
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tailoring nanoporous aluminosilicates with well-defined Al(OSi)4 sites using dimethylsilanol-modified cage siloxanes and ammonium cations†
Takuya Hikino a,
Hikaru Mochizukib,
Takamichi Matsunobcd,
Kazuyuki Kurodabc and
Atsushi Shimojima*bcd
a,
Hikaru Mochizukib,
Takamichi Matsunobcd,
Kazuyuki Kurodabc and
Atsushi Shimojima*bcd
aDepartment of Advanced Science and Engineering, Faculty of Science and Engineering, Waseda University, 3-4-1 Okubo, Shinjuku-ku, Tokyo 169-8555, Japan
bDepartment of Applied Chemistry, Faculty of Science and Engineering, Waseda University, 3-4-1 Okubo, Shinjuku-ku, Tokyo 169-8555, Japan. E-mail: shimojima@waseda.jp
cKagami Memorial Research Institute for Materials Science and Technology, Waseda University, 2-8-26 Nishiwaseda, Shinjuku-ku, Tokyo 169-0051, Japan
dWaseda Research Institute for Science and Engineering, Waseda University, 3-4-1 Okubo, Shinjuku-ku, Tokyo 169-8555, Japan
First published on 4th April 2025
Abstract
The precise synthesis of nanoporous aluminosilicates with well-defined Al(OSi)4 sites was achieved using rigid cage siloxanes. Organic ammonium cations served not only as counterions for the negatively charged framework but also as modulators of pore characteristics. Treatment with an aqueous NH4Cl solution generated Brønsted acid sites. This work paves the way for designing nanoporous aluminosilicate materials.
Porous aluminosilicates, such as zeolites and Al-containing mesoporous silica, possess a variety of potential applications, including catalysis, adsorption, and ion exchange.1,2 The AlO4 tetrahedra in the frameworks provide Brønsted acid sites, and their environment, location, and distribution are key factors, especially for catalytic functions.3,4 Zeolites possess well-defined crystalline frameworks; however, precise control of the Al sites in the frameworks is challenging, mainly because of the complex crystallization mechanism under hydrothermal conditions. Extensive research has also been conducted on the preparation of mesoporous aluminosilicates,5–7 typically by the sol–gel process using silica and alumina precursors with surfactant templates;6,7 however, most of the conventional materials have amorphous frameworks with uncontrolled Al sites. Additionally, six-coordinated, extra-framework Al species are easily formed due to the rearrangement of the Si–O–Si and Si–O–Al bonds upon calcination at a high temperature.8
Non-hydrolytic condensation reactions are useful for the selective formation of Si–O–Al bonds. Well-defined Al(OSi)4 sites can be formed through the reaction of silanol groups with alkylaluminum or aluminum chloride in the presence of amines.9,10 Imaizumi et al. reported the synthesis of zeolite11 and amorphous porous aluminosilicate12 using a bicyclic aluminosilicate precursor, (PyH+)[Al{Ph2Si(OSiPh2O)2}2]− (PyH+ = pyridinium cation). In these reports, however, it is unclear whether the local structure of the Al sites in the precursor was retained in the final products because of the hydrothermal treatment at high pH conditions and/or calcination at a high temperature to cleave the Si–C bonds for intermolecular linking. To overcome these obstacles, the design of building block molecules and the development of methods to construct porous frameworks under milder conditions are essential.
Rigid cage-type siloxane structures, such as double n-membered ring (dnr) structures (n = 4 and 6), are useful building blocks for constructing nanoporous materials with molecularly designed siloxane frameworks.13–16 Al-incorporated cage siloxanes have attracted attention as precursors of porous aluminosilicate.17,18 Recently, a d4r-type aluminosilicate compound, TMA4[Al4Si4O12(OH)8]·13H2O (TMA = tetramethylammonium cation), was used for the synthesis of an LTA-type zeolite with a Si/Al ratio of 1.18 However, the formation process is not entirely clear as it still relies on hydrothermal treatment similar to the conventional zeolite synthesis. Additionally, cage-type aluminosilicate compounds available as building blocks are very limited, as compared to Al-free cage-type silicates, making variation of the framework structures difficult.
