
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDinuclear iridium complexes ligated by lithium-ion endohedral fullerene Li+@C60†
Chinari
Fukushi
 ,
Takashi
Komuro
* and
Hisako
Hashimoto
*
,
Takashi
Komuro
* and
Hisako
Hashimoto
*
Department of Chemistry, Graduate School of Science, Tohoku University, Sendai 980-8578, Japan. E-mail: takashi.komuro.c5@tohoku.ac.jp; hisako.hashimoto.b7@tohoku.ac.jp
First published on 2nd January 2025
Abstract
The diiridium complexes of lithium-ion endohedral fullerene Li+@C60 were synthesised in high yields. X-ray crystallography revealed the η2:η2-coordination of Li+@C60 and the disorder of the Li+ ion over two sites close to the coordinated carbons. 13C NMR study suggested the presence of dynamic behaviour via haptotropic rearrangements. UV/Vis and CV characteristics were also investigated experimentally and theoretically.
Donor–acceptor complexes consisting of a transition metal (TM) donor and an organic acceptor have gained significant attention owing to their wide range of applications in materials engineering,1 molecular electronics,2 catalytic chemistry, etc.3 Adducts of electron-rich TM units with fullerene C60 can be viewed as donor–acceptor molecules and are potential candidates for excellent building blocks of such materials.4 However, developing highly functional materials based on C60–TM complexes faces several challenges. For instance, the addition of multi-metal centres to the C60 shell is likely to result in the formation of multiple regioisomers because the entire surface of C60 can act as a reaction site.5 Moreover, electrically neutral C60 tends to easily aggregate because of its low solubility in various organic solvents,6 leading to low yields of C60–TM complexes and difficulties in isolation/characterisation. Therefore, it is necessary to develop a method for the selective synthesis of highly soluble single complexes.
From this perspective, lithium-ion endohedral fullerene Li+@C607 is a promising molecule for selective synthesis because Li+@C60 has higher electron acceptability relative to C60 due to the electrostatic interaction between the Li+ ion and the C60 carbons.8 Consequently, Li+@C60 readily reacts with electron-rich TM fragments to form Li+@C60–TM complexes as demonstrated using mononuclear metal fragments [Fig. 1(a), A–C].9 These complexes exhibit greater stability compared with the corresponding C60 analogues due to the formation of strong C60–TM bonds based on the stronger π-back-donation from TM (donor) to C60 (acceptor) [Fig. 1(b)].10 Moreover, the ionic nature of Li+@C60 allows the improvement of its solubility, modifying counter anions with high affinity toward organic solvents.11 Therefore, Li+@C60–TM complexes could offer access to unique molecular structures and properties that are inaccessible by use of C60 analogues. In this context, we recently became interested in Li+@C60–TM complexes with multi-metal units. In these complexes, the donor–acceptor interaction between the TM and fullerene units and the electrostatic interaction between the C60 cage and endohedral Li+-ion should be further modulated by π-back-donation from the multi-metal centres.
 | ||
| Fig. 1 (a) Previously-reported Li+@C60–TM complexes.9 (b) Schematic diagram of the electrostatic interaction between the C60 cage and metal fragments MLn. (c) Illustration of the Li+@C60–[Ir2] complexes in this work. | ||
As the first example of a multi-metallic C60–TM complex, the tetrairidium complex C60{Ir2Cl2(cod)2}2 (D) (cod = 1,5-cyclooctadiene) has been previously obtained as a low-solubility product.12 However, no isolated yield or detailed properties, except the crystal structure, have been reported. Inspired by this example, we recently performed reactions of Li+@C60 with three halogen-bridged diiridium complexes, Ir2X2(cod)2 (X = Cl, Br and I). Herein, we report the synthesis of dinuclear Li+@C60–TM complexes, [{μ-η2:η2-(Li+@C60)}{Ir2X2(cod)2}](NTf2−) [X = Cl (1), Br (2) and I (3); Tf = CF3SO2], and their solid-state structures and spectroscopic properties in solution [Fig. 1(c)].
