
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
DBU as base and ligand in phosphine-free ruthenium complexes for hydrogenation of CO2†
Andrew Z.
Preston
 a,
Alexander S.
Phearman
a,
Manuel
Quiroz
a,
Sarah E.
Flowers
a,
Nilakshi
Devi
b,
Christopher M.
Zall
b,
Eric S.
Wiedner
*a,
Aaron M.
Appel
a and
John C.
Linehan
*a
a,
Alexander S.
Phearman
a,
Manuel
Quiroz
a,
Sarah E.
Flowers
a,
Nilakshi
Devi
b,
Christopher M.
Zall
b,
Eric S.
Wiedner
*a,
Aaron M.
Appel
a and
John C.
Linehan
*a
aInstitute for Integrated Catalysis, Pacific Northwest National Laboratory, P.O. Box 999, Richland, Washington 99352, USA. E-mail: eric.wiedner@pnnl.gov; john.linehan@pnnl.gov
bDepartment of Chemistry, Sam Houston State University, Huntsville, Texas 77341, USA
First published on 19th June 2025
Abstract
In the presence of DBU base, ruthenium piano stool complexes catalyze the hydrogenation of CO2 to formate at room temperature without phosphine ligands. Thermodynamic and kinetic studies reveal the active catalyst is formed by binding DBU as a ligand.
The catalytic hydrogenation of carbon dioxide is of interest for production of organic hydrogen carriers using an abundant and renewable carbon feedstock. Compared to molecular hydrogen, products such as formic acid, formate salts, and methanol formed from the hydrogenation of CO2 are safer and easier to transport,1–5 making them more suitable for temporary storage of hydrogen generated from intermittent energy sources.
Transition-metal complexes are effective catalysts for hydrogenation of CO2 to formate/formic acid,6–9 though many contain phosphine ligands that are prone to oxidation and are often challenging to synthesize. Some of the most active catalysts for hydrogenation of CO2 to formate are the phosphine-free [Cp*Ir(L)(H2O)]+ (L = N,N-bidentate ligand) family of complexes.10–12 Other phosphine-free catalysts include complexes of Ru,13–16 Co,17,18 Fe,19 and Mn,20 typically possessing either pyridyl or N-heterocyclic carbene ligands.
Watari and Ikariya demonstrated that simple copper salts of the amidine base DBU (1,8-diazabicyclo[5.4.0]undec-7-ene) are catalysts for hydrogenation of CO2 to formate (Fig. 1a).21,22 In this example, DBU serves as both the ligand as well as the more typical role of exogenous base.23–25 Subsequent reports showed that DBU binding to copper phosphine complexes, Cu(PCP)(DBU)+ and Cu(triphos)(DBU)+, also afforded catalysts for CO2 hydrogenation.26–29 Examples of DBU binding during catalytic reactions are sparse and typically lead to inhibition or decomposition of the catalyst, such as in C–N30–32 and C–C33,34 cross-coupling reactions. Therefore, these copper catalysts are notable for their ability to operate in the presence of DBU binding. However, relative to typical phosphine-based catalysts, the phosphine-free catalyst, Cu(DBU)2X, exhibits a very low activity (TOF ∼ 1 h−1 at 80 °C).22 As a result, it remains unclear whether DBU is an effective supporting ligand for catalysis.
