
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective hydrogenation of nitrocyclohexane to cyclohexanone oxime over a Cu12Ag17(SR)12(PPh3)4 cluster catalyst†
Jinzhi
Lu
,
Danyun
Yao
,
Shisi
Tang
,
Xiao
Cai
,
Weiping
Ding
 and
Yan
Zhu
*
and
Yan
Zhu
*
State Key Laboratory of Coordination Chemistry, Key Laboratory of Mesoscopic Chemistry of Ministry of Education, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing 210093, China. E-mail: zhuyan@nju.edu.cn
First published on 17th February 2025
Abstract
Atomically precise metal cluster catalysts with crystallographically solved structures have been documented to be promising alternatives to tackle the challenge of selective hydrogenation of organics into high value products. Here we report that a Cu12Ag17(SR)12(PPh3)4 (SR = 1,3-benzenedithiol) cluster as a heterogeneous catalyst can achieve a highly catalytic selectivity of cyclohexanone oxime in catalytic hydrogenation of nitrocyclohexane, due to the unique synergy in the bimetallic cluster.
It is of paramount importance to develop sustainable green chemical processes to replace traditional chemical processes. Cyclohexanone oxime (CHO) is a key intermediate in the generation of caprolactam, which is an important precursor in the production of nylon fibers, engineering plastics and plastic films.1,2 Currently, the industrial production of CHO mainly relies on the reaction of cyclohexanone (CHone) and hydroxylamine salt, while the process has many problems such as low atomic utilization, high energy consumption and serious environmental pollution.3,4 Therefore, there is an urgent requirement to develop green, environmentally friendly and efficient technologies for CHO production. The direct reduction of nitrocyclohexane (NCH) to CHO has attracted extensive research due to its high atom economy, easy operation and environmental friendliness.5–7 The difficulties of this above technology include the design of high-performance catalysts and the resolution of the contradiction between the conversion of the reactants and the selectivity of the target products.4 Initially, the hydrogenation of NCH to CHO was dominated by noble metal catalysts, while the high price of noble metals and the scarcity of raw materials limit the industrialization.8,9 Consequently, non-noble metals like Cu and Ni are preferred.3,10 For example, Liu et al. synthesized MOF-derived Ni@C catalysts containing rich defects, in which strong metal–support interaction between the defect-rich carbon and Ni was demonstrated to enhance the dispersion of Ni nanoparticles and create electron-deficient Ni sites, thereby attenuating the adsorption of CHO and leading to high selectivity for CHO.2 Furthermore, the addition of a second metal has also been utilized to modulate the electronic and chemical properties of the metals to improve their activities. Luo et al. prepared a series of activated carbon supported bimetallic Ni-based catalysts, in which the supported CuNi catalyst exhibited highly catalytic performances, since the Cu could effectively improve the reduction of nickel oxides and the dispersion of nickel, and finally CuNi could effectively activate the N–O bond in NCH and facilitate the desorption of CHO as compared with Ni.11 What's more, N doping was utilized to improve the basic properties of the catalysts, which in turn led to an enhanced activity.6 Besides, the ethylenediamine (EDA)-modified Pt/In2O3 catalyst gave rise to 100% selectivity in EDA solvent.9 However, 100% CHO selectivity has not yet been attained by non-Pt-group metal catalysts.
Atomically precise metal clusters with exact atomic numbers and crystal structures have been demonstrated to be remarkable in understanding fundamental catalysis, exhibiting unique catalytic performances in many selective hydrogenation reactions.12–16 These metal clusters with small sizes are typically different from conventional metal nanoparticles or metal complex catalysts and they have been demonstrated to tackle the challenge of selective hydrogenation processes into high value products.17–19 For example, Au36(SR)24 cluster catalysed the hydrogenation of nitrobenzene in low sulfuric acid content medium to produce exclusively p-aminophenol.19 In this work, we utilized a Cu12Ag17(SR)12(PPh3)4 (SR = 1,3-benzenedithiol; PPh3 = triphenylphosphine) cluster to catalyse the hydrogenation of NCH to CHO.
The Cu12Ag17(SR)12(PPh3)4 cluster, denoted as Cu12Ag17, was synthesized based on the previously reported method and its atomic-pattern structure is displayed in Fig. 1a.20,21 The total structure of Cu12Ag17 is composed of an Ag13 icosahedral kernel, four Cu3(SR)3 units and four AgPPh3 units. In the Cu3(SR)3 units, one S atom in each bidentate ligand is attached to a Cu atom, constituting a staple conformation of S–Cu–S–Cu–S–Cu. Each PPh3 ligand is connected to an Ag atom and the Ag atom is connected to the second S atom in the bidentate ligand to form a tetrahedrally oriented cage-like wrapped Ag13 icosahedron.
 | ||
| Fig. 1 (a) Total structure of the Cu12Ag17(SR)12(PPh3)4 cluster. Color codes: green = Cu; pink = Ag; yellow = S; pale blue = P; gray = C. The H atoms are omitted for clarity. (b) Comparison of Cu12Ag17 with recently reported catalysts based on the CHO selectivity. | ||
The hydrogenation process of NCH to CHO mainly involves the hydrogenation of NCH, removing one molecule of H2O to produce nitrosocyclohexane (NSCH), and transferring the α-H to produce CHO. We first explored the catalytic performances of Cu12Ag17 cluster catalysts for the hydrogenation of NCH performed in various solvents. It was found that CHO could give rise to a ∼100% selectivity of CHO in the EDA solvent (Fig. S1, ESI†). CHone was generally obtained by hydrolysis of CHO, suggesting that EDA can promote the oximization and inhibit the hydrolysis of CHO.9 Moreover, as shown in Fig. 2a–c, Cu12Ag17 always exhibited ∼100% selectivity for CHO product performed in a series of reaction conditions, including the reaction temperature, reaction time and hydrogen pressure. This is the first time that ∼100% selectivity of CHO has been achieved on non-Pt-group metal catalysts (Fig. 1b and Table S1, ESI†). Additionally, in the catalytic test, the Cu12Ag17 cluster catalyst was filtered out after the reaction was completed and reused in a new reaction. As shown in Fig. 2d, the recycled cluster catalysts exhibited similar catalytic performances after the third test and the nitrocyclohexane conversion slightly increased during the second and third tests, possibly as the adsorption of ethylenediamine on the catalyst led to basicity increase of the catalyst, improving the reaction.
 | ||
| Fig. 2 Catalytic performances of Cu12Ag17 catalysts for the hydrogenation of NCH under different reaction conditions: (a) reaction temperature, (b) reaction time and (c) H2 pressure. (d) Recyclability of Cu12Ag17 catalysts in terms of the activity and CHO selectivity. Reaction conditions: 3 mg catalyst, 0.3 mmol NCH, 5 mL EDA, 3 MPa H2, 100 °C for 20 h. All the data were the average of the three reactions. | ||
We next investigated the potential reaction mechanism to reveal the reasons for highly selective hydrogenation of NCH to CHO over the Cu12Ag17 cluster catalyst. The UV-vis spectrum of the spent cluster catalyst showed that the peak at 365 nm disappeared and the peak at 441 nm shifted to 447 nm (Fig. S2, ESI†), compared to that of fresh clusters. The spent catalyst after the catalytic reaction was washed several times with methanol to obtain spent Cu12Ag17 washed catalyst. For the sample washed, its UV-vis spectrum showed that the peak at 365 nm appeared and the peak at 447 nm shifted to 443 nm (Fig. S2, ESI†). These observations showed that the Cu12Ag17 clusters were robust during the reaction. Furthermore, the UV-vis spectra of Cu12Ag17 clusters in EDA solvent over time were carried out, which suggested that, as shown in Fig. 3a, the interaction of EDA and the cluster possibly led to changes in the difference of UV-vis spectra between fresh and spent catalysts. Time-resolved in situ infrared Fourier transform (FTIR) spectra of Cu12Ag17 clusters performed in a gas–solid-phase reaction cell at 100 °C were also carried out, which showed that the Cu12Ag17 clusters had an excellent structure stability (Fig. 3b).
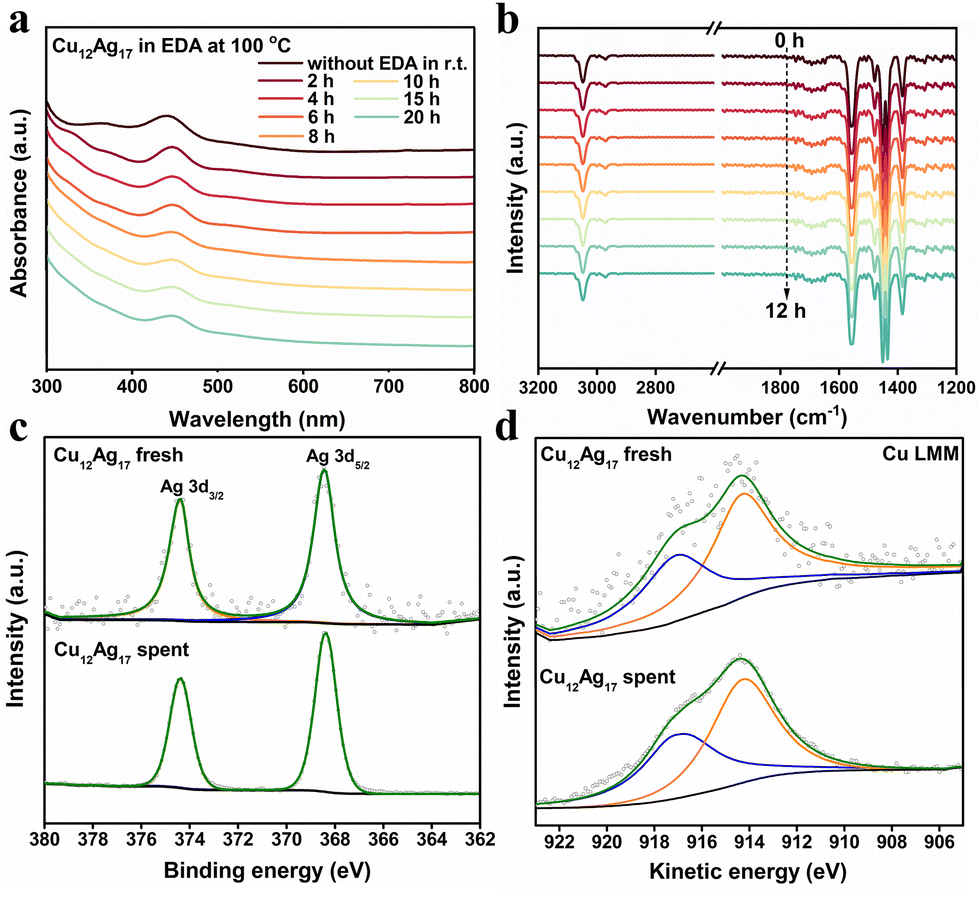 | ||
| Fig. 3 (a) UV-vis spectra of Cu12Ag17 clusters at 100 °C in EDA solution. (b) In situ FTIR spectra of Cu12Ag17 clusters at 100 °C in N2. (c) Ag 3d XPS profiles of fresh and spent Cu12Ag17 clusters. (d) Cu LMM XAES of fresh and spent Cu12Ag17 clusters. | ||
Notably, EDA was adsorbed onto the surface of the Cu12Ag17 catalyst during the reaction, which was confirmed by FTIR studies. Deduced from the FTIR spectra of fresh Cu12Ag17 and spent Cu12Ag17 catalysts, as shown in Fig. S3 (ESI†), the band at 1597 cm−1 assigned to NH2 bending vibration appeared on the spent Cu12Ag17 sample,22 suggesting that EDA was adsorbed on the catalyst after the reaction. The N 1s XPS data showed that there were N-containing species from the reaction system adsorbed on the spent catalyst (Fig. S4, ESI†). It was noteworthy that the band at 1434 cm−1 assigned to the stretching vibration of triphenylphosphine ligand of the Cu12Ag17 catalyst almost disappeared after the reaction (Fig. S3b, ESI†),23 while the peak of P 2p was almost invisible on the X-ray photoelectron spectra (XPS) of the spent Cu12Ag17 catalyst (Fig. S5, ESI†). These results suggested that PPh3 was detached from the catalyst during the reaction. Since Ag sites were exposed after the PPh3 ligands were removed from the cluster, it was likely that EDA adsorbed onto the Ag sites. There was no significant change in the binding energy of Ag 3d of the spent Cu12Ag17 catalyst, compared to the fresh sample (Fig. 3c), implying that the Ag sites might be coordinated by ligands or EDA.24 It was reported that the N adsorption onto the catalyst surface might enhance the Lewis basicity of the catalyst, thereby promoting CHO synthesis.6 For the Cu LMM XAES (X-ray induced Auger electron spectra) of the catalysts (Fig. 3d), the broad peaks were divided into two peaks located at around 917.10 eV and 914.31 eV, which were assigned to Cu0 and Cu+ species, respectively.25 There was a slight increase in Cu0 species on the spent Cu12Ag17 catalyst, as listed in Table S2 (ESI†), compared to the fresh one.
The formation of CHO by α-H transfer on NSCH and the formation of cyclohexylamine by hydrogenation of NSCH are the two competing pathways during NCH hydrogenation.3In situ FTIR spectra of the NCH hydrogenation over the Cu12Ag17 cluster catalyst with 3 MPa H2 for 60 min via a gas–solid-phase reaction cell are shown in Fig. 4a and b. The bands at 1374 and 1545 cm−1 were assigned to the asymmetric stretching vibration of the NO2 group of nitrocyclohexane, which were blue-shifted after 60 min of the reaction, indicating that the nitrocyclohexane adsorbed onto the cluster catalyst was being converted (Fig. 4d).3 As the reaction proceeded, the bands of nitrocyclohexane gradually weakened and a new small peak appeared at 1690 cm−1 belonging to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching vibration of the oxime, suggesting that the CHO was produced (Fig. 4a and b).3,26 It was noted that cyclohexylamine and cyclohexanone can be condensed to form condensation by-products, while IR signals from the condensation species were not detected, ruling out the generation of condensation by-products.2,10 In addition, no other by-products were detected. The results confirmed that the Cu12Ag17 catalyst was able to adsorb and activate NCH and promoted the isomerization of NSCH, which led to an excellent selectivity of CHO. The in situ FTIR spectra for the interaction of the Cu12Ag17 catalyst with H2 at room temperature are shown in Fig. 4c, where a peak assigned to a Cu–H signal at 1044 cm−1 appeared and gradually intensified with the increase of the contact time with H2.27 The peak remained visible after 30 min of N2 purging, suggesting that the Cu atoms of the Cu12Ag17 catalyst served as the active sites for H2 adsorption and activation. This could possibly account for more Cu0 species on the catalyst after the reaction (Table S2, ESI†). Therefore, the Cu sites in Cu12Ag17 were responsible for the activation of hydrogen, while the adsorption of EDA on the Ag sites promoted the conversion of NCH, constituting a two-site synergy of Cu and Ag in this bimetallic cluster.
N stretching vibration of the oxime, suggesting that the CHO was produced (Fig. 4a and b).3,26 It was noted that cyclohexylamine and cyclohexanone can be condensed to form condensation by-products, while IR signals from the condensation species were not detected, ruling out the generation of condensation by-products.2,10 In addition, no other by-products were detected. The results confirmed that the Cu12Ag17 catalyst was able to adsorb and activate NCH and promoted the isomerization of NSCH, which led to an excellent selectivity of CHO. The in situ FTIR spectra for the interaction of the Cu12Ag17 catalyst with H2 at room temperature are shown in Fig. 4c, where a peak assigned to a Cu–H signal at 1044 cm−1 appeared and gradually intensified with the increase of the contact time with H2.27 The peak remained visible after 30 min of N2 purging, suggesting that the Cu atoms of the Cu12Ag17 catalyst served as the active sites for H2 adsorption and activation. This could possibly account for more Cu0 species on the catalyst after the reaction (Table S2, ESI†). Therefore, the Cu sites in Cu12Ag17 were responsible for the activation of hydrogen, while the adsorption of EDA on the Ag sites promoted the conversion of NCH, constituting a two-site synergy of Cu and Ag in this bimetallic cluster.
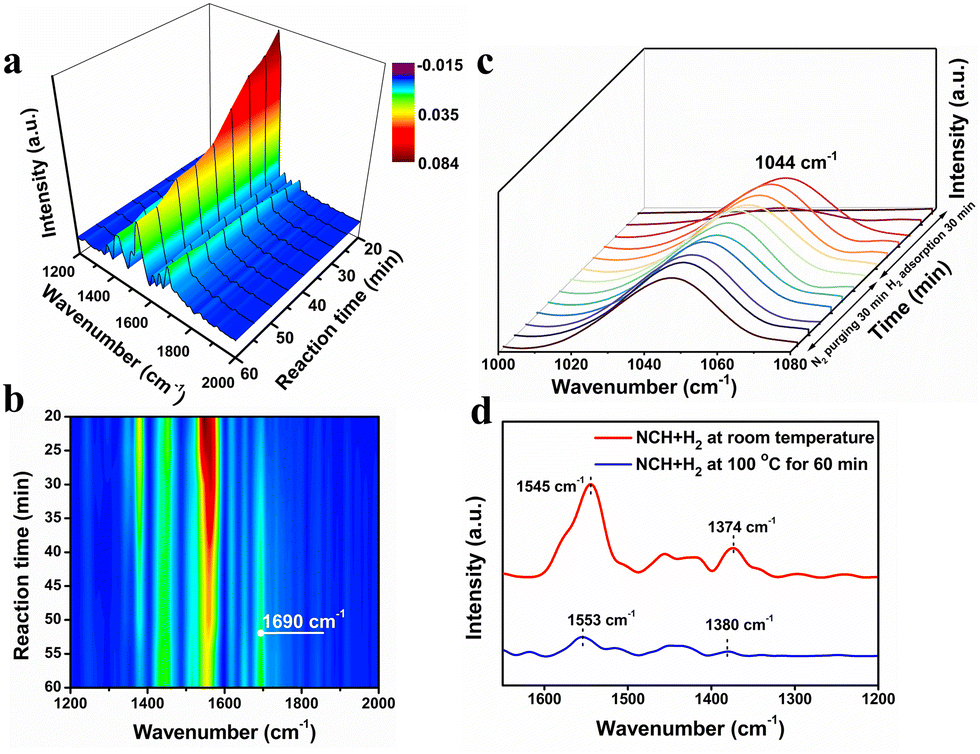 | ||
| Fig. 4 (a) and (b) Time-resolved in situ FTIR spectra of the intermediates and products formed on Cu12Ag17 clusters for NCH hydrogenation with 3 MPa H2 at 100 °C. (c) FTIR spectra collected in the Cu12Ag17 clusters with H2 at room temperature and then N2 purging for 30 min. (d) FTIR spectra of NCH with H2 at room temperature and NCH with H2 at 100 °C for 60 min on the Cu12Ag17 clusters, respectively. | ||
In summary, we report that an atomically precise Cu12Ag17(SR)12(PPh3)4 cluster as a heterogeneous catalyst can exhibit an excellent catalytic performance in the hydrogenation of NCH to yield CHO, which can be comparable to the high-performance Pt-group metal catalysts. The high selectivity of CHO from the hydrogenation of NCH is mainly attributed to the unique synergy in this Cu12Ag17(SR)12(PPh3)4 cluster, in which the Ag sites might bind with the EDA molecules and the Cu sites may be responsible for the adsorption and activation of hydrogen. This work can provide a distinct insight into catalytic hydrogenation of atomically precise cluster catalysts, which is different from conventional metal nanoparticles. Atomically precise metal catalysts are promising alternatives to tackle the challenge of selective hydrogenation processes into high value products.
We acknowledge financial support from the National Natural Science Foundation of China (22125202, 92461312, U24A20487, 92361201) and the Natural Science Foundation of Jiangsu Province (BK20220033).
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- P. Serna, M. Lopez-Haro, J. Calvino and A. Corma, J. Catal., 2009, 263, 328–334 Search PubMed.
- P. Yuan, X. Liao, H. Cui, F. Hao, W. Xiong, H. Luo, Y. Lv and P. Liu, ACS Catal., 2023, 13, 3224–3241 CrossRef CAS.
- Y. Zhang, X. Liao, H. Cui, H. Luo, Y. Lv and P. Liu, J. Colloid Interface Sci., 2024, 678, 353–365 CrossRef PubMed.
- E. Kowalewski and A. Srebowata, Catal. Sci. Technol., 2022, 12, 5478–5487 RSC.
- F. Yao, S. Liu, H. Cui, Y. Lv, Y. Zhang, P. Liu, F. Hao, W. Xiong and H. Luo, ACS Sustainable Chem. Eng., 2021, 9, 3300–3315 CrossRef CAS.
- P. Yuan, X. Liao, H. Cui, F. Hao, W. Xiong, H. Luo, P. Liu and Y. Lv, Chem. Eng. J., 2023, 455, 115824 Search PubMed.
- Q. Zhang, J. Dong, Y. Liu, Y. Cao, H. He and Y. Wang, Chem. Commun., 2017, 53, 2930–2933 RSC.
- M. Liqiu, L. Xing, L. Guang, Y. Dulin, Y. Kui and L. He, Chem. Ind. Eng. Prog., 2009, 28, 1024–1026 Search PubMed.
- Q. Wu, W. Zhou, H. Shen, R. Qin, Q. Hong, X. Yi and N. Zheng, CCS Chem., 2023, 5, 1215–1224 CrossRef CAS.
- Y. Zhang, X. Liao, H. Cui, H. Luo, Y. Lv and P. Liu, ACS Sustainable Chem. Eng., 2024, 12, 595–609 CrossRef CAS.
- F. Yao, S. Liu, H. Cui, Y. Lv, Y. Zhang, P. Liu, F. Hao, W. Xiong and H. Luo, ACS Sustainable Chem. Eng., 2021, 9, 3300–3315 CrossRef CAS.
- J. Dong, J. Robinson, Z. Gao and L. Wang, J. Am. Chem. Soc., 2022, 144, 12501–12509 CrossRef CAS PubMed.
- C. Liu, H. Abroshan, C. Yan, G. Li and M. Haruta, ACS Catal., 2016, 6, 92–99 CrossRef CAS.
- G. Li, D. Jiang, S. Kumar, Y. Chen and R. Jin, ACS Catal., 2014, 4, 2463–2469 CrossRef CAS.
- G. Li, C. Zeng and R. Jin, J. Am. Chem. Soc., 2014, 136, 3673–3679 CrossRef CAS PubMed.
- G. Li and R. Jin, J. Am. Chem. Soc., 2014, 136, 11347–11354 CrossRef CAS PubMed.
- T. Song, Z. Yao, G. Li, X. Cai, X. Liu, Y. Wang, W. Ding and Y. Zhu, ACS Catal., 2023, 13, 10878–10886 CrossRef CAS.
- D. Yang, W. Pei, S. Zhou, J. Zhao, W. Ding and Y. Zhu, Angew. Chem., Int. Ed., 2020, 59, 1919–1924 CrossRef CAS PubMed.
- J. Lu, K. Tang, G. Qi, C. Juan, J. Xu, Z. Cai, D. Li, X. Cai, X. Liu, M. Chen, W. Ding and Y. Zhu, Chem. Sci., 2024, 15, 15617–15624 RSC.
- X. Kang, H. Abroshan, S. Wang and M. Zhu, Inorg. Chem., 2019, 58, 11000–11009 CrossRef CAS PubMed.
- G. Li, J. Hou, X. Lei, D. Li, E. Yu, W. Hu, X. Cai, X. Liu, M. Chen and Y. Zhu, Angew. Chem., Int. Ed., 2023, 62, e202216735 CrossRef CAS PubMed.
- S. Korpayev, C. Kavakli, S. Colak and A. Kavakli, Cellulose, 2018, 25, 813–828 CrossRef.
- X. Cai, W. Hu, S. Xu, D. Yang, M. Chen, M. Shu, R. Si, W. Ding and Y. Zhu, J. Am. Chem. Soc., 2020, 142, 4141–4153 CrossRef CAS.
- G. Chen, C. Xu, X. Huang, J. Ye, L. Gu, G. Li, Z. Tang, B. Wu, H. Yang, Z. Zhao, Z. Zhou, G. Fu and N. Zheng, Nat. Mater., 2016, 15, 564–569 CrossRef CAS PubMed.
- J. Pang, M. Zheng, C. Wang, X. Yang, H. Liu, X. Liu, J. Sun, Y. Wang and T. Zhang, ACS Catal., 2020, 10, 13624–13629 CrossRef CAS.
- S. Ramalingam, M. Karabacak, S. Periandy, N. Puviarasan and D. Tanuja, Spectrochim. Acta, Part A, 2012, 96, 207–220 CrossRef CAS PubMed.
- C. Lousada, R. Fernandes, N. Tarakina and I. Soroka, Dalton Trans., 2017, 46, 6533–6543 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc06745b |
| This journal is © The Royal Society of Chemistry 2025 |