
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Redox-induced dimerisations of a phosphacyclic biradicaloid†
David
Zuber
a,
Oksana
Storcheva
a,
Karsten Paul
Lüdtke
 b,
Leonidas
Brunk
a and
Peter
Coburger
*a
b,
Leonidas
Brunk
a and
Peter
Coburger
*a
aTechnical University of Munich, Department of Chemistry, Lichtenbergstr. 4, D-85747, Garching, Germany. E-mail: peter.coburger@tum.de
bUniversity of Rostock, Institute of Chemistry, Albert-Einstein-Straße 3a, D-18059, Rostock, Germany
First published on 30th December 2024
Abstract
Despite the first examples being isolated more than two decades ago, little is known about the redox chemistry of stable phosphacyclic biradicaloids. Here, we demonstrate that a biradicaloid featuring a diphosphaindenyl backbone is able to undergo both oxidation and reduction reactions. One-electron oxidation results in the formation of a dicationic cage compound structurally related to an isomer of hypostrophene (C10H10). Reduction of PPIPh with [Co2(CO)8] results in the formation of the bimetallic complex 2, which contains a bis(benzodiphosphole) ligand.
Biradicaloids are open-shell singlet species in which two spatially separated radical centers couple antiferromagnetically. Consequently, such species often exhibit high reactivity and are believed to be essential intermediates in bond breaking and making processes.1 However, starting with the pioneering work of Niecke, a range of phosphacyclic biradicaloids was isolated.2,3 Some selected members are shown in Fig. 1a.4–9 Their accessibility enabled systematic reactivity studies,3 as illustrated in Fig. 1b: As expected, such species function as biradicals, exemplified in the concerted addition of dihydrogen leading to secondary bis(phosphines) (VII).7,8 Also, the radical centres can react independently.10,11 This is demonstrated in the reactivity of II toward BrCCl3 which involves two consecutive radical abstraction reactions leading to VIII. Additionally, biradicaloids can also act as charge-separated zwitterionic compounds as is, among others,12,13 demonstrated by the isolation of gold complex IX. Recent studies have also revealed that phosphacyclic biradicaloids can serve as π-donating ligands, forming half-sandwich complexes such as X.14–16 In these complexes, the biradicaloid ligands exhibit redox non-innocent behavior.15,16 Compared to this rich follow-up chemistry, the redox chemistry of phosphacyclic biradicaloids is somewhat limited. Notable examples are the reduction of I with an excess of lithium,5 which yields III and the one-electron oxidations of IV and IDP (V) with ferrocenium and silver salts, resulting in the formation of radical cations XI and XII. Due to the steric protection provided by their bulky substituents, these radical cations are stable and can be isolated. Based on these observations, we hypothesized that biradicaloids with reduced steric hindrance may serve as sources of radicals upon reduction or oxidation, which may undergo subsequent follow-up reactions.
