
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of α-arylacetophenone derivatives by Grignard reactions and transformations of arynes via C–C bond cleavage†
Yukitaka
Hoshi
,
Shinya
Tabata
and
Suguru
Yoshida
 *
*
Department of Biological Science and Technology, Faculty of Advanced Engineering, Tokyo University of Science, 6-3-1 Niijuku, Katsushika-ku, Tokyo 125-8585, Japan. E-mail: s-yoshida@rs.tus.ac.jp
First published on 3rd February 2025
Abstract
Divergent synthesis of α-arylacetophenones by 1,2-addition and subsequent transformations via aryne intermediates is disclosed. The choice of solvent played a critical role in the efficient synthesis of benzocyclobutenols by the Grignard reaction. The generation of arynes from the resulting alcohols via the C–C bond cleavage facilitated the synthesis of a diverse array of α-arylacetophenones.
Diverse α-aryl ketones such as zaltoprofen1a and chilenine1b are of great importance in broad research fields including pharmaceutical sciences and natural product chemistry (Fig. 1A). Despite continuous efforts to develop novel efficient synthesis methods for α-aryl ketones such as palladium-catalyzed α-arylation of ketones (Fig. 1B), it is still challenging to prepare highly functionalized α-aryl ketones from simple starting materials due to the poor functional group tolerance under harsh conditions.2 Herein, we disclose efficient preparation of α-aryl ketones through the Grignard reactions of benzocyclobutenones and subsequent aryne reactions via C–C bond cleavage.
 | ||
| Fig. 1 (A) Significant α-aryl ketones. (B) Palladium-catalyzed synthesis of α-aryl ketones. (C) Li's work. (D) Our previous studies. (E) This work. | ||
Transformations through aryne intermediates via C–C bond cleavage were independently developed by the Li group and our group.3–5 In 2017, Li and coworkers accomplished the synthesis of α-arylacetic acid esters via the generation of aryne intermediates through the C–C cleavage (Fig. 1C).4 We succeeded in transformations of α-aryl-ketone-type arynes generated via the C–C bond cleavage.5 Benzocyclobutenones used as the aryne precursors were easily synthesized from 1,3-bis(triflyloxy)-2-iodobenzene via 3-(triflyloxy)benzyne by our previously developed method (Fig. 1D).6 Particularly, in 2017, we found that treatment of benzocyclobutenones with organolithiums in the presence of arynophiles such as furan furnished α-aryl ketones by the three-component coupling, in which 1,2-addition of ketones with organolithiums and aryne generation from the resulting alkoxide intermediates took place smoothly.5a To avoid the poor functional group tolerance of organolithium reagents, we conceived that diverse alcohols can be synthesized from benzocyclobutenones 2 with organomagnesiums prepared using the turbo Grignard reagent7,8 (Fig. 1E). Since the turbo Grignard reagent have led to synthesis of divergent products that contain various nucleophile-susceptive moieties such as esters, we expected that synthesizing alcohols from 2 and aryl halides 1 followed by aryne reactions via the C–C bond cleavage will enable us to prepare a wide variety of α-aryl ketones under the good transformability of aryne intermediates. Due to the weak C–C bond of benzocyclobutene derivatives4,5 and strict limitation in choosing solvents for the halogen–magnesium exchange and following 1,2-addition,7,8 it is challenging to accomplish the facile synthesis of alcohols.
