
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA new strategy to synthesize degradable polystyrene of ultra-high molecular weight†
Jianhan
Li
a,
Hongjun
Yang
 a,
Qimin
Jiang
a,
Jiang
Li
a,
Bibiao
Jiang
*a,
Xiaoqiang
Xue
a,
Sridhar
Komarneni
*b and
Wenyan
Huang
*ab
a,
Qimin
Jiang
a,
Jiang
Li
a,
Bibiao
Jiang
*a,
Xiaoqiang
Xue
a,
Sridhar
Komarneni
*b and
Wenyan
Huang
*ab
aKey Laboratory of Environmentally Friendly Polymeric Materials, School of Materials Science and Engineering, Jiangsu Collaborative Innovation Centre of Photovoltaic Science and Engineering, Changzhou University, Changzhou, 213164, Jiangsu, P. R. China. E-mail: jiangbibiao@cczu.edu.cn; ellahwy@cczu.edu.cn
bMaterials Research Institute and Department of Ecosystem Science and Management, Materials Research Laboratory, The Pennsylvania State University, University Park, Pennsylvania 16802, USA. E-mail: komarneni@psu.edu
First published on 6th February 2025
Abstract
Degradation or recycling of styrene-copolymers whose main chains are saturated carbon chains is very difficult under natural conditions. Here, we synthesized a new styrene-copolymer with degradable groups in the main chain using an emulsion polymerization process at room temperature by first synthesizing a functional monomer (tert-butyl lipoate) with cyclic disulfide bonds.
Polystyrene (PS) is one of the main sources of plastic products in the world, because of its good durability, easy processing, and hydrolysis stability. It is widely used in the packaging, insulation, construction and food processing industries.1 Ultrahigh molecular weight polystyrene (UHMWPS) has better mechanical properties than low molecular weight polystyrene.2–4 High entanglement greatly improves its overall performance of wear resistance, mechanical and thermal stability and chemical resistance.5,6 However, since the main chain of polystyrene is composed of saturated carbons and the side groups consist of benzene rings,7–9 it is difficult to achieve degradation of this polymer under natural conditions. The extent of pollution caused by PS waste to the environment cannot be underestimated, and it has become the main pollutant of soil, rivers, lakes, and oceans.10,11 Therefore, the development of easily degradable ultra-high molecular weight polystyrene is urgently needed.
The degradation and recycling of polystyrene are conducted primarily through photocatalysis, pyrolysis, microbial degradation, and chemically.12–17 Synthesizing easily degradable polystyrene is also an effective way to solve polystyrene plastic pollution. The design and commercialization of new recyclable or biodegradable polymers can be accelerated by the introduction of weak links or degradable groups such as carbonyl or ester bonds in the main or side chains of the all-carbon chain, thus directly addressing the waste problem of plastics.18,19 The polymer can be broken down into smaller pieces by using this method at the end of its useful life for recycling, industrial composting or degradation in the environment.20–23 Some studies have shown that copolymerization of cycloketoacetals (CKAs) with vinyl monomers can introduce cleavable ester bonds in each polymer chain.21,24–26 Despite their utility, CKAs have a number of disadvantages, including (i) the competitive side reactions of CKA resulting in non-cleavable C–C bonds, (ii) poor reactivity with styrene, and so on.27,28 Other cyclic monomers may overcome these challenges. For example, Johnson and Guillaneuf et al. conducted free radical copolymerization of styrene with cyclic thiolactone monomers, and the polystyrene could be degraded by alkaline hydrolysis, aminohydrolysis, oxidative hydrolysis or methanolysis by interposing the thioester bond in the polymer chain.29,30 However, cyclic thiolactones require expensive, multi-step synthesis, and because polystyrene chains contain a large number of ester bonds, the thermal properties of polystyrene materials will be greatly affected. Therefore, an ideal degradable copolymeric monomer should be able to be used on a large scale, but also easy to copolymerize with styrene monomer, and will not affect the performance of polystyrene.