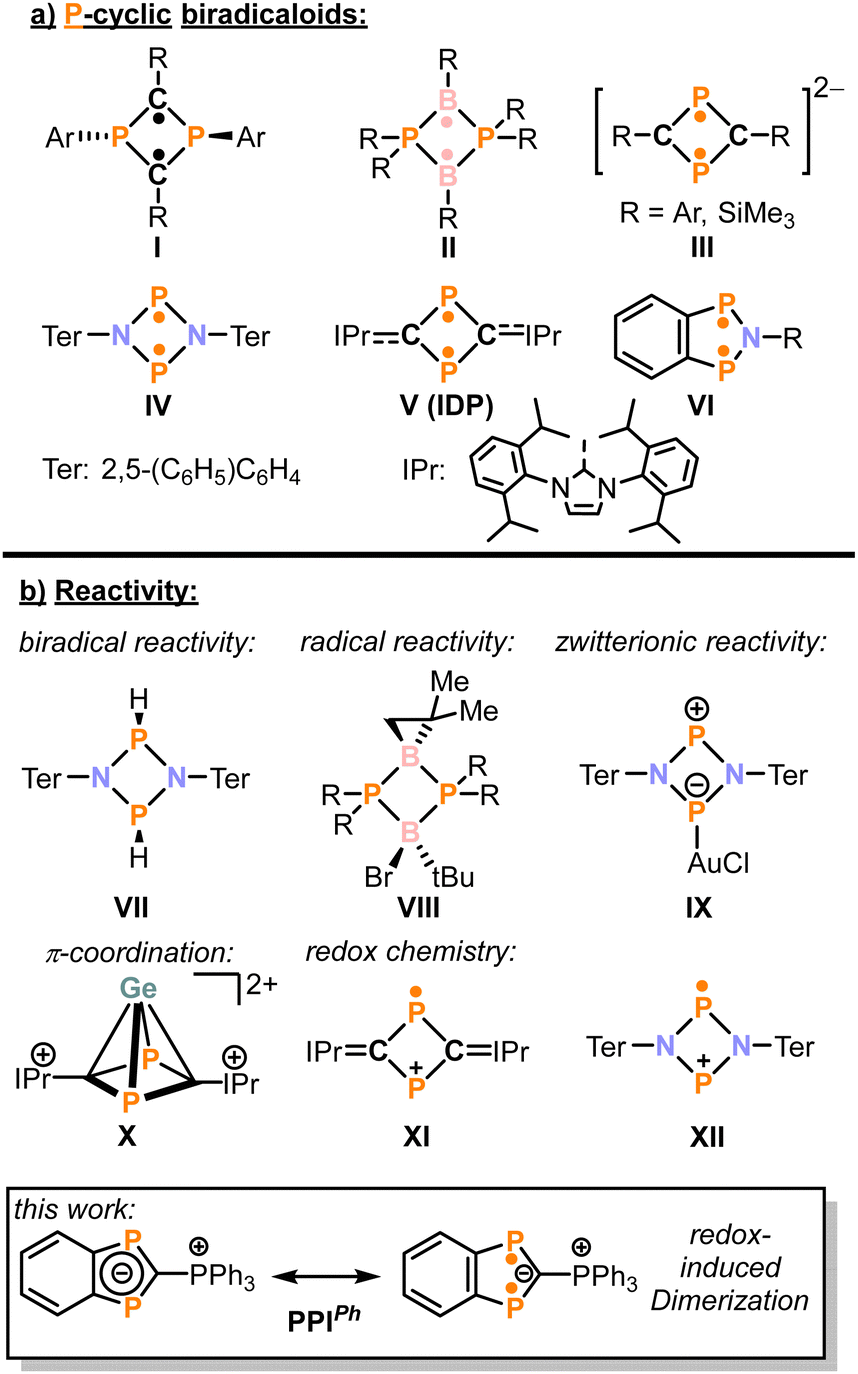 | ||
| Fig. 1 (a) Selected examples of phosphacyclic biradicaloids; (b) reactivity of phosphacyclic biradicaloids. For clarity, only the biradical resonance structure of all biradicaloids is shown, except for PPI. | ||
Recently, we reported on the synthesis of PPIp-tol, a phosphonium-substituted diphosphaindenylide exhibiting a notable biradical character (see Fig. 1a).17 Here, the superscript denotes the substituents at the phosphonium moiety (p-tol: para-tolyl). A CV study revealed the presence of two irreversible redox events, an oxidation at E = −0.30 V and a reduction at E = −2.95 V vs. Fc/Fc+ (peak potentials at a scan rate of 100 mV s−1, [nBu4N]PF6 as electrolyte, THF).17 Consequently, we identified PPI as an appropriate candidate for further investigation into the redox chemistry of phosphacyclic biradicaloids.
