier, L. Madegard, A. Rachet, A. Delaunay-Moisan, F. Vinchon, P. Thuéry, G. Chiappetta, P. A. Champagne, G. Pieters, D. Audisio and F. Taran, J. Am. Chem. Soc., 2023, 145, 2219–2229 CrossRef
- J. Baudet, E. Lesur, M. Ribéraud, A. Chevalier, T. D’Anfray, P. Thuéry, D. Audisio and F. Taran, Chem. Commun., 2024, 60, 3657–3660 RSC
- S. Mummel, F. Lederle, E. G. Hübner, J. C. Namyslo, M. Nieger and A. Schmidt, Angew. Chem., Int. Ed., 2021, 60, 18882–18887 CrossRef CAS PubMed
- J. M. Ruxer, J. Mauger, D. Bénard and C. Lachoux, J. Heterocycl. Chem., 1995, 32, 643–654 CrossRef CAS
- I. A. Cherepanov and V. N. Kalinin, Mendeleev Commun., 2000, 10, 181–182 Search PubMed
- N. V. Kalganova, A. F. Smolyakov, S. K. Moiseev, M. A. Cherevatskaya and I. A. Cherepanov, Russ. Chem. Bull., 2023, 72, 1688–1700 CrossRef CAS
- I. A. Cherepanov, L. H. Kusaeva, I. A. Godovikov and V. N. Kalinin, Russ. Chem. Bull., 2009, 58, 2474–2477 CrossRef CAS
- I. A. Cherepanov, E. D. Savin, N. G. Frolova, M. O. Shishkova, I. A. Godovikov, K. Yu Suponitsky, K. A. Lyssenko and V. N. Kalinin, Mendeleev Commun., 2009, 19, 320–321 Search PubMed
- T. Freese, J. C. Namyslo, M. Nieger and A. Schmidt, RSC Adv., 2019, 9, 4781–4788 Search PubMed
- I. A. Cherepanov, S. N. Lebedev, A. S. Samarskaya, I. A. Godovikov, Y. V. Nelyubina and V. N. Kalinin, Mendeleev Commun., 2009, 19, 322–323 CrossRef CAS
- I. A. Cherepanov, A. S. Samarskaya, R. G. Nosov, I. A. Godovikov, Y. V. Nelyubina and V. N. Kalinin, Mendeleev Commun., 2014, 24, 386–387 CrossRef CAS
- V. N. Kalinin, S. N. Lebedev, I. A. Cherepanov, I. A. Godovikov, K. A. Lyssenko and E. Hey-Hawkins, Polyhedron, 2009, 28, 2411–2417 CrossRef CAS
- M. Goetz, K. Grozinger and J. T. Oliver, J. Med. Chem., 1973, 16, 671–673 Search PubMed
- M. Feng, L. Madegard, M. Riomet, M. Louis, P. A. Champagne, G. Pieters, D. Audisio and F. Taran, Chem. Commun., 2022, 58, 8500–8503 RSC
- M. Riomet, E. Decuypere, K. Porte, S. Bernard, L. Plougastel, S. Kolodych, D. Audisio and F. Taran, Chem. – Eur. J., 2018, 24, 8535–8541 CrossRef CAS PubMed
- H. Liu, D. Audisio, L. Plougastel, E. Decuypere, D.-A. Buisson, O. Koniev, S. Kolodych, A. Wagner, M. Elhabiri, A. Krzyczmonik, S. Forsback, O. Solin, V. Gouverneur and F. Taran, Angew. Chem., Int. Ed., 2016, 55, 12073–12077 CrossRef CAS
- H. U. Daeniker and J. Druey, Helv. Chim. Acta, 1962, 45, 2426–2441 CrossRef CAS
- V. Z. Gorkin, V. G. Yashunskii, M. D. Mashkovskii, R. A. Al’tshuler, I. V. Verevkina and L. E. Kholodov, J. Med. Chem., 1971, 14, 1013–1015 CrossRef CAS PubMed
- A. Nortcliffe, A. G. Ekstrom, J. R. Black, J. A. Ross, F. K. Habib, N. P. Botting and D. O’Hagan, Bioorg. Med. Chem., 2014, 22, 756–761 CrossRef CAS PubMed
- Y. Asahi, K. Shinozaki and M. Nagaoka, Chem. Pharm. Bull., 1971, 19, 1079–1088 CrossRef CAS
