
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structural insights of mechanochemically amorphised MIL-125-NH2†
Emily V.
Shaw
 a,
Celia
Castillo-Blas
*a,
Timothy
Lambden
a,
Beatriz
de Santos
b,
Bethan
Turner
c,
Giulio I.
Lampronti
a,
Joonatan E. M.
Laulainen
a,
Georgina P.
Robertson
ad,
Ashleigh M.
Chester
a,
Chumei
Ye
ae,
Shaoliang
Guan
ae,
Joshua K. G.
Karlsson
a,
Valentina
Martinez
f,
Ivana
Brekalo
f,
Bahar
Karadeniz
f,
Silvia
Cabrera
gh,
Lauren N.
McHugh
c,
Krunoslav
Užarević
f,
Jose
Alemán
bh,
Alberto
Fraile
bh,
Rachel C.
Evans
a,
Paul A.
Midgley
a,
David A.
Keen
i,
Xavier
Moya
a and
Thomas D.
Bennett
*j
a,
Celia
Castillo-Blas
*a,
Timothy
Lambden
a,
Beatriz
de Santos
b,
Bethan
Turner
c,
Giulio I.
Lampronti
a,
Joonatan E. M.
Laulainen
a,
Georgina P.
Robertson
ad,
Ashleigh M.
Chester
a,
Chumei
Ye
ae,
Shaoliang
Guan
ae,
Joshua K. G.
Karlsson
a,
Valentina
Martinez
f,
Ivana
Brekalo
f,
Bahar
Karadeniz
f,
Silvia
Cabrera
gh,
Lauren N.
McHugh
c,
Krunoslav
Užarević
f,
Jose
Alemán
bh,
Alberto
Fraile
bh,
Rachel C.
Evans
a,
Paul A.
Midgley
a,
David A.
Keen
i,
Xavier
Moya
a and
Thomas D.
Bennett
*j
aDepartment of Materials Science & Metallurgy, University of Cambridge, 27 Charles Babbage Road, Cambridge, UK. E-mail: cc2078@cam.ac.uk
bOrganic Chemistry Department, Science Faculty, Universidad Autónoma de Madrid, C/ Francisco Tomás y Valiente, 7, 28049 Madrid, Spain
cDepartment of Chemistry, University of Liverpool, Crown Street, Liverpool L69 7ZD, UK
dDiamond Light Source Ltd., Diamond House, Harwell Campus, Didcot, Oxfordshire, UK
eMaxwell Centre, Cavendish Laboratory, University of Cambridge, J.J. Thomson Avenue, Cambridge, CB3 0HE, UK
fDivision of Physical Chemistry, Ruđer Bošković Institute, Zagreb, Croatia
gInorganic Chemistry Department, Science Faculty, Universidad Autónoma de Madrid, C/ Francisco Tomás y Valiente, 7, 28049 Madrid, Spain
hInstitute for Advanced Research in Chemical Science (IAdChem), Universidad Autónoma de Madrid, C/ Francisco Tomás y Valiente, 7, 28049 Madrid, Spain
iISIS Facility, Rutherford Appleton Laboratory, Harwell Campus, Didcot, Oxfordshire OX11 0QX, UK
jSchool of Physical and Chemical Sciences, University of Canterbury, Private Bag 4800, Christchurch 8140, New Zealand. E-mail: thomas.bennett@canterbury.ac.nz
First published on 28th February 2025
Abstract
In this work, we investigated the response of the metal–organic framework MIL-125-NH2 to ball-milling. Both localised and bulk analyses revealed prolongued ball-milling results in a complete loss of long-range structural order. Investigation of this disorder revealed partial retention of the local bonding of the secondary building unit, suggesting structure collapse progressed primarily through metal–linker bond breakage. We explored the photocatalytic performance of the materials, and examined the materials’ band gap using UV-Vis reflectance spectroscopy.