Lipoic acid can be polymerized to synthesize polylipoic acid, which contains dynamic disulfide bonds on the main chain and carboxyl groups on the side group by ring-opening polymerization (radical ring-opening or cationic ring-opening).31 This linear polydisulfide can be depolymerized in a dilute solution of sodium hydroxide and re-converted into lipoic acid monomer so as to achieve chemical closed-loop recovery.32 And it can also be biodegraded to biocompatible small molecules by disulfide bond reduction. Lipoic acid and its derivatives can be copolymerized not only with various vinyl monomers,33–35 but also with aromatic olefins such as styrene.36,37 However, it has not been reported that lipoic acid and styrene are chemically bonded to the same molecular chain through free radical copolymerization. Pioneering work by Tsarevsky's group35 reported that the free radical copolymerization of ethyl lipoate and ethyl acrylate produces disulphide bonds in lipoate-lipoate diads, and this disulfide bond can be dissociated under reducing conditions. However, due to the low glass transition temperature of ethyl lipoate,33 the thermal properties of polystyrene will be decreased when ethyl lipoate is copolymerized with styrene. Here, we first synthesized a functional monomer (tert-butyl lipoate, t-BLp) with cyclic disulfide bonds. Then, potassium persulfate (KPS) was used as an initiator, and t-BLp was used for ring-opening and copolymerized with styrene by free radical emulsion polymerization at room temperature. Thus, we developed a new process for the preparation of degradable ultra-high molecular weight styrene-copolymers.
Firstly, the functional monomer, tert-butyl lipoate (t-BLp), was synthesized using lipoic acid (α-LpA) and tert-butanol, and its reaction process is shown in Fig. 1A. The purity of the functional monomer t-BLp was determined to be 96.9% by HPLC (Fig. 1E). The infrared spectrum shows a stretching vibration peak at 1728 cm−1 (Fig. 1C), which belongs to the ester bond formed during the synthesis. The 1H-NMR spectrum (Fig. 1D) also reveals the formation of t-BLp. A degradable ultra-high molecular weight styrene-copolymer with linear disulfide bonds was prepared with t-BLp using free radical emulsion polymerization of styrene (St) at 25 °C that was initiated by potassium persulfate (KPS).
 | ||
| Fig. 1 (A) The synthesis of tert-butyl lipoate. (B) FT-IR of lipoic acid (LpA). (C) FT-IR of tert-butyl lipoate (t-BLp). (D) 1H-NMR of tert-butyl lipoate. (E) HPLC of tert-butyl lipoate. | ||
To reveal the polymerization mechanism and confirm the copolymer structure, the 1H-NMR, FT-IR and Raman spectra of the copolymer prepared with the feed ratio of [St]0/[t-BLp]0/[KPS]0 = 100/2/2 were determined. Fig. 2A presents the 1H-NMR spectrum of the copolymer obtained after the polymerization of St and t-BLp at 25 °C. Signals at a chemical shift of 3.67 ppm are ascribed to the methylene proton (–S–CH–C–, a) that is connected with sulfur on t-BLp. Signals at a chemical shift of 3.17 ppm are ascribed to the methylene proton (–S–CH2–C–, b) that is connected to styrene by a sulfur atom on t-BLp after polymerization. Signals at a chemical shift of 3.83 ppm could be ascribed to the methylene proton (–S–CH–C–, c) that is connected to the styrene by the sulfur atom on t-BLp after the polymerization of St and t-BLp. Signals at a chemical shift of 1.43 ppm are ascribed to a methyl proton (–O–C–(CH3)3, d) of tert-butyl on the t-BLp. Signals at a chemical shift of 6.27–7.28 ppm could be ascribed to the benzene ring proton (e) of polystyrene. FT-IR spectra of the copolymer polymerized by St and t-BLp at 25 °C (Fig. 2B) showing the stretching vibration peak belonging to the ester bond (–C(O)–O–) at 1728 cm−1, which is absent in the styrene homopolymers (Fig. 2C). Fig. 2D is the Raman spectrum of the copolymer obtained after the polymerization of St and t-BLp at 25 °C, which shows the peak belonging to the linear disulfide bond (–S–S–) at 480.3 cm−1. Therefore, the above results show that the styrene-copolymer with linear disulfide bonds in the backbone has been successfully prepared.
 | ||
| Fig. 2 (A) 1H-NMR of PS-co-tBLp-1. (B) FT-IR of PS-co-tBLp-1. (C) FT-IR of PS. (D) Raman spectrum of PS-co-tBLp-1. (E) Raman spectrum of PS. (PS-co-tBLp-1: [St]100/[t-BLP]2/[KPS]2, at 25 °C. PS: [St]100/[KPS]2, at 25 °C). (F) Molecular weight differential distribution curves of partially representative PS-co-tBLp. (PS-co-tBLp-3: [St]100/[t-BLP]1/[KPS]1, at 25 °C. PS-co-tBLp-5: [St]100/[t-BLP]0.4/[KPS]1, at 25 °C. PS-co-tBLp-7: [St]100/[t-BLP]0.2/[KPS]1, at 25 °C. PS-co-tBLp-10: [St]100/[t-BLP]1/[KPS]0.2, at 80 °C.) | ||
To reveal the polymerization mechanism and confirm the molecular weight and molecular weight distribution of the copolymer, the reaction conditions such as the dosage of t-BLp or KPS and temperature were studied, as shown in Table S1 (see ESI†). Fig. 2F shows the molecular weight differential distribution curves of the copolymers obtained after the copolymerization of St and t-BLp, which shows Mw.GPC > 106 g mol−1. At 25 °C, regardless of the ratio of the polymerization reaction, the conversion of styrene was not high (less than 75% in 24 h), and extending the polymerization time did not continue to increase the conversion, which may be due to the formation of sulfhydryl groups after the ring-opening of t-BLp, and the chain transfer reaction occurs during the polymerization process. Therefore, when the amount of t-BLp is higher than 1% m.St, the relative weight average molecular weight of the obtained PS-co-tBLp is not high. When the amount of t-BLp is less than 1% m.St, the relative weight average molecular weight of the obtained PS-co-tBLp can reach more than millions (Table S1, ESI†), which also verifies the above hypothesis. However, when the polymerization temperature was increased to more than 60 °C, the conversion of styrene could reach 100%. To explain the reason for this phenomenon, the polymerization of lipoic acid (LpA) with different monomers was taken as an example, and the polymerization mechanism was studied, as shown in Scheme 1. Under normal circumstances, the sulfate free radicals generated by the decomposition of KPS will lead to a chain initiation reaction, but lipoic acid is a natural free radical scavenger, which will react with sulfate free radicals and consume a part of the sulfate free radicals. The reaction rate constant of lipoic acid and the sulfate free radical is 2.3 × 109 L mol−1 s−1, while the decomposition rate constant of KPS is kd,![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) KPS,25°C = 8.3 × 10−8 s−1 at 25 °C and kd,KPS,80°C = 4.8 × 10−5 s−1 at 80 °C, and the chain growth rate constant of styrene is kp,St = 102–104 L mol−1 s−1. However, there are many more primary radicals in the system at 80 °C than at 25 °C, which is more conducive to the free radical polymerization (as shown in Table S1, ESI†). Similarly, the same conclusion was obtained in the system of tert-butyl lipoate with a similar structure.
KPS,25°C = 8.3 × 10−8 s−1 at 25 °C and kd,KPS,80°C = 4.8 × 10−5 s−1 at 80 °C, and the chain growth rate constant of styrene is kp,St = 102–104 L mol−1 s−1. However, there are many more primary radicals in the system at 80 °C than at 25 °C, which is more conducive to the free radical polymerization (as shown in Table S1, ESI†). Similarly, the same conclusion was obtained in the system of tert-butyl lipoate with a similar structure.
 | ||
| Scheme 1 The copolymerization process of tert-butyl lipoate and styrene. | ||
The degradation reaction process of synthesized PS-co-tBLp copolymer is shown in Scheme 2. The disulfide bonds on the backbone of the obtained PS-co-tBLp copolymer can be cleaved to form polystyrene with a sulfhydryl end group (–SH) by irradiation with UV light at 365 nm at room temperature or by the action of dithiothreitol (DTT) at 80 °C. Fig. S1 (see ESI†) is the molecular weight differential distribution curve of the PS-co-tBLp copolymers before and after degradation, and it can be seen that the molecular weight of the PS-co-tBLp copolymer decreased significantly whether it was degraded by dithiothreitol (DTT) at 80 °C or irradiated by a 365 nm ultraviolet lamp, indicating that we have successfully prepared a degradable ultra-high molecular weight styrene-copolymer with disulfide bonds in the backbone.
 | ||
| Scheme 2 Degradation process of PS-co-tBLp. | ||
To reveal the degradation reaction mechanism and confirm the degraded structure of the PS-co-tBLp copolymers, the 1H-NMR, FT-IR and Raman spectrum of the degradation products (degraded by DTT at 80 °C) of the copolymer prepared with the feed ratio of [St]0/[t-BLp]0/[KPS]0 = 100/2/2 at 25 °C were analyzed. Fig. 3A is the 1H-NMR spectrum of the product after thermal degradation of the PS-co-tBLp copolymer. The attribution of each peak has not changed, but new signals at a chemical shift of 1.53 ppm were produced, which could be ascribed to a sulfhydryl group (–SH, f) belonging to the end group. Fig. 3B is the FT-IR spectrum of the obtained PS-co-tBLp copolymer after thermal degradation, which reveals that the stretching vibration peak belonging to the ester bond at 1728 cm−1 did not disappear, indicating that the chemical degradation of the PS-co-tBLp copolymer and the reduction in molecular weight are not due to the ester bond of t-BLp. The Raman spectrum (Fig. 3C) of the copolymer after thermal degradation reveals that the peak belonging to the linear disulfide bond (–S–S–) at 480.3 cm−1 disappeared completely. Therefore, the above results indicate that the ultra-high molecular weight polystyrene formed with linear disulfide bonds in the backbone could be degraded because the disulfide bond confers degradable properties to the styrene-copolymer.
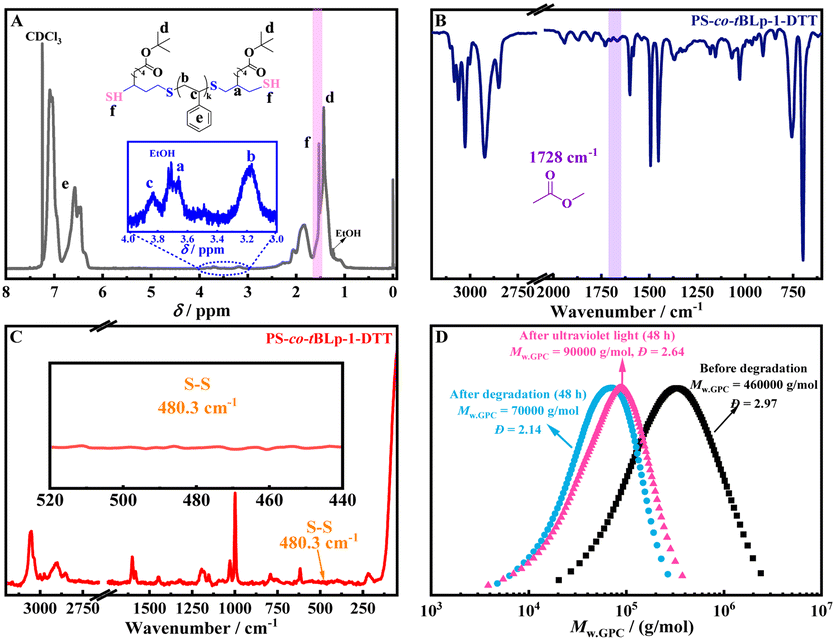 | ||
| Fig. 3 (A) 1H-NMR of PS-co-tBLp after degradation. (B) FT-IR of PS-co-tBLp after degradation. (C) Raman spectrum of PS-co-tBLp after degradation. (D) Molecular weight differential distribution curves of PS-co-tBLp-1 before and after ultraviolet photodegradation and DTT catalytic thermal degradation. (PS-co-tBLp-1: [St]100/[t-BLP]2/[KPS]2). | ||
The DSC and TGA of the PS-co-tBLp copolymer were studied to determine the effect of t-BLp on the thermal properties of polystyrene (Fig. S2, ESI†). The results indicate that the addition of a small amount of t-BLp will not affect the secondary structure of the obtained copolymer, but excessive t-BLp will reduce the glass transition temperature of the obtained copolymer. See ESI,† for detailed analysis.
Here, a functional monomer with cyclic disulfide bonds was synthesized first. Then ultra-high molecular weight styrene-copolymer with a disulfide bond was synthesized successfully through a green polymerization method. It is shown by GPC, FTIR and Raman that the disulfide bond of the UHMW styrene-copolymer can be broken under DTT catalysis or UV conditions.
National Natural Science Foundation of China (22271025, 22301022, U2241286), a Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions supported this work and all the above support is gratefully acknowledged.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- K. S. Khuong, W. H. Jones, W. A. Pryor and K. N. Houk, J. Am. Chem. Soc., 2005, 127, 1265–1277 CrossRef CAS PubMed.
- L. Jiang, Z. Shen, Y. Yang and Y. Zhang, Polym. Int., 2001, 50, 63–66 CrossRef CAS.
- L. Wu, B. Li and B. Li, Acta Polymerica Sinica, 2001, 5, 633–638 Search PubMed.
- D. Yang, J. Jin, C. Qi and Y. Liu, Chem. J. Chin. Univ., 1993, 14, 1627–1628 CAS.
- G. Rui, Q. Lv, J. Lu, T. Wu, S. Zhao, R. Huang, B. Han and W. Yang, Polymer, 2021, 214, 12345 CrossRef.
- C. Con, R. Dey, M. Ferguson, J. Zhang, R. Mansour, M. Yavuz and B. Cui, Microelectron. Eng., 2012, 98, 254–257 CrossRef CAS.
- S. Banerjee, T. Maji, T. K. Paira and T. K. Mandal, Macromol. Chem. Phys., 2014, 215, 440–451 CrossRef CAS.
- M. Ciftci, M. Arslan, M. Buchmeiser and Y. Yagci, ACS Macro Lett., 2016, 5, 946–949 CrossRef CAS PubMed.
- D. Hyun Kim