
Understanding the Ag–S interface stability and electrocatalytic activity of CO2 electroreduction in atomically precise Ag25 nanoclusters†
Yuting
Ye‡
a,
Xia
Zhou‡
b,
Yuping
Chen
a,
Likai
Wang
 *b and
Qing
Tang
*a
*b and
Qing
Tang
*a
aSchool of Chemistry and Chemical Engineering, Chongqing Key Laboratory of Chemical Theory and Mechanism, Chongqing University, Chongqing 401331, China. E-mail: qingtang@cqu.edu.cn
bSchool of Chemistry and Chemical Engineering, Shandong University of Technology, Zibo, Shandong 255049, China. E-mail: lkwangchem@sdut.edu.cn
First published on 9th January 2025
Abstract
Elucidating the catalytic properties of metal nanoclusters (NCs) with essentially the same structure but different core metals is of fundamental interest. Our current studies have demonstrated that the thiolated Ag25(SR)18 NC exhibits SR ligand leaching dynamics and electrocatalytic activity in CO2 reduction distinct from those of its Au25(SR)18 NC structural analogue.
In recent years, small metal nanoclusters (NCs) made with atomic precision have attracted great attention in various fields such as catalysis, chemical sensing and bio-applications.1–6 The use of atomically precise metal NCs could eliminate concerns of inhomogeneity, and their ultrasmall size (1–2 nm) minimizes the material gap between experiments and modelling, thus holding great promise in fundamental catalysis research.7,8 In the large family of metal NCs protected by various ligands, the thiolate-protected metal NCs have attracted extensive research interest, wherein Au and Ag NCs are two extensively studied groups.9–12 Thiolated Au NCs have been widely studied in many catalytic applications.5,6,13,14 For example, gold NCs have been recognized for their exceptional electrocatalytic activities in electrocatalytic reduction of CO2 to CO.15 The effects of cluster size, shape, surface ligands and heterometal doping have been extensively employed to modulate the electrocatalytic activity and selectivity of gold NCs.16–19
Compared to the widely studied gold NCs, thiolated Ag NCs have been less extensively investigated, possibly due to their moderate stability, leading to a limited understanding of their catalytic properties,20,21 In particular, there are few recent studies on Ag NCs for photocatalytic CO2RR.22,23 Investigating Ag-based catalysts is of great importance for uncovering new catalytic mechanisms and utilizing metals that are less expensive than Au. Note that the catalytic properties of metal NCs are highly sensitive to the packing of atomic structures, and a subtle disturbance to the structure, composition and morphology may cause a significant change in the electronic and catalytic behaviors. In this regard, a direct comparison of catalytic performances of materials with essentially identical structures but distinct metal cores is of particular interest but has been hardly been pursued. In the large family of thiolate-protected metal NCs, the well-characterized Ag25(SR)18 provides an ideal model to study the effect of the core since this cluster and Au25(SR)18 share essentially the same structure. They both feature an icosahedral M13 core protected by six V-shaped motifs arranged in a quasi-Th symmetry (Fig. S1, ESI†),10 thus enabling a direct comparison of their catalytic activities.10,24 Of note, recent studies have demonstrated that the Au–S interface in Au25(SR)18 NCs is unstable under electrochemical conditions, and would be attacked by water from electrolyte to lead to stripping of some of the thiolate ligands and hence expose under-coordinated Au atoms to the surface.5,25,26 Since the Ag–S bond is expected to be stronger and more ionic than the Au–S bond based on the difference in electronegativity between S and these two metals, it would be interesting to determine whether the Ag–S interface would become destabilized under electrochemical environments. If the thiolate ligands in Ag25(SR)18 can be eliminated from the Ag–S interface, then it would be of great importance to investigate the electrocatalytic response of the exposed Ag sites in comparison to Au in the dethiolated Au25 counterpart.