- K. Kikuchi, M. Hirata, A. Nagaoka and Y. Aramaki, Jpn. J. Pharmacol., 1970, 20, 23–43 CrossRef CAS PubMed
- S. Tanayama, T. Fujita, Y. Shirakawa and Z. Suzuoki, Jpn. J. Pharmacol., 1970, 20, 413–423 CrossRef CAS PubMed
- H. Bohn and K. Schönafinger, J. Cardiovasc. Pharmacol., 1989, 14, S6 CrossRef CAS
- S. Acharya, P. Rogers, R. R. Krishnamoorthy, D. L. Stankowska, H. V. R. Dias and T. Yorio, Bioorg. Med. Chem. Lett., 2016, 26, 1490–1494 CrossRef CAS
- D. L. Stankowska, A. Dibas, L. Li, W. Zhang, V. R. Krishnamoorthy, S. H. Chavala, T. P. Nguyen, T. Yorio, D. Z. Ellis and S. Acharya, Invest Ophthalmol. Visual Sci., 2019, 60, 3064–3073 CrossRef CAS
- K. Szőke, A. Czompa, I. Lekli, P. Szabados-Fürjesi, M. Herczeg, M. Csávás, A. Borbás, P. Herczegh and Á. Tósaki, Eur. J. Pharm. Sci., 2019, 131, 159–166 CrossRef
- N. Saffon-Merceron, C. Lherbet and P. Hoffmann, Molbank, 2024, M1886 CrossRef CAS
- C. L. Wainwright and P. A. Martorana, J. Cardiovasc. Pharmacol., 1993, 22(Suppl 7), S44–S50 CrossRef CAS
- K. Ivanova, M. Schaefer, C. Drummer and R. Gerzer, Eur. J. Pharmacol., Mol. Pharmacol. Sect., 1993, 244, 37–47 CrossRef CAS
- I. A. Cherepanov, E. V. Shevaldina, D. A. Lapshin, Y. Ya Spiridonov, V. A. Abubikerov and S. K. Moiseev, J. Organomet. Chem., 2021, 943, 121841 CrossRef CAS
- N. V. Kalganova, N. G. Frolova, I. A. Godovikov, A. F. Smol’yakov, D. A. Lapshin and I. A. Cherepanov, J. Organomet. Chem., 2024, 1005, 122975 CrossRef CAS
- Yu. Ya Spiridonov, I. A. Cherepanov, V. A. Abubikerov, I. Yu. Spiridonova, N. V. Kalganova, N. G. Frolova and S. K. Moiseev, Russ. Agric. Sci., 2022, 48, S63–S68 CrossRef
- E. V. Shevaldina, V. A. Tsyganov, N. V. Kalganova, A. F. Smol’yakov, N. G. Frolova and I. A. Cherepanov, Appl. Organomet. Chem., 2023, 37, e6981 CrossRef CAS
- N. V. Kalganova, N. G. Frolova, I. A. Godovikov, A. F. Smol’yakov, E. G. Kononova and I. A. Cherepanov, Appl. Organomet. Chem., 2024, 38, e7471 CrossRef CAS
- V. Bashkatova, A.-M. Mathieu-Kia, C. Durand and J. Penit-Soria, Ann. N. Y. Acad. Sci., 2002, 965, 180–192 CrossRef CAS
- J. M. Witkin, N. Savtchenko, M. Mashkovsky, M. Beekman, P. Munzar, M. Gasior, S. R. Goldberg, J. T. Ungard, J. Kim, T. Shippenberg and V. Chefer, J. Pharmacol. Exp. Ther., 1999, 288, 1298–1310 CrossRef CAS
- S. L. Erdö, B. Kiss and B. Rosdy, Pol. J. Pharmacol. Pharm., 1981, 33, 141–147 CrossRef
- S. Aggarwal, M. H. Cheng, J. M. Salvino, I. Bahar and O. V. Mortensen, Biomedicines, 2021, 9, 634 CrossRef CAS PubMed
- S. Huguenin, J. Fleury-Feith, L. Kheuang, M.-C. Jaurand, M. Bolla, J.-P. Riffaud, D. K. Chopin and F. Vacherot, Prostate, 2004, 61, 132–141 CrossRef CAS PubMed
- A. Nortcliffe, A. G. Ekstrom, J. R. Black, J. A. Ross, F. K. Habib, N. P. Botting and D. O’Hagan, Bioorg. Med. Chem., 2014, 22, 756–761 CrossRef CAS PubMed
- L. Soulère, P. Hoffmanna and F. Bringaud, J. Heterocycl. Chem., 2003, 40, 943–947 CrossRef
- D. Cordova, E. A. Benner, M. E. Schroeder, C. W. Holyoke, W. Zhang, T. F. Pahutski, R. M. Leighty, D. R. Vincent and J. C. Hamm, Insect Biochem. Mol. Biol., 2016, 74, 32–41 CrossRef CAS PubMed
- S. Du, X. Hu, M. Li, X. Jiang, X. Xu, J. Cheng and X. Qian, Bioorg. Med. Chem. Lett., 2021, 46, 128120 CrossRef CAS
- I. A. Cherepanov and V. N. Kalinin, Mendeleev Commun., 2000, 10, 181–182 CrossRef
- T. Freese, A.-L. Lücke, C. A. S. Schmidt, M. Polamo, M. Nieger, J. C. Namyslo and A. Schmidt, Tetrahedron, 2017, 73, 5350–5357 CrossRef CAS
- A. Liske, K. Verlinden, H. Buhl, K. Schaper and C. Ganter, Organometallics, 2013, 32, 5269–5272 CrossRef CAS
- D. E. Vita, M. A. Schiel, M. J. Lo Fiego and G. F. Silbestri, Organometallics, 2024, 43, 191–201 CrossRef CAS
- X. Bantreil, N. Pétry and F. Lamaty, Dalton Trans., 2019, 48, 15753–15761 Search PubMed
- I. A. Cherepanov and V. N. Kalinin, Mendeleev Commun., 2000, 10, 181–182 CrossRef
- A.-L. Lücke, L. Pruschinski, T. Freese and A. Schmidt, ARKIVOC, 2020, 2020, 94–104 Search PubMed
- R. J. Huisgen, Org. Chem., 1976, 41, 403 CrossRef CAS
- V. F. Vasil’eva and B. G. Yashunskii, Zh. Obshch. Khim., 1964, 34, 702 Search PubMed
- K. T. Potts, S. Usain and S. Husai, J. Chem. Soc. D, 1970, 1360–1361 RSC
- S. Bernard, D. Audisio, M. Riomet, S. Bregant, A. Sallustrau, L. Plougastel, E. Decuypere, S. Gabillet, R. A. Kumar, J. Elyian, M. N. Trinh, O. Koniev, A. Wagner, S. Kolodych and F. Taran, Angew. Chem., Int. Ed., 2017, 56, 15612–15616 CrossRef CAS PubMed
- X. Ji, Z. Pan, B. Yu, L. K. De La Cruz, Y. Zheng, B. Ke and B. Wang, Chem. Soc. Rev., 2019, 48, 1077–1094 RSC
- M. Riomet, E. Decuypere, K. Porte, S. Bernard, L. Plougastel, S. Kolodych, D. Audisio and F. Taran, Chem. – Eur. J., 2018, 34, 8535–8541 CrossRef
- L. Plougastel, O. Koniev, S. Specklin, E. Decuypere, C. Créminon, D.-A. Buisson, A. Wagner, S. Kolodych and F. Taran, Chem. Commun., 2014, 50, 9376–9378 RSC
- M. Ribéraud, K. Porte, A. Chevalier, L. Madegard, A. Rachet, A. Delaunay-Moisan, F. Vinchon, P. Thuéry, G. Chiappetta, P. Alexandre Champagne, G. Pieters, D. Audisio and F. Taran, J. Am. Chem. Soc., 2023, 145, 2219–2229 CrossRef
- M. Louis, G. Force, T. D’Anfray, E. Bourgeat, E. Romero, P. Thuéry, D. Audisio, A. Sallustrau and F. Taran, Chem. – Eur. J., 2024, e202302713 CrossRef CAS PubMed
- S. Saidjalolov, E. Braud, Z. Edoo, L. Iannazzo, F. Rusconi, M. Riomet, A. Sallustrau, F. Taran, M. Arthur, M. Fonvielle and M. Etheve-Quelquejeu, Chem. – Eur. J., 2021, 27, 7687–7695 CrossRef CAS