A promising approach to address the aforementioned issues is the use of cage siloxane building blocks having functional groups that can selectively form Si–O–Al bonds under mild conditions. This makes the formation process clearer, as the bond cleavage that occurs under hydrothermal conditions can be avoided, allowing precise control of the aluminosilicate framework. Recently, Kejik et al. reported the reaction of a trimethyltin-modified d4r siloxane (Si8O12(OSnMe3)8) with a pyridine–trimethylaluminum complex (Py·AlMe3) at 100 °C to form porous networks with Si–O–Al bonds.19 The resulting materials were primarily composed of 4-coordinated Al(OSi)4 sites with some Al(OSi)3Py and unreacted SiOSnMe3 groups. The structure of the Al sites was not fully defined due to uncertainty regarding the number of coordinated cages.
Herein, we report a direct synthetic approach to nanoporous aluminosilicates with well-defined Al(OSi)4 sites through the non-hydrolytic condensation reaction of dimethylsilanol-modified d4r siloxane compound (D4R-SiMe2OH) with AlMe3 in the presence of various amines at 40 °C (Scheme 1). The type of amine was found to affect both the Al environment and pore characteristics. Using piperidine, a porous aluminosilicate with a pore size ranging from micro to meso scale and with a higher surface area than the previously reported materials12,19 was obtained. Moreover, ion-exchange treatment of the resulting porous materials with NH4+ suggested the formation of Brønsted acid sites. These findings pave the way for the truly modular synthesis of nanoporous aluminosilicates with controlled framework structures.
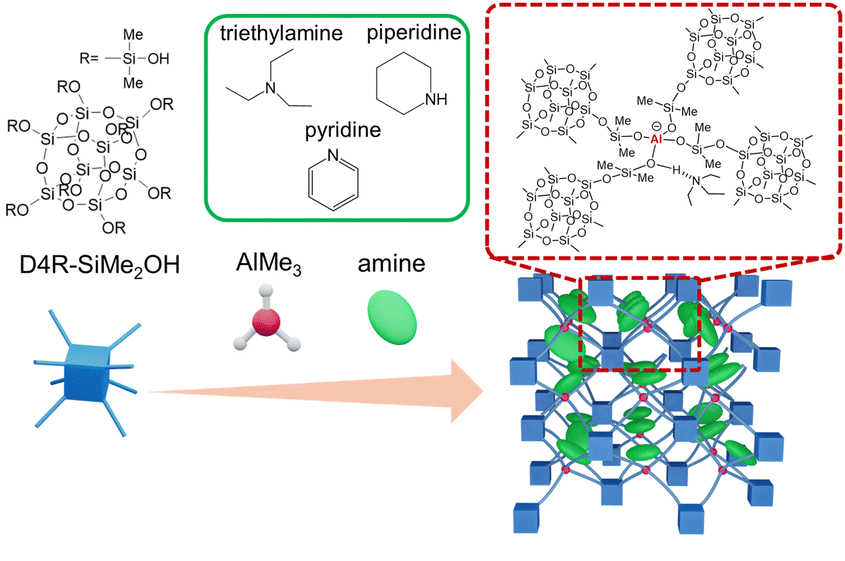 | ||
| Scheme 1 Synthesis of nanoporous aluminosilicates through a condensation reaction of dimethylsilanol-modified cage siloxane with AlMe3 in the presence of amines. In the dotted frame, triethylamine is shown as an example. | ||
D4R-SiMe2OH was synthesized by oxidation of dimethylsilylated d4r siloxane (Si8O12(OSiMe2H)8) (see ESI† for details).13 AlMe3 was added to the mixture of D4R-SiMe2OH and amines in THF under N2 atmosphere. The molar ratio of D4R-SiMe2OH to AlMe3 and amines was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:2, ensuring that all SiOH groups can form Si–O–Al bonds. The amines used were triethylamine (TEA), pyridine (Py), and piperidine (Pip). After the addition of AlMe3, gelation occurred, and the volatile components were removed under reduced pressure, resulting in white solids. The samples were denoted as Al-D4R-TEA, Al-D4R-Py, and Al-D4R-Pip.