Amorphous metal–organic frameworks (aMOFs) have gained increased attention thanks to their facile synthesis and promising physical and chemical properties.1 These aMOFs preserve the interconnected nature of the parent crystalline material, whilst lacking any correlated long-range order.1,2 Formation of aMOFs can be achieved through several methodologies, including direct synthesis, thermal treatment or application of mechanical shear stress, frequently resulting in a non-reversible transition to a disordered structure.3
The high concentration of intrinsic defects in aMOFs often enhances the functionalities, acting as the active sites for both catalysis and gas sorption applications.4,5 However, amorphisation typically reduces their porosity compared to the crystalline parent materials. Despite this, aMOFs have been utilised as slow drug release systems, also displaying improved gas selectivity and separation, compared to crystalline equivelents.6 Therefore, understanding the structural effects of amorphisation has become crucial for predicting the properties of these amorphous materials for their potential applications.
Ball-milling has been shown to reduce crystallite size and introduce structural defects, which, at critical concentrations, leads to structural distortion and eventual collapse into a highly disordered, amorphous state.7,8 Several MOFs, e.g., Zn-ZIF-8, Al-ndc and Zn-MOF-74, retain their extended X-ray absorption fine structure (EXAFS) profiles upon mechanical amorphisation, indicating a retention of the short-range order (SRO) and secondary building unit (SBU).8 In contrast, ball-milled UiO-66 exhibits distortion of its Zr6O4(OH)4 SBU during amorphisation,9 which progresses via the breakage of metal–linker (M–L) bonds, as suggested by X-ray pair distribution (XPDF) anaylsis.10
Here we present a structural study of mechanically amorphised MIL-125-NH2 using powder X-ray diffraction (PXRD), scanning electron diffraction (SED) and X-ray pair distribution function (XPDF) analysis. The optical properties of this photoactive material were evaluated using ultraviolet-visible (UV-Vis) spectroscopy. MIL-125-NH2, [Ti8O8OH4(H2N-bdc)6] (bdc = benzene-1,4-dicarboxylic acid, C8H7NO4) is a titanium-MOF isostructural to MIL-125 (Fig. 1a), consisting of a porous three-dimensional quasi cubic tetragonal structure with fcu topology possessing tetrahedral and octahedral cages.11 The SBU is defined as an Ti8O8(OH)4 octameric ring of eight titanium oxo-clusters with corner and edge-sharing TiO6 octahedral units.
 | ||
Fig. 1 (a) Structure of MIL-125, highlighting the pore cavities with two diameters (blue = 6.1 Å, yellow = 12.5 Å). Titanium polyhedra, oxygen and carbon are represented in purple, red and black, respectively. Hydrogen atoms are omitted for clarity. Structure calculated from a reported crystal structure.12 (b) PXRD patterns of MIL-125-NH2 as a function of duration of ball milling. Wavelength λ = 1.5418![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Å. Å. | ||
MIL-125-NH2 was prepared following a modified reported synthetic procedure (see ESI,† methods).13 Phase purity was confirmed by PXRD and Pawley refinement (Fig. S2 and Table S1, ESI†). Upon ball-milling of crystalline MIL-125-NH2, rapid loss of sharp Bragg intensities was observed in the PXRD (Fig. 1b and Fig. S3 and Table S2, ESI†). Amorphisation was complete after 15 minutes, with this sample referred to as amMIL-125-NH2 (am = mechanochemically amorphised). In situ mechanochemical PXRD studies, performed at the P.02.1 beamline (DESY) (Fig. S4, ESI†), confirmed the timescale of this collapse. Sequential Rietveld refinements indicated that the unit cell parameters remained largely unchanged until ∼6 minutes, at which point the crystallite size decreased to approximately the unit cell size, Fig. S5 (ESI†). Phase quantification suggested that amorphisation initially progressed through a reduction in crystallite size, with a delayed onset of amorphisation until ∼8 minutes. Scanning electron microscopy (SEM) confirmed this decrease in crystallite size, and subsequent formation of aggregate clusters (Fig. S6, ESI†).