Optimization of the reaction conditions for 1,2-addition to ketone 2a by an arylmagnesium intermediate prepared from 1a enabled the synthesis of alcohol 3a in a practical manner (Table 1). First, we treated 1a with isopropylmagnesium chloride lithium chloride complex in tetrahydrofuran (THF) at −40 °C followed by the addition of 2a at −20 °C (entry 1). After aqueous workup of the resulting mixture, we succeeded in the synthesis of alcohol 3a albeit in low yield, which formed without aryne generation via the C–C bond cleavage. Given that ketone 2a was consumed and a complex mixture of side products was obtained, this result suggested the challenge in synthesizing alcohol 3a owing to the unstable structure of the alkoxide intermediate. Changing the solvent significantly affected the reaction efficiency. Although the reaction using cyclopentyl methyl ether (CPME) instead of THF resulted in a slight improvement of the yield (entry 2), we accomplished the synthesis of alcohol 3a in good yield when using diethyl ether as a solvent (entry 3). Moreover, we found that both the I–Mg exchange and subsequent Grignard reaction smoothly took place in toluene without side reactions owing to the labile C–C bonds, allowing us to prepare alcohol 3a in high yield leaving the ester moiety intact (entry 4). It is worth noting that a gram-scale synthesis of alcohol 3a was achieved in an excellent yield (entry 5). In contrast, the desired alcohol 3a was not obtained by the reaction in acetonitrile since the I–Mg exchange did not proceed owing to the deprotonation from the solvent (entry 6).

We accomplished the synthesis of a wide range of benzocyclobutenols 3a–3h from ketone 2a and various aryl halides 1 (Fig. 2A and B). For example, the I–Mg exchange of ethyl 3-iodobenzoate (1b) with the turbo Grignard reagent followed by the 1,2-addition to ketone 2a proceeded smoothly to afford alcohol 3b in high yield. Also, we realized the synthesis of alcohol 3c bearing cyano group through the preparation