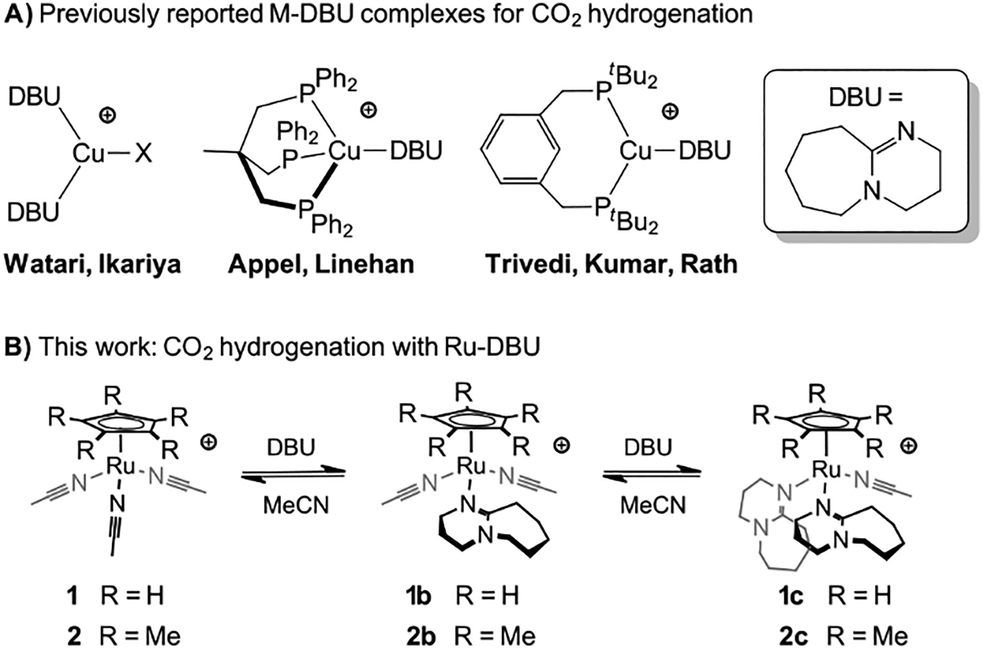 | ||
| Fig. 1 (A) Examples of previously reported phosphine-free catalysts for CO2 hydrogenation. (B) This work shows that Ru piano-stool complexes generate active CO2 hydrogenation catalysts upon binding DBU as ligand. | ||
We report the hydrogenation of CO2 to formate by [CpRu(MeCN)3]+ (1) and [Cp*Ru(MeCN)3]+ (2), and demonstrate the essential role of DBU binding in this process. Both 1 and 2 are commercially available and forego phosphine ligands common for ruthenium-based CO2 hydrogenation catalysts. Kinetic, thermodynamic, and spectroscopic experiments suggest that the precatalysts are activated by the reversible binding of up to two DBU ligands (Fig. 1b), resulting in highly active phosphine-free catalysts under mild conditions.
Complexes 1 and 2 were screened for activity in the catalytic hydrogenation of CO2 with DBU. Addition of DBU (0.6 M) to a CD3CN solution of 1 or 2 resulted in an immediate color change from bright yellow to red-orange. This solution was transferred to a custom PEEK NMR tube,35,36 pressurized with H2/CO2, and the reaction was monitored using high-pressure operando1H NMR spectroscopy. At 25 °C and 34 atm of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 H2/CO2, both catalysts exhibited a steady rate of conversion to formate (Fig. 2a), with 1 exhibiting a higher rate (0.45 M h−1) than 2 (0.1 M h−1).
:1 H2/CO2, both catalysts exhibited a steady rate of conversion to formate (Fig. 2a), with 1 exhibiting a higher rate (0.45 M h−1) than 2 (0.1 M h−1).