Scanning electron diffraction (Fig. 2), was utilised to probe the order at the nanoscale. This was operated at a low electron flux (10e− A−2 s−1) to reduce beam-induced damage commonly observed in MOFs and hybrid materials.14 The crystalline Bragg spots observed in MIL-125-NH2, Fig. 2d, are lost upon amorphisation of the sample, instead producing diffuse rings of scattering, Fig. 2e. This was quantified through the normalised variance as a function of reciprocal distance, Fig. 2f.
 | ||
| Fig. 2 SED of MIL-125-NH2 (a) virtual bright-field image of crystalline MIL-125-NH2, scale bar = 500 nm. Virtual high-angle annular dark-field (HAADF) images of (b) nanocrystal of crystalline MIL-125-NH2 and (c) nanocrystal of amMIL-125-NH2, scale bars = 50 nm. Summed area diffraction maps of these crystals are shown in (d) and (e), respectively, with scale bar = 2 nm−1. (f) Normalised variance, measured azimuthally, of the diffraction maps in (d) (blue) and (e) (orange) with the central beam excluded. The observed peaks were assigned to (i) (002) (211), (ii) (112), (iii) (330) (iv) (004) crystal planes. | ||
X-ray total scattering data were collected at P02.1 beamline of PETRA III (DESY) to investigate the SRO upon mechanical amorphisation. Bragg peak intensities decreased upon increased ball-milling time, consistent with SED, at a similar rate to PXRD (Fig. S7, ESI†). Signals >10 Å, indicating LRO, decreased with ball-milling time and were absent in amMIL-125-NH2, (Fig. S8, ESI†). The XPDF of crystalline MIL-125-NH2 showed long-range correlations, corresponding to order extending between SBUs, suggestive of an extended ordered structure (Fig. S8, ESI†).
Correlations of the SRO were calculated using an isolated SBU with PDFgui (Fig. S9, ESI†).15 Comparing the SRO of the ball-milled versus crystalline samples revealed some retention of peak intensity and position, with deviations highlighted in the difference pattern (Fig. 3b). Identifying the structural origin of the changes upon amorphisation is challenging due to the complexity of the SBU structure. Partial PDFs were used (Fig. S10, ESI†) to investigate the origin of the contributions of each peak. Correlations involving hydrogen were excluded as they contributed minimally to the scattering. Key peak contributions are outlined in Table S3 (ESI†) and Fig. 3c. The loss of peak intensity for peaks at longer length scale confirmed the reduction of correlations extending across the linker (Fig. S12, ESI†). Changes in peak area are qualitatively illustrated in Fig. S12a (ESI†). SRO analysis revealed peak movement and broadening (Fig. S12b, ESI†), suggesting linker-SBU bond breakage (Fig. S9, ESI†), consistent with observations in UiO-66, MIL-100 and several ZIF species.10,16,17
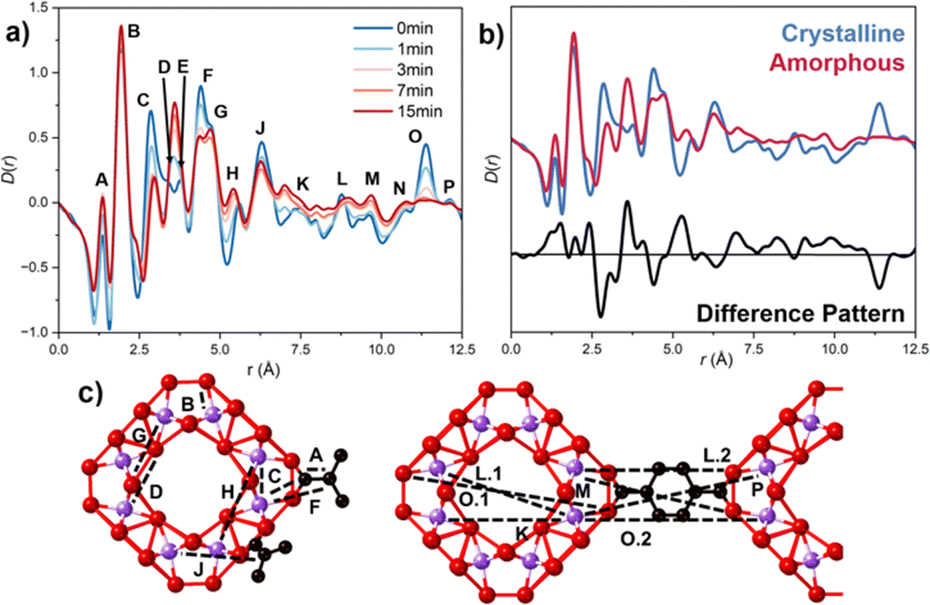 | ||
| Fig. 3 (a) Pair distribution functions (PDF) of the ball-milled materials. Key shows the duration of milling in minutes. Labels correspond to some key correlations, marked in (c). (b) Comparison of SRO region of PDFs of crystalline and amMIL-125-NH2. (c) SBU and MIL-125-NH2 structure. Ti, O and C are denoted in purple, red and black, respectively. | ||