:2:2, ensuring that all SiOH groups can form Si–O–Al bonds. The amines used were triethylamine (TEA), pyridine (Py), and piperidine (Pip). After the addition of AlMe3, gelation occurred, and the volatile components were removed under reduced pressure, resulting in white solids. The samples were denoted as Al-D4R-TEA, Al-D4R-Py, and Al-D4R-Pip.
In the Fourier transform infrared (FT-IR) spectra of Al-D4R-TEA, Al-D4R-Py, and Al-D4R-Pip (Fig. S1, ESI†), the intensity of the Si–OH vibration band at 890 cm−1 decreased, and a new band assignable to Si–O–Al bonds appeared at 960 cm−1,10,20 suggesting that the reaction between the silanol groups and AlMe3 progressed. The 29Si magic-angle spinning (MAS) NMR spectra of the samples (Fig. 1(A)) showed signals corresponding to the Q4 [Si(OSi)4] site at around −107 ppm, with no signal attributed to the Q3 [Si(OSi)3OH] site, confirming the retention of the cage siloxane structure. The signal at −20 ppm is assignable to the D2(1Al) site [SiOSiMe2OAl] as its chemical shift is slightly upfield from that of the D2(0Al) site [SiOSiMe2OSi] (−17.5 ppm).15 A similar difference in chemical shifts was also reported for the [SiOSiPh2OAl] and [SiOSiPh2OSi] sites.9,21,22 A small D1 signal of unreacted dimethylsilanol groups [SiOSiMe2OH] (−10 ppm) were observed for Al-D4R-TEA. Fig. 1(B) shows the 27Al MAS NMR spectra. In all samples, signals attributed to 4-coordinated Al species (AlO4) and 6-coordinated Al species (AlO6) were observed at 57 and 5 ppm, respectively. The AlO4/(AlO4 + AlO6) ratios were 0.95, 0.87, and 0.91 for Al-D4R-TEA, Al-D4R-Py, and Al-D4R-Pip, respectively. The differences are discussed later in relation to the elemental analysis.
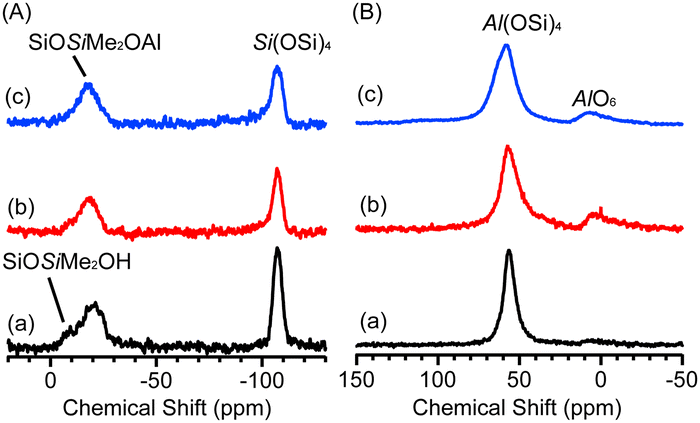 | ||
| Fig. 1 (A) 29Si MAS NMR spectra and (B) 27Al MAS NMR spectra of (a) Al-D4R-TEA, (b) Al-D4R-Py, and (c) Al-D4R-Pip. | ||
The 13C cross-polarization (CP)/MAS NMR spectra of Al-D4R-TEA, Al-D4R-Py, and Al-D4R-Pip (Fig. S2, ESI†) showed the signals derived from TEA, Py, and Pip, respectively. In the 1H MAS NMR spectra (Fig. S3, ESI†), and the protons of ammonium cations were observed at 7.9 ppm23 for all samples. FT-IR spectra of Al-D4R-TEA and Al-D4R-Pip (Fig. S1, ESI†) showed broad bands around 1450 cm−1, which might be attributed to the N–H bending vibration of organic ammonium cations.24 Focusing on the bands derived from Py, the two bands around 1450 and 1550 cm−1 in the spectrum of Al-D4R-Py can be attributed to the ring vibration of Py coordinated to Lewis acid sites and the N–H bending vibration of Py adsorbed on Brønsted acid sites, respectively.25,26
The elemental analysis (Table 1) confirmed that the Al/Si ratios of the samples were consistent with the starting mixtures (Al/Si = 0.125). The higher Al/N ratios (>1.0) are possibly due to the formation of 4-coordinated Al species having two bridging siloxy groups (di-μ-oxo Al species)27 and 6-coordinated Al species, followed by volatilization of residual amines during the drying process. The presence of di-μ-oxo Al species was supported by the high AlO4/(AlO4 + AlO6) ratios in the 27Al NMR spectra, despite the high Al/N ratios, which is based on the assumption that the di-μ-oxo Al species exhibit a similar 27Al NMR chemical shift to the Al(OSi)4 species. The lowest Al/N ratio in Al-D4R-Py was attributed to undergoing stoichiometric reactions and/or the formation of AlMe3 and a pyridine complex,28 which might be less prone to volatilization during the drying process.