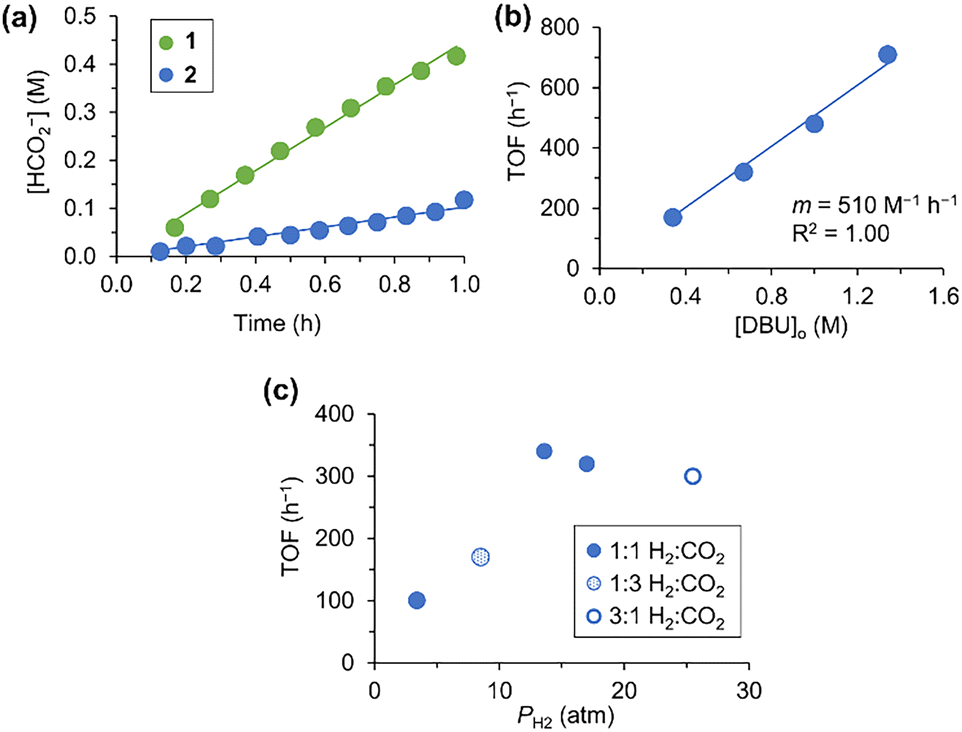 | ||
| Fig. 2 (a) Conversion of CO2 to formate by 1 and 2. Conditions: 0.5 mM 1/2, 600 mM DBU, 17 atm H2/17 atm CO2 at 25 °C in CD3CN. (b) Plot of the DBU dependence on the TOF for 2 (17 atm H2/17 atm CO2, 25 °C). (c) Plot of the H2 dependence on the TOF for 2 ([DBU]o = 0.61–0.67 M, 25 °C). | ||
To determine the catalytic rate law for 2, the initial turnover frequency (TOF) for catalysis was measured under varying conditions (Table S1, ESI†). Doubling [2] from 0.45 mM to 0.9 mM led to a 2× increase in the initial rate of formate production and no change in the TOF, consistent with a first-order dependence in the catalyst concentration. The dependence on base was determined using four different initial concentrations of DBU (0.3 M, 0.6 M, 0.9 M, 1.2 M) at 25 °C and 34 atm of 1:1 H2/CO2. A linear correlation was observed between the TOF and the initial DBU concentration (Fig. 2b), indicating a first-order dependence on DBU.
The reaction order in H2 and CO2 was tested by performing catalysis with 2 at varying pressures and ratios of the two gases with 0.6 M DBU at 25 °C.37 The resulting TOF values showed saturation behavior in PH2, where the TOF is dependent on H2 at low pressures and independent at higher pressures (Fig. 2c). No apparent dependence on PCO2 was observed, with the 1:3 H2/CO2 gas mixture giving the second lowest TOF despite having the highest PCO2 of 30 atm.
The possibility of DBU binding to Ru was first explored for 1 since the Cp ligand resonances are well-separated from those for DBU in the 1H NMR spectrum. Upon mixing a sample of 1 and DBU (<50 mM) in CD3CN, the formation of a second cyclopentadienyl species was observed by 1H, 13C{1H}, and gHMBC NMR spectroscopy (Fig. 3 and Fig. S13–S18, ESI†).
