
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electrochemical oxidative dearomatization of electron-deficient phenols using Br+/Br− catalysis†
Kai
Matsui
,
Muhammet
Uyanik
 * and
Kazuaki
Ishihara
*
* and
Kazuaki
Ishihara
*
Graduate School of Engineering, Nagoya University, Furo-cho, Chikusa, Nagoya 464-8603, Japan. E-mail: muha@chembio.nagoya-u.ac.jp; ishihara@cc.nagoya-u.ac.jp
First published on 6th January 2025
Abstract
An electrochemical method for the oxidative dearomatization of electron-deficient phenols by employing tetrabutylammonium bromide as a mediator under aqueous biphasic conditions is reported. This approach represents a safer alternative to the use of stoichiometric chemical oxidants and enables oxidative dearomative spirolactonization and spiroetherification reactions. Compared to previous approaches based on direct electrolysis, this strategy expands the substrate scope to electron-deficient phenols. Cyclic-voltammetry analysis suggests that the bromide ions might be oxidized to Br2 or Br3− ions that are in equilibrium with the catalytically active hypobromite under aqueous conditions.
The dearomatization of arenols represents a powerful strategy for the synthesis of three-dimensional complex molecules.1 Halogens, especially iodine-based oxidants or catalysts, have emerged as desirable reagents to accomplish such oxidative transformations without relying on toxic or expensive transition metals.2 We have previously developed a quaternary ammonium hypoiodite catalysis2c,3 for the oxidative dearomative spirocyclization of arenols using hydrogen peroxide or oxone as a stoichiometric chemical oxidant (Scheme 1a).4 However, the scope of these reactions is restricted to electron-rich arenols that bear electron-donating groups (EDGs). To address this limitation, we have recently developed a catalytic approach using ammonium hypobromite,5 which offers a higher oxidation potential than hypoiodite, thereby expanding the scope to include phenols substituted with electron-withdrawing groups (EWGs) (Scheme 1b).6
 | ||
| Scheme 1 (Electro)oxidative dearomatization of arenols. | ||
Despite these advances, the development of safer oxidation methods that minimize the risks associated with high-energy and potentially hazardous chemical oxidants remains a pressing challenge. In this context, electrochemical approaches have garnered attention as inherently safer alternatives.7,8 For instance, Kalek and colleagues have recently reported an anodic oxidative dearomative spirolactonization and spiroetherification of arenols tethered to a carboxylic acid or alcohol as an internal nucleophile, respectively, which generates hydrogen as the sole side product (Scheme 1c).9 This method involves the direct electrolysis without a mediator under nonaqueous conditions, and the phenols are oxidized directly at the anode through a single-electron-transfer (SET) mechanism coupled with a proton transfer.8 In this process, a phenoxyl radical is initially generated, subsequently oxidized to a phenoxenium ion, and then trapped by an internal nucleophile, leading to the desired product. However, the phenoxyl radical intermediate is prone to side reactions, such as dimerization,8,9b especially when the second oxidation step is slow. This limitation in chemoselectivity potentially restricts the direct electrolysis method to highly reactive, electron-rich arenols, such as 1- or 2-naphthols or phenols with electron-donating groups (EDGs).9
Meanwhile, recent studies have highlighted the potential of anodic oxidations employing halides as mediators (i.e., indirect electrolysis), particularly for enhancing substrate compatibility and chemoselectivity.10 Notably, several advancements have been made in catalytic halide mediators for various transformations.10 However, to the best of our knowledge, no examples of halide-mediated electrooxidative dearomative transformations have been reported so far. Inspired by these halide-mediated anodic oxidation reactions, we sought to integrate anodic oxidation into our hypobromite catalysis,6 allowing us to leverage the advantages of indirect electrolysis in dearomatization reactions as a safer alternative to stoichiometric chemical oxidants (Scheme 1d). This approach differs fundamentally from Kalek's method; rather than the phenols being directly oxidized at the anode, preferential oxidation of bromide to Br+ species (i.e., Bu4N+BrO−) would occur. These active Br+ species would subsequently oxidize the phenol substrate through a two-electron transfer process,6 thus bypassing the formation of radical intermediates. We envisioned that this mechanism would suppress undesired side reactions, thereby improving the chemoselectivity and potentially broadening the substrate compatibility of this transformation. We focused specifically on the oxidative dearomative spirolactonization and spiroetherification (C–O coupling) of phenols substituted with EWGs in order to expand the substrate scope beyond the electron-rich arenols commonly employed in previous direct electrolysis methods.9
To further minimize the direct oxidation of phenols at the anode, especially under catalytic conditions, we designed a biphasic organic solvent/water system for hypobromite electrocatalysis in which the electrodes are located in the aqueous phase (Scheme 1d).11 This setup physically separates the phenol substrate from the electrodes, thus reducing the likelihood of its direct electrolysis. Moreover, this biphasic electrolysis system allows for the use of inexpensive inorganic salts as electrolytes in the aqueous phase, eliminating the need for the more costly ammonium salts (e.g., Bu4N+PF6−) that are typically used, thereby enhancing the practicality of the method.
We commenced our investigation by examining the electrooxidative spirolactonization of 4-bromo-substituted phenol 1a as a model substrate using a catalytic amount of tetrabutylammonium bromide as a mediator (Table 1). Platinum and stainless steel (SST) were chosen as the anode and cathode materials, respectively, and the electrolysis was conducted in an undivided cell using the inexpensive inorganic electrolyte sodium tetrafluoroborate (for details, see Table S1, ESI†). To meet the specific requirements of our system, we employed a dichloroethane (DCE)/water biphasic solvent, in which the denser organic layer remains beneath the aqueous phase (Scheme 1d). This arrangement is essential to ensure that electrolysis occurs in the upper aqueous layer containing the electrolyte and electrodes to generate Br+ species. These catalytically active species then migrate into the organic layer to facilitate the desired reaction.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | x (mol%) | T (°C) | 1a, Conv.b (%) | 2a, Yieldb (%) |
| a Unless otherwise noted, the reactions were carried out using 1a (0.2 mmol), Bu4NBr (40 or 20 mol%), and NaBF4 (2.0 equiv.) under constant-current electrolysis using a Pt anode (10 mm × 10 mm × 0.2 mm) and an SST cathode (10 mm × 10 mm × 0.15 mm). The distance between electrodes was 10 mm. b Determined by 1H NMR analysis of the crude product using methyl 3,5-dibromobenzoate as an internal standard. c NaHSO4 (2.0 equiv.) was used as an additive. d CCE (1.0 mA); reaction time: 13.9 h. e Isolated yield. f 1a (1.0 mmol), NaBF4 (0.8 equiv.) under CCE (1.5 mA) using a Pt anode (10 mm × 15 mm × 0.2 mm) and an SST cathode (10 mm × 15 mm × 0.15 mm), 46.5 h. g Bu4NPF6 (20 mol%) was used instead of Bu4NBr. h 6-Bromo-8-(tert-butyl)chroman-2-one generated by intramolecular dehydration of 1a was obtained in 9% as the main side product. | |||||
| 1 | DCE | 20 | 25 | 84 | 72 |
| 2c | DCE | 20 | 25 | 73 | 60 |
| 3 | DCE | 20 | 5 | 78 | 71 |
| 4 | DCE | 40 | 5 | 95 | 92 |
| 5 | CH2Cl2 | 40 | 5 | 83 | 78 |
| 6 | C6F5CF3 | 40 | 5 | 88 | 83 |
| 7 | CH3NO2 | 40 | 5 | 18 | 5 |
| 8d | DCE | 40 | 5 | 94 | 92e |
| 9f | DCE | 40 | 5 | >99 | 99e |
| 10 | DCE | 0g | 25 | 12h | <5 |
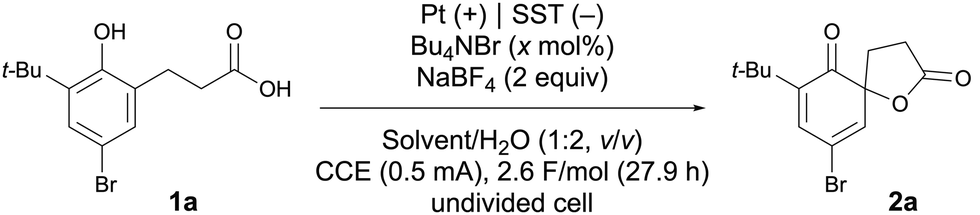
Electrolysis was conducted at a constant current of 0.5 mA at room temperature until a total charge of 2.6 F mol−1 had been passed, which afforded the desired spirolactone 2a in 72% yield (entry 1). Drawing on our previous studies, which showed that acidic conditions could accelerate hypohalite catalysis,4,6 we tested the use of NaHSO4 as an acidic additive (entry 2). However, the yield of 2a decreased in that case, likely due to the accelerated generation6 and subsequent escape of Br2 from the reaction mixture, as evidenced by the presence of brown droplets on the rubber septum and electrodes. To improve mass balance, we lowered the reaction temperature to 5 °C (entry 3). Furthermore, a higher loading of Bu4NBr (40 mol%) was found to significantly improve the yield of 2a to 92% (entry 4). Alternative denser-than-water solvents, such as dichloromethane, trifluorotoluene, and nitromethane, were also examined; however, neither improved the yield (entries 5–7). Finally, the reaction time was reduced by half by increasing the current from 0.5 to 1.0 mA without compromising the yield of 2a, effectively suppressing the competitive oxidation of water (entry 8). Additionally, we have successfully scaled up the reaction to a 1 mmol scale (entry 9). Notably, when Bu4NPF6 was used instead of Bu4NBr, only trace amounts of product were observed, underscoring the crucial role of bromide as a mediator under the biphasic electrolysis conditions (entry 10).
Then, several electron-deficient phenols bearing EWGs were examined under the optimal conditions (Scheme 2). The chemoselective oxidative dearomatization of 4-acetylphenol derivative 1b afforded the desired product (2b) in 71% yield; no side product corresponding to carbonyl α-bromination was observed.12 In addition, substrates containing silyl (1d) or additional benzylic methylene (1e–g) groups, which are potentially sensitive to Br+ or Br˙ species,13 were well-tolerated under the reaction conditions, providing the corresponding cyclohexadienone spirolactones in good yield. Moreover, in addition to halogens (1a, 1d–g, and 1i–m) and acyl (1b and 1c) groups, a cyano group (1h) was also tolerated as an electron-withdrawing group in this Br+ electrocatalysis. The dearomative 5- or 6-membered spirolactonization of dihalo-substituted phenol derivatives 1i and 1j gave the corresponding (4+2)-cyclodimers (3i and 3j) as single diastereomers via the in situ generation of the less-hindered and unstable cyclohexadienones 2.4,6,14 Dearomative spirolactonization at the para-position was also achieved, i.e., 2k was obtained quantitatively from the oxidation of 1k. In addition to spirolactonization, spiroetherification reactions were also performed. Using alcohol and phenol as the internal nucleophiles, the corresponding spiroethers (2l and 2m) were obtained in moderate-to-high yield.
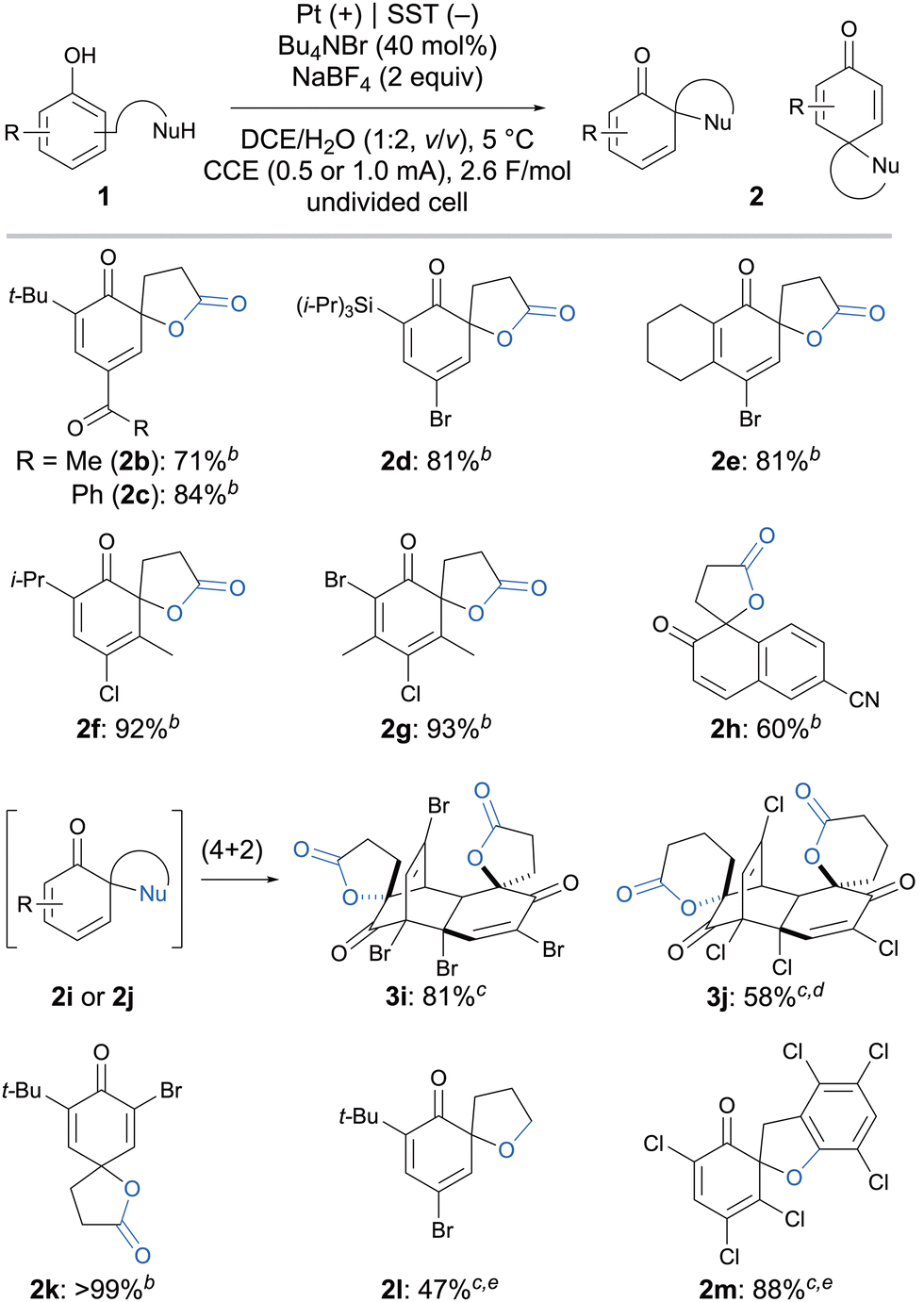 | ||
Scheme 2 Substrate scope. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 1 (0.2 mmol), Bu4NBr (40 mol%), and NaBF4 (2.0 equiv.) under constant-current electrolysis using a Pt anode (10 mm × 10 mm × 0.2 mm) and SST cathode (10 mm × 10 mm × 0.15 mm) at 5 °C. The distance between electrodes was 10 mm. b 1 (0.2 mmol), Bu4NBr (40 mol%), and NaBF4 (2.0 equiv.) under constant-current electrolysis using a Pt anode (10 mm × 10 mm × 0.2 mm) and SST cathode (10 mm × 10 mm × 0.15 mm) at 5 °C. The distance between electrodes was 10 mm. b![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) CCE (1.0 mA). cCCE (0.5 mA). dAfter the electrolysis was completed, the reaction mixture was further stirred at 25 °C for 14 h to complete the (4+2) cyclodimerization. eThe reaction was performed at 25 °C. CCE (1.0 mA). cCCE (0.5 mA). dAfter the electrolysis was completed, the reaction mixture was further stirred at 25 °C for 14 h to complete the (4+2) cyclodimerization. eThe reaction was performed at 25 °C. | ||
To elucidate the reaction mechanism, cyclic-voltammetry (CV) measurements were performed under anhydrous and aqueous conditions using CH3CN and CH3CN/H2O (9:1, v/v) as the solvent, respectively, and Bu4NPF6 (0.1 M) as the electrolyte (Fig. 1). The CV curve for Bu4NBr in CH3CN showed two distinct oxidation peaks at 0.40 V and 0.71 V vs. Ag/Ag+ (Fig. 1a, curve i), which corresponded to the 3Br− → Br3− and 2Br3− → 3Br2 oxidation processes, respectively, in agreement with previous reports.15 In comparison, 1a showed a higher oxidation potential of 1.21 V vs. Ag/Ag+ (Fig. 1a, curve ii), indicating that Br− oxidation remained more favorable. When Bu4NBr and 1a were combined, the resulting CV curve displayed two peaks resembling those of Bu4NBr (Fig. 1b, curve iii). Notably, the absence of an increase in the oxidation current suggests that the direct involvement of Br˙, Br3−, or Br2 in the oxidation of 1a would be unlikely.
 | ||
| Fig. 1 Cyclic voltammetry measurements under non-aqueous (a) and aqueous conditions (b) at room temperature at a scan rate of 100 mV s−1. | ||
In contrast, an obvious catalytic current was observed in the CV curve of Bu4NBr in the presence of 1a, indicating that Bu4NBr acted as a redox mediator (Fig. 1b, curve vi). These findings align with our previous study6 and strongly support the proposed mechanism (Scheme 1d), in which electrochemically generated bromine-based intermediates, such as Br2 or Br3−, are in equilibrium with hypobromite (BrO−) or hypobromous acid (BrOH) under aqueous conditions.15a,16 These active species would then migrate into the organic phase, facilitating the oxidative dearomatization of 1avia a two-electron oxidation process6 to yield the desired product. Taken together, these findings emphasize the role of hypobromite as a catalytically active species and highlight the importance of aqueous conditions in modulating both the reactivity and selectivity of bromine-based catalysis under biphasic electrochemical conditions (for further discussion, see Schemes S1–S3, ESI†).
In summary, we have developed an electrochemical approach for the oxidative dearomatization of electron-deficient phenols using tetrabutylammonium bromide as a catalytic mediator under aqueous biphasic conditions. This method offers greater safety by eliminating the use of stoichiometric chemical oxidants while maintaining the efficiency of our previously reported hypobromite catalysis.6 Compared to direct electrolysis methods, this approach expands the substrate scope to include electron-deficient phenols, enabling oxidative dearomative spirolactonization and spiroetherification reactions. Mechanistic studies using cyclic voltammetry suggested that bromide ions are oxidized at the anode to form bromine-based intermediates that are in equilibrium with hypobromite species, which ultimately mediate the oxidative dearomatization reaction.
Financial support for this project was partially provided by JSPS KAKENHI grants 23H05467 (to K. I.), 21H01932 (to M. U.), and 24KJ1261 (to K. M. and K. I.), as well as the Nagoya University Graduate Program of Transformative Chem-Bio Research (GTR) (to K. M.). We are deeply grateful to Prof. S. Suga and Assist. Prof. E. Sato (Okayama University) for their invaluable support in assembling the electrochemical reaction system. We thank Dr T. Kato for performing additional control experiments.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) D. Magdziak, S. J. Meek and T. R. R. Pettus, Chem. Rev., 2004, 104, 1383 CrossRef CAS PubMed; (b) S. P. Roche and J. A. PorcoJr, Angew. Chem., Int. Ed., 2011, 50, 4068 CrossRef CAS PubMed; (c) C.-X. Zhuo, W. Zhang and S.-L. You, Angew. Chem., Int. Ed., 2012, 51, 12662 CrossRef PubMed; (d) S.-L. You, Asymmetric Dearomatization Reactions, Wiley-VCH, Weinheim, 2016 CrossRef; (e) W. C. Wertjes, E. H. Southgate and D. Sarlah, Chem. Soc. Rev., 2018, 47, 7996 RSC; (f) M. Uyanik and K. Ishihara, in Comprehensive Chirality, ed. J. Cossy, Elsevier: Academic Press, 2024, vol. 7, pp. 243 Search PubMed.
- For selected recent reviews, see: (a) L. Pouységu, D. Deffieux and S. Quideau, Tetrahedron, 2010, 66, 2235 CrossRef; (b) Y. Kita and T. Dohi, Chem. Rec., 2015, 15, 886 CrossRef CAS PubMed; (c) K. Ishihara and K. Muñiz, Iodine Catalysis in Organic Synthesis, Wiley-VCH, Weinheim, 2022 CrossRef; (d) C. Rocq, M. Denis and S. Canesi, Chem. Commun., 2023, 59, 6495 RSC; (e) F. V. Singh, S. E. Shetgaonkar, M. Krishnan and T. Wirth, Chem. Soc. Rev., 2022, 51, 8102 RSC.
- (a) M. Uyanik, H. Okamoto, T. Yasui and K. Ishihara, Science, 2010, 328, 1376 CrossRef CAS PubMed; (b) M. Uyanik and K. Ishihara, ChemCatChem, 2012, 4, 177 CrossRef CAS; (c) P. Finkbeiner and B. J. Nachtsheim, Synthesis, 2013, 979 CAS; (d) R. Chen, J. Chen, J. Zhang and X. Wan, Chem. Rec., 2018, 18, 1292 CrossRef CAS PubMed.
- (a) M. Uyanik, N. Sasakura, E. Kaneko, K. Ohori and K. Ishihara, Chem. Lett., 2015, 44, 179 CrossRef; (b) M. Uyanik, T. Kato, N. Sahara, O. Katade and K. Ishihara, ACS Catal., 2019, 9, 11619 CrossRef CAS; (c) M. Uyanik, N. Sahara, O. Katade and K. Ishihara, Org. Lett., 2020, 22, 560 CrossRef CAS PubMed.
- (a) K. Moriyama, K. Ishida and H. Togo, Chem. Commun., 2012, 48, 8574 RSC; (b) Z. Li and R. Tong, J. Org. Chem., 2016, 81, 4847 CrossRef CAS PubMed; (c) J. Xu, L. Liang, H. Zheng, Y. R. Chi and R. Tong, Nat. Commun., 2019, 10, 4754 CrossRef PubMed; (d) C. Mairhofer and M. Waser, Adv. Synth. Catal., 2023, 365, 2757 CrossRef CAS.
- T. Kato, N. Sahara, S. Akagawa, M. Uyanik and K. Ishihara, Org. Lett., 2024, 26, 7255 CrossRef CAS PubMed.
- (a) M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230 CrossRef CAS PubMed; (b) J. Yoshida, A. Shimizu and R. Hayashi, Chem. Rev., 2018, 118, 4702 CrossRef CAS PubMed; (c) M. Elsherbini and T. Wirth, Chem. – Eur. J., 2018, 24, 13399 CrossRef CAS PubMed; (d) K. Yamamoto, M. Kuriyama and O. Onomura, Acc. Chem. Res., 2020, 53, 105 CrossRef CAS PubMed; (e) C. Schotten, T. P. Nicholls, R. A. Bourne, N. Kapur, B. N. Nguyen and C. E. Willans, Green Chem., 2020, 22, 3358 RSC.
- (a) S. Quideau, L. Pouységu and D. Deffieux, Curr. Org. Chem., 2004, 8, 113 CrossRef CAS; (b) S. R. Waldvogel, S. Lips, M. Selt, B. Riehl and C. Kampf, Chem. Rev., 2018, 118, 6706 CrossRef CAS PubMed; (c) T. Yamamoto, T. Saitoh, Y. Einaga and S. Nishiyama, Chem. Rec., 2021, 21, 2254 CrossRef CAS PubMed; (d) F. Medici, S. Resta, A. Puglisi, S. Rossi, L. Raimondi and M. Benaglia, Molecules, 2021, 26, 6968 CrossRef CAS PubMed.
- (a) S. S. Beigbaghlou, R. S. Yafele and M. Kalek, Synthesis, 2023, 4173 Search PubMed; (b) I. Tomczyk and M. Kalek, Chem. – Eur. J., 2024, 30, e202303916 CrossRef CAS PubMed.
- For selected reviews, see: (a) K. Liu, C. Song and A. Lei, Org. Biomol. Chem., 2018, 16, 2375 RSC; (b) H.-T. Tang, J.-S. Jia and Y.-M. Pan, Org. Biomol. Chem., 2020, 18, 5315 RSC; (c) L. F. T. Novaes, J. Liu, Y. Shen, J. M. Meinhardt and S. Lin, Chem. Soc. Rev., 2021, 50, 7941 RSC; (d) F. Lian, K. Xu and C. Zeng, Chem. Rec., 2021, 21, 2290 CrossRef CAS PubMed; (e) K. Mitsudo, Y. Okumura, E. Sato and S. Suga, Eur. J. Org. Chem., 2023, e202300835 CrossRef CAS; (f) J. Du, Y.-L. Du and Q.-W. Gui, Synthesis, 2023, 2799 CAS; (g) K. Yamamoto, M. Kuriyama and O. Onomura, Isr. J. Chem., 2024, 64, e202300068 CrossRef.
- (a) V. P. Kashparova, V. A. Klushin, D. V. Leontyeva, N. V. Smirnova, V. M. Chernyshev and V. P. Ananikov, Chem. – Asian J., 2016, 11, 2578 CrossRef CAS PubMed; (b) K. Kulangiappar, M. Ramaprakash, D. Vasudevan and T. Raju, Synth. Commun., 2016, 46, 145 CrossRef CAS; (c) X. Tan, Q. Wang and J. Sun, Nat. Commun., 2023, 14, 357 CrossRef CAS PubMed; (d) Y. Duan and S. Luo, Angew. Chem., Int. Ed., 2024, 63, e202319206 CrossRef CAS PubMed.
- S. Liang, K. Xu, C.-C. Zheng, H.-Y. Tian and B.-G. Sun, Adv. Synth. Catal., 2018, 360, 4266 CrossRef CAS.
- (a) B. Saxena, R. I. Patel and A. Sharma, RSC Sustainable, 2024, 2, 2169 RSC; (b) Z. Qing, P. Mao, T. Wang and H. Zhai, J. Am. Chem. Soc., 2022, 144, 10640 CrossRef CAS PubMed.
- J. Gagnepain, R. Méreau, D. Dejugnac, J.-M. Léger, F. Castet, D. Deffieux, L. Pouységu and S. Quideau, Tetrahedron, 2007, 63, 6493 CrossRef CAS.
- (a) T. Takiguchi and T. Nonaka, Bull. Chem. Soc. Jpn., 1987, 60, 3137 CrossRef CAS; (b) B. Bennett, J. Chang and A. J. Bard, Electrochim. Acta, 2016, 219, 1 CrossRef CAS; (c) K. Mitsudo, R. Matsuo, T. Yonezawa, H. Inoue, H. Mandai and S. Suga, Angew. Chem., Int. Ed., 2020, 59, 7803 CrossRef CAS PubMed; (d) L. G. Gombos and S. R. Waldvogel, Sustainable Chem., 2022, 3, 430 CrossRef CAS; (e) Q.-L. Yang, W.-W. Li, Z.-X. Zhang, H.-M. Zhang, X.-J. Lian and H.-M. Guo, Org. Chem. Front., 2023, 10, 5369 RSC.
- Y. Zheng, Y. T. Cheung, L. Liang, H. Qiu, L. Zhang, A. Tsang, Q. Chen and R. Tong, Chem. Sci., 2022, 13, 10479 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental protocols, nuclear magnetic resonance spectra. See DOI: https://doi.org/10.1039/d4cc06472k |
| This journal is © The Royal Society of Chemistry 2025 |