
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photodynamic ROS inducers delivered via electrostatic antibody targeted (ELART) nanocarriers incorporate into tumour cells and inhibit colony growth†
Andreas
Faust‡
 a,
Andrew F.
Berdel‡
b,
Evelyn
Bruns‡
b,
Nicole
Bäumer
b,
Timo
Krassuski
b,
Lisa
Wittmann
b,
Linnea
Franke
b,
Iván
Maisuls
cd,
Stephan
Niland
e,
Johannes A.
Eble
e,
Cristian A.
Strassert
cd,
Georg
Lenz
b,
Wolfgang E.
Berdel
b and
Sebastian
Bäumer
b
a,
Andrew F.
Berdel‡
b,
Evelyn
Bruns‡
b,
Nicole
Bäumer
b,
Timo
Krassuski
b,
Lisa
Wittmann
b,
Linnea
Franke
b,
Iván
Maisuls
cd,
Stephan
Niland
e,
Johannes A.
Eble
e,
Cristian A.
Strassert
cd,
Georg
Lenz
b,
Wolfgang E.
Berdel
b and
Sebastian
Bäumer
b
aEuropean Institute for Molecular Imaging (EIMI), University of Münster, Röntgenstraße 16, Münster, Germany. E-mail: faustan@uni-muenster.de
bDepartment of Medicine A, Hematology/Oncology, University Hospital Münster, Albert-Schweitzer Campus 1, Münster, Germany. E-mail: baumers@uni-muenster.de
cInstitut fuer Anorganische und Analytische Chemie, Universität Munster, Corrensstraße 28/30, 48149 Münster, Germany
dCiMIC, SoN, CeNTech, Universität Münster, Heisenbergstraße 11, 48149 Münster, Germany
eInstitute of Physiological Chemistry and Pathobiochemistry, University of Münster, Waldeyerstraße 15, 48149 Münster, Germany
First published on 6th May 2025
Abstract
We designed and synthesised an anionic small-molecular photosensitizer (aPSM-Cy3.5) for incorporation into electrostatic antibody targeted (ELART) vesicles, consisting of a protamine-coupled antibody as targeting unit, free protamine and aPSM-Cy3.5. These nanocarriers specifically internalize into different solid tumour cell lines. Upon illumination, the aPSM component initiates the production of reactive oxygen species (ROS). Tumour cells from lung, colorectal and pancreatic cancer with internalized aPSM show decreased growth in colony forming assay. This supports the development of systemically applicable anionic ROS inducers capable of specifically targeting tumour cells.
Reactive oxygen species (ROS) have a variety of physiological functions in the cell, such as signalling and host defence.1 However, in high concentrations they can damage a wide range of intracellular biomolecules, causing irreversible damage and inducing cell death.2 Photodynamic therapy (PDT) triggers ROS production upon light exposure near cancer cells.3,4 The main advantage of PDT is the ability to focus ROS induced damage locally to the desired site of action, minimizing collateral damage to healthy tissue, making it minimally invasive in contrast to surgery or radiation. Moreover, elevation of ROS levels was shown to be the mode of action of several anti-cancer therapies.5 Because of its unique mechanisms, little to no resistance to PDT is known to date, offering the possibility of repeated applications. Considering the short lifetime of ROS (nano- to microsecond range), their diffusion range in cells is restricted to nanometre range, so the subcellular distribution of the PS defines its photodamage properties. Nonetheless, PDT has some significant intrinsic limitations. Hampered dosimetry due to varying PS distribution, limited inclusion of tumour margins and distant metastases, and the depth of light-penetration limit the applicability of PDT. Off-target phototoxicity may occur if the PS is not selectively targeted to the cell type of interest. Lung cancer (LC) and colorectal cancer (CRC) each account for approx. 2 million deaths per year worldwide.6,7 Pancreatic cancer (PC) is rarely diagnosed in its early stages, as symptoms often occur only after metastasis or local invasive growth. With more than 400