Principal component analysis (PCA) of D(r)'s have previously been used to study structural distortions in aMOFs, such as Fe-BTC, as well as to investigate amorphisation by melting in TIF-4 material.18 In this work, PCA of the PDF data at different ball-milling times (Fig. S13 and Tables S4, S5, ESI†) showed PC1 represents an average of the data sets, primarily containing information of the SRO. PC2 highlights the structural distortion upon amorphisation, shown by the equivalence to the difference pattern, Fig. S13b (ESI†). The significance of the structural distortion was illustrated by the high variance contribution, 8.83%. Peaks at high-r values indicated a reduction in LRO as ball-milling time increased, while low-r peaks suggested potential structural distortions. Strong peaks in PC2 located at peak F (4.35 Å) and between 5.68–6.46 Å (peaks H and J), are consistent with loss of Ti–C correlations, consistent with amorphisation progressing through metal–linker bond breakage, and the partial loss of linker. A prominent peak at 2.74 Å (peak C), showed rapid loss of intensity supporting this.
Several of the observed changes, however, cannot be explained for solely by M–L bond breakage, or the loss of long-range order. The coalescence of peaks D and E during amorphisation, which contributes to the observed increase in peak intensity, suggests a distortion within the SBU of the structure (Fig. S12b, ESI†). The observed increase in intensity could also be attributed to contributions from peak F, through compression of the Ti-SBU, or from peak C, through an increase of Ti–C bond length as a result of linker monocoordination. While initial decrease in intensity of peak H was attributed to the loss of Ti–C interactions, the shift to shorter distances is supportive of a lateral collapse of the SBU structure. The subsequent increase in peak intensity is likely as a result of both this, and the broadening of peak G. Beyond this point, it deconvolution of structural changes from the loss of LRO was not possible.
X-ray photoelectron spectroscopy was used to investigate this potential change further (Fig. S15, ESI†). The deconvolution of O 1s, N 1s, C 1s and Ti 2p signals were consistent with literature (Fig. S16 and S17, ESI†). The binding energies showed little variation in the binding environments between the crystalline and amorphous samples. The Ti–O contribution was reduced upon amorphisation, Table S6 (ESI†), consistent with the breakage of these SBU-based bonds. This might indicate the formation of alternative SBU structures rather than just distortions, and potential formation of TiO2 nanodomains. The increased O–H signal upon amorphisation supports the idea of M–L bond breakage. Simulated PDFs for other Ti-based MOF containing different SBUs (e.g., MOF-901 (Ti6O6), MIL-177 (Ti6O9), COK-69 (Ti3O6) or NTU-9) showed similarities to amMIL-125-NH2, Fig. S14 (ESI†), though further analysis is needed to confirm specific structures. Comparisons were also made to TiO2, both rutile and anatase (Fig. S14d and e, ESI†), which showed limited correlation with the peaks in amMIL-125-NH2, past what is defined by Ti–O bond length. This does not, however, rule out the formation of small clusters of Ti–O, separate to that held within the SBU.
Fourier-transform infrared (FTIR) spectroscopy confirmed changes in linker-binding mode upon amorphisation (Fig. S18 and Table S7, ESI†). Significant broadening of carbonyl vibration peaks, both symmetric and asymmetric, suggests a mix of divalent, monovalent, and uncoordinated linkers, indicating distortion in the linker binding environments.19 Increased peak intensity at 1689 cm−1 and 1567 cm−1 were characteristic of the presence of uncoordinated and monocoordinated linker, respectively.20,21 These observations suggest that the amorphisation involves M–L bond breakage and the formation of a high concentration of linker-based defects. Broadening in the 400–800 cm−1 range indicates changes in the O–Ti–O bonds, consistent with SBU collapse or rearrangement.12
Thermogravimetric analysis (TGA) was used to investigate missing-linker defects (Fig. S19, ESI†).22 TGA of two samples of amMIL-125-NH2, one with prolonged exposure to air, showed increased weight loss from solvent evaporation, suggesting rapid coordination of atmospheric water capping missing-linker defects.20 Few compositional changes were detected (Tables S8, S9 and Fig. S19, ESI†), likely due to missing linkers being trapped in the pores or through mono-coordination. Washing amMIL-125-NH2 removed the trapped linkers, with an SBU:linker ratio of 1:2.97 from 1:4.99 being detected, confirming the presence of a high concentration of missing linker defects, (Fig. S20, ESI†). CHN confirmed no large quantity of TiO2 had been produced.
These missing-linker defects could be valuable for aMOF applications. MIL-125-NH2 has shown promising photocatalytic activity due to linker-to-metal charge transfer (LMCT) under visible light exposure.23 The localisation of the LUMO and HOMO, on the metal cluster and organic linker, respectively, inhibits fast electron–hole recombination, enhancing photocatalytic efficiency.23 Since amorphisation alters M–L bonds and increases the number of accessible Ti active sites, it likely affects photocatalytic performance.23,24 This is supported by the notable shift in colour observed upon amorphisation, indicative of a change in band gap energy, Fig. S21 (ESI†). This could present a way of tuning the band gap for specific applications through the controlled introduction of these defects. While band gap engineering has focused on crystal facet growth and defect control, the impact of combined defects and disorder on photocatalysis remains underexplored.23,24
Reflectance UV-Vis (RUV-Vis) spectroscopy, employed to investigate this colour change, showed a key absorbance at ∼380 nm, attributed to the LMCT band (Fig. 4a and Fig. S22, ESI†).23,24 A Kubelka–Munk transformation of the RUV-Vis data extracted a band gap value for the crystalline material consistent with the literature (Fig. 4b).23,24 Amorphisation caused a red-shift in the band gap from 2.80 eV to 2.72 eV, aligning with the observed colour change and absorbance shift.
 | ||
| Fig. 4 (a) UV-Vis diffuse reflectance spectra of MIL-125-NH2 (blue) and amMIL-125-NH2 (red). Due to the absorbance of glass, features below 300 nm have been subtracted as background. The feature observed at ∼400 nm was attributed to a change between detectors, required to measure across the whole wavelength range. (b) Tauc plot. | ||
The photocatalytic activity of the crystalline and amorphous materials was tested via the oxidative coupling of amines to imines (Scheme S1, ESI†). Although the amorphous sample showed higher absorbance at the irradiated wavelengths (Fig. S24, ESI†), the photocatalytic activity was slightly lower (Table S10, ESI†). This suggested that factors beyond band gap energy, that are affected by the amorphisation process, e.g. particle size, morphology, structural arrangements and/or porosity, also influence the photocatalytic performance of amMIL-125-NH2.3,13
N2 and CO2 adsorption isotherms (Fig. S25, ESI†) showed a significant drop in sorption capacity for the amorphous material. The BET surface area of MIL-125-NH2 decreased from 785 m2 g−1 to 51 m2 g−1 after amorphisation. As photocatalytic reactions rely on a large accessible surface area to reach defects sites, balancing defect concentration and porosity is crucial for optimal activity. Whilst a decrease in overall photocatalytic activity is observed, relative to the substantial reduction in surface area, amorphisation was found to enhance the intrinsic photocatalytic activity of the material. Solvent-assisted stabilisation could help retain some of the original porosity while introducing defects.16
In conclusion, ball-milling amorphisation in MIL-125-NH2 occurs via missing-linker defects, M–L bond breakage, and partial SBU distortion. Further analysis of short-range order is needed to identify the dominant factor. These structural changes affect the band gap, which, with other parameters, could be tuned to influence photocatalytic properties. The results highlight ball-milling as a promising method for defect engineering in MOFs.
C. C. B. and T. D. B. designed the project. E. V. S. wrote the original manuscript with inputs from all authors. E. V. S. preprepared and characterized the materials by PXRD, SEM, TGA and FTIR. TEM data was collected by J. E. M. L. and P. A. M., with T. L. performing the analysis. Total scattering data were collected by C. C. B., G. P. R., D. A. K. and A. M. C., and analysed by E. V. S., with inputs from C. C. B. and D. A. K. S. G. collected the XPS data. C. Y., V. M., K. U. and B. K. collected the in situ mechanochemical PXRD, with analysis by G. I. L. B. T. and L. N. M. collected the N2 and CO2 isotherms. B. S., S. C., A. F. and J. A. performed the photocatalytic tests. UV-Vis data were collected by J. K. G. K., and analysed by E. V. S., with supervision from R. C. E.
Authors thank the Leverhulme Trust (RPG-2020-005) (T. D. B., C. C. B., A. M. C.), the Royal Society for both a University Research Fellowship (URF\R\211013) (T. D. B.) and a research grant (RGS\R2\212221) (T. D. B.), UKRI and Diamond light source (STU0366) (G.P.R.), the EPSRC Cambridge NanoDTC (EP/S022953/1) (T. L.), St Edmunds College (C. Y.), the Croatian Science Foundation (IP-2020-02-4702) (V. M., I. B., B. K. and K. U.), the Spanish MICINN (PID2021-122299NB-100) (B. S., S. C., J. A. and A. F), the University of Liverpool (L. N. M., B. T.), and the ERC European Union's Horizon 2020 research and innovation programme (Grant Agreement No. 818762) (R. C. E., J. K. G. K.). We acknowledge DESY (Hamburg, Germany), a member of the Helmholtz Association HGF, for the provision of experimental facilities. Parts of this research were carried out at PETRA III and we would like to thank M. Etter for assistance in using beamline P02.1. Beamtime was allocated for proposals I-20221330 EC and I-20230031 EC.
Data availability
The data supporting this article have been included as part of the ESI,† for scanning electron microscopy and transmission electron microscopy characterisation and photocatalysis tests. Data for this article, including DSC, PDF and X-ray analysis, FTIR, gas sorption isotherms, TGA, XPS and UV-vis spectroscopy are available at Open Science Framework at DOI https://doi.org/10.17605/OSF.IO/3UQYT.Conflicts of interest
There are no conflicts to declare.References
- T. D. Bennett and A. K. Cheetham, Acc. Chem. Res., 2014, 47, 1555–1562 CrossRef CAS PubMed.
- J. Fonseca, T. Gong, L. Jiao and H.-L. Jiang, J. Mater. Chem. A, 2021, 9, 10562–10611 RSC.
- E. V. Shaw, A. M. Chester, G. P. Robertson, C. Castillo-Blas and T. D. Bennett, Chem. Sci., 2024, 15, 10689–10712 RSC.
- J. Fonseca and S. Choi, Catal. Sci. Technol., 2020, 10, 8265–8282 RSC.
- H. Wang, Q. Yang, N. Zheng, X. Zhai, T. Xu, Z. Sun, L. Wu and M. Zhou, Nano Res., 2023, 16, 4107–4118 CrossRef CAS.
- T. D. Bennett, P. J. Saines, D. A. Keen, J.-C. Tan and A. K. Cheetham, Chem. – Eur. J., 2013, 19, 7049–7055 CrossRef CAS PubMed.
- Y. Shan, G. Zhang, Y. Shi and H. Pang, Cell Rep. Phys. Sci., 2023, 4, 101301 CrossRef CAS.
- A. M. Belenguer, A. A. L. Michalchuk, G. I. Lampronti and J. K. M. Sanders, Beilstein J. Org. Chem., 2019, 15, 1226–1235 CrossRef CAS PubMed.
- T. Panda, S. Horike, K. Hagi, N. Ogiwara, K. Kadota, T. Itakura, M. Tsujimoto and S. Kitagawa, Angew. Chem., Int. Ed., 2017, 56, 2413–2417 CrossRef CAS PubMed.
- T. D. Bennett, T. K. Todorova, E. F. Baxter, D. G. Reid, C. Gervais, B. Bueken, B. Van de Voorde, D. De Vos, D. A. Keen and C. Mellot-Draznieks, Phys. Chem. Chem. Phys., 2016, 18, 2192–2201 RSC.
- B. Kim, Y.-R. Lee, H.-Y. Kim and W.-S. Ahn, Polyhedron, 2018, 154, 343–349 CrossRef CAS.
- M. Dan-Hardi, C. Serre, T. Frot, L. Rozes, G. Maurin, C. Sanchez and G. Férey, J. Am. Chem. Soc., 2009, 131, 10857–10859 CrossRef CAS PubMed.
- C. Zlotea, D. Phanon, M. Mazaj, D. Heurtaux, V. Guillerm, C. Serre, P. Horcajada, T. Devic, E. Magnier, F. Cuevas, G. Férey, P. L. Llewellyn and M. Latroche, Dalton Trans., 2011, 40, 4879–4881 RSC.
- A. F. Sapnik, C. Sun, J. E. M. Laulainen, D. N. Johnstone, R. Brydson, T. Johnson, P. A. Midgley, T. D. Bennett and S. M. Collins, Commun. Chem., 2023, 6, 92 CrossRef CAS PubMed.
- C. L. Farrow, P. Juhas, J. W. Liu, D. Bryndin, E. S. Božin, J. Bloch, T. Proffen and S. J. L. Billinge, J. Condens. Matter Phys., 2007, 19, 335219 CrossRef CAS PubMed.
- A. F. Sapnik, D. N. Johnstone, S. M. Collins, G. Divitini, A. M. Bumstead, C. W. Ashling, P. A. Chater, D. S. Keeble, T. Johnson, D. A. Keen and T. D. Bennett, Dalton Trans., 2021, 50, 5011–5022 RSC.
- T. D. Bennett, S. Cao, J. C. Tan, D. A. Keen, E. G. Bithell, P. J. Beldon, T. Friscic and A. K. Cheetham, J. Am. Chem. Soc., 2011, 133, 14546–14549 CrossRef CAS PubMed.
- A. F. Sapnik, I. Bechis, A. M. Bumstead, T. Johnson, P. A. Chater, D. A. Keen, K. E. Jelfs and T. D. Bennett, Nat. Commun., 2022, 13, 2173 CrossRef CAS PubMed.
- N. J. Castellanos, Z. Martinez Rojas, H. A. Camargo, S. Biswas and G. Granados-Oliveros, Transition Met. Chem., 2019, 44, 77–87 CrossRef CAS.
- K. Tan, H. Pandey, H. Wang, E. Velasco, K.-Y. Wang, H.-C. Zhou, J. Li and T. Thonhauser, J. Am. Chem. Soc., 2021, 143, 6328–6332 CrossRef CAS PubMed.
- Z. Su, Y.-R. Miao, G. Zhang, J. T. Miller and K. S. Suslick, Chem. Sci., 2017, 8, 8004–8011 RSC.
- G. C. Shearer, S. Forselv, S. Chavan, S. Bordiga, K. Mathisen, M. Bjørgen, S. Svelle and K. P. Lillerud, Top. Catal., 2013, 56, 770–782 CrossRef CAS.
- V. Kavun, E. Uslamin, B. van der Linden, S. Canossa, A. Goryachev, E. E. Bos, J. Garcia Santaclara, G. Smolentsev, E. Repo and M. A. van der Veen, ACS Appl. Mater. Interfaces, 2023, 15, 54590–54601 CrossRef CAS PubMed.
- L. Pukdeejorhor, S. Wannapaiboon, J. Berger, K. Rodewald, S. Thongratkaew, S. Impeng, J. Warnan, S. Bureekaew and R. A. Fischer, J. Mater. Chem. A, 2023, 11, 9143–9151 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc06320a |
| This journal is © The Royal Society of Chemistry 2025 |