The powder X-ray diffraction (XRD) patterns showed broad peaks at d = 1.20 nm (Fig. S4, ESI†), which possibly correspond to the average distance between the adjacent d4r units in the cross-linked networks. Similar peaks are generally observed for the porous materials prepared by cross-linking of d4r siloxane compounds.14,15
Fig. 2(A) shows the N2 adsorption–desorption isotherms of the samples. The pre-treatment of the sample did not affect the organic species in the samples, as revealed by the FT-IR spectra of Al-D4R-Py (Fig. S5, ESI†). The use of Py and Pip resulted in higher Brunauer–Emmett–Teller (BET) areas and larger pore volumes in comparison to the use of TEA (Table 1). The BET area values were higher than the previously reported Al-containing d4r-based porous materials (126 m2 g−1).19 The pore size distributions of Al-D4R-Py and Al-D4R-Pip, calculated by the Saito–Foley (SF) method (Fig. 2(B)), indicate the presence of micropores and those calculated using the Barrett–Joyner–Halenda (BJH) method (Fig. 2(C)) reveal the existence of mesopores. A potential correlation exists between the porosity and structural characteristics of the amines. Py and Pip are more rigid than TEA in terms of the number of rotational bonds, which may have facilitated the formation of voids between the cage siloxane units.
 | ||
| Fig. 2 (A) N2 adsorption–desorption isotherms of (a) Al-D4R-TEA, (b) Al-D4R-Py, and (c) Al-D4R-Pip. (B) and (C) Pore size distributions of (a) Al-D4R-Py and (b) Al-D4R-Pip (B: SF method, C: BJH method). | ||
The ion exchange behaviors of these porous materials for replacing organic ammonium cations with NH4+ were investigated as a potential step toward the formation of Brønsted acid sites. Al-D4R-TEA, Al-D4R-Py, and Al-D4R-Pip were stirred in a biphasic mixture of a saturated aqueous solution of ammonium chloride and diethyl ether, recovered by filtration, and air-dried at 100 °C to yield white powders (denoted as Al-D4R-TEA-ex, Al-D4R-Py-ex, and Al-D4R-Pip-ex, respectively). The 1H MAS NMR and 13C CP/MAS NMR analyses revealed that the triethylammonium cation was decreased in Al-D4R-TEA-ex and that PyH+ and piperidinium cations disappeared in Al-D4R-Py-ex and Al-D4R-Pip-ex, respectively (Fig. S6 and S7, ESI†). On the other hand, the signal of the proton from the ammonium cations was still observed at ∼8 ppm by 1H NMR.23 These results indicate the replacement of the protonated amines with NH4+. The progress of ion-exchange was also supported by elemental analysis and 29Si MAS NMR measurement (calculation details are shown in ESI,† Section 3).