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 deaths annually worldwide, the 5-year survival rate for PC remains below 20%.8 In this study, we aimed to overcome the limitations of PS delivery for the treatment of solid tumours by achieving higher cargo efficiency and targeted tumour cell specificity. To this end, we modified our established ELART nanocarrier system, originally designed for the transport of anionic siRNA (electrostatic antibody siRNA targeting therapy).9–13 This was achieved through auto-assembly driven complexation of the drug into an electrostatically stabilised spheroidal nanocarriers. These nanocarriers consist of a modified photosensitizing molecule (PSM) with exposed anionic charged moieties, the cationic natural peptide protamine, and a targeting antibody chemically conjugated to protamine. We successfully incorporated aPSM-Cy3.5 into these nanocarriers, which were internalised by LC, CRC and PC cell lines. Upon exposure to light, the internalised nanocarriers generated ROS and significantly disrupted anchorage-independent growth of cancer cells. Targeting the PSM is of great interest to enhance the therapeutic efficacy of PDT by improving selective cellular uptake and overall efficacy. The complexation of a PSM with protamine is crucial for the formation of ELART nanocarriers (Fig. 1A). To achieve this, we have converted a specific PSM into a strongly anionic compound (aPSM) by modification with the dye Cy3.5 (Scheme 1 and S1, ESI†). The photosensitizing component of the molecule consists of an asymmetric and hydrophilic Zn(II) phthalocyanine, synthesised according to literature.14 The resulting azido-functionalised PSM was conjugated with the anionic cyanine dye sulfo-Cy3.5 alkyne via Huisgen cycloaddition (see ESI† for details). This modification enabled the aPSM to bind the αEGFR-mAb-protamine and free protamine to form an electrostatic antibody–drug complex (Fig. 1A) and additionally facilitated easy cellular tracking via red fluorescence, as previously demonstrated by our group with ibrutinib-Cy3.5 (Fig. 1A and scheme S2, ESI†).13 The UV-vis absorption spectrum of aPSM-Cy3.5 in H2O showed the characteristic band of the Cy3.5 (Fig. S1 and S2, ESI†) and less prominent bands in the 650–700 nm, attributed to the phthalocyanine moiety, likely aggregated in H2O. The emission spectrum in H2O showed only the characteristic Cy3.5 band (Fig. S4, ESI†), while in dimethylformamide (DMF) a characteristic phthalocyanine fluorescence was detected with a λmax = 695 nm (Fig. S4, ESI†). Interestingly, the UV-vis absorption spectrum of nanocarriers suspended in H2O shows spectral characteristics similar to those measured for aPSM-Cy3.5 in DMF (Fig. S1 and S3, ESI†), suggesting that aggregation is being partially prevented (Fig. S5, ESI†). Thus, a similar behavior is observed in the fluorescence spectrum, where the phthalocyanine band can be traced (Fig. S6, ESI†). To further investigate the role of ROS, the phosphorescence from photosensitized singlet dioxygen (1O2) was demonstrated in DMF for aPSM-Cy3.5 (detection in H2O was not feasible due to the rapid decay of 1O2). The 1O2 band at 1275 nm was clearly observed (Fig. 1B), confirming that 1O2 is formed upon photoexcitation of aPSM-Cy3.5. Due to the spectral similarities observed for the nanocarriers in H2O, these results are consistent with what is expected upon cellular uptake. To form ELART nanocarriers, αEGFR-mAb-P/P electrostatically complexed with aPSM-Cy3.5 (αEGFR-mAb-P/P-aPSM-Cy3.5) was analysed for its loading capacity by electromobility shift assay and for vesicle formation by fluorescence microscopy (Fig. 1C and D). The band-shift assay confirmed electrostatical binding of aPSM-Cy3.5 to αEGFR-mAb-P/P, thereby visualising the nanocarriers’ loading capacity. The optimal loading