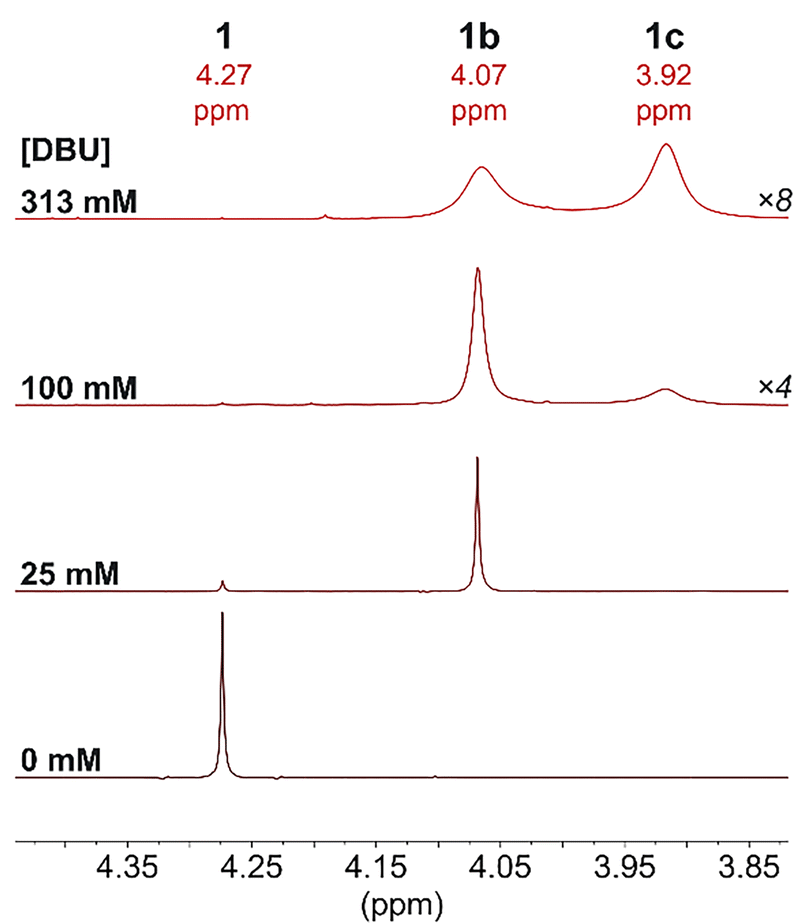 | ||
| Fig. 3 1H NMR spectrum (500 MHz, CD3CN, 25 °C) of the Cp resonance of 1 (25 mM) with varying [DBU]. Spectra with 100 and 313 mM DBU are magnified ×4 and ×8, respectively, due to significant broadening of the Cp signals. | ||
Additionally, new DBU resonances were observed downfield of the small resonances for free DBU, integrating to 1 DBU per new Cp-containing species by 1H and quantitative 13C{1H} NMR spectroscopy. No NH resonance for DBU(H)+ was observed. These spectra are consistent with 91% conversion to [CpRu(DBU)(MeCN)2]+ (1b) with 25 mM DBU.
Higher concentrations of DBU (100 or 313 mM) resulted in the disappearance of 1 and the appearance of [CpRu(DBU)2(MeCN)]+ (1c) (Fig. 3 and Fig. S19, ESI†). In 1H NMR spectra at 25 °C, the Cp resonances for 1b and 1c were noticeably broadened as [DBU] increased, consistent with ligand exchange that is occurring on the NMR time scale. At lower temperatures of −35 °C to −10 °C, sharp well-defined peaks are observed for 1b and 1c (Fig. S42, ESI†). Quantitative 13C{1H} NMR spectroscopy with 1.3 M DBU at −35 °C confirmed the presence of two magnetically equivalent DBU ligands per Cp ring in 1c (Fig. S20, ESI†). The exchange rate between 1b and 1c increases with the temperature and concentration of DBU, and the species coalesce into a single peak by 25 °C with 1.3 M DBU. An absolute rate of 29 s−1 at 25 °C was calculated for exchange between 1b and 1c using the peak shapes (Fig. S21–S30 and Table S2, ESI†). This rate of ligand exchange is very fast relative to the timescale of catalytic CO2 hydrogenation (0.03–0.2 s−1 at 25 °C).
Similar to 1, a solution of 2 (25 mM) and DBU (25 mM) gave rise to shifted Cp* and DBU resonances. 1-D and 2-D 1H and 13C NMR spectroscopy and quantitative 13C{1H} NMR spectroscopy experiments are consistent with 83% conversion to [Cp*Ru(DBU)(MeCN)2]+ (2b) at 25 °C (Fig. S31 and S32, ESI†). With 100 mM DBU, 97% conversion to 2b is observed by 1H NMR spectroscopy. Higher [DBU] obscures the Cp* peak in the 1H NMR spectrum, but 13C{1H} NMR spectra with higher [DBU] show full conversion of 2 to 2b (Fig. S32–S36, ESI†). Compared to the CpRu speciation at the same [DBU], the Cp*Ru species requires higher [DBU] to fully form [Cp*Ru(DBU)(MeCN)2]+. No formation of [Cp*Ru(DBU)2(MeCN)]+ (2c) is observed until much higher DBU concentrations are used (Fig. S37 and S38, ESI†). Quantitative 13C{1H} NMR spectra of 2 in a mixture of 6.04 M DBU and 2.14 M CD3CN at −35 °C, necessary to reduce broadening from ligand exchange, show 45% conversion to 2c.