Therefore, to gain a better understanding of the catalytic mechanism of Ag NCs, we selected Ag25(SR)18 as our research model, and first performed density functional theory (DFT) calculations to investigate its dynamic stability in electrochemical conditions.27–30 To reduce the computational cost, the SR ligands were simplified to be –SCH3.5 The computational details are provided in ESI.† To simulate the cluster/water interface, Ag25 and Au25 were placed in respective periodic boxes, each with dimensions of 20 × 20 × 30 Å3 and surrounded by 90 explicit water molecules with a density of about 1 g cm−3 (Fig. S2, ESI†). The potential of the entire system was regulated by introducing different amounts of Na atoms into the aqueous layer while the overall system was kept charge neutral. In this way, URHE = (Φ – 4.44)/e + 0.0592 × pH would give a different applied potential U depending on the value of the work function (Φ) for each model.31 Under a neutral pH condition (pH = 7), the addition of 1, 2 and 3 Na atoms into the Ag25/water system would thus yield potentials of, respectively, 0.33 V, −0.84 V and −1.10 V vs. reversible hydrogen electrode (RHE).
According to the previous studies,5,26 the desorption of thiolate ligands from the Au25(SR)18 surface is promoted by H2O attacking the S atom to initiate the weakening and breakage of Au–S bonds. In the case of Ag25(SCH3)18 NCs, the –SR ligand would be expected to undergo a similar leaching process, and the corresponding control variable (CV) for H adsorption (step 1) and Ag–S breakage (step 2) are defined schematically in Fig. S3 (ESI†). Our constrained ab initio molecular dynamics (AIMD) simulations at 300 K indicated the dynamic behavior of the Ag–S interface to be strongly dependent on the applied potential (Fig. 1a). At a positive potential of 0.33 V (Fig. S4, ESI†), the Ag–S interface is very stable: an attack of water from the solution onto the ligated S atom to form an adsorbed S–H bond and solvated OH− species would have to overcome a very high barrier of 1.28 eV. For the potential shifted negatively to −0.84 V, the barrier for the water attack forming an S–H bond decreases to 1.04 eV (Fig. 1b). Note that the water molecule prefers to attack the S atom from the exterior thiolate ligand initially coordinated to two staple Ag atoms (Fig. 1a). This picture is quite different from that for the previously reported Au25(SR)18 NC,5 where the water molecule tends to attack the interior thiolate ligand initially coordinated to one staple Au and one icosahedral surface Au. Formation of the S–H bond is accompanied by a weakening of and hence more easily breakable Ag–S bond (Fig. 1c). Note that at URHE = −0.84 V, only one staple Ag–S bond is broken with a small free energy barrier of 0.16 eV, while the other staple Ag still strongly interacts with the coordinated S atom (Fig. 1a). For the potential further decreased to URHE = −1.10 V, one of the staple Ag–S bonds first breaks spontaneously after the structural relaxation at 0.8 ps, followed by formation of an S–H bond with a moderate barrier of 0.67 eV, and the other staple Ag–S bond can be easily broken with a negligibly small barrier of 0.08 eV, which ultimately results in the complete removal of the –SCH3 ligand, forming a free HSCH3 molecule dissolved in solution and exposing two dethiolated Ag sites (exposed Ag–Ag bond length of 2.74 Å). These simulations indicated that the applied potential can significantly affect the leaching dynamics of the Ag–S interface, and a lower reduction potential could kinetically promote the removal of thiolate ligands.