capacity was determined to be up to 30 moles of aPSM-Cy3.5 per mole of αEGFR-mAb-P/P. Higher concentrations of aPSM-Cy3.5 resulted in unbound aPSM-Cy3.5, visible as bands in the shift assay (Fig. 1C). Bright-field microscopy revealed the formation of spheroidal αEGFR-mAb-P/P-aPSM-Cy3.5 nanocarriers of 200–500 nm diameter (Fig. 1B, left), which also displayed red fluorescence (Fig. 1B, right). These results confirm successful nanocarrier formation with the newly designed aPSM-Cy3.5 and αEGFR-mAb-P/P. Receptor-dependent internalisation is the critical first step to ensure a cell-specific effect. Internalisation of αEGFR-mAb-P/P-aPSM-Cy3.5 was confirmed on EGFR-positive cell lines Panc1 and MiaPaCa2 (PC; Fig. S8, ESI†), A549 and SK-LU1 (LC; Fig. S9, ESI†), as well as DLD1 and SW480 (CRC; Fig. S10, ESI†). These cell lines were treated either with PBS as negative control, untargeted aPSM-Cy3.5 or αEGFR-mAb-P/P-aPSM-Cy3.5 nanocarriers. In fluorescence microscopy, treatment with αEGFR-mAb-P/P-aPSM-Cy3.5 resulted in more intense and dispersed distribution of red fluorescent signals with higher perinuclear accumulation (Fig. S7–S9C, F, I and L, ESI†), while treatment with non-targeted aPSM-Cy3.5 resulted in vesicular accumulation mainly inside the cytoplasm and only partial perinuclear localization (Fig. S7–S9B, E, H and K, ESI†). Laser scanning microscopy of Panc1 (Fig. 2) and A549 cells (Fig. S10, ESI†) after treatment with αEGFR-mAb-P/P-aPSM-Cy3.5 further confirmed internalisation and dispersed distribution of Cy3.5 fluorescence in the cytoplasm and demonstrated strong perinuclear and lysosomal colocalisation. This internalisation was dependent on the concentration of αEGFR-mAb-P/P-aPSM-Cy3.5 as quantified by flow cytometry (Fig. S11, ESI†). These findings demonstrate efficient and predominantly EGFR-dependent internalisation across all tested cell lines.
000 deaths annually worldwide, the 5-year survival rate for PC remains below 20%.8 In this study, we aimed to overcome the limitations of PS delivery for the treatment of solid tumours by achieving higher cargo efficiency and targeted tumour cell specificity. To this end, we modified our established ELART nanocarrier system, originally designed for the transport of anionic siRNA (electrostatic antibody siRNA targeting therapy).9–13 This was achieved through auto-assembly driven complexation of the drug into an electrostatically stabilised spheroidal nanocarriers. These nanocarriers consist of a modified photosensitizing molecule (PSM) with exposed anionic charged moieties, the cationic natural peptide protamine, and a targeting antibody chemically conjugated to protamine. We successfully incorporated aPSM-Cy3.5 into these nanocarriers, which were internalised by LC, CRC and PC cell lines. Upon exposure to light, the internalised nanocarriers generated ROS and significantly disrupted anchorage-independent growth of cancer cells. Targeting the PSM is of great interest to enhance the therapeutic efficacy of PDT by improving selective cellular uptake and overall efficacy. The complexation of a PSM with protamine is crucial for the formation of ELART nanocarriers (Fig. 1A). To achieve this, we have converted a specific PSM into a strongly anionic compound (aPSM) by modification with the dye Cy3.5 (Scheme 1 and S1, ESI†). The photosensitizing component of the molecule consists of an asymmetric and hydrophilic Zn(II) phthalocyanine, synthesised according to literature.14 The resulting azido-functionalised PSM was conjugated with the anionic cyanine dye sulfo-Cy3.5 alkyne via Huisgen cycloaddition (see ESI† for details). This modification enabled the aPSM to bind the αEGFR-mAb-protamine and free protamine to form an electrostatic antibody–drug complex (Fig. 1A) and