- I. Dovgan, A. Hentz, O. Koniev, A. Ehkirch, S. Hessmann, S. Ursuegui, S. Delacroix, M. Riomet, F. Taran, S. Cianférani, S. Kolodych and A. Wagner, Chem. Sci., 2020, 11, 1210–1215 RSC
- V. Lehot, O. Lidicky, J. Most, S. Erb, I. Dovgan, A. Osypenko, O. Koniev, S. Kolodych, L. Kotrchová, G. Chaubet, S. Cianférani, T. Etrych and A. Wagner, ACS Omega, 2023, 8, 40508–40516 CrossRef CAS PubMed
- E. Decuypère, M. Riomet, A. Sallustrau, S. Bregant, R. Thai, G. Pieters, G. Clavier, D. Audisio and F. Taran, Chem. Commun., 2018, 54, 10758–10761 Search PubMed
- M. Riomet, K. Porte, A. Wijkhuisen, D. Audisio and F. Taran, Chem. Commun., 2020, 56, 7183–7186 RSC
- W. Xu, Z. Shao, C. Tang, C. Zhang, Y. Chen and Y. Liang, Org. Biomol. Chem., 2022, 20, 5953–5957 RSC
- F. Liu, D. I. Danylchuk, B. Andreiuk and A. S. Klymchenko, Chem. Sci., 2022, 13, 3652–3660 RSC
- J. Baudet, E. Lesur, M. Ribéraud, A. Chevalier, T. D’Anfray, P. Thuéry, D. Audisio and F. Taran, Chem. Commun., 2024, 60, 3657–3660 Search PubMed
-
(a) Q. Fu, S. Shen, P. Sun, Z. Gu, Y. Bai, X. Wang and Z. Liu, Chem. Soc. Rev., 2023, 52, 7737–7772 RSC
- Z. Shao, W. Liu, H. Tao, F. Liu, R. Zeng, P. A. Champagne, Y. Cao, K. N. Houk and Y. Liang, Chem. Commun., 2018, 54, 14089–14092 RSC
- W. Xu, H. Yu, R. Zhao and Y. Liang, Bioorg. Med. Chem. Lett., 2023, 81, 129129 CrossRef CAS PubMed
- M. Feng, L. Madegard, M. Riomet, M. Louis, P. A. Champagne, G. Pieters, D. Audisio and F. Taran, Chem. Commun., 2022, 58, 8500–8503 Search PubMed
- K. Porte, B. Renoux, E. Péraudeau, J. Clarhaut, B. Eddhif, P. Poinot, E. Gravel, E. Doris, A. Wijkhuisen, D. Audisio, S. Papot and F. Taran, Angew. Chem., Int. Ed., 2019, 58, 6366–6370 CrossRef CAS PubMed
- L. Madegard, M. Girard, B. R. Yaw, K. Porte, D. Audisio, S. Papot and F. Taran, Chem. – Eur. J., 2023, 29, e202301359 CrossRef CAS
- L. Madegard, M. Girard, E. Blochouse, B. Riss Yaw, A. Palazzolo, M. Laquembe, D. Audisio, P. Poinot, S. Papot and F. Taran, Angew. Chem., Int. Ed., 2025, e202422627 CAS
- X. Yan, K. Li, T.-Q. Xie, X.-K. Jin, C. Zhang, Q.-R. Li, J. Feng, C.-J. Liu and X.-Z. Zhang, Angew. Chem., Int. Ed., 2024, 63, e202318539 CrossRef CAS PubMed
- M. Ribéraud, E. Porret, A. Pruvost, F. Theodoro, A. L. Nguyen, S. Specklin, D. Kereselidze, C. Denis, B. Jego, P. Barbe, M. Keck, T. D’Anfray, B. Kuhnast, D. Audisio, C. Truillet and F. Taran, Chem. Sci., 2024, 15, 18825–18831 RSC
- See for example:
(a) R. Rossin, R. M. Versteegen, J. Wu, A. Khasanov, H. J. Wessels, E. J. Steenbergen, W. ten Hoeve, H. M. Janssen, A. H. A. M. van Onzen, P. J. Hudson and M. S. Robillard, Nat. Commun., 2018, 9, 1484–1495 CrossRef PubMed
- J. M. McFarland, M. Aleĉković, G. Coricor, S. Srinivasan, M. Tso, J. Lee, T.-H. Nguyen and J. M. Mejia Oneto, ACS Cent. Sci., 2023, 9(7), 1400–1408 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2025 |