To get preliminary insights, the spin densities of the anticipated one-electron oxidation and reduction products [PPI]+ and [PPI]− were calculated using the ORCA program package (Fig. 2, see the ESI,† for details).18 These calculations highlight the zwitterionic nature of PPI: inspection of the spin-density plot and the Mulliken spin populations indicate that the one-electron oxidation of PPI primarily takes place at the endocyclic phosphorus atoms which carry a spin population of 0.45e and 0.47e, respectively. In contrast, the one-electron reduction is expected to occur at the cationic phosphonium moiety (Fig. 2b). With these DFT results in hand, we proceeded to investigate the chemical redox behaviour of PPI. Note that, as we encountered difficulties with crystallisations using the already reported PPIp-tol, we prepared the phenyl-substituted derivative PPIPh to isolate and characterise the novel species in this study (see the ESI,† for the analytical data of PPIPh).
 | ||
| Fig. 2 Spin-density plots of [PPIPh]˙+ (a) and [PPIPh]˙− (b). | ||
Indeed, treatment of PPIPh with one equivalent of ferrocenium tetrafluoroborate or triflate in polar solvents such as acetonitrile, THF or 1,2-dimethoxyethane (DME) results in the facile and selective formation of a novel polyphosphorus compound [1]2+ as indicated by 31P{1H} NMR spectroscopy (Scheme 1 and Fig. 3b, vide infra). Therefore, presumably due to the absence of significant steric shielding, the initially formed radical cation [PPIPh]+ dimerises. The salts [1](OTf)2·dme and[1](BF4)2·dme were obtained as colourless crystals in 59% or 90% yield, respectively. X-ray diffraction measurements revealed that [1]2+ is a benzannulated C2P4 cage compound which consists of two PPIPh fragments fused via two C–P bonds and one P–P bond resulting (Fig. 3a). The P4–P5 distance in the bicyclic C2P4 cage falls within the range expected for single bonds (2.248(1) Å). In contrast, the C–P distances correspond to elongated single bonds (ϕC–P: 1.91 Å).19
 | ||
| Scheme 1 Redox chemistry of PPIPh. For a proposed mechanism on the formation of 2, see Scheme 2. | ||
 | ||
| Fig. 3 (a) Solid-state structure of [1]2+; (b) section of the 31P{1H} NMR spectrum of [1](OTf)2 in CD2Cl2; (c) solid-state structure of 2; (d) MAS 31P solid-state NMR spectrum of 2 at a MAS frequency of 13.5 kHz, spinning side bands are marked with a star (*). For all solid-state structures, ellipsoids are drawn at the 50% probability level, hydrogen atoms and counterions are omitted for clarity and PPh3 moieties and part of the indenyl systems are drawn as wireframe for clarity. Selected distances [Å]: [1]2+: C1–P1 1.819(3), C1–P2 1.905(3), C1–P3 1.894(3), C1–P5 1.916(3), C2–P2 1.900(3), C2–P3 1.924(3), C2–P4 1.902(3), C2–P6 1.821(3), P4–P5 2.248(1); 2: C1–P1 1.811(5), C1–P2 1.780(4), C1–P3 1.698(4), C2–P1 1.836(5), C3–P2 1.830(5), P2–P2′ 2.248(2), Co1–P1 2.468(2), Co1–P2′ 2.226(2). | ||
These values are similar to the ones observed in the tetraphosphabicyclo[2.1.1]hexane (tBuC)2(PR)4 (P–P: 2.2334(6) Å, ϕC–P: 1.89 Å, R = Cl and Cy).20 Additionally, the exocyclic C–P distances correspond to single bonds and are thus indicative of the cationic phosphonium character of the PPh3 moieties (ϕC–P: 1.82 Å). Altogether, [1]2+ is best described as a phosphonium-substituted tetraphospha-derivative of an isomer of hypostrophene (C10H10).21 Notably, the related hexaphospha-derivative of this hypostrophene isomer was synthesised recently via the oxidation of the triphospholide [tBu2C2P3]−.22
In a CD2Cl2 solution, [1]2+ is C2-symmetric as indicated by the presence of three multiplets with an approximate integral ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 in the 31P{1H} NMR spectrum. In line with DFT calculations, scalar couplings between the 31P nuclei within the C2P4 cage are negligible, and thus only couplings to the exocyclic phosphonium moieties are observed (see the ESI†). This results in well-resolved first-order multiplets at 26.9 (P1, P6), 29.6 (P4, P5) and 45.7 (P2, P3) ppm (numbering according to Fig. 3a, see Fig. 3b for the respective coupling constants).