The identity of 2b was further confirmed by X-ray diffraction studies. Single crystals of 2b were grown by vapor diffusion of Et2O into a DBU/MeCN solution of the Ru species (Fig. 4). This is the first reported structure of a Ru–DBU complex.38
 | ||
| Fig. 4 Solid state structure of 2b as determined by X-ray crystallography. Hydrogen atoms and the PF6 anion are omitted for clarity. Thermal ellipsoids are shown at 50% probability. | ||
Thermodynamic parameters for binding the DBU ligands were determined from Van’t Hoff plots (Table S7, Fig. S40–S44, ESI†). The binding enthalpy of the first DBU  was found to be nearly identical for 1 (−6.9 kcal mol−1) and 2 (−7.2 kcal mol−1), while the entropy of binding
was found to be nearly identical for 1 (−6.9 kcal mol−1) and 2 (−7.2 kcal mol−1), while the entropy of binding  was lower for 2 (−4.6 cal mol−1 K−1) than for 1 (0.2 cal mol−1 K−1). As a result, K1 at 25 °C is larger for 1 (1.2 × 105) than for 2 (1.8 × 104). These results indicate the first DBU binding is enthalpically driven with 2b having a slight entropic penalty, potentially due to restricted motion of the DBU ligand caused by steric interactions with Cp*.
was lower for 2 (−4.6 cal mol−1 K−1) than for 1 (0.2 cal mol−1 K−1). As a result, K1 at 25 °C is larger for 1 (1.2 × 105) than for 2 (1.8 × 104). These results indicate the first DBU binding is enthalpically driven with 2b having a slight entropic penalty, potentially due to restricted motion of the DBU ligand caused by steric interactions with Cp*.
The equilibrium for binding the second DBU exhibited a more pronounced difference between 1b and 2b. The binding enthalpy for the second DBU 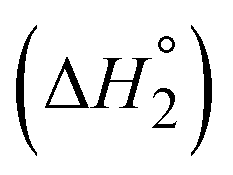 is exothermic for 1b (−4.1 kcal mol−1) but endothermic for 2b (3.7 kcal mol−1). In addition, the entropy of binding
is exothermic for 1b (−4.1 kcal mol−1) but endothermic for 2b (3.7 kcal mol−1). In addition, the entropy of binding  changes from negative for 1b (−5.5 cal mol−1 K−1) to positive for 2b (12 cal mol−1 K−1). At 25 °C, K2 is nearly two orders of magnitude larger for 1b (66) than for 2b (0.9). These large differences in
changes from negative for 1b (−5.5 cal mol−1 K−1) to positive for 2b (12 cal mol−1 K−1). At 25 °C, K2 is nearly two orders of magnitude larger for 1b (66) than for 2b (0.9). These large differences in 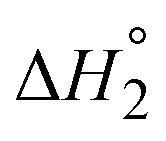 and
and  suggest that the CH3CN ligand of 2c may be much more labile than in 1c (see ESI†); however, this could not be confirmed with the available data due to the low equilibrium population of 2c.
suggest that the CH3CN ligand of 2c may be much more labile than in 1c (see ESI†); however, this could not be confirmed with the available data due to the low equilibrium population of 2c.
The measured binding constants indicate 2 is a precatalyst that rapidly binds DBU to generate a mixture of 2b (97%) and 2c (3%) under catalytic conditions. The kinetic data is most consistent with 2b being the active catalyst form according to the mechanism in Scheme 1. From the 2b resting state, catalysis proceeds by pre-equilibrium binding of H2 to give a dihydrogen complex, [Cp*Ru(H2)(DBU)(CH3CN)]+ (2b-H2). Rate-limiting deprotonation by exogenous DBU to form Cp*Ru(H)(DBU)(CH3CN) (2b-H) and a protonated base then occurs. The cycle is completed by rapid hydride transfer from 2b-H to CO2 to yield 2b and formate.
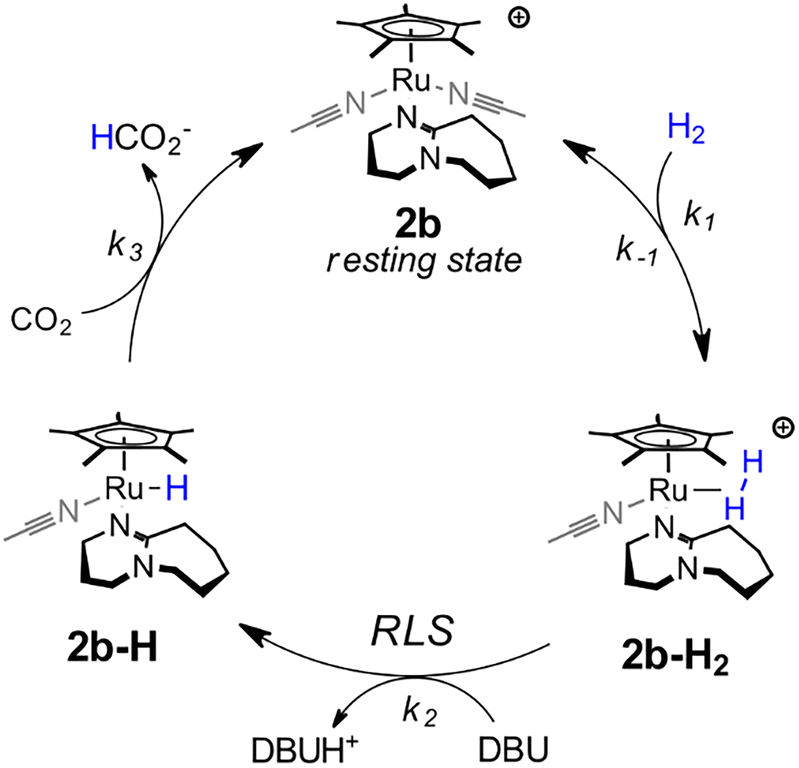 | ||
| Scheme 1 Proposed catalytic cycle for hydrogenation of CO2 to formate by 2. | ||
Catalysis could also occur through an alternative cycle involving two DBU ligands, but the first-order dependence on [DBU] indicates such a cycle is not kinetically competitive for 2. This could be due to its unfavorable equilibrium for binding a second DBU ligand. Under catalytic conditions, 2c constitutes only 3–7% of the total Ru species. It is noteworthy that 1 exhibits both a higher initial population of the bis-DBU adduct (70%) and a higher TOF, potentially consistent with a secondary catalytic cycle involving coordination of two DBU ligands.
Phosphine-free molecular catalysts for CO2 hydrogenation are relatively uncommon and typically require high temperatures and pressures.6,7 The most active phosphine-free catalysts, [Cp*Ir(L)(H2O)]+ (L = N,N-bidentate ligand), display initial TOFs as high as 200 h−1 under ambient conditions (25 °C, 1 atm) in aqueous NaHCO3 solution.39 Catalyst 2 displays a comparable TOF of 100 h−1 under slightly more forcing conditions (25 °C, 6.8 atm) using DBU as the base in MeCN solution.
Higher TOFs are obtained for phosphine-based ruthenium catalysts at elevated temperatures and pressures. For example, a Ru(PNP) pincer complex exhibits a TOF of 36000 h−1 (65 °C, 40 atm, 1.1 M DBU).40 A more direct comparison with 2 can be made by extrapolating the temperature-dependent TOF of the Ru(PNP) catalyst to a value of 9400 h−1 at 25 °C with 40 atm of 3:1 H2:CO2 (Fig. S45, ESI†).41 Under similar conditions (1.3 M DBU, 34 atm 1:1 H2:CO2), 2 affords a TOF of 710 h−1. Normalizing for the difference in pressure suggests 2 is within an order of magnitude of the activity of Ru(PNP). This modest difference is quite remarkable since 2 does not rely on phosphine ligands, but rather uses the simple DBU base as a ligand.
In conclusion, we observed that the phosphine-free complexes [CpRu(MeCN)3]+ and [Cp*Ru(MeCN)3]+ are active precatalysts for the hydrogenation of CO2 to formate with DBU base. Mechanistic studies reveal the key role of coordination of DBU as an ancillary ligand in the active species. Detailed studies of the catalytic mechanism revealed the rate determining step is the heterolytic cleavage of a Ru–H2 complex to form the subsequent hydride and a protonated base.
This work was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, Chemical Sciences, Geosciences & Biosciences Division, Catalysis Science program, FWP 47319. Work by C. M. Z. and N. D. was supported by the U.S. Department of Energy, Office of Science, Office of Workforce Development for Teachers and Scientists (WDTS) under the Visiting Faculty Program (VFP), as well as by Sam Houston State University through the Faculty Research Grant.
Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for 2b has been deposited at the CCDC under the number 2329534.†Notes and references
- A. Kumar, P. Daw and D. Milstein, Chem. Rev., 2022, 122, 385–441 CrossRef CAS PubMed.
- R. Sen, A. Goeppert and G. K. Surya Prakash, Angew. Chem., Int. Ed., 2022, 61, e202207278 CrossRef CAS PubMed.
- S. Chatterjee, I. Dutta, Y. Lum, Z. Lai and K.-W. Huang, Energy Environ. Sci., 2021, 14, 1194–1246 RSC.
- K. Grubel, H. Jeong, C. W. Yoon and T. Autrey, J. Energy Chem., 2020, 41, 216–224 CrossRef.
- S. Enthaler, J. von Langermann and T. Schmidt, Energy Environ. Sci., 2010, 3, 1207–1217 RSC.
- W.-H. Wang, Y. Himeda, J. T. Muckerman, G. F. Manbeck and E. Fujita, Chem. Rev., 2015, 115, 12936–12973 CrossRef CAS PubMed.
- K. Sordakis, C. Tang, L. K. Vogt, H. Junge, P. J. Dyson, M. Beller and G. Laurenczy, Chem. Rev., 2018, 118, 372–433 CrossRef CAS PubMed.
- P. G. Jessop, T. Ikariya and R. Noyori, Chem. Rev., 1995, 95, 259–272 CrossRef CAS.
- W. Wang, S. Wang, X. Ma and J. Gong, Chem. Soc. Rev., 2011, 40, 3703–3727 RSC.
- J. F. Hull, Y. Himeda, W. H. Wang, B. Hashiguchi, R. Periana, D. J. Szalda, J. T. Muckerman and E. Fujita, Nat. Chem., 2012, 4, 383–388 CrossRef CAS PubMed.
- R. Kanega, N. Onishi, D. J. Szalda, M. Z. Ertem, J. T. Muckerman, E. Fujita and Y. Himeda, ACS Catal., 2017, 7, 6426–6429 CrossRef CAS.
- A. Nijamudheen, R. Kanega, N. Onishi, Y. Himeda, E. Fujita and M. Z. Ertem, ACS Catal., 2021, 11, 5776–5788 CrossRef CAS.
- S. Ogo, R. Kabe, H. Hayashi, R. Harada and S. Fukuzumi, Dalton Trans., 2006, 4657–4663 RSC.
- H. Hayashi, S. Ogo and S. Fukuzumi, Chem. Commun., 2004, 2714–2715 RSC.
- A. Weilhard, S. P. Argent and V. Sans, Nat. Commun., 2021, 12, 231 CrossRef CAS PubMed.
- A. Weilhard, K. Salzmann, M. Navarro, J. Dupont, M. Albrecht and V. Sans, J. Catal., 2020, 385, 1–9 CrossRef CAS.
- Y. M. Badiei, W.-H. Wang, J. F. Hull, D. J. Szalda, J. T. Muckerman, Y. Himeda and E. Fujita, Inorg. Chem., 2013, 52, 12576–12586 CrossRef CAS PubMed.
- W. Yao, G. Olajide, C. M. Boudreaux, M. M. Wysocki, M. K. Ahmed, F. Qu, T. Szilvási and E. T. Papish, Organometallics, 2024, 43, 1447–1458 CrossRef CAS.
- S. Coufourier, Q. Gaignard Gaillard, J.-F. Lohier, A. Poater, S. Gaillard and J.-L. Renaud, ACS Catal., 2020, 10, 2108–2116 CrossRef CAS.
- A. Dubey, L. Nencini, R. R. Fayzullin, C. Nervi and J. R. Khusnutdinova, ACS Catal., 2017, 7, 3864–3868 CrossRef CAS.
- R. Watari, S. Kuwata and Y. Kayaki, Chem. Lett., 2020, 49, 252–254 CrossRef CAS.
- R. Watari, Y. Kayaki, S. Hirano, N. Matsumoto and T. Ikariya, Adv. Synth. Catal., 2015, 357, 1369–1373 CrossRef CAS.
- S. Kostera, S. Weber, M. Peruzzini, L. F. Veiros, K. Kirchner and L. Gonsalvi, Organometallics, 2021, 40, 1213–1220 CrossRef CAS PubMed.
- C. M. Hert, J. B. Curley, S. P. Kelley, N. Hazari and W. H. Bernskoetter, Organometallics, 2022, 41, 3332–3340 CrossRef CAS.
- J. Hu, Q. J. Bruch and A. J. M. Miller, J. Am. Chem. Soc., 2021, 143, 945–954 CrossRef CAS PubMed.
- M. Trivedi, A. Kumar, A. Husain and N. P. Rath, Inorg. Chem., 2021, 60, 4385–4396 CrossRef CAS PubMed.
- R. R. Persaud, Z. Fang, C. M. Zall, A. M. Appel and D. A. Dixon, J. Phys. Chem. A, 2021, 125, 6600–6610 CrossRef CAS PubMed.
- C. M. Zall, J. C. Linehan and A. M. Appel, J. Am. Chem. Soc., 2016, 138, 9968–9977 CrossRef CAS PubMed.
- C. M. Zall, J. C. Linehan and A. M. Appel, ACS Catal., 2015, 5, 5301–5305 CrossRef CAS.
- L. Fan and O. V. Ozerov, Chem. Commun., 2005, 4450–4452 RSC.
- N. Kumagai, S. Matsunaga and M. Shibasaki, J. Am. Chem. Soc., 2004, 126, 13632–13633 CrossRef CAS PubMed.
- N. Kumagai, S. Matsunaga and M. Shibasaki, Tetrahedron, 2007, 63, 8598–8608 CrossRef CAS.
- R. Y. Liu, J. M. Dennis and S. L. Buchwald, J. Am. Chem. Soc., 2020, 142, 4500–4507 CrossRef CAS PubMed.
- J. M. Dennis, N. A. White, R. Y. Liu and S. L. Buchwald, ACS Catal., 2019, 9, 3822–3830 CrossRef CAS PubMed.
- C. R. Yonker and J. C. Linehan, Prog. Nucl. Magn. Reson. Spectrosc., 2005, 47, 95–109 CrossRef CAS.
- C. R. Yonker and J. C. Linehan, J. Organomet. Chem., 2002, 650, 249–257 CrossRef CAS.
- Z. K. Lopez-Castillo, S. N. V. K. Aki, M. A. Stadtherr and J. F. Brennecke, Ind. Eng. Chem. Res., 2008, 47, 570–576 CrossRef CAS.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS PubMed.
- R. Kanega, M. Z. Ertem, N. Onishi, D. J. Szalda, E. Fujita and Y. Himeda, Organometallics, 2020, 39, 1519–1531 CrossRef CAS.
- G. A. Filonenko, R. van Putten, E. N. Schulpen, E. J. M. Hensen and E. A. Pidko, ChemCatChem, 2014, 6, 1526–1530 CrossRef CAS.
- G. A. Filonenko, E. J. M. Hensen and E. A. Pidko, Catal. Sci. Technol., 2014, 4, 3474–3485 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental data, kinetic data, spectral data, additional crystallographic data. CCDC 2329534. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5cc00512d |
| This journal is © The Royal Society of Chemistry 2025 |