The FT-IR spectra of Al-D4R-TEA-ex, Al-D4R-Py-ex, and Al-D4R-Pip-ex (Fig. S8, ESI†) showed the bands of the d4r units (around 560 cm−1).20 The bands derived from N–H bending vibration at 1450 cm−1 also supported the presence of NH4+.29 The 29Si MAS NMR spectra of these samples exhibited small Q3 signals (shoulders at around −100 ppm), indicative of partial cleavage of the Si–O–Si bonds (Fig. S9, ESI†). The D/Q ratio of Al-D4R-Py-ex and Al-D4R-Pip-ex decreased after ion exchange (Table S2, ESI†). This phenomenon can be attributed to nucleophilic attack on Si by water, resulting in the formation and subsequent volatilization of low molecular weight D unit oligomers. The 27Al MAS NMR spectra (Fig. S10, ESI†) indicated that the AlO4/(AlO4 + AlO6) ratios became lower after the ion-exchange treatment (Table S3, ESI†), which was attributed to the partial cleavage of the Al(OSi)4 sites by hydrolysis.
From the results of elemental analysis, the Al/Si ratios slightly increased after ion-exchange (Table S1, ESI†), which is consistent with the decrease of the D units in the framework. The AlO4/N ratio (Table S4, ESI†) increased after the ion-exchange treatment, which might be due to ion-exchange between the organic ammonium cation and H+ in the weakly acidic ammonium chloride solution. Evidence for the formation of Brønsted acid sites was demonstrated by the adsorption of trimethylphosphine oxide (TMPO). The 31P MAS NMR spectra of the TMPO-adsorbed samples (Fig. S11, ESI†) showed signals at approximately 66 and 44 ppm, which can be attributed to TMPO adsorbed at a Brønsted acid site and physisorbed TMPO, respectively.30,31 The signal at 43 ppm (Fig. S11(a), ESI†) might be attributed to crystalline TMPO.32 The chemical shift of 66 ppm is comparable to that observed for zeolite HY and Al-SBA-15.33,34 The 1H MAS NMR spectra of the TMPO-adsorbed samples are shown in Fig. S12 (ESI†). The methyl groups of TMPO appear at 2.2 ppm, while signals corresponding to Brønsted acid sites and those covered with TMPO35 were observed at around 3 and 5 ppm, respectively, for Al-D4R-Py-ex and Al-D4R-Pip-ex. The 27Al MAS NMR spectra of TMPO-adsorbed samples (Fig. S13, ESI†) showed the formation of three-coordinated Al species, likely due to the cleavage of SiOHAl(OSi)3 into SiOH and Al(OSi)3. This suggests the inherent weakness of the Al–O bond in SiOHAl, enabling its dissociation upon TMPO adsorption. The XRD patterns of the ion-exchange samples are shown in Fig. S14, ESI.† The broadening of the peaks reflecting d4r arrangements indicates a potential correlation with structural disorder, as evidenced by NMR analysis (Fig. S9, ESI†). The N2 adsorption–desorption isotherms of ion-exchanged samples (Fig. S15 and Table S5, ESI†) demonstrated that the BET areas and pore volumes decreased approximately 40–60% upon the replacement of the organic ammonium cation with NH4+. The pore size distributions of Al-D4R-Py-ex and Al-D4R-Pip-ex exhibited a reduction of the micropores and mesopores (Fig. S16, ESI†). This may result from the rearrangement of the framework via Si–O–Si(Al) bond cleavage to reduce voids between the cage units. Although optimization of the ion-exchange conditions is still required, nanoporous aluminosilicates with Brønsted acid sites were successfully synthesized via the building block approach.
The significance of the d4r units in the construction of the porous framework is investigated using another type of siloxane-based oligomer, Si(OSiMe2OH)4 (QDOH4), which contains fewer Q-type Si atoms (SiO4 units) than D4R-SiMe2OH. Such a tetrahedrally-branched unit is universally present in the zeolite frameworks. The reaction of QDOH4 with AlMe3 in the presence of TEA or Py was conducted (see ESI† for details), resulting in the formation of cross-linked materials (denoted as Al-QD4-TEA and Al-QD4-Py, respectively). However, the formation of a porous structure was not confirmed for these products (Table S6, ESI†). These results suggest that the rigid and bulky cage structure of the oligosiloxane building blocks is a crucial factor in the formation of the porous structure.
In conclusion, we have developed a novel synthetic approach to nanoporous aluminosilicates with well-defined Al sites using silanol-modified d4r siloxanes. The porous structure can be tailored by the choice of amines. Additionally, the introduced organic ammonium cations can be exchanged for NH4+, a process that appears to simultaneously form Brønsted acid sites, possibly through H+ exchange. The rigidity of the oligosiloxane building blocks is important for the construction of porous materials. This approach will allow fine control over the aluminosilicate framework and pore structures by using other silanol-modified dnr siloxane cages, which will lead to the development of novel functional catalysts.
This work was based on results obtained from a project commissioned by the New Energy and Industrial Technology Development Organization (NEDO) (JPNP06046). This work was also supported in part by JSPS KAKENHI (grant no. 23H02051 and 23K26744) and JSPS Research Fellowship for Young Scientists (grant no. 22J22262). This work was the result of using research equipment (JEOL JNM-ECZ500R (C1028), JEOL JNM-ECA400 (C1026), Quantachrome Autosorb-iQ (G1038), and RINT-Ultima III (G1035)) shared in the MEXT Project for promoting public utilization of advanced research infrastructure (program for supporting construction of core facilities) (grant number JPMXS0440500024).
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.References
- M. Shamzhy, M. Opanasenko, P. Concepción and A. Martínez, Chem. Soc. Rev., 2019, 48, 1095–1149 RSC.
- Y. Li, L. Li and J. Yu, Chem, 2017, 3, 928–949 CAS.
- T. Yokoi, H. Mochizuki, S. Namba, J. N. Kondo and T. Tatsumi, J. Phys. Chem. C, 2015, 119, 15303–15315 CrossRef CAS.
- J. Dědeček, E. Tabor and S. Sklenak, ChemSusChem, 2019, 12, 556–576 CrossRef PubMed.
- M. R. Agliullin, I. G. Danilova, A. V. Faizullin, S. V. Amarantov, S. V. Bubennov, T. R. Prosochkina, N. G. Grigor’eva, E. A. Paukshtis and B. I. Kutepov, Microporous Mesoporous Mater., 2016, 230, 118–127 CrossRef CAS.
- C. T. Kresge, M. E. Leonowicz, W. J. Roth, J. C. Vartuli and J. S. Beck, Nature, 1992, 359, 710–712 CrossRef CAS.
- Y. Li, W. Zhang, L. Zhang, Q. Yang, Z. Wei, Z. Feng and C. Li, J. Phys. Chem. B, 2004, 108, 9739–9744 CrossRef CAS.
- H. Kosslick, G. Lischke, B. Parlitz, W. Storek and R. Fricke, Appl. Catal., A, 1999, 184, 49–60 CrossRef CAS.
- M. D. Skowronska-Ptasinska, R. Duchateau, R. A. van Santen and G. P. A. Yap, Eur. J. Inorg. Chem., 2001, 133–137 CrossRef CAS.
- Y. K. Gun’ko, R. Reilly and V. G. Kessler, New J. Chem., 2001, 25, 528–530 RSC.
- A. Imaizumi, A. Nakada, T. Matsumoto and H.-C. Chang, CrystEngComm, 2020, 22, 5862–5870 RSC.
- A. Imaizumi, A. Nakada, T. Matsumoto, T. Yokoi and H.-C. Chang, Inorg. Chem., 2022, 61, 13481–13496 CrossRef CAS PubMed.
- N. Sato, Y. Kuroda, H. Wada, A. Shimojima and K. Kuroda, Chem. – Eur. J., 2018, 24, 17033–17038 CrossRef CAS PubMed.
- T. Hikino, K. Fujino, N. Sato, H. Wada, K. Kuroda and A. Shimojima, Chem. Lett., 2021, 50, 1643–1647 CrossRef CAS.