of the 4-cyanophenylmagnesium intermediate at 0 °C and following the Grignard reaction. Of note, the preparation of a broad variety of benzocyclobutenols 3d–3f having iodo, fluoro, and bromo groups was achieved using the corresponding aryl halides 1d–1f. Selective magnesiation of dibromides 1g and 1h and subsequent 1,2-addition to ketone 2a resulted in the efficient synthesis of alcohols 3g and 3h, respectively, by the heteroarylations without damaging remaining bromo groups. These good functional group tolerances are obvious advantages over our previous study using organolithium reagents.5a
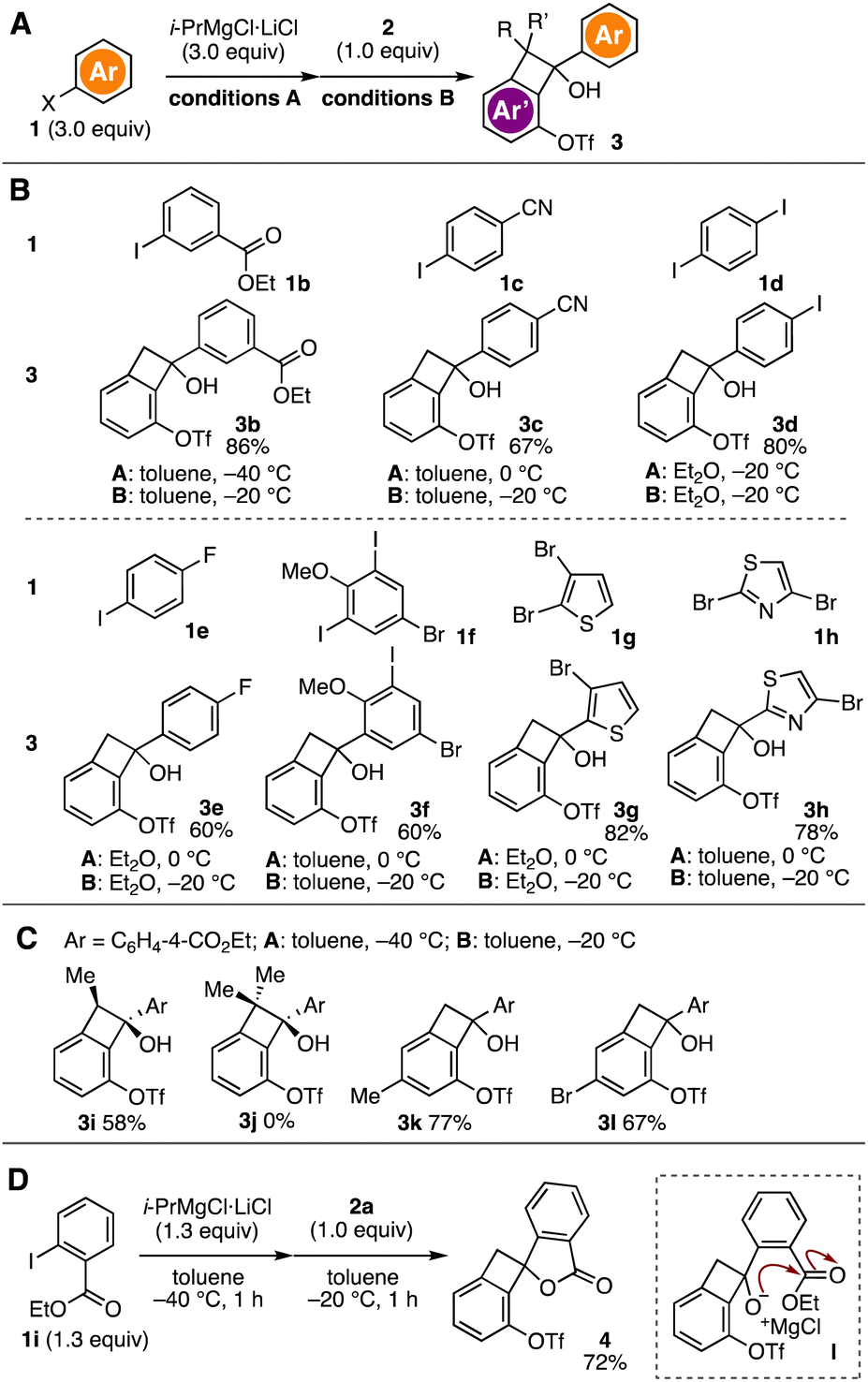 | ||
| Fig. 2 Synthesis of benzocyclobutenol derivatives. (A) General scheme. (B) Synthesis using various aryl halides 1. (C) Synthesis using various ketones 2. See the ESI,† for the structures of 2b–2e. (D) Synthesis of lactone 4. | ||
Various benzocyclobutenones 2 successfully participated in the alcohol synthesis with ethyl 4-iodobenzoate (1a) (Fig. 2A and C). For instance, after I–Mg exchange of 1a, treatment of α-methyl-substituted ketone 2b with the resulting arylmagnesium intermediate provided the alcohol 3i in a diastereoselective manner. The stereochemistry was confirmed by the nuclear Overhauser effect spectroscopy (NOESY) experiment.9 Unfortunately, arylation of α,α-dimethyl-substituted ketone 2c did not take place probably due to the steric hindrance. We accomplished the synthesis of alcohols 3k and 3l from ketones 2d and 2e, which were prepared from 5-methyl- and 5-bromo-substituted 1,3-bis(triflyloxy)-2-iodobenzene by [2+2] cycloaddition of 3-(triflyloxy)aryne intermediates.6
We achieved the synthesis of spirocyclic lactone 4 from ethyl 2-iodobenzoate (1i) and ketone 2a (Fig. 2D). Indeed, treatment of 1i with the turbo Grignard reagent followed by the addition of ketone 2a furnished lactone 4 in good yield. The lactone formation would involve the 1,2-addition and following cyclization of the resulting alkoxide intermediate I.