additionally facilitated easy cellular tracking via red fluorescence, as previously demonstrated by our group with ibrutinib-Cy3.5 (Fig. 1A and scheme S2, ESI†).13 The UV-vis absorption spectrum of aPSM-Cy3.5 in H2O showed the characteristic band of the Cy3.5 (Fig. S1 and S2, ESI†) and less prominent bands in the 650–700 nm, attributed to the phthalocyanine moiety, likely aggregated in H2O. The emission spectrum in H2O showed only the characteristic Cy3.5 band (Fig. S4, ESI†), while in dimethylformamide (DMF) a characteristic phthalocyanine fluorescence was detected with a λmax = 695 nm (Fig. S4, ESI†). Interestingly, the UV-vis absorption spectrum of nanocarriers suspended in H2O shows spectral characteristics similar to those measured for aPSM-Cy3.5 in DMF (Fig. S1 and S3, ESI†), suggesting that aggregation is being partially prevented (Fig. S5, ESI†). Thus, a similar behavior is observed in the fluorescence spectrum, where the phthalocyanine band can be traced (Fig. S6, ESI†). To further investigate the role of ROS, the phosphorescence from photosensitized singlet dioxygen (1O2) was demonstrated in DMF for aPSM-Cy3.5 (detection in H2O was not feasible due to the rapid decay of 1O2). The 1O2 band at 1275 nm was clearly observed (Fig. 1B), confirming that 1O2 is formed upon photoexcitation of aPSM-Cy3.5. Due to the spectral similarities observed for the nanocarriers in H2O, these results are consistent with what is expected upon cellular uptake. To form ELART nanocarriers, αEGFR-mAb-P/P electrostatically complexed with aPSM-Cy3.5 (αEGFR-mAb-P/P-aPSM-Cy3.5) was analysed for its loading capacity by electromobility shift assay and for vesicle formation by fluorescence microscopy (Fig. 1C and D). The band-shift assay confirmed electrostatical binding of aPSM-Cy3.5 to αEGFR-mAb-P/P, thereby visualising the nanocarriers’ loading capacity. The optimal loading capacity was determined to be up to 30 moles of aPSM-Cy3.5 per mole of αEGFR-mAb-P/P. Higher concentrations of aPSM-Cy3.5 resulted in unbound aPSM-Cy3.5, visible as bands in the shift assay (Fig. 1C). Bright-field microscopy revealed the formation of spheroidal αEGFR-mAb-P/P-aPSM-Cy3.5 nanocarriers of 200–500 nm diameter (Fig. 1B, left), which also displayed red fluorescence (Fig. 1B, right). These results confirm successful nanocarrier formation with the newly designed aPSM-Cy3.5 and αEGFR-mAb-P/P. Receptor-dependent internalisation is the critical first step to ensure a cell-specific effect. Internalisation of αEGFR-mAb-P/P-aPSM-Cy3.5 was confirmed on EGFR-positive cell lines Panc1 and MiaPaCa2 (PC; Fig. S8, ESI†), A549 and SK-LU1 (LC; Fig. S9, ESI†), as well as DLD1 and SW480 (CRC; Fig. S10, ESI†). These cell lines were treated either with PBS as negative control, untargeted aPSM-Cy3.5 or αEGFR-mAb-P/P-aPSM-Cy3.5 nanocarriers. In fluorescence microscopy, treatment with αEGFR-mAb-P/P-aPSM-Cy3.5 resulted in more intense and dispersed distribution of red fluorescent signals with higher perinuclear accumulation (Fig. S7–S9C, F, I and L, ESI†), while treatment with non-targeted aPSM-Cy3.5 resulted in vesicular accumulation mainly inside the cytoplasm and only partial perinuclear localization (Fig. S7–S9B, E, H and K, ESI†). Laser scanning microscopy of Panc1 (Fig. 2) and A549 cells (Fig. S10, ESI†) after treatment with αEGFR-mAb-P/P-aPSM-Cy3.5 further confirmed internalisation and dispersed distribution of Cy3.5 fluorescence in the cytoplasm and demonstrated strong perinuclear and lysosomal colocalisation. This internalisation was dependent on the concentration of αEGFR-mAb-P/P-aPSM-Cy3.5 as quantified by flow cytometry (Fig. S11, ESI†). These findings demonstrate efficient and predominantly EGFR-dependent internalisation across all tested cell lines.