:1:1 in the 31P{1H} NMR spectrum. In line with DFT calculations, scalar couplings between the 31P nuclei within the C2P4 cage are negligible, and thus only couplings to the exocyclic phosphonium moieties are observed (see the ESI†). This results in well-resolved first-order multiplets at 26.9 (P1, P6), 29.6 (P4, P5) and 45.7 (P2, P3) ppm (numbering according to Fig. 3a, see Fig. 3b for the respective coupling constants).
Having explored the oxidation chemistry of PPIPh, we shifted our focus to its reactivity toward reducing agents. Treatment of PPIPh with one equivalent of potassium graphite, KC8, yields an intractable product mixture with triphenylphosphine being a major product (see the ESI†). Thus, in agreement with our DFT calculations, the reduction might take place at the phosphonium moiety resulting in the radical anion [PPIPh]−. This radical species might decompose to PPh3 and a diphosphaindenyl radical which then undergoes further unselective reactions. In light of these findings, we reasoned that a reductant capable of simultaneously acting as a coordinating agent might improve the selectivity of the reduction reaction by effectively trapping the generated radical anions. And indeed, treatment of PPIPh with [Co2(CO)8] leads to the formation of the bimetallic complex 2 as a yellow insoluble product which was isolated in 70% yield (Scheme 2). Single-crystal X-ray diffraction studies revealed the molecular structure of 2 (Fig. 3c): here, two PPIPh units are linked via a P–P single bond (P2–P2′: 2.248(2) Å) and the resulting dimeric ligand chelates two Co(CO)3 units. Within the phosphorus ligand, all C–P distances correspond to single bonds (ϕC–P: 1.81 Å).19
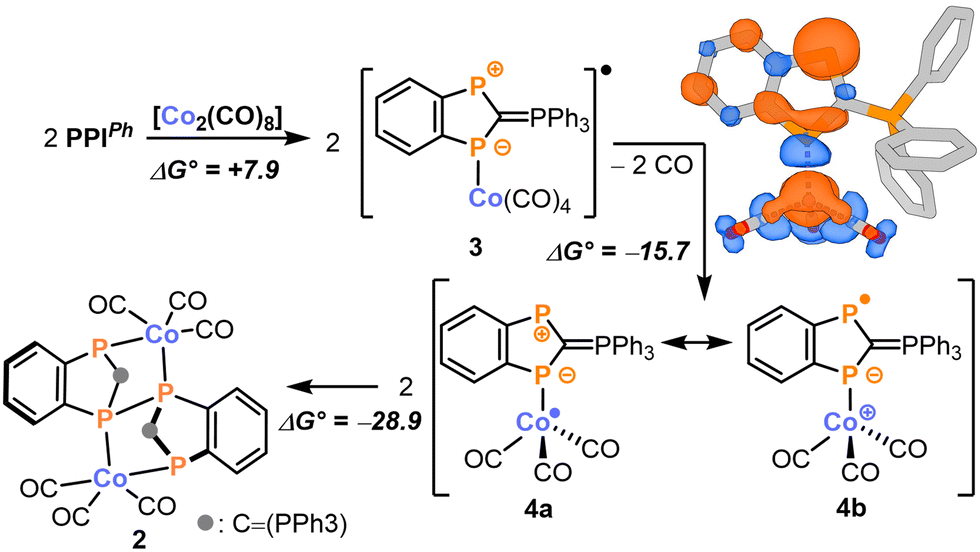 | ||
| Scheme 2 Tentative mechanism for the formation of 2 together with the calculated spin density of 4 (top right corner). Energy values are given in kcal mol−1. | ||
In contrast to the oxidation product [1]2+, the C–P bond formed between the C3P2 ring and the exocyclic PPh3 moiety is a double bond indicating the presence of a neutral C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) PPh3 phosphorane moiety (C1–P3 1.698(4) Å).23 Furthermore, the cobalt atoms in 2 adopt a trigonal-bipyramidal coordination environment with the phosphorus atoms occupying axial (P1, P1′) and equatorial positions (P2, P2′). The Co1–P2′ distance of 2.226(2) Å lies within the range typically found for Co–P single bonds in species with multiple Co–P bonds.24–27 In contrast, the Co1–P1 distance is notably elongated with 2.468(2) Å, presumably due to the structural constraints provided by the chelating ligand. Due to its insolubility in common organic solvents, 2 was further characterised via elemental analysis, powder X-ray diffraction and magic-angle spinning (MAS) 31P solid-state NMR spectroscopy (see the ESI,† for details). In line with the solid-state structure, the 31P NMR spectrum shows three resonances at 15.5 (P3, P3′), 92.7 (P1, P1′) and 181.5 (P2, P2′) ppm (assigned using DFT calculations, see the ESI,† for details). Notably, the coupling of the 59Co nucleus to P2/P2′ is well-resolved (I = 7/2, 100% natural abundance, see the insert of Fig. 3d). After fitting, a coupling constant of 1JP–Co = −535 Hz was obtained, consistent with values observed in other cobalt carbonyl complexes with phosphine ligands (see the ESI†).28,29 In the ATR-IR spectrum, 2 shows three intense CO stretching vibrations at 2017, 1973 and 1939 cm−1. These values fall within the range reported for cobalt(I) carbonyl complexes with phosphine ligands.30,31 Therefore, consistent with the assumption of a reduction of PPI during the reaction with cobalt carbonyl, the IR data allow to characterise 2 as a cobalt(I) complex with a chelating dianionic bis(benzodiphosphole)-diide ligand.
PPh3 phosphorane moiety (C1–P3 1.698(4) Å).23 Furthermore, the cobalt atoms in 2 adopt a trigonal-bipyramidal coordination environment with the phosphorus atoms occupying axial (P1, P1′) and equatorial positions (P2, P2′). The Co1–P2′ distance of 2.226(2) Å lies within the range typically found for Co–P single bonds in species with multiple Co–P bonds.24–27 In contrast, the Co1–P1 distance is notably elongated with 2.468(2) Å, presumably due to the structural constraints provided by the chelating ligand. Due to its insolubility in common organic solvents, 2 was further characterised via elemental analysis, powder X-ray diffraction and magic-angle spinning (MAS) 31P solid-state NMR spectroscopy (see the ESI,† for details). In line with the solid-state structure, the 31P NMR spectrum shows three resonances at 15.5 (P3, P3′), 92.7 (P1, P1′) and 181.5 (P2, P2′) ppm (assigned using DFT calculations, see the ESI,† for details). Notably, the coupling of the 59Co nucleus to P2/P2′ is well-resolved (I = 7/2, 100% natural abundance, see the insert of Fig. 3d). After fitting, a coupling constant of 1JP–Co = −535 Hz was obtained, consistent with values observed in other cobalt carbonyl complexes with phosphine ligands (see the ESI†).28,29 In the ATR-IR spectrum, 2 shows three intense CO stretching vibrations at 2017, 1973 and 1939 cm−1. These values fall within the range reported for cobalt(I) carbonyl complexes with phosphine ligands.30,31 Therefore, consistent with the assumption of a reduction of PPI during the reaction with cobalt carbonyl, the IR data allow to characterise 2 as a cobalt(I) complex with a chelating dianionic bis(benzodiphosphole)-diide ligand.
Further insight into the electronic structure of 2 was obtained from DFT and CASSCF calculations (see the ESI†). These calculations reveal a 3d8 configuration of the cobalt centres together with dative P2′ → Co1 bonding interactions. The P–Co bonding orbitals involving P1/P1′ exhibit a high covalency and are formed by interaction of a p-type orbital on P1 and a d-type orbital on Co. This covalent bond is polarised toward P1 as indicated by the occupation of the p-type orbital of 1.27e (see the ESI,† for a detailed discussion). Thus, our calculations support a formal oxidation state of +1 for the cobalt centers in 2.