The reaction of [Li+@C60](NTf2−) with 1 equiv of Ir2Cl2(cod)2 in 1,1,2,2-tetrachloroethane (TCE) for 5 min at room temperature afforded almost quantitatively [{μ-η2:η2-(Li+@C60)}{Ir2Cl2(cod)2}](NTf2−) (1). Complex 1 was isolated as black crystals in 89% yield (Scheme 1). Using Ir2Br2(cod)2 and Ir2I2(cod)2 instead of Ir2Cl2(cod)2, the corresponding complexes 2 and 3 were isolated in 86% and 90% yields, respectively. Complexes 1–3 showed good solubility in various organic solvents [CH2Cl2, THF, 1,2-dichlorobenzene (o-DCB), etc.], which enabled us to collect multiple spectroscopic data in solution, including 1H, 7Li and 13C NMR, as well as UV/Vis and cyclic voltammetry (CV), in addition to their solid-state structures.
 | ||
| Scheme 1 Synthesis of Li+@C60-diiridium complexes 1–3. | ||
The crystal structures of complexes 1–3 were determined using single-crystal X-ray diffraction (SC-XRD). As shown in Fig. 2(a) for 1, and Fig. S2 and S3 (ESI†) for 2 and 3, respectively, it was unambiguously revealed that each complex had an [Ir2] unit coordinated by the Li+@C60 shell in an η2:η2-fashion. For example, the four Ir–C(C60) distances of 1 are 2.170(4), 2.162(4), 2.169(4) and 2.170(4) Å [Fig. 2(b)], which indicate the η2:η2-coordination to the two iridium atoms at the [6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6] positions of the C60 shell. The average values of the four bond distances, 1 [av. 2.168(2) Å] is comparable with those of 2 [av. 2.169(2) Å] and 3 [av. 2.183(4) Å] (Table S2, ESI†), which are significantly shorter than that of non-Li+ encapsulated C60-iridium complex D [av. 2.218(3) Å].12 Moreover, the three C–C bond distances at the C1–C2–C3–C4 moiety of 1 are lengthened by 6–8% upon coordination, relevant to the corresponding values of Li+@C60 (1.39 and 1.45 Å at the [6:6] and [6:5] positions, respectively) [Fig. 2(b)]. These C–C distances at the C1–C2–C3–C4 moiety were almost the same on average among the three complexes [av. 1.506(3) (1), av. 1.508(3) (2) and av. 1.511(6) (3) Å], and are significantly longer than that of D [av. 1.487(6) Å].12 Thus, the π-back-donation from the two Ir centres to the C60 shell in 1–3 is estimated to be much stronger than that of related neutral D, owing to the presence of the Li+ ion.
:6] positions of the C60 shell. The average values of the four bond distances, 1 [av. 2.168(2) Å] is comparable with those of 2 [av. 2.169(2) Å] and 3 [av. 2.183(4) Å] (Table S2, ESI†), which are significantly shorter than that of non-Li+ encapsulated C60-iridium complex D [av. 2.218(3) Å].12 Moreover, the three C–C bond distances at the C1–C2–C3–C4 moiety of 1 are lengthened by 6–8% upon coordination, relevant to the corresponding values of Li+@C60 (1.39 and 1.45 Å at the [6:6] and [6:5] positions, respectively) [Fig. 2(b)]. These C–C distances at the C1–C2–C3–C4 moiety were almost the same on average among the three complexes [av. 1.506(3) (1), av. 1.508(3) (2) and av. 1.511(6) (3) Å], and are significantly longer than that of D [av. 1.487(6) Å].12 Thus, the π-back-donation from the two Ir centres to the C60 shell in 1–3 is estimated to be much stronger than that of related neutral D, owing to the presence of the Li+ ion.