 | ||
| Fig. 1 (A) ELART nanocarriers are composed of an antibody (here: anti-EGFR-mAb) chemically linked via sulfo-SMCC to the cationic protein protamine, unbound (MCC)-protamine and the anionic small molecule aPSM-Cy3.5. Fully assembled, their goal is to specifically target and kill cancer cell lines upon light-induced ROS production (Created in BioRender. Berdel, A. (2025) https://BioRender.com/d98q114). (B) 1O2 (singlet dioxygen) phosphorescence spectra (λex = 525 nm, λmax = 1275 nm) photogenerated by aPSM-Cy3.5 in DMF at 298 K. (C) Bandshift assay showing the electrostatic loading capacity of αEGFR-mAb-P/P conjugated with aPSM-Cy3.5. M = marker. (D) Nanoparticle formation of αEGFR-mAb-P/P (60 nM) with aPSM-Cy3.5 (1800 nM). Fluorescence imaging and bright field imaging of αEGFR-mAb-P/P-aPSM-Cy3.5 stability after auto-assembly, overnight incubation and mounting on glass slides. Cy3.5 signal is shown in red. Bars 10 μm. | ||
 | ||
| Scheme 1 Synthesis of aPSM-Cy3.5 and preparation of the tumour-targeting nanocarrier: (a) Zn(OAc)2, NEt3, EtOH, sealed tube, 120 °C, 6 h, 49%; (b) MsCl, NEt3, CH2Cl2, 4 °C, 1 h; (c) NaN3, DMF, 100 °C, 16 h; (d) sulfo-cyanine 3.5-alkyne, CuSO4·5H2O, Na ascorbate, CH2Cl2, rt, 16 h, (e) AcOHglac, 70 °C, 1 h, 26% (over four steps); (f) αEGFR-mAb-protamine, rt, 1 h. In part (coloured icons) created in BioRender. Berdel, A. (2024) https://BioRender.com/k99v398. | ||
 | ||
| Fig. 2 Laser scanning microscopy (LSM) analysis of internalisation. Panc1 cells pre-treated with αEGFR-mAb-P/P (180 nM) electrostatically complexed with aPSM-Cy3.5 (5400 nM). (A) Mitochondria stained with MitoView Fix 640 are shown in purple. (B) Green fluorescence displays lysosomes stained with LysoTracker Green DND-26. (C) Red fluorescence displays aPSM-Cy3.5. (D) Blue fluorescence displays nuclei stained with Hoechst 33342. (E) Merged channels of (A)–(D) displaying subcellular localisation of ELART nanocarriers. | ||
After confirmation of internalisation, intracellular ROS generation upon illumination was confirmed in vitro by luminometric detection, showing a significant increase in ROS levels in Panc1 and A549 cells treated with αEGFR-mAb-P/P-aPSM-Cy3.5 compared to the PBS control (Fig. 3 and Fig. S12, ESI†). To evaluate the cytotoxic potential of these ROS, colony formation assays were performed, depicting anchorage-independent clonal tumour cell growth as a measure for tumourigenicity of tumour cells as well as for toxicity of a treatment. Cancer cells were illuminated after internalisation of αEGFR-mAb-P/P-aPSM-Cy3.5. In all cancer cell lines tested, a significant inhibition of clonal growth was observed after 10 min or less of illumination (Fig. 4 Panc1; Fig. S13A, ESI†). Controls showed no significant effect for illumination alone. Free aPSM-Cy3.5 significantly reduced colony formation only in LC cell lines A549 and SK-LU1 (Fig. S13B, ESI†), while electrostatic complexation of aPSM-Cy3.5 into a nanocarrier boosted efficacy in SW480, MiaPaca and DLD1 (Fig S13B, ESI†).
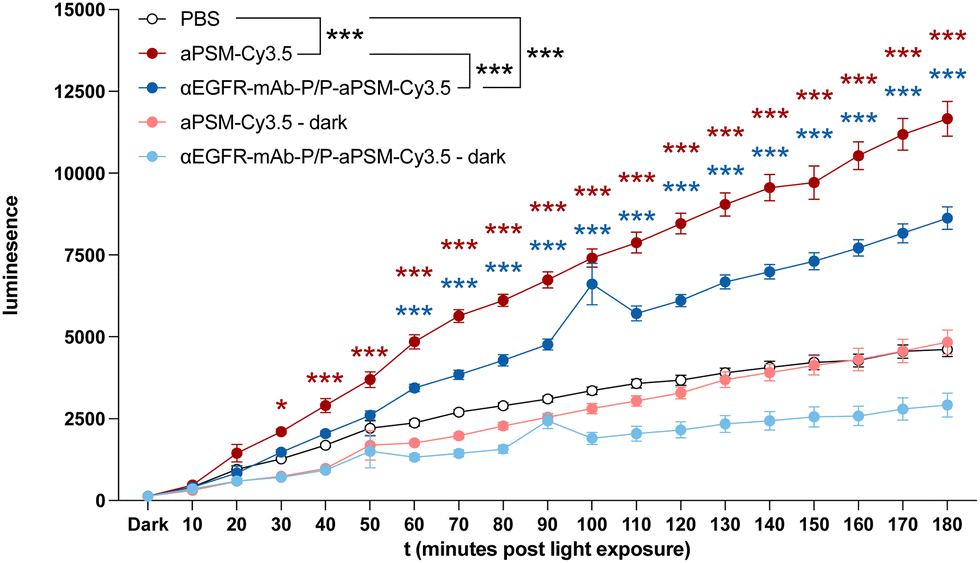 | ||
| Fig. 3 ROS detection assay of Panc1 cells pre-treated either with αEGFR-mAb-P/P (1800 nM) electrostatically complexed with aPSM-Cy3.5 (5400 nM) or free aPSM-Cy3.5 (5400 nM) compound, and light-activated for the indicated time frames. Dark = no illumination. Data are presented as means ± standard error mean (SEM). Pair-wise comparisons were performed using two-sample t-test for independent samples (two-sided) between PBS and aPSM-Cy3.5 (red asterisk) or αEGFR-mAb-P/P-aPSM-Cy3.5 (blue asterisk) at individual timepoints. Group comparison (black asterisk) was performed using two-way analysis of variance (ANOVA) (*P < 0.05, **P < 0.01, and ***P < 0.001). | ||
 | ||
| Fig. 4 Colony formation of Panc1 cells treated with αEGFR-mAb-P/P (60 nM) electrostatically complexed with aPSM-Cy3.5 (1800 nM) and illuminated for indicated time frames (mean of 3 independent experiments in triplicates). Data are presented as means ± SEM. Pair-wise comparisons were performed using two-sample t-test for independent samples (two-sided). Group comparisons were performed using regular one-way ANOVA (*P < 0.05, **P < 0.01, and ***P < 0.001). | ||