A wide range of α-arylacetophenone derivatives were synthesized by the base-promoted aryne generation from alcohol 3a through the C–C bond cleavage (Fig. 3A). Among various bases screened, potassium carbonate facilitated the aryne generation to afford cycloadduct 6 with 2,5-dimethylfuran (5) in good yield. Furthermore, we found cycloadduct 6 was efficiently synthesized with cesium fluoride as an activator.9 With optimized conditions for aryne generation in hand, we succeeded in the preparation of highly functionalized α-arylacetophenones via the aryne intermediate (Fig. 3B and C). For example, N-arylation of the aryne generated from alcohol 3a proceeded smoothly to provide 8a–8c when using N-methylaniline (7a), morpholine (7b), or 4-(ethoxycarbonyl)piperidine (7c), where meta adducts were obtained as major isomers.10 We succeeded in the synthesis of cycloadduct 10 by the [2+3] cycloaddition of the aryne intermediates with azide 9, in which the regioisomer was not observed.11 The preparation of silyl acetal 12 was realized by the aryne-generation-[2+2]-cycloaddition sequence from 3a and ketene silyl acetal 11 without the regioisomer.12 To our surprise, side products generated via the desilylation from 12 were not observed although cesium fluoride was used as an activator. In addition, the amination of the aryne intermediate generated from methyl-substituted benzocyclobutenol 3i took place efficiently using morpholine (7b) as an arynophile. Moreover, we achieved the synthesis of acridine 14 from alcohol 3a and 2-benzoylaniline (13) in one-step via nucleophilic attack of aniline 13 to the aryne intermediate, cyclization of the resulting carbanion, and dehydration (Fig. 3D).13 In the reaction, meta-aminated side product 15 was obtained in low yield, showing protonation of the anionic intermediate also took place.
 | ||
Fig. 3 (A) Aryne generation from 3a. (B) General scheme for transformations of 3via aryne intermediates. (C) Synthesis of various α-arylacetophenone derivatives. See the ESI,† for the structures of arynophiles. (D) Synthesis of acridine 14. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Yields based on 1H NMR analysis. bIsolated yields. Yields based on 1H NMR analysis. bIsolated yields. | ||
We have developed a modular synthetic route to α-arylacetophenone derivatives from 3-(triflyloxy)aryne precursors, aryl halides, and arynophiles via two types of aryne intermediates such as II and III (Fig. 4). First, treatment of aryne precursor 16 with (trimethylsilyl)methylmagnesium chloride in the presence of ketene silyl acetal 11 and following formation of the carbonyl group with boron trifluoride diethyl ether complex provided the corresponding benzocyclobutenone via aryne II in one-pot fashion according to our previous study.6b Second, benzocylobutenol synthesis was accomplished using 4-iodobenzonitrile (1c) through the I–Mg exchange and subsequent Grignard reaction, wherein the structural integrity of hydroxy, methyl, triflyloxy, and cyano groups remained intact. Third, we succeeded in the synthesis of benzotriazole 17 through the generation of aryne intermediate III followed by [2+3] cycloaddition with azide 9. Thus, the modular synthesis allowed preparing α-arylacetophenone 17 from four components 16, 11, 1c, and 9 in short steps. Due to the great transformability of aryne intermediates, this modular synthetic route will serve in the construction of α-arylacetophenone derivatives by combinatorial chemistry.
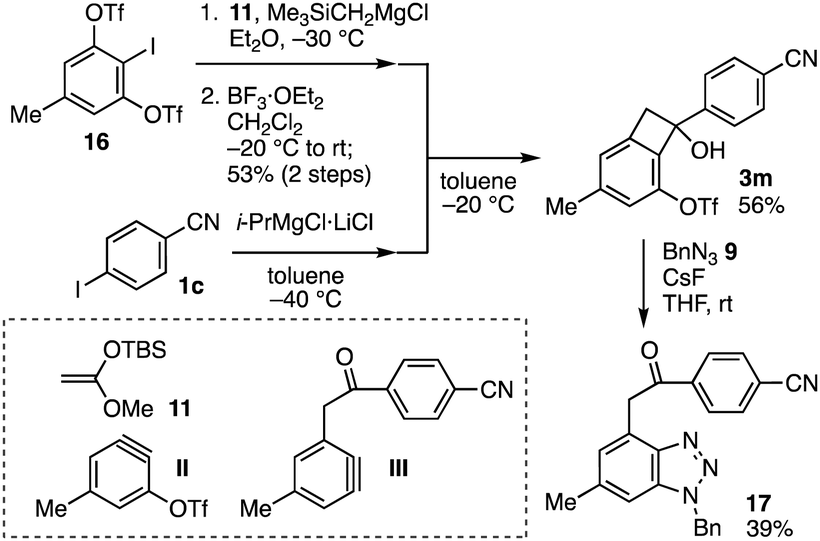 | ||
| Fig. 4 Modular synthesis of α-arylacetophenone derivative 17. | ||
A synthetic advantage for highly functionalized α-arylacetophenone derivatives was demonstrated by benzo[de]chromene synthesis from alcohol 3a in only two steps (Fig. 5A). Indeed, acylalkylation of the aryne intermediate IV generated from alcohol 3a with acetylacetone (18) in boiling THF efficiently proceeded to afford hemiacetal 19 in moderate yield without damaging ester moiety along with a complex mixture of side products (Fig. 5A and B).14 The unique straightforward formation of the benzo[de]chromene skeleton would be realized via acylalkylated product 21 and following twice cyclizations of intermediates V and VI. Further dehydration of 19 with triflic acid smoothly occurred to provide benzo[de]chromene 20 in good yield. Since benzo[de]chromene was synthesized from 3-(triflyloxy)benzyne precursor, ketene silyl acetal 11, ethyl 4-iodobenzoate (1a), and acetyl acetone (18) in short steps, a wide variety of benzo[de]chromene derivatives would be prepared by changing simple modules, whereby will serve in broad disciplines including natural product chemistry and pharmaceutical sciences.15
 | ||
| Fig. 5 (A) Synthesis of benzo[de]chromene derivatives. (B) Plausible reaction mechanism. Ar = C6H4-4-(CO2Et). | ||
In summary, we have developed a facile synthetic route to α-arylacetophenone derivatives by the Grignard reaction of ketones 2 and subsequent transformations of aryne intermediates through C–C bond cleavage. While changing solvents significantly affected the efficiency of alcohol synthesis via the I–Mg exchange and subsequent 1,2-addition, we succeeded in the facile synthesis of alcohols 3 using toluene as a solvent without side reactions through the cleavage of labile C–C bonds. Good functional group tolerance of the synthesis with a turbo Grignard reagent and divergence of aryne intermediates will contribute to the synthesis of various molecules that contain α-aryl ketone scaffolds. Further studies including diverse α-arylacetophenone synthesis using various arynophiles16 and detailed mechanistic studies on the unique aryne generation via the C–C cleavage are ongoing in our research group.
The authors thank Central Glass Co., Ltd. for providing Tf2O and TfOH. This work was supported by JSPS KAKENHI Grant Number JP23K23354 (S. Y.) and Tokuyama Science Foundation (S. Y.).
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) K. Hirate, A. Uchida and Y. Ogawa, J. Neurosci. Res., 2006, 54, 288 CrossRef CAS PubMed; (b) V. Fajardo, V. Elango, B. K. Cassels and M. Shamma, Tetrahedron Lett., 1982, 23, 39 CrossRef CAS.
- (a) J. M. Fox, X. Huang, A. Chieffi and S. L. Buchwald, J. Am. Chem. Soc., 2000, 122, 1360 CrossRef CAS; (b) A. Ehrentraut, A. Zapf and M. Beller, Adv. Synth. Catal., 2002, 344, 209 CrossRef CAS.
- For selected reviews of aryne chemistry, see: (a) P. M. Tadross and B. M. Stoltz, Chem. Rev., 2012, 112, 3550 CrossRef CAS PubMed; (b) S. Yoshida and T. Hosoya, Chem. Lett., 2015, 44, 1450 CrossRef CAS; (c) A. E. Goetz, T. K. Shah and N. K. Garg, Chem. Commun., 2015, 51, 34 RSC; (d) J.-A. García-López and M. F. Greaney, Chem. Soc. Rev., 2016, 45, 6766 RSC; (e) F. I. M. Idiris and C. R. Jones, Org. Biomol. Chem., 2017, 15, 9044 RSC; (f) H. Takikawa, A. Nishii, T. Sakai and K. Suzuki, Chem. Soc. Rev., 2018, 47, 8030 RSC; (g) T. Roy and A. T. Biju, Chem. Commun., 2018, 54, 2580 RSC; (h) T. Matsuzawa, S. Yoshida and T. Hosoya, Tetrahedron Lett., 2018, 59, 4197 CrossRef CAS; (i) J. Shi, L. Li and Y. Li, Chem. Rev., 2021, 121, 3892 CrossRef CAS PubMed; (j) H. Hazarika and P. Gogoi, Org. Biomol. Chem., 2021, 19, 8466 RSC; (k) R. Zhang, X. Peng and J. Tan, Synthesis, 2022, 5064 CAS; (l) N. Kim, M. Choi, S.-E. Suh and D. M. Chenoweth, Chem. Rev., 2024, 124, 11435 CrossRef CAS PubMed.
- J. Shi, H. Xu, D. Qiu, J. He and Y. Li, J. Am. Chem. Soc., 2017, 139, 623 CrossRef CAS PubMed.
- (a) K. Uchida, S. Yoshida and T. Hosoya, Org. Lett., 2017, 19, 1184 CrossRef CAS PubMed; (b) K. Uchida, Y. Minami, S. Yoshida and T. Hosoya, Org. Lett., 2019, 21, 9019 CrossRef CAS PubMed.
- (a) S. Yoshida, K. Uchida, K. Igawa, K. Tomooka and T. Hosoya, Chem. Commun., 2014, 50, 15059 RSC; (b) K. Uchida, S. Yoshida and T. Hosoya, Synthesis, 2016, 4099 CAS.
- A. Krasovskiy and P. Knochel, Angew. Chem., Int. Ed., 2004, 43, 3333 CrossRef CAS PubMed.
- For selected reviews of turbo Grignard reagents, see: (a) R. L.-Y. Bao, R. Zhao and L. Shi, Chem. Commun., 2015, 51, 6884 RSC; (b) D. S. Ziegler, B. Wei and P. Knochel, Chem. – Eur. J., 2019, 25, 2695 CrossRef CAS PubMed.
- See the ESI† for details.
- Z. Liu and R. C. Larock, Org. Lett., 2003, 5, 4673 CrossRef CAS PubMed.
- F. Shi, J. P. Waldo, Y. Chen and R. C. Larock, Org. Lett., 2008, 10, 2409 CrossRef CAS PubMed.
- (a) R. V. Stevens and G. S. Bisacchi, J. Org. Chem., 1982, 47, 2393 CrossRef CAS; (b) T. Hosoya, T. Hasegawa, Y. Kuriyama, T. Matsumoto and K. Suzuki, Synlett, 1995, 177 CrossRef CAS.
- D. C. Rogness and R. C. Larock, J. Org. Chem., 2010, 75, 2289 CrossRef CAS PubMed.
- H. Yoshida, M. Watanabe, J. Ohshita and A. Kunai, Chem. Commun., 2005, 3292 RSC.
- For example of benzochromene synthesis, see: P. Hu, Y. Zhang, Y. Xu, S. Yang, B. Liu and X. Li, Org. Lett., 2018, 20, 2160 CrossRef CAS PubMed.
- When using aniline, dodecane thiol, thiophenol, or p-cresol as an arynophile under the conditions shown in Fig. 3C, a complex mixture of products were obtained.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization for new compounds including NMR spectra. See DOI: https://doi.org/10.1039/d4cc06769j |
| This journal is © The Royal Society of Chemistry 2025 |