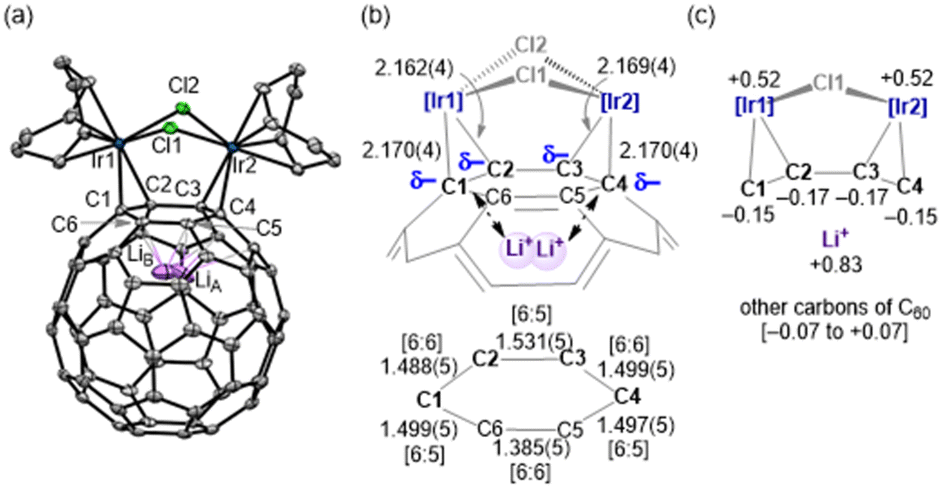 | ||
| Fig. 2 (a) Molecular structure of the cationic part of 1 with thermal ellipsoids at the 50% probability level. All hydrogen atoms are omitted for clarity. The Li atom is disordered in the two sites (LiA: LiB = 53%: 47%). (b) Selected bond distances (Å) and schematic electronic interaction between the C60 shell and the encapsulated Li+-ion. [6:6] and [6:5] denote the positions in the C60 shell. (c) Selected NPA charge calculated by DFT. | ||
The endohedral Li+-ion is disordered in two positions in dinuclear 1–3, in contrast to mononuclear A–C, where the position of the Li+-ion is fixed in a single site close to the metal (1.54–1.57 Å away from the C60 centroid).9 The averaged position is shifted to the metal fragment from the C60 centroid by av. 1.56 (1), 1.55 (2) and 1.58 (3) Å, reflecting the considerable electrostatic interaction between the Li+ ion and the C60 cage [Fig. 2(b) and (c)]. The disordering of the Li+ ion in two positions can be caused by the presence of two sets of the negatively charged C–C bonds attached to the diiridium unit and/or the electrostatic effect of the counter ion (The closest distance between LiA and atoms (F3B and O3C) of NTf2− for 1 is 5.6 Å). In other words, the disordering of the Li+-ion of 1–3 was restricted to only near the two metal sites, in contrast to the endohedral Li+-ion in the Li+@C60 salts, which were disordered at multiple sites.7a,b Relevant restricted disordering of the Li+-ion has also been reported for Li+@C60–Cu4 polymer.13 The occurrence of such disordering is an interesting issue in the chemistry of Li+@C607,14 because the positional change of the Li+ ion by the action of external perturbation is expected to apply to molecular electronics, such as the recently reported multiple-conductivity switching of Li+@C60.15
It should be noted that the two halogen ligands, X1 and X2, are inequivalently bridged over the two iridium atoms in complexes 1–3: X1 and X2 are located at the endo- and exo-positions, respectively, with respect to the coordinated six-membered ring of C60. The Ir–X2 distances [av. 2.5433(7) (1), 2.6459(4) (2) and 2.7676(4) (3) Å] are longer than those of Ir–X1 [av. 2.3990(6) (1), 2.5262(4) (2) and 2.6803(5) (3) Å] by ca. 0.1 Å. The average Ir–X1/X2 bond distances are elongated by about 0.1–0.2 Å in the order of Cl < Br < I, in accordance with the ionic radii of the halogens (Cl = 1.81, Br = 1.96 and I = 2.20 Å).16 Oppositely, the Ir–X–Ir bond angle decreases in this order (Table S2, ESI†). These structural differences are small but may influence their electronic structures.
The high solubility of complexes 1–3 in CD2Cl2 allowed us to investigate their properties in solution. In the 7Li NMR spectra of 1–3, all the signals of the Li+ ion were observed exactly at δ −11.0, which is shifted up-field by 0.9 ppm from that of [Li+@C60](NTf2−) (δ −10.1), due to the coordination of the Li+@C60 cage to the metal fragment. This up-field shift of the signal is comparable with those reported for mononuclear complexes A (δ −11.6), B (δ −11.0) and C (δ −10.8).9 The 1H NMR spectra of 1–3 at room temperature (300 K) showed multiple broadened signals assignable to the cod ligand in the region of δ 1.5–5.5 [Fig. 3(a)]. The two cod ligands were observed to be equivalent, indicating that the complex had a Cs-symmetric structure with a pseudo-mirror plane passing through the two halogen atoms and the Li+-ion in solution. In the 13C NMR spectrum at 300 K [Fig. 3(b)], a very broad signal was observed in the C60 carbon region (δ 140–150), implying fluxional behaviour. Upon decreasing the temperature to 200 K, the broad signal was separated into 30 singlets among the expected 32 signals in theory (28 singlets at δ 135–155 and two singlets at δ 55–85), two of which must overlap (δ 145.4 and 143.4). The two signals observed at δ 83.3 and 57.5, which are considerably shifted up-field compared with those of other C60 and are assigned to the carbons bonded to the Ir atoms. A similar up-field shift was observed for B (δ 76.8) and C (δ 76.1).9 The signal change of the C60-carbons between 300 and 200 K was likely driven by haptotropic rearrangements, where the diiridium unit migrated over the surface of the fullerene cage by changing the positions of the coordination bonds with C60 carbons between intra- or inter-rings [Fig. 3(c)].9,17
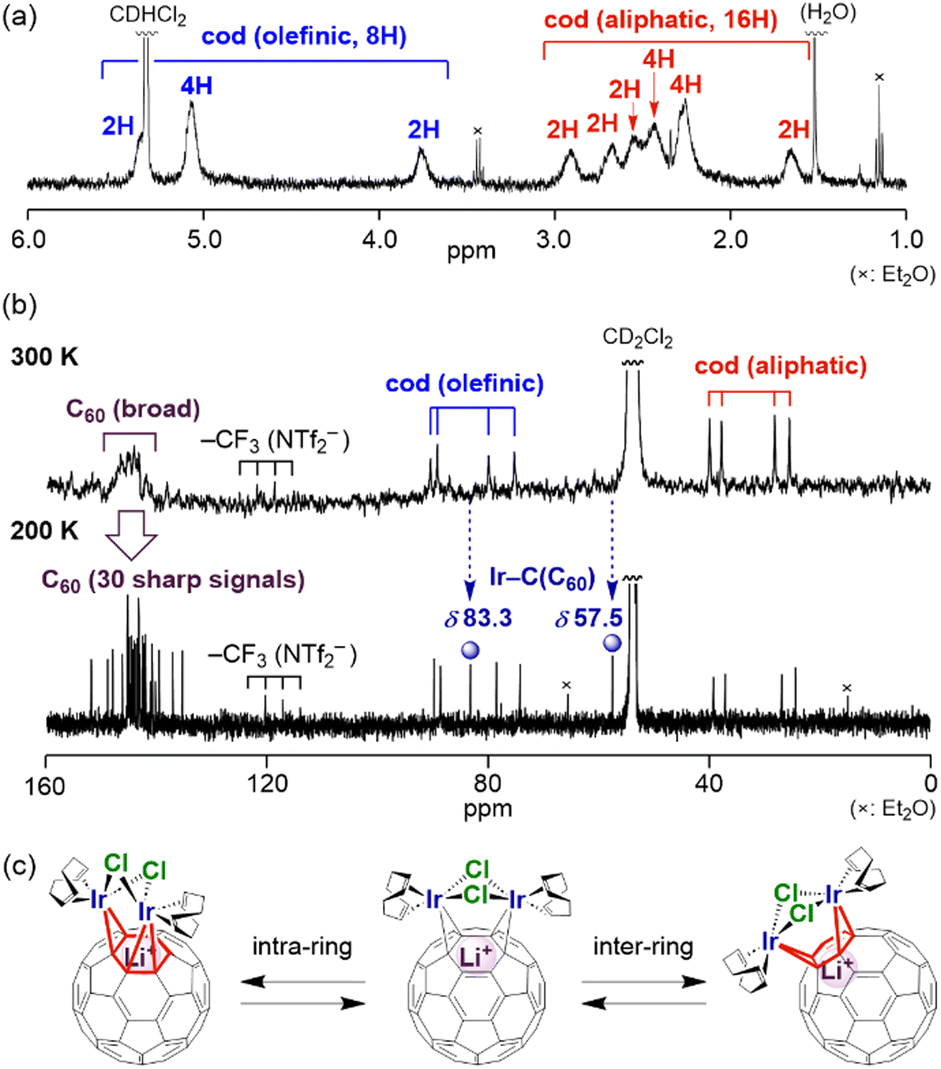 | ||
| Fig. 3 (a) 1H NMR spectrum of 1 in CD2Cl2 (400 MHz, 300 K). (b) 13C{1H} NMR spectra of 1 in CD2Cl2 at 300 and 200 K (101 MHz). (c) A possible mechanism for the dynamic behaviour of 1via haptotropic rearrangements. | ||
The UV/Vis spectra of 1–3 measured in CH2Cl2 exhibited three extensive absorption bands (at ca. 330, 420 and 530 nm), together with a very weak shoulder-like band extending to ca. 800 nm in all cases (see Fig. S20 in ESI†). As a representative example, the two bands at 327 and 530 nm of 1 are assigned to π–π* transitions, although some contributions from the Ir d-orbitals are present, according to TD-DFT calculations (calcd. λmax = 331 nm, f = 0.109 and 541 nm, f = 0.032 for 1, see ESI†). These two bands were comparable to those observed for Ir complex A (λmax = 331 and 582 nm).9 The other band at 416 nm was assigned to a metal-to-ligand charge transfer (MLCT) transition (calcd. λmax = 424 nm, f = 0.082). The last very weak band is assignable to HOMO → LUMO (vide infra), which should be an almost forbidden transition (calcd. λmax = 689 nm, f = 0.002 for 1). Interestingly, the absorption coefficients of the bands at longer wavelengths (ca. 420, 530 and 700 nm) slightly increased and red-shifted in descending order of the halogen atoms, suggesting some contribution from the halogen orbitals to the transitions (vide infra).
The cyclic voltammograms of 1–3 were recorded in o-DCB (Fig. S21 and S22, ESI†). All the complexes exhibited one set of oxidation peaks with slight cathodic shifts in the oxidation potentials from 1 (E1/2 = +1.00 V) to 2 (E1/2 = +0.84 V) and 3 (E1/2 = +0.86 V). In contrast, 1–3 displayed five reduction peaks accompanied by peaks originating from free Li+@C60 (Fig. S22, ESI†), implying the occurrence of a dissociation equilibrium between the fullerene cage and metal units in solution under reductive conditions.
To obtain further insight into the electronic structures of 1–3, we performed orbital analysis of their cationic parts using DFT calculations [B3PW91-D3/SDD for Ir and I, 6-311+G(d,p) for others//B3PW91/Lanl2DZ for Ir and I, and 6-31G(d) for others]. As depicted in Fig. S26–S28 in the ESI,† the three complexes have analogous orbitals. Thus, the important orbitals of chlorido complex 1 are shown in Fig. 4 as representatives. Both the HOMO and HOMO–1 of 1 consist of a C60 π-orbital combined with the d-orbitals of two iridium atoms in a bonding fashion and the p-orbital of one of the two bridging halogen atoms (Cl2 atom). In contrast, the LUMO of 1 represents the π*orbitals localised in the C60 shell. The LUMO+1 consists of the C60 π*-orbital combined with the d-orbitals of two Ir atoms in an antibonding fashion, accompanied by a slight contribution from the bridging halogen orbitals. The participation of the Ir d-orbitals and halogen p-orbitals in the fullerene π system is also found in other bonding and antibonding orbitals, which can be responsible for the aforementioned spectral changes among 1–3 in the UV/Vis and CV measurements, reflecting the energy differences and orbital sizes of 3p (Cl), 4p (Br) and 5p (I) of the halogen atoms. Moreover, from these frontier orbital features, it is understood that oxidation reduces the electron density of the entire molecule. In contrast, reduction occurs initially in the fullerene cage. However, over-reduction weakened the coordination of the fullerene cage to the Ir2 unit, as observed by CV measurements.
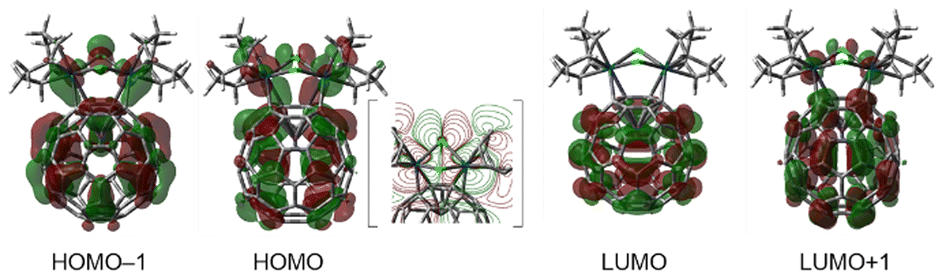 | ||
| Fig. 4 Selected Kohn–Sham orbitals of 1 (isovalue = 0.02). The contour map of HOMO is inserted in the square brackets. | ||
In addition, the NPA charge distribution supported the presence of strong Ir-to-C60 π-back-donation in 1–3 (Fig. S29–S31, ESI†). In the case of 1, the four carbon atoms bonded to Ir atoms are much more negatively charged (−0.15 to −0.17), relevant to the remaining carbons of C60 (−0.07 to +0.07), as illustrated in Fig. 2(c).
In summary, three diiridium complexes coordinated with Li+-ion endohedral fullerene were successfully synthesised as soluble products in high yields (∼90%). SC-XRD, DFT calculations and multiple spectroscopic techniques revealed their fundamental properties. The SC-XRD study confirmed the η2:η2 coordination of the Li+@C60 cage to the Ir2 unit and the disordered location of the Li+-ion at two positions adjacent to the two metal atoms. The latter observation indicated the existence of enhanced electrostatic interactions between the Li+ cation and the negatively charged carbon atoms caused by strong π-back-donation from the two metal centres to the C60 cage, which was supported by DFT calculations. The 7Li NMR spectra supported the coordination of the Li+@C60 cage to the metal fragment. 1H and VT 13C NMR studies revealed dynamic behaviours, including haptotropic rearrangements. Moreover, UV/Vis and CV characteristics and DFT calculations suggest significant orbital interactions among the C60 π-system, the d-orbitals of diiridium, and the p-orbitals of the bridging halogen atoms. The development of electron transporting materials based on complexes 1–3 is currently underway.
This research was supported by JSPS KAKENHI Grants JP22K05123 and JP22H02088 from the Japan Society for the Promotion of Science (JSPS). We thank Dr H. Sato (Rigaku Corporation, Tokyo, Japan) for his support in XRD study and acknowledge the Research and Analytical Center for Giant Molecules, Tohoku University, for high-resolution mass spectroscopy and elemental analysis. The computation was performed at the Research Center for Computational Science, Okazaki, Japan (Project: 23-IMS-C246 and 24-IMS-C260). The authors are grateful to Idea International Co, Ltd, for providing the Li+@C60 salt sample. One of the authors (C. F.) thanks the Tohoku University Division for Interdisciplinary Advanced Research and Education (DIARE) for scholarship support.
Data availability
The data supporting this article have been included in the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) O. Maury and H. L. Bozec, Acc. Chem. Res., 2005, 38, 691–704 CrossRef CAS PubMed; (b) W. C. H. Choy, W. K. Chan and Y. Yuan, Adv. Mater., 2014, 26, 5368–5399 CrossRef CAS.
- (a) X. Wan, C. Li, M. Zhang and Y. Chen, Chem. Soc. Rev., 2020, 49, 2828–2842 RSC; (b) Y. Tanaka, Dalton Trans., 2024, 53, 8512–8523 RSC.
- (a) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed; (b) F. Glaser and O. S. Wenger, Coord. Chem. Rev., 2020, 405, 213129 CrossRef CAS.
- (a) A. L. Balch and M. M. Olmstead, Chem. Rev., 1998, 98, 2123–2166 CrossRef CAS PubMed; (b) M. A. Lebedeva, T. W. Chamberlain and A. N. Khlobystov, Chem. Rev., 2015, 115, 11301–11351 CrossRef CAS PubMed; (c) A. L. Balch and K. Winkler, Chem. Rev., 2016, 116, 3812–3882 CrossRef CAS; (d) D. V. Konarev and R. N. Lyubovskaya, Russ. Chem. Rev., 2016, 85, 1215–1228 CrossRef CAS; (e) A. L. Balch and K. Winkler, Coord. Chem. Rev., 2021, 438, 213623 CrossRef CAS; (f) D. V. Konarev, S. S. Khasanov, A. F. Shestakov, M. Ishikawa, A. Otsuka, H. Yamochi, G. Saito and R. N. Lyubovskaya, J. Am. Chem. Soc., 2016, 138, 16592–16595 CrossRef CAS; (g) D. V. Konarev, A. V. Kuzmin, S. S. Khasanov, S. I. Troyanov, A. Otsuka, H. Yamochi, H. Kitagawa and R. N. Lyubovskaya, Organometallics, 2017, 36, 4032–4037 CrossRef CAS; (h) T. Tanase, K. Nakamae, Y. Kitagawa and T. Nakajima, Chem. Eur. J., 2021, 27, 12953–12958 CrossRef CAS; (i) S.-Z. Zhan, G.-H. Zhang, J.-H. Li, J.-L. Liu, S.-H. Zhu, W. Lu, J. Zheng, S. W. Ng and D. Li, J. Am. Chem. Soc., 2020, 142, 5943–5947 CrossRef CAS PubMed; (j) S.-Z. Zhan, Y.-L. Liu, H. Cai, M.-D. Li, Q. Huang, X.-D. Wang, M. Li, L. Dang, S. W. Ng, W. Lu and D. Li, Angew. Chem., Int. Ed., 2023, 62, e202312698 CrossRef CAS; (k) Y. Hashikawa, H. Kawasaki and Y. Murata, Organometallics, 2022, 41, 354–359 CrossRef CAS; (l) Y. Hashikawa and Y. Murata, Organometallics, 2024, 43, 227–232 CrossRef CAS.
- (a) A. Hirsch, I. Lamparth and H. R. Karfunkel, Angew. Chem., Int. Ed. Engl., 1994, 33, 437–438 CrossRef; (b) A. L. Balch, J. W. Lee, B. C. Noll and M. M. Olmstead, Inorg. Chem., 1994, 33, 5238–5243 CrossRef CAS.
- N. O. Mchedlov-Petrossyan, Chem. Rev., 2013, 113, 5149–5193 CrossRef CAS PubMed.
- (a) S. Aoyagi, E. Nishibori, H. Sawa, K. Sugimoto, M. Takata, R. Kitaura, H. Shinohara, H. Okada, T. Sakai, Y. Ono, K. Kawachi, K. Yokoo, S. Ono, K. Omote, Y. Kasama, S. Ishikawa, T. Komuro and H. Tobita, Nat. Chem., 2010, 2, 678–683 CrossRef CAS PubMed; (b) S. Aoyagi, Y. Sado, E. Nishibori, H. Sawa, H. Okada, H. Tobita, Y. Kasama, R. Kitaura and H. Shinohara, Angew. Chem., Int. Ed., 2012, 51, 3377–3381 CrossRef CAS PubMed.
- H. Okada, T. Komuro, T. Sakai, Y. Matsuo, Y. Ono, K. Omote, K. Yokoo, K. Kawachi, Y. Kasama, S. Ono, R. Hatakeyama, T. Kaneko and H. Tobita, RSC Adv., 2012, 2, 10624–10631 RSC.
- T. Watanabe, M. F. Itoh, T. Komuro, H. Okada, T. Sakai, Y. Ono, K. Kawachi, Y. Kasama and H. Tobita, Organometallics, 2014, 33, 608–611 CrossRef CAS.
- (a) M.-C. Yang, A. K. Sharma, W. M. C. Sameera, K. Morokuma and M.-D. Su, J. Phys. Chem. A, 2017, 121, 2665–2673 CrossRef CAS; (b) M.-C. Yang and M.-D. Su, ACS Omega, 2019, 4, 3105–3113 CrossRef CAS PubMed.
- H. Okada and Y. Matsuo, Fullerenes, Nanotubes Carbon Nanostruct., 2014, 22, 262–268 CrossRef CAS.
- M. Rasinkangas, T. T. Pakkanen, T. A. Pakkanen, M. Ahlgrén and J. Rouvinen, J. Am. Chem. Soc., 1993, 115, 4901 CrossRef CAS.
- Y. Shen, M. Cui, S. Takaishi, H. Kawasoko, K. Sugimoto, T. Tsumuraya, A. Otsuka, E. Kwon, T. Yoshida, N. Hoshino, K. Kawachi, Y. Kasama, T. Akutagawa, T. Fukumura and M. Yamashita, Nat. Commun., 2022, 13, 495 CrossRef CAS.
- (a) S. Aoyagi, A. Tokumitu, K. Sugimoto, H. Okada, N. Hoshino and T. Akutagawa, J. Phys. Soc. Jpn., 2016, 85, 094605 CrossRef; (b) S. Aoyagi, K. Miwa, H. Ueno, H. Okada, Y. Matsuo and K. Kokubo, R. Soc. Open Sci., 2018, 5, 180337 CrossRef PubMed; (c) H. Suzuki, M. Ishida, M. Yamashita, C. Otani, K. Kawachi, Y. Kasama and E. Kwon, Phys. Chem. Chem. Phys., 2016, 18, 31384–31387 RSC; (d) H. Suzuki, M. Ishida, C. Otani, K. Kawachi, Y. Kasama, E. Kwon, Y. Miyazaki and M. Nakano, Phys. Chem. Chem. Phys., 2019, 21, 16147–16153 RSC; (e) H. Ando and Y. Nakao, Phys. Chem. Chem. Phys., 2021, 23, 9785–9803 RSC; (f) H. Ando and Y. Nakao, Phys. Chem. Chem. Phys., 2023, 25, 8446–8462 RSC.
- (a) H. J. Chandler, M. Stefanou, E. E. B. Campbell and R. Schaub, Nat. Commun., 2019, 10, 2283 CrossRef PubMed; (b) A. K. Ismael, ACS Omega, 2023, 8, 19767–19771 CrossRef CAS PubMed.
- R. D. Shannon, Acta Crystallogr. A, 1976, 32, 751–767 CrossRef.
- (a) M. L. H. Green and A. H. H. Stephens, Chem. Commun., 1997, 793–794 RSC; (b) I. D. Gridnev, Coord. Chem. Rev., 2008, 252, 1798–1818 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic and experimental procedures, characterisation data, details of X-ray crystallographic analysis, NMR spectra and theoretical calculations. CCDC 2389980 (for 1), 2389981 (for 2) and 2389982 (for 3). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc05485g |
| This journal is © The Royal Society of Chemistry 2025 |