- M. Kikuchi, T. Hayashi, T. Matsuno, K. Kuroda and A. Shimojima, Dalton Trans., 2024, 53, 6256–6263 RSC.
- T. Nishitoba, T. Matsumoto, F. Yagihashi, J. Satou, T. Kikuchi, K. Sato and M. Igarashi, Chem. Mater., 2024, 36, 10198–10204 CrossRef CAS.
- G. Fu, C. A. Fyfe, W. Schwieger and G. T. Kokotailo, Angew. Chem., Int. Ed. Engl., 1995, 34, 1499–1502 CrossRef CAS.
- A. Imaizumi, Y. Ohnishi, A. Nakada, A. Honda, T. Matsumoto, K. Katayama and H.-C. Chang, Bull. Chem. Soc. Jpn., 2024, uoae060 CrossRef CAS.
- M. Kejik, J. Brus, L. Jeremias, L. Simonikova, Z. Moravec, L. Kobera, A. Styskalik, C. E. Barnes and J. Pinkas, Inorg. Chem., 2024, 63, 2679–2694 CrossRef CAS PubMed.
- W. Mozgawa, M. Handke and W. Jastrzębski, J. Mol. Struct., 2004, 704, 247–257 CrossRef CAS.
- M. Veith, M. Jarczyk and V. Huch, Angew. Chem., Int. Ed. Engl., 1997, 36, 117–119 CrossRef CAS.
- B. X. Mayer, P. Zöllner and H. Kählig, J. Chromatogr. A, 1999, 848, 251–260 CrossRef CAS.
- W. P. J. H. Jacobs, J. W. de Haan, L. J. M. van de Ven and R. A. van Santen, J. Phys. Chem., 1993, 97, 10394–10402 CrossRef CAS.
- T. Miyazawa, T. Shimanouchi and S. Mizushima, J. Chem. Phys., 1958, 29, 611–616 CrossRef CAS.
- A. Vimont, F. Thibault-Starzyk and J. C. Lavalley, J. Phys. Chem. B, 2000, 104, 286–291 CrossRef CAS.
- M. Castellà-Ventura, Y. Akacem and E. Kassab, J. Phys. Chem. C, 2008, 112, 19045–19054 CrossRef.
- R. N. Kerber, A. Kermagoret, E. Callens, P. Florian, D. Massiot, A. Lesage, C. Copéret, F. Delbecq, X. Rozanska and P. Sautet, J. Am. Chem. Soc., 2012, 134, 6767–6775 CrossRef CAS PubMed.
- K. Korona, I. Justyniak, J. Pogrebetsky, M. Lemieszka, P. Bernatowicz, A. Pietrzykowski, A. Kubas and J. Lewiński, Chem. Commun., 2024, 60, 9392–9395 RSC.
- A. Zecchina, L. Marchese, S. Bordiga, C. Pazè and E. Gianotti, J. Phys. Chem. B, 1997, 101, 10128–10135 CrossRef CAS.
- Q. Zhao, W.-H. Chen, S.-J. Huang, Y.-C. Wu, H.-K. Lee and S.-B. Liu, J. Phys. Chem. B, 2002, 106, 4462–4469 CrossRef CAS.
- C. Bornes, C. F. G. C. Geraldes, J. Rocha and L. Mafra, Microporous Mesoporous Mater., 2023, 360, 112666 CrossRef CAS.
- S. Hayashi, Anal. Sci., 2009, 25, 133–136 CrossRef CAS PubMed.
- E. F. Rakiewicz, A. W. Peters, R. F. Wormsbecher, K. J. Sutovich and K. T. Mueller, J. Phys. Chem. B, 1998, 102, 2890–2896 CrossRef CAS.
- W. Hu, Q. Luo, Y. Su, L. Chen, Y. Yue, C. Ye and F. Deng, Microporous Mesoporous Mater., 2006, 92, 22–30 CrossRef CAS.
- S. Hayashi, Chem. Lett., 2009, 38, 960–961 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cc00004a |
| This journal is © The Royal Society of Chemistry 2025 |