In summary, the anionic modification of a PSM enabled the successful complexation into our electrostatic-nanocarrier system for cell specific targeting. This modification preserved its ability to produce ROS upon illumination, resulting in a robust cytotoxic effect across multiple carcinoma types. Our targeted ELART nanocarrier system effectively transports both short interfering RNA and small molecules specifically into a variety of cancer cells, and thereby elicits a strong antitumour effect both in vitro and in vivo.9–13,15 Besides essential physiological roles, ROS harbour a significant cytotoxic potential,2 that is subject to investigation in several cancer therapy strategies.16 One such approach is PDT through application of a PSM and a light-source to induce ROS-production in the illuminated area.4,17 To overcome some of these problems, PS can be conjugated to targeting moieties that specifically bind to receptors expressed on tumour cells, leading to improved internalisation at the site of interest and decreasing unwanted off-target effects. The epidermal growth factor receptor (EGFR) serves as such a targeting receptor in various applications, including PS delivery to tumour cells. Examples comprise the use of the EGFR ligands, EGFR-binding peptides, EGFR-interfering small molecules and αEGFR-antibodies as targeting compounds.17,18 Targeted delivery of PS has reached FDA approval and clinical use.19 As with all antibody–drug conjugates (ADCs), the carrier-to-cargo ratio is crucial. In classic chemically conjugated ADCs the linear architecture, comprising antibody-linker-effector, at present typically defines the conjugation ratio at 1:8 or lower.20 This limits delivery of any effector to the target cell. In this study, we successfully modified aPSM with Cy3.5 to form the anionic aPSM-Cy3.5, which was then electrostatically integrated into a modified version of our established nanocarrier system. These nanocarriers transport at least 30 mol aPSM-Cy3.5 per mol of antibody, therefore hundreds to thousands of aPSM-Cy3.5 per nanocarrier. This approach enabled the targeted delivery of aPSM specifically into various carcinoma cells, resulting in a robust ROS-mediated cytotoxic effect upon illumination. Since our nanocarrier system is suitable for systemic application, this new formulation has the potential to overcome two of the three main limitations of PDT: systemic delivery and cell type specific targeting. An unsolved limitation is the dependence on a light-source. Notably, the red-shifted absorption of our phthalocyanine photosensitizer enhances the tissue penetration of the excitation light. However, to better mitigate this limitation of PDT, the development of a chemical luminescence source that can be applied systemically, ideally through our ELART nanocarrier system in parallel with aPSM-Cy3.5 to concentrate in the tumour environment, would be ideal. These chemoluminescent agents are currently discussed3 and will be subject of our further studies. Another possible mitigation strategy worth exploring is exchanging the aPSM with a light-independent ROS inducer, as several small molecules can induce ROS upon a variety of stimuli or even independently, eliciting a cytotoxic effect.16 Beyond photosensitizers, ultrasound can be used with sonosensitizers to induce ROS, enabling deep tissue penetration.21 Another promising class are direct ROS-inducers, i.e. the natural estradiol-metabolite 2-methoxyestradiol, which has been shown to induce apoptosis in cancer cells through ROS-production.22 These light-independent ROS inducers may be modified for electrostatic integration into our nanocarriers, potentially overcoming remaining limitations, and are subject of further investigation.