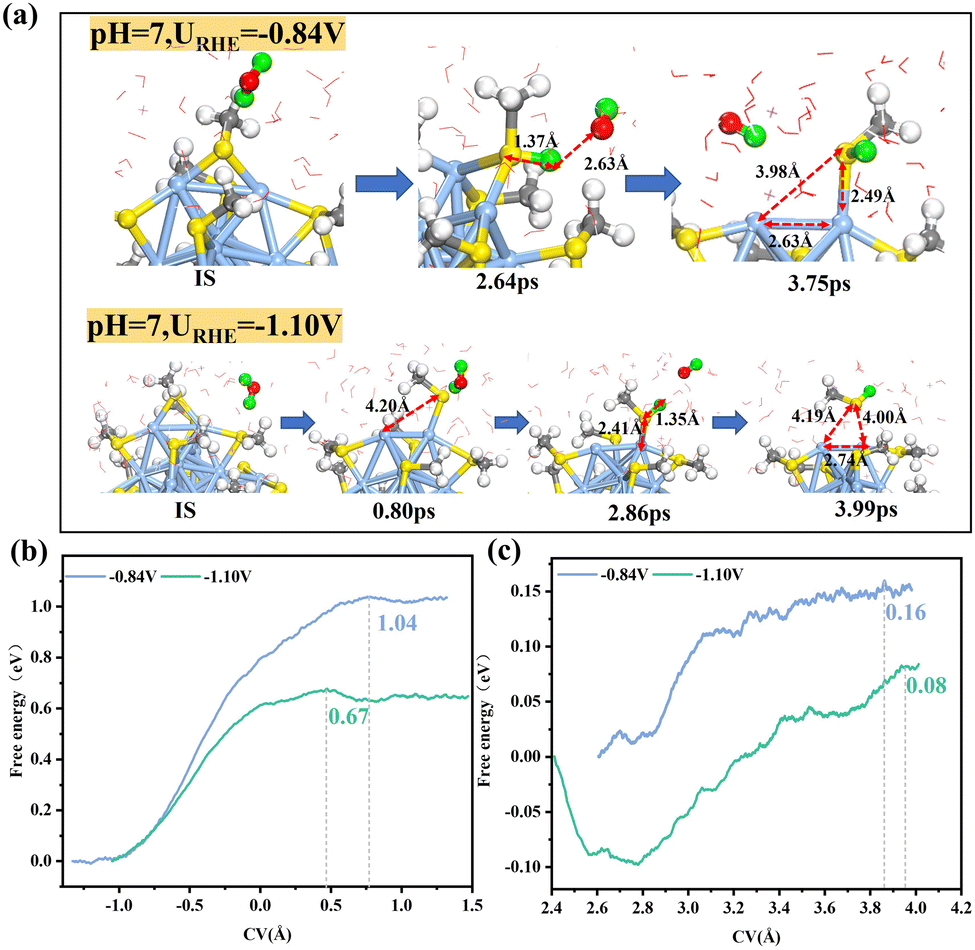 | ||
| Fig. 1 (a) Schematic of local structures formed during adsorption of *H onto the S site and during Ag–S bond breaking in Ag25(SR)18 at URHE = −0.84 V and −1.10 V. (b) Integral free energy curves for *H adsorption and (c) Ag–S bond breaking at URHE = −0.84 V and −1.10 V, respectively. The Ag, S, C, O and H atoms are shown as pale blue, yellow, grey, red and white spheres, respectively. The H from the attacking H2O is colored in green. | ||
As a comparison, we also performed AIMD simulations on the Au25(SCH3)18 analogue in the same neutral environment. In this case, additions of one and two Na atoms into the water layer correspond to applied potential URHE values of 0.11 V and −0.60 V, respectively. Note that addition of 3 Na atoms would lead to severe deformation of the Au25 NC due to an especially low potential produced (URHE = −1.30 V, Fig. S5, ESI†), which is beyond our consideration. Similar to the case in Ag25, the ligand leaching in Au25 was also found to be dependent on potential. At URHE = 0.11 V, adsorption of water-dissociation-derived H onto the S atom from the interior thiolate ligand has to overcome a high barrier of 1.13 eV (Fig. S6, ESI†). For the potential lowered to −0.60 V (Fig. 2), the attack of S by water becomes much more facile, and the formation of the S–H bond is accompanied by the consecutive breaking of the surface Au–S bond and the staple Au–S bond. Note the difference between the water attack site in Au25 (interior thiolate) and that in Ag25 (exterior thiolate). To understand this difference, we analysed the charge states of the two possible S sites. The Bader charge analysis carried out (Fig. S7, ESI†) revealed that in Ag25, the exterior S site carries a more negative charge of −0.37|e| as compared to the interior S site (−0.35|e|); conversely, in Au25, the analysis indicated the interior S to be more negatively charged (−0.30|e|) than the exterior S site (−0.12|e|). The S site bearing a more negative charge is prone to be attacked by H2O and is hence more reactive. At 1.16 ps, the two breaking Au–S bonds reach an equilibrium length of greater than 3 Å, at which point the –SCH3 ligand can be considered to be completely removed, and the two exposed Au atoms are bonded to each other with a shortened bond length of 2.71 Å. In particular, the negligible barrier of 0.07 eV (Fig. 2c) indicates that the thiolate leaching in Au25 occurs spontaneously at a negative reduction potential. Notably, at a comparable reduction potential (−0.84 V in Ag25vs. −0.60 V in Au25), the leaching of thiolate from the Ag–S interface is kinetically more difficult than that from the Au–S interface, which is closely related to the difference between their M–S bond strengths.
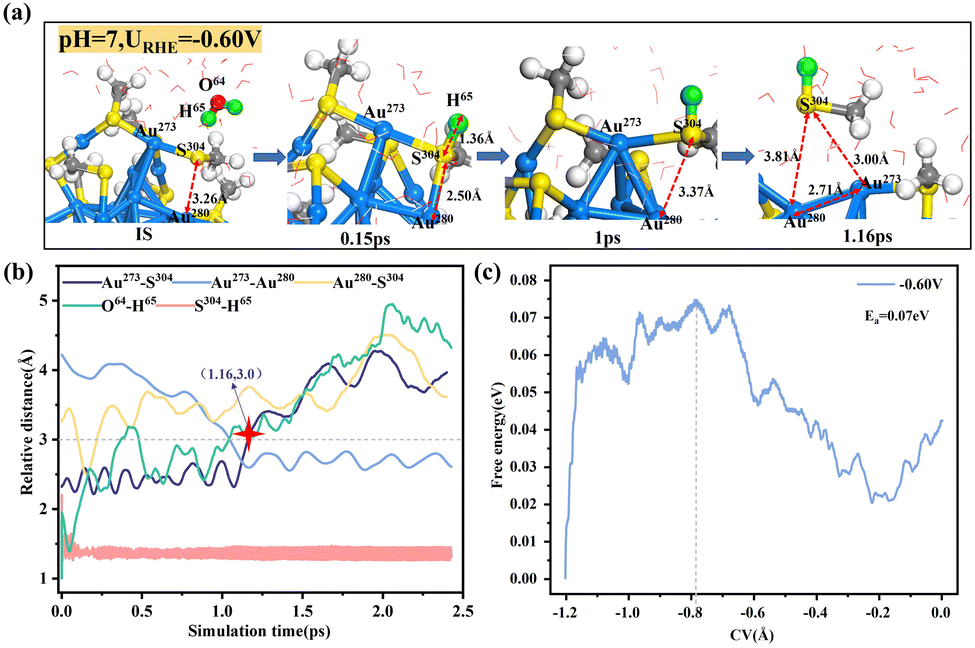 | ||
| Fig. 2 (a) Schematic of the local structures formed during *H adsorption and Au–S breaking in Au25(SCH3)18 at URHE = −0.60 V. (b) Statistics of the relative distances between representative atoms and (c) the integral free energy curve during constrained DFT-MD simulations at URHE = −0.60 V. | ||
After the removal of–SR ligands, the dethiolated metal atoms can serve as the catalytically active sites. Our Bader charge analysis (Table S1, ESI†) revealed that the exposed two Ag atoms are positively charged (0.06|e| and 0.03|e|), while the exposed two Au atoms are negatively charged (−0.09|e| and −0.13|e|), with this difference between the exposed Ag and Au atoms implying that their catalytic properties could be different. We thus proceeded to investigate the dethiolated Ag25 and Au25 in the electrocatalytic CO2 reduction reaction (CO2RR). Fig. 3 shows the calculated integral free energy curves of the four elementary reaction steps for Ag25 and Au25 under the same neutral condition with two Na atoms added into the water layer. As shown in Fig. 3a, the CO2 reactant can be effectively captured and activated at the exposed metal sites with very low kinetic barriers (0.22 eV in Ag25 and 0.13 eV in Au25). In Ag25, the CO2 is bonded to two staple Ag atoms in a bridging manner, while in Au25, it is only singly bonded to the staple Au atom. After the activation step, the nearest water molecule would approach *CO2 to form *COOH and a solvated OH− species (Fig. 3b). This step has to overcome a relatively high barrier of about 0.93 eV in Ag25, as compared to 0.71 eV in Au25. Subsequently, the approach of another H2O molecule to the O atom at the O–H end of *COOH leads to formation of adsorbed *CO and H2O (Fig. 3c). This step needs to overcome a high barrier of 0.81 eV in Au25, while in Ag25, the barrier is only 0.68 eV. The final step corresponds to *CO desorption, forming CO gas and exposing the reactive metal sites (Fig. 3d). This step is very facile, and the desorption barrier is very low, about 0.23 eV in Ag25 and 0.28 eV in Au25. The above analysis indicated a different rate-determining step (RDS) of the CO2RR for Ag25 than for Au25. In Ag25, the highest potential barrier corresponds to the formation of *COOH, whereas in Au25, the most difficult step is the formation of *CO. This analysis has thus demonstrated that changing the metal to another one with sufficiently different properties can change the RDS. We also examined the distributions of water density and hydrogen bonds at the cluster/water interface during the CO2RR for Au25 and Ag25 (Fig. S8, ESI†). The results showed slightly denser water density at the Au25/water interface, accompanied by more hydrogen bonds in Au25 than in Ag25. This observation suggested more water molecules to be accessible around the electrochemical interface of the Au25 NC, thus facilitating the reaction kinetics for water and proton transfer during the CO2RR.
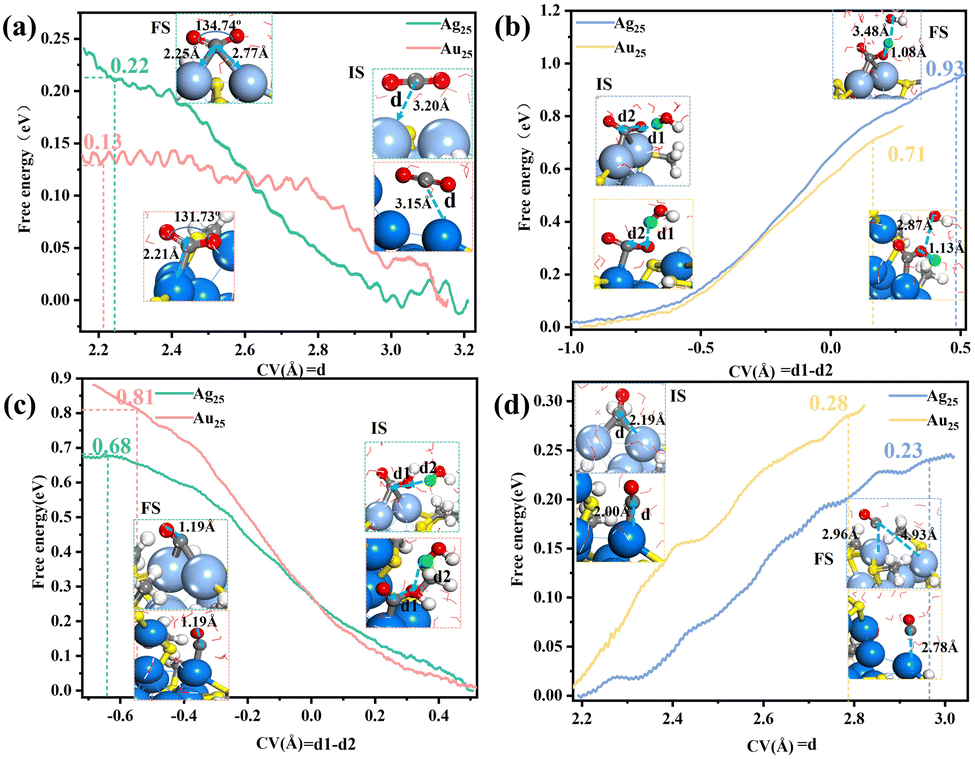 | ||
| Fig. 3 Comparison of the integral free energy curves between Ag25 and Au25 in (a) CO2 activation, (b) *COOH formation, (c) *CO formation and (d) CO desorption as determined from constrained AIMD simulations. The local structures of the initial state (IS), final state (FS) and the defined collective variable (CV, d1–d2 or d) are shown in the insets. | ||
Furthermore, to probe the CO2RR selectivity, we also investigated its competitive hydrogen evolution reaction (HER). In the first Volmer step (Fig. S9a, ESI†), the dissociation of H2O to form an *H intermediate requires a high barrier, of 1.45 eV in Ag25 and 1.07 eV in Au25, to be overcome. In the subsequent Heyrovsky step, the adsorbed *H would then combine with another H from water to produce H2 and a solvated OH− species. This step has to overcome a high barrier of 1.34 eV in Au25 and 1.27 eV in Ag25 (Fig. S9b, ESI†). Such a high barrier effectively inhibits HER at the exposed metal sites, thus ensuring the high selectivity of electroreduction of CO2 to CO in both Ag25 and Au25.
To further probe whether ligand simplification would affect the reaction kinetics, we replaced the simplified –SCH3 near the exposed metal site with the longer –SC6H13 ligand and calculated the corresponding free energy barriers of the CO2RR (Fig. S10, ESI†) and HER (Fig. S11, ESI†). In contrast to the simplified –SCH3, the long-chain –SC6H13 ligand facilitates CO2 activation and *CO formation in Ag25 as well as *COOH formation in Au25. However, upon replacing the ligand, the energy barrier for the RDS (*COOH formation in Ag25 and *CO formation in Au25) remains almost unchanged for both NCs. In the HER process, the presence of the long-chain –SC6H13 greatly decreases the barrier for the Volmer and Heyrovsky steps, yet the barrier for the RDS is still higher than that in the CO2RR, suggesting the higher selectivity for CO2 electroreduction. These results indicated that the predicted reaction kinetics and selectivity based on the simplified –SCH3 should be qualitatively reliable.
Furthermore, we conducted experiments to verify the prediction. As shown in Fig. S12 (ESI†), the Ag25(SphMe2)18 NC (SphMe2 = 2,4-dimethylbenzenethiol) sample exhibits characteristic absorption peaks at 390 nm, 490 nm, and 675 nm, while Au25(PET)18 (PET = phenylethylmercaptan) NC sample exhibits characteristic absorption peaks at 400 nm, 450 nm, and 670 nm, consistent with those reported in the literature.24,32 Their performance in the CO2RR was first evaluated by performing linear sweep voltammetry (LSV). As shown in Fig. 4a, the current density for each of the NCs is larger in CO2-saturated electrolyte than in N2-saturated electrolyte, suggesting greater involvement of current in the CO2RR to CO. The CO faradaic efficiency (FECO) measurements (Fig. 4b) showed Au25(PET)18 achieving an FECO higher than that of Ag25(SphMe2)18. The current density (jCO) of Au25(PET)18 is also higher than that of Ag25(SphMe2)18 (Fig. 4c). Moreover, from Fig. 4d, it is evident that the turnover frequency (TOF) value for Au25(PET)18 NCs is greater than that of Ag25(SphMe2)18. These measurements suggested a better intrinsic activity and higher catalytic rate for Au25(PET)18 than for Ag25(SphMe2)18. Additionally, the stability tests (Fig. 4e and f) indicated better FECO stability and current density stability demonstrated by Au25(PET)18 during continuous operation at −0.38 V for 16 hours. Evidently, these results agreed well with the theoretical predictions, where the inferior performance of Ag25 could be correlated to its poorer ligand etching kinetics and higher RDS barrier in the CO2RR process.
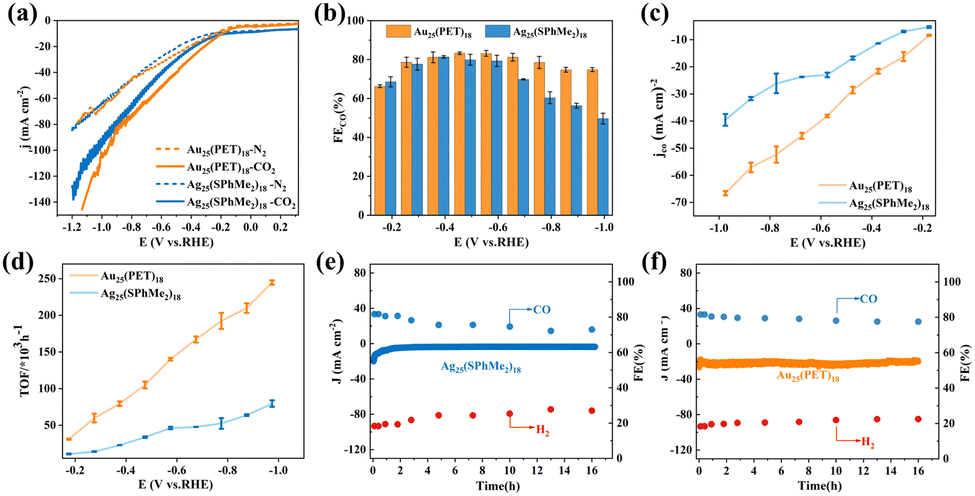 | ||
| Fig. 4 Electrocatalytic CO2RR performance in a CO2 flow cell with 1 M KOH electrolyte. (a) LSV curves of Ag25(SphMe2)18 and of Au25(PET)18, each in N2-saturated or CO2-saturated 1 M KOH electrolyte. (b) Faradaic efficiency of CO (FECO), (c) fractional current density of CO (jCO), and (d) turnover frequency (TOF) of CO for Ag25 and Au25 at indicated potentials. (e) and (f) Tests of long-term stability of (e) Ag25 and (f) Au25 in the CO2RR at −0.38 V. | ||
In summary, our studies have demonstrated distinct interface dynamics and the CO2RR mechanisms for Au25 and Ag25, which would be expected to inspire the development of functional nanostructures with enhanced catalytic properties through metal core engineering.
This work was supported by National Natural Science Foundation of China (No. 22473017 and No. 21805170) and Chongqing Science and Technology Commission (CSTB2024NSCQ-MSX0250).
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- S. Chen, S. Wang, J. Zhong, Y. Song, J. Zhang, H. Sheng, Y. Pei and M. Zhu, Angew. Chem., Int. Ed., 2015, 127, 3188–3192 CrossRef.
- Z. Chen, A. G. Walsh, X. Wei, M. Zhu and P. Zhang, J. Phys. Chem. C, 2022, 126, 12721–12727 CrossRef.
- Y. Du, H. Sheng, D. Astruc and M. Zhu, Chem. Rev., 2019, 120, 526–622 CrossRef PubMed.
- J. H. Lee, S. Kattel, Z. Xie, B. M. Tackett, J. Wang, C. J. Liu and J. G. Chen, Adv. Funct. Mater., 2018, 28, 1804762 CrossRef.
- F. Sun, L. Qin, Z. Tang, G. Deng, M. S. Bootharaju, Z. Wei, Q. Tang and T. Hyeon, Chem. Sci., 2023, 14, 10532–10546 RSC.
- Q. Yao, Z. Wu, Z. Liu, Y. Lin, X. Yuan and J. Xie, Chem. Sci., 2021, 12, 99–127 RSC.
- R. Jin, G. Li, S. Sharma, Y. Li and X. Du, Chem. Rev., 2020, 121, 567–648 CrossRef PubMed.
- S. Li, A. V. Nagarajan, Y. Li, D. R. Kauffman, G. Mpourmpakis and R. Jin, Nanoscale, 2021, 13, 2333–2337 RSC.
- M. Hesari and Z. Ding, Acc. Chem. Res., 2017, 50, 218–230 CrossRef PubMed.
- C. P. Joshi, M. S. Bootharaju, M. J. Alhilaly and O. M. Bakr, J. Am. Chem. Soc., 2015, 137, 11578–11581 CrossRef PubMed.
- C. Liu, T. Li, H. Abroshan, Z. Li, C. Zhang, H. J. Kim, G. Li and R. Jin, Nat. Commun., 2018, 9, 744 CrossRef PubMed.
- X. Zhu, L. Chen, Y. Liu and Z. Tang, POM, 2023, 2, 9140031 Search PubMed.
- P. Zhang, J. Phys. Chem. C, 2014, 118, 25291–25299 CrossRef.
- F. Sun, C. Deng, S. Tian and Q. Tang, ACS Catal., 2021, 11, 7957–7969 CrossRef.
- D. R. Kauffman, D. Alfonso, C. Matranga, H. Qian and R. Jin, J. Am. Chem. Soc., 2012, 134, 10237–10243 CrossRef PubMed.
- S. Li, A. V. Nagarajan, D. R. Alfonso, M. Sun, D. R. Kauffman, G. Mpourmpakis and R. Jin, Angew. Chem., Int. Ed., 2021, 60, 6351–6356 CrossRef PubMed.
- S. Li, D. Alfonso, A. V. Nagarajan, S. D. House, J. C. Yang, D. R. Kauffman, G. Mpourmpakis and R. Jin, ACS Catal., 2020, 10, 12011–12016 CrossRef.
- X. Liu, E. Wang, M. Zhou, Y. Wan, Y. Zhang, H. Liu, Y. Zhao, J. Li, Y. Gao and Y. Zhu, Angew. Chem., Int. Ed., 2022, 134, e202207685 CrossRef.
- J. Wang, F. Xu, Z. Y. Wang, S. Q. Zang and T. C. Mak, Angew. Chem., Int. Ed., 2022, 61, e202207492 CrossRef.
- R. Jin, S. Zhao, Y. Xing and R. Jin, CrystEngComm, 2016, 18, 3996–4005 RSC.
- S. Yoo, S. Yoo, G. Deng, F. Sun, K. Lee, H. Jang, C. W. Lee, X. Liu, J. Jang, Q. Tang, Y. J. Hwang, T. Hyeon and M. S. Bootharaju, Adv. Mater., 2024, 36, 2313032 CrossRef.
- Y. Feng, F. Fu, L. Zeng, M. Zhao, X. Xin, J. Liang, M. Zhou, X. Fang, H. Lv and G. Y. Yang, Angew. Chem., Int. Ed., 2024, 63, e202317341 CrossRef.
- Y. Xiong, H. Chen, Y. Hu, S. Yang, X. Xue, L. He, X. Liu, J. Ma and Z. Jin, Nano Lett., 2021, 21, 8693–8700 CrossRef.
- M. Zhu, W. T. Eckenhoff, T. Pintauer and R. Jin, J. Phys. Chem. C, 2008, 112, 14221–14224 CrossRef.
- H. Seong, V. Efremov, G. Park, H. Kim, J. S. Yoo and D. Lee, Angew. Chem., Int. Ed., 2021, 133, 14684–14691 CrossRef.
- F. Sun, L. Qin, Z. Tang and Q. Tang, Chem. Sci., 2024, 15, 16142–16155 RSC.
- H. Cao, Z. Zhang, J.-W. Chen and Y.-G. Wang, ACS Catal., 2022, 12, 6606–6617 CrossRef.
- I. V. Chernyshova and S. Ponnurangam, Phys. Chem. Chem. Phys., 2019, 21, 8797–8807 RSC.
- J. A. Gauthier, M. Fields, M. Bajdich, L. D. Chen, R. B. Sandberg, K. Chan and J. K. Nørskov, J. Phys. Chem. C, 2019, 123, 29278–29283 CrossRef.
- S. Li, A. V. Nagarajan, X. Du, Y. Li, Z. Liu, D. R. Kauffman, G. Mpourmpakis and R. Jin, Angew. Chem., Int. Ed., 2022, 134, e202211771 CrossRef.
- K. Xie, F. Wang, F. Wei, J. Zhao and S. Lin, J. Phys. Chem. C, 2022, 126, 5180–5188 CrossRef.
- X. Liu, J. Yuan, C. Yao, J. Chen, L. Li, X. Bao, J. Yang and Z. Wu, J. Phys. Chem. C, 2017, 121, 13848–13853 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc06185c |
| ‡ These two authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |