
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelf-encapsulation or picket-fence? An answer to molecular designs of highly solid-state luminescent conjugated polymers†
Tsuneaki
Sakurai
 *a,
Kyota
Ohkura
a,
Teruyoshi
Ikeda
a,
Rin
Terao
a,
Yoshiki
Kamimura
a,
Mitsuo
Hara
b and
Masaki
Shimizu
a
*a,
Kyota
Ohkura
a,
Teruyoshi
Ikeda
a,
Rin
Terao
a,
Yoshiki
Kamimura
a,
Mitsuo
Hara
b and
Masaki
Shimizu
a
aFaculty of Molecular Chemistry and Engineering, Kyoto Institute of Technology, Hashikami-cho, Matsugasaki, Sakyo-ku, Kyoto 606-8585, Japan. E-mail: sakurai@kit.ac.jp
bFaculty of Engineering and Design, Kagawa University, 2217-20, Hayashi-cho, Takamatsu 761-0396, Japan
First published on 4th February 2025
Abstract
Both self-encapsulation and picket-fence molecular designs are individually applied to three types of phenylene-based conjugated polymers for reducing the interchain interactions and enhancing the solid-state luminescence. We discovered that all the picket-fence polymers show higher photoluminescence quantum yields in solid films than the corresponding self-encapsulated analogues despite their less bulky substituents.
Conjugated polymers (CPs) in the solid state transport both charges and excitons within and between the main chains, which has encouraged their application to electronic and sensing devices.1 Some CPs exhibit fluorescence even in the solid state, making them useful candidates as emissive layers in solution-processable organic light-emitting diodes and transistors.2 However, most CPs in the solid state are weakly emissive due to the interchain exciton transport and their eventual deactivation through strong interactions of the main chains.3 Thus, particular care is needed for the molecular design of solid-state luminescent CPs.
Self-encapsulation is one of the well-known, innovative molecular designs to reduce the interchain interactions.4 This design represents that the conjugated chain is covered with the insulated alkylene straps or molecular hosts, separating the main chains even in the solid state and thus reducing their interactions (Fig. 1a). Several examples of self-encapsulated CPs have been reported to exhibit excellent photoluminescence quantum yields (Φ) in films. Sugiyasu, Takeuchi and coworkers developed a self-encapsulated phenylene unit, which transformed into a series of alternating copolymers showing efficient luminescence.5 The same type of encapsulated structure was further employed by Bronstein et al.6 Terao and coworkers reported solid-state photoluminescent cyclodextrin-encapsulated CPs.7 Meanwhile, “picket fence” is another type of design concept that prevents π–π stacking by using bulky or vertical groups (Fig. 1a), as represented by 2,6-disubstituted phenyl groups, attached to a horizontal π-conjugated plane.8 Its fence-like shape aligns vertically with respect to the π-plane due to the high rotational barrier of the C–C bond between the group and π-plane. Although not clearly denoted as “picket-fence” polymers in the literature, several highly photoluminescent CPs can be classified in this category.9
 | ||
| Fig. 1 (a) Schematic illustrations and (b) molecular structures of self-encapsulated and picket-fenced conjugated polymers. | ||
We were strongly interested in the question of which is more effective for efficient solid-state luminescence, the self-encapsulated design or the picket-fence one (Fig. 1a). However, there has been no research that systematically studies and compares the photophysical properties of both designs of CPs derived from a common main chain. To answer this question, here we report the synthesis and evaluation of self-encapsulated and picket-fence CPs from three phenylene-based one-dimensional (1D) chains – poly(p-phenylene) (PP), poly(p-phenylene vinylene) (PPV) and poly(p-phenylene butadiynylene) (PPB) (Fig. 1b and Fig. S1 in ESI†). The encapsulating unit was two octylene chains, while the picket-fence group is the methyl analogue (Fig. 1b). We disclosed that the picket-fence design is more effective, in spite of the less bulkiness, allowing higher Φ in solid films than the corresponding self-encapsulated CPs. Additionally, we found the unprecedented notion that encapsulating chains, arranged along the 1D conjugated chain, sometimes promote aggregation of CPs via multiple van der Waals interactions of strapping chains and decrease the solid-state luminescence efficiency. This aggregation is more enhanced by the polymers with a higher degree of polymerization (DP).
The full synthetic scheme of self-encapsulated monomers SE-M1, SE-M2 and SE-M3 and picket-fence monomers PF-M1, PF-M2 and PF-M3 (Fig. 2) is shown in Fig. S2 (ESI†). Boronic acids 1 and 2 are the picket-fence unit and precursor of the self-encapsulating unit, respectively, while 3 is a precursor of the diethynyl monomer. The monomers for PPs, SE-M1 and PF-M1, were synthesized according to the earlier report5 except shortening the steps for SE-M1 by using direct Williamson ether synthesis (see the protocols in ESI†). SE-M2 was synthesized via a Suzuki–Miyaura cross-coupling reaction of diethyl 2,5-dibromoterephthalate with 2, followed by reduction of the ethoxycarbonyl groups, deprotection of methoxymethyl groups, strapping with octylene chains, and transformation of the hydroxymethyl groups to bromomethyl groups (Fig. S2, ESI†). PF-M2 was synthesized similarly using compound 1 instead of 2. SE-M3 was synthesized via a Suzuki–Miyaura cross coupling reaction between 2 and 3, followed by the deprotection of the methoxymethyl groups, strapping with octylene chains and deprotection of the triisopropylsilyl groups (Fig. S2, ESI†). PF-M3 was synthesized similarly starting from 1 and 3. The NMR charts of these monomers are listed in Fig. S3–S21 (ESI†).
 | ||
| Fig. 2 Synthetic schemes with isolated yields of self-encapsulated and picket-fenced polymers from corresponding monomers. R = C12H25. | ||
SE-PP and PF-PP (Fig. 1b) were prepared via Suzuki-coupling polymerizations of SE-M1 and PF-M1, respectively, with phenylene-1,4-diboronic acid (Fig. 2). Although the same reaction conditions were employed, the average size of the polymers was larger for PF-PP, judging from the retention time in analytical size-exclusion chromatography (SEC) of the crude polymers. This difference is probably due to the different reactivity of monomers originating from the larger steric effect by the encapsulating units for SE-M1. Thus, SE-PP was purified by preparative SEC to remove the oligomeric small molecular products. As a result, SE-PP and PF-PP were isolated with comparable average molecular weights (Mn and Mw) (Table 1). The Gilch polymerization of SE-M2 and PF-M2 in the presence of tBuOK in PhCl at 110 °C yielded SE-PPV and PF-PPV, respectively (Fig. 2). The molecular weight of PF-PPV is much larger than that of SE-PPV, probably due to the smaller steric effect of picket-fences than encapsulating chains. After reprecipitation, we collected PF-PPV in two fractions by preparative SEC and denoted them as PF-PPVL and PF-PPVS, with relatively large (L) and small (S) Mn, respectively (Table 1). The Mn of SE-PPV is even much smaller than that of PF-PPVS. The PPBs were synthesized via Eglinton coupling of SE-M3 or PF-M3 in the presence of excess Cu(OAc)2 in pyridine (Fig. 2). SE-PPB with enough DP was obtained by using PhCl as a co-solvent and elevating the reaction temperature at 80 °C. PF-PPBL and PF-PPBS with relatively large and small average molecular weights were obtained by more mild conditions using PhCl/pyridine at 60 °C and pyridine at 25 °C, respectively. To our surprise, these PPBs, though having two dodecyl chains per monomer unit and bulky encapsulating or picket-fence units, show low solubility in CHCl3 and PhCl, implying the strong interpolymer interactions. The SEC charts of the polymers are shown in Fig. S22–S29 (ESI†) and the average molecular weights, Mn and Mw, are estimated based on polystyrene standards (Table 1).
| M n/Mw | λ abs/nm | λ em/nm | Φ | ||
|---|---|---|---|---|---|
| SE-PP | 5800/8600 | CHCl3 | 271 | 386 | 0.49 |
| Film | 269 | 387 | 0.32 | ||
| PF-PP | 5800/7900 | CHCl3 | 272 | 384 | 0.61 |
| Film | 272 | 386 | 0.39 | ||
| SE-PPV | 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000/18000 000/18000 |
CHCl3 | 431 | 493, 523 | 0.58 |
| Film | 435 | 496, 531 | 0.28 | ||
| PF-PPVL | 130000/186000 |
CHCl3 | 431 | 484, 515 | 0.79 |
| Film | 441 | 494, 528 | 0.25 | ||
| PF-PPVS | 61000/94000 |
CHCl3 | 431 | 484, 514 | 0.83 |
| Film | 441 | 493, 528 | 0.30 | ||
| SE-PPB | 17800/47200 |
PhCl | 403, 413, 441 | 430, 445, 474, 490 | 0.25 |
| Film | 401, 411, 440 | 446, 475, 491 | 0.09 | ||
| PF-PPBL | 20000/67600 |
PhCl | 404, 414, 441 | 434, 449, 477, 493 | 0.33 |
| Film | 405, 415, 441 | 449, 479, 495 | 0.12 | ||
| PF-PPBS | 7400/13600 |
PhCl | 390, 409 | 437, 461 | 0.38 |
| Film | 401, 414, 439 | 446, 477, 494 | 0.13 |
The absorption and fluorescence spectra of the polymers are shown in Fig. 3, and the related parameters are summarized in Table 1 and Table S1 (ESI†). The fluorescence spectra of SE-PP in CHCl3 and the spin-coated film are basically close to each other, but the spectrum in the film shows a shoulder over 450 nm (Fig. 3a). PF-PP shows a further higher degree of spectral similarity between the solution and film states (Fig. 3b). The observed spectral similarities indicate that the excited-state interactions among the main chains are suppressed in their films. In fact, both SE-PP and PF-PP recorded high Φ values of 0.32 and 0.39 in films. Of interest here, the value of PF-PP is higher than that of SE-PP. SE-PPV exhibited a large Φ of 0.58 in CHCl3 and 0.28 in the film state, where the absorption and emission maxima are almost the same in the solution and film (Fig. 3c). PF-PPVL and PF-PPVS show remarkably bright luminescence in solutions with Φ of 0.79 and 0.83, respectively. These values are much larger than that of the conventional PPV derivatives such as MEH-PPV (poly[2-methoxy-5-(2′-ethylhexyloxy)-1,4-phenylenevinylene])9c (Φ = 0.33 in our measurements). The high DP (Mn = 130000 and 61000) and picket-fence structures leading to the highly planar conformation main chain would contribute to this remarkable luminescence efficiency (see the large radiative rate constant value (kr) in Table S1, ESI†). The values of Φ in a film dropped compared with those in solution and exhibited small red shifts in the absorption and emission maxima (Fig. 3d and e). However, the Φ value of 0.30 observed for the film of PF-PPVS is still higher than those of well-known MEH-PPV9c (Φ ∼ 0.14) and Super Yellow10 (Φ ∼ 0.28) (Fig. S30 and Table S1, ESI†) as well as that of SE-PPV. Our observations so far revealed a new insight – the picket-fence design has an advantage over self-encapsulation on the luminescence efficiency in solid films.
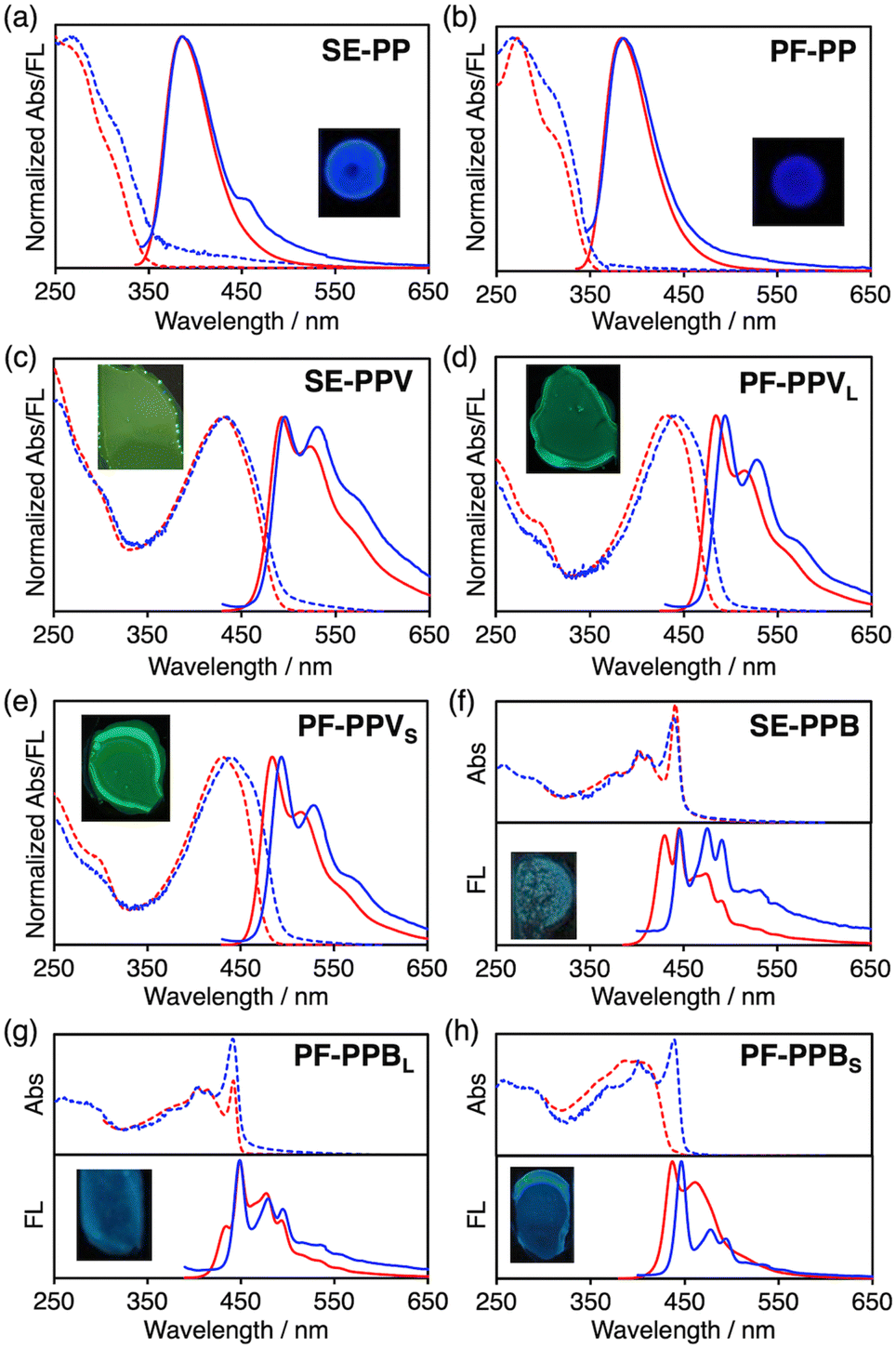 | ||
| Fig. 3 Electronic absorption (dashed) and fluorescence (solid) spectra of (a) SE-PP, (b) PF-PP, (c) SE-PPV, (d) PF-PPVL, (e) PF-PPVS, (f) SE-PPB, (g) PF-PPBL and (h) PF-PPBS in solution (red) and film (blue). λex = 320, 405 and 365 nm for PPs, PPVs and PPBs, respectively. CHCl3 was used for PPs and PPVs, while PhCl for PPBs as solvents. The concentration of the solutions is 2.0 × 10−5 M. Inset shows the photographs of the spin-coated films under 365 nm light. | ||
All the films of SE-PPB, PF-PPBL and PF-PPBS showed Φ values around 0.10. These lower values compared with PP and PPV derivatives originate from both the significant decrease of kr and large nonradiative rate constant (knr) (Table S2, ESI†). The interdigitated aggregates formed in the films may cause the cancellation of the transition dipole moments along the main chain direction and thus decrease kr.11 The large knr can be partly explained by the strong contribution of the intersystem crossing for diyne-containing structures.12 Taking the unexpected low solubility of the PPB derivatives into account, we focused on the aggregation of polymers in diluted solutions. In PhCl solutions, SE-PPB and PF-PPBL display obvious vibronic structures both in their absorption and fluorescence spectra (Fig. 3f and g). The characteristic sharp absorption at around 440 nm corresponds to the highly coplanar conjugated main chains. This absorption band has been observed for the β phase of polyfluorenes and low-temperature solution phase of PPEs,13 where the planarization of the main chains and their inter-chain aggregation work cooperatively. To confirm the aggregation behavior of these two polymers in diluted solutions, dynamic light scattering (DLS) measurements were carried out. These polymers in PhCl give strong light scattering and suggest aggregates with an approximate hydrodynamic radius of 1–10 nm (Fig. 4a and b). Moreover, the absorption band at ∼440 nm decreased with increasing the temperature and eventually disappeared at 100 °C (Fig. 4c and d), while atomic force microscopy confirms the aggregates with a height of 4–10 nm (Fig. S31, ESI†). The observed aggregation behavior has not been expected because we thought the encapsulating chains and picket-fence groups must suppress the π–π stacking. In contrast, PF-PPBS does not show the sharp absorption at ∼440 nm and the vibronic structure is absent (Fig. 3h and 4e). Namely, the value of DP, i.e. the average chain length, significantly affects the strength of interchain interactions. We consider that the aggregates are formed by the interdigitation of the periodically-arranged encapsulating or picket-fence units. As shown in Fig. 4f and Fig. S32 (ESI†), the space between adjacent encapsulating or picket-fence units can be filled with those from other chains. Consequently, the highly planar PPB chains are formed without direct π–π stackings. The stronger aggregation for SE-PPB than PF-PPBL is most likely due to the larger total van der Waals contacts given by the interdigitation of the straps arranged along the long main chain.
 | ||
| Fig. 4 Hydrodynamic radius distributions of (a) SE-PPB and (b) PF-PPBL in PhCl at 2.0 × 10−5 M at 25 °C. VT-absorption spectra of (c) SE-PPB, (d) PF-PPBL and (e) PF-PPBS in PhCl at 30–100 °C on heating. (f) Packing model of two adjacent chains of trimer segments for the SE-PPB aggregate. | ||
The aggregated structures of the polymers were investigated by the powder X-ray diffraction (PXRD) of the reprecipitated polymer solids and wide-angle X-ray scattering (WAXS) of the dropcast films (Fig. S33, ESI†). Some of the observed peaks can be assigned as a periodicity of the interdigitation for PPBs (Fig. S34, ESI†). Meanwhile, PPs and PPVs are revealed to be amorphous, where the aggregates do not have clear structural periodicity. Our studies so far suggest a new insight into the importance of DPs on the solid-state luminescence. Large DP tends to induce strong polymer aggregation via multiple van der Waals contacts, especially stronger for the self-encapsulated design, thus decreasing the solid-state luminescence efficiency.
In summary, a series of 1D CPs with self-encapsulated or picket-fence structures were synthesized, and their solution and solid-state luminescence were characterized. We clarified that the picket-fence derivatives show higher Φ in films even though the steric effect from the picket-fence dimethyl units is smaller than the encapsulating octylene chain. Considering the easier synthesis of picket-fence CPs over self-encapsulated analogues, this finding is quite encouraging. We propose that a dense arrangement of picket-fence units is important to avoid the unfavourable interdigitation of the polymer chains, while moderate DPs possibly enhance the kr by full conjugation of the main chains and avoid significant increase of knr due to the strong parallel stacking of the chains. The highly emissive picket-fence polymers can be synthesized via only 5–7 steps with a sufficient yield in each step. The picket-fence unit used in this study has an advantage in terms of its applicability to other CPs, which will potentially boost future development of solution-processable efficient solid-state emitters from CPs with controlled DPs.
The authors thank Drs Yusuke Tsutsui and Wookjin Choi in Kyoto University for AFM measurements. T. S. acknowledges support from Grant-in-Aid for Scientific Research (No. 23K26628, 23H01935, and 22H04540) from JSPS, and research grants from Kansai Research Foundation for Promotion of Technology and Toshiaki Ogasawara Memorial Foundation.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- S. Rochat and T. M. Swager, ACS Appl. Mater. Interfaces, 2013, 5, 4488 CrossRef CAS PubMed.
- (a) E. Smarsly, D. Daume, R. Tone, L. Veith, E. R. Curticean, I. Wacker, R. R. Schröder, H. M. Sauer, E. Dörsam and U. H. F. Bunz, ACS Appl. Mater. Interfaces, 2019, 11, 3317 CrossRef CAS PubMed; (b) B. Van der Zee, Y. Li, G.-J. A. H. Wetzelaer and P. W. M. Blom, Adv. Mater., 2022, 34, 2108887 CrossRef CAS; (c) Z. Qin, H. Gao, H. Dong and W. Hu, Adv. Mater., 2021, 33, 2007149 CrossRef CAS.
- A. Ruseckas, M. Theander, L. Valkunas, M. R. Andersson, O. Inganäs and V. Sundström, J. Lumin., 1998, 76, 474 CrossRef.
- (a) M. J. Frampton and H. L. Anderson, Angew. Chem., Int. Ed., 2007, 46, 1028 CrossRef CAS PubMed; (b) C. Pan, C. Zhao, M. Takeuchi and K. Sugiyasu, Chem. – Asian J., 2015, 10, 1820 CrossRef CAS PubMed; (c) J. Royakkers and H. Bronstein, Macromolecules, 2021, 54, 1083 CrossRef CAS.
- C. Pan, K. Sugiyasu, Y. Wakayama, A. Sato and M. Takeuchi, Angew. Chem., Int. Ed., 2013, 52, 10775 CrossRef CAS.
- J. Royakkers, A. Minotto, D. G. Congrave, W. Zeng, A. Hassan, A. Leventis, F. Cacialli and H. Bronstein, Chem. Mater., 2020, 32, 10140 CrossRef CAS.
- J. Terao, S. Tsuda, Y. Tanaka, K. Okoshi, T. Fujihara, Y. Tsuji and N. Kambe, J. Am. Chem. Soc., 2008, 131, 16004 CrossRef.
- (a) J. P. Collman, R. R. Gagne, C. Reed, T. R. Halbert, G. Lang and W. T. Robinson, J. Am. Chem. Soc., 1975, 97, 1427 CrossRef CAS PubMed; (b) C. Pan, K. Sugiyasu, J. Aimi, A. Sato and M. Takeuchi, Angew. Chem., Int. Ed., 2014, 53, 8870 CrossRef CAS; (c) H. J. Cho, S. W. Kim, S. Kim, S. Lee, J. Lee, Y. Cho, Y. Lee, T.-W. Lee, H.-J. Shin and C. Song, J. Mater. Chem. C, 2020, 8, 17289 RSC.
- (a) C. H. Zhao, A. Wakamiya and S. Yamaguchi, Macromolecules, 2007, 40, 3898 CrossRef CAS; (b) A. Rose, Z. Zhu, C. F. Madigan, T. M. Swager and V. Bulović, Nature, 2005, 434, 876 CrossRef CAS; (c) M. R. Andersson, G. Yu and A. J. Heeger, Synth. Met., 1997, 85, 1275 CrossRef CAS; (d) D. A. Vithanage, A. L. Kanibolotsky, S. Rajbhandari, P. P. Manousiadis, M. T. Sajjad, H. Chun, G. E. Faulkner, D. C. O’Brien, P. J. Skabara, I. D. W. Samuel and G. A. Turnbull, J. Mater. Chem. C, 2017, 5, 8916 RSC.
- H. Spreitzer, H. Becker, E. Kluge, W. Kreuder, H. Schenk, R. Demandt and H. Schoo, Adv. Mater., 1998, 10, 1340 CrossRef CAS.
- T. Hirose, Y. Tsunoi, Y. Fujimori and K. Matsuda, Chem. Eur. J., 2015, 21, 1637 CrossRef CAS.
- P. Baronas, R. Komskis, E. Tankelevičiutė, P. Adomėnas, O. Adomėnienė and S. Juršėnas, J. Phys. Chem. Lett., 2021, 12, 6827 CrossRef CAS PubMed.
- (a) M.-N. Yu, H. Soleimaninejad, J.-Y. Lin, Z.-Y. Zuo, B. Liu, Y.-F. Bo, L.-B. Bai, Y.-M. Han, T. A. Smith, M. Xu, X.-P. Wu, D. E. Dunstan, R.-D. Xia, L.-H. Xie, D. D. C. Bradley and W. Huang, J. Phys. Chem. Lett., 2018, 9, 364 CrossRef CAS PubMed; (b) Y. Hattori, N. Nishimura, Y. Tsutsui, S. Ghosh, T. Sakurai, K. Sugiyasu, M. Takeuchi and S. Seki, Chem. Commun., 2019, 55, 13342 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental methods, synthetic details and additional data. See DOI: https://doi.org/10.1039/d4cc06672c |
| This journal is © The Royal Society of Chemistry 2025 |