The formation of 2 can be explained by the proposed mechanism outlined in Scheme 2 (see the ESI,† for details). Initially, the reaction between two equivalents of PPIPh and [Co2(CO)8] might form the 19-valence electron complex 3 in an endergonic reaction (ΔG° = 7.9 kcal mol−1). From there, loss of carbon monoxide generates the 17-valence electron complex 4 in an exergonic reaction (ΔG° = −15.7 kcal mol−1). Two mesomeric resonance structures can be formulated to describe the electronic structure of 4 (Scheme 2). In 4a, the unpaired electron is located at the cobalt centre making it a Co0 complex. In contrast, 4b is a CoI complex with a reduced PPIPh ligand carrying an unpaired electron on the non-coordinated endocyclic P atom (Scheme 2). The considerable spin population of 0.33e of this phosphorus atom indicates a substantial contribution of 4b to the overall electronic structure of 4 (see Scheme 2 for a depiction of the spin density). Consequently, 4 can dimerise upon formation of a P–P single bond to form 2 in an exergonic reaction (ΔG° = −28.9 kcal mol−1).
In summary, the zwitterionic structure of PPIPh promotes a diverse redox chemistry, i.e. facile one-electron oxidation and reduction reactions.
The lack of steric hindrance allows the initially generated radical species to undergo subsequent reactions. In the case of oxidation, this leads to the selective formation of [1]2+. In contrast, reduction with potassium graphite results in the unselective formation of multiple products. Gratifyingly, [Co2(CO)8] as reductant effectively redirects the reduction event to the C2P3 ring rather than to the phosphonium moiety leading to the formation of complex 2, which features a reduced bis(benzodiphosphole) ligand. Therefore, using other bimetallic complexes for the reduction of PPIPh may yield a range of novel coordination compounds. Notably, the significant elongation of one Co–P bond in complex 2 suggests potential hemilabile behaviour, making it promising for catalytic applications. We are now focusing on improving the solubility of bimetallic complexes like 2 and to further assess their reactivity.
DZ: investigation (experimental study), writing (original draft). OS: investigation (SS-NMR spectroscopy). KPL: investigation (provision of starting materials). LB: investigation (powder diffraction). PC: theoretical calculations, conceptualisation, supervision, funding acquisition, writing (review and editing).
This work was supported by the Emmy-Noether programme of the DFG (grant for P. C.) and the FCI (Phd scholarship for D. Z.). P. C. thanks Thomas Fässler and the Chair of Inorganic Chemistry with Focus on New Materials for continuous support and access to the X-ray diffractometers.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- F. Breher, Coord. Chem. Rev., 2007, 251, 1007–1043 CrossRef.
- W. W. Schoeller, Eur. J. Inorg. Chem., 2019, 1495–1506 CrossRef.
- A. Hinz, J. Bresien, F. Breher and A. Schulz, Chem. Rev., 2023, 123, 10468–10526 CrossRef PubMed.
- E. Niecke, A. Fuchs, F. Baumeister, M. Nieger and W. W. Schoeller, Angew. Chem., Int. Ed. Engl., 1995, 34, 555–557 CrossRef.
- M. Sebastian, M. Nieger, D. Szieberth, L. Nyulászi and E. Niecke, Angew. Chem., Int. Ed., 2004, 43, 637–641 CrossRef PubMed.
- D. Scheschkewitz, H. Amii, H. Gornitzka, W. W. Schoeller, D. Bourissou and G. Bertrand, Science, 2002, 295, 1880–1881 CrossRef PubMed.
- T. Beweries, R. Kuzora, U. Rosenthal, A. Schulz and A. Villinger, Angew. Chem., Int. Ed., 2011, 50, 8974–8978 CrossRef PubMed.
- Z. Li, X. Chen, D. M. Andrada, G. Frenking, Z. Benkö, Y. Li, J. R. Harmer, C.-Y. Su and H. Grützmacher, Angew. Chem., Int. Ed., 2017, 56, 5744–5749 CrossRef.
- J. Bresien, D. Michalik, A. Schulz, A. Villinger and E. Zander, Angew. Chem., Int. Ed., 2021, 60, 1507–1512 CrossRef.
- H. Amii, L. Vranicar, H. Gornitzka, D. Bourissou and G. Bertrand, J. Am. Chem. Soc., 2004, 126, 1344–1345 CrossRef.
- J. Rosenboom, L. Chojetzki, T. Suhrbier, J. Rabeah, A. Villinger, R. Wustrack, J. Bresien and A. Schulz, Chem. – Eur. J., 2022, 28, e202200624 CrossRef PubMed.
- A. Hinz, A. Schulz and A. Villinger, Chem. Commun., 2016, 52, 6328–6331 RSC.
- Z. Li, Y. Hou, Y. Li, A. Hinz and X. Chen, Chem. – Eur. J., 2018, 24, 4849–4855 CrossRef.
- Z. Li, X. Chen, L. L. Liu, M. T. Scharnhölz and H. Grützmacher, Angew. Chem., Int. Ed., 2020, 59, 4288–4293 CrossRef PubMed.
- M. T. Scharnhölz, P. Coburger, L. Gravogl, D. Klose, J. J. Gamboa-Carballo, G. Le Corre, J. Bösken, C. Schweinzer, D. Thöny, Z. Li, K. Meyer and H. Grützmacher, Angew. Chem., Int. Ed., 2022, 61, e202205371 CrossRef.
- P. Coburger, F. Masero, J. Bösken, V. Mougel and H. Grützmacher, Angew. Chem., Int. Ed., 2022, 61, e202211749 CrossRef.
- P. Coburger, D. Zuber, C. Schweinzer and M. Scharnhölz, Chem. – Eur. J., 2024, 30, e202302970 CrossRef PubMed.
- F. Neese, F. Wennmohs, U. Becker and C. Riplinger, J. Chem. Phys., 2020, 152, 224108 CrossRef PubMed.
- F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, J. Chem. Soc., Perkin Trans. 2, 1987, S1–S19 RSC.
- C. Jones, P. C. Junk, A. F. Richards and M. Waugh, New J. Chem., 2002, 26, 1209–1215 RSC.
- B. M. Gimarc and M. Zhao, J. Org. Chem., 1995, 60, 1971–1974 CrossRef.
- M. K. Uttendorfer, G. Hierlmeier, G. Balázs and R. Wolf, Dalton Trans., 2024, 53, 10113–10119 RSC.
- R. Appel, Pure Appl. Chem., 1987, 59, 977–982 CrossRef.
- D. M. Jenkins and J. C. Peters, J. Am. Chem. Soc., 2005, 127, 7148–7165 CrossRef PubMed.
- P. Coburger, S. Demeshko, C. Rödl, E. Hey-Hawkins and R. Wolf, Angew. Chem., Int. Ed., 2017, 56, 15871–15875 CrossRef PubMed.
- P. Coburger, J. Leitl, D. J. Scott, G. Hierlmeier, I. G. Shenderovich, E. Hey-Hawkins and R. Wolf, Chem. Sci., 2021, 12, 11225–11235 RSC.
- R. Franz, D. Gál, C. Bruhn, Z. Kelemen and R. Pietschnig, Adv. Sci., 2024, 11, 2306805 CrossRef PubMed.
- D. Massiot, F. Fayon, M. Capron, I. King, S. Le Calvé, B. Alonso, J.-O. Durand, B. Bujoli, Z. Gan and G. Hoatson, Magn. Reson. Chem., 2002, 40, 70–76 CrossRef.
- G. Szalontai, in Current Developments in Solid State NMR Spectroscopy, ed. N. Müller and P. K. Madhu, Springer, Vienna, 2003, pp. 95–106 Search PubMed.
- P. Rigo, M. Bressan and A. Morvillo, J. Organomet. Chem., 1976, 105, 263–269 CrossRef.
- H. Schumann, M. Meissner and H.-J. Kroth, Z. Naturforsch., B, 1978, 33, 1489–1490 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2389673–2389675. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc05656f |
| This journal is © The Royal Society of Chemistry 2025 |