In conclusion, the developed nanocarrier system enables cell specific targeting and killing of carcinoma cells through intracellular ROS-production following illumination. This can be translated into a new modality of cancer therapy.
This study was supported by Deutsche Krebshilfe No. 70114814, the IZKF of the Medical Faculty of the University of Münster (UM) No. Bäu2/009/19 both given to S. B. A. F. Berdel was supported by a research grant from the Innovative Medical Research Fund (IMF; BE122204) of the UM Faculty of Medicine and the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation – 493624047 (Clinician Scientist CareerS Münster)). S. Niland and J. A. Eble were supported by a research grant from the DFG (EB177-19/1).
Data availability
All data of this study are within the publication and its ESI.†Conflicts of interest
NB, LW, AF, GL, WEB, SB have filed patent applications on electrostatic nanocarrier technology and founded ELVESCA Biotherapeutics GmbH. All other authors disclose no conflict of interest.References
- M. Schieber and N. S. Chandel, Curr. Biol., 2014, 24, R453–462 CrossRef CAS PubMed.
- M. Redza-Dutordoir and D. A. Averill-Bates, Biochim. Biophys. Acta, 2016, 1863, 2977–2992 CrossRef CAS PubMed.
- X. Wang, et al., Cancers, 2021, 13, 2992–3011 CrossRef CAS PubMed.
- G. M. Cramer, et al., Cancer Res., 2022, 82, 534–536 CrossRef CAS PubMed.
- B. Perillo, et al., Exp. Mol. Med., 2020, 52, 192–203 CrossRef CAS PubMed.
- G. Roshandel, et al., Cancers, 2024, 16, 1530–1552 CrossRef PubMed.
- K. C. Thandra, et al., Contemp. Oncol., 2021, 25, 45–52 CAS.
- O. Partyka, et al., Cancers, 2023, 15, 3634–3645 CrossRef CAS PubMed.
- N. Bäumer, et al., Nat. Protoc., 2016, 11, 22–36 CrossRef PubMed.
- N. Bäumer, et al., PLoS One, 2018, 13, e0200163 CrossRef PubMed.
- N. Bäumer, et al., J. Hematol. Oncol., 2022, 15, 171 CrossRef PubMed.
- S. Bäumer, et al., Clin. Cancer Res., 2015, 21, 1383–1394 CrossRef PubMed.
- A. Faust, et al., Angew. Chem., Int. Ed., 2022, 61, e202109769 CrossRef CAS PubMed.
- Y. Zorlu, et al., Tetrahedron Lett., 2010, 51, 6615–6618 CrossRef CAS.
- N. Bäumer, et al., Mol. Pharmaceutics, 2017, 14, 1339–1351 CrossRef PubMed.
- W. Yang, et al., Research, 2022, 9897464 CAS.
- L. Ulfo, et al., Pharmaceutics, 2022, 14, 241–285 CrossRef CAS PubMed.
- M. Mitsunaga, et al., Nat. Med., 2011, 17, 1685–1691 CrossRef CAS PubMed.
- D. M. Cognetti, et al., Head Neck, 2021, 43, 3875–3887 CrossRef PubMed.
- J. M. Sasso, et al., Bioconjugate Chem., 2023, 34, 1951–2000 CrossRef CAS PubMed.
- M. Niedre, et al., Photochem. Photobiol., 2002, 75, 382–391 CrossRef CAS PubMed.
- T. Hagen, et al., Biochem. Biophys. Res. Commun., 2004, 322, 923–929 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic procedures, UV-vis absorption and emission spectra, 1O2 phosphorescence spectra, fluorescence microscopy, ROS detection and colony forming assays, NMR and MS spectra. See DOI: https://doi.org/10.1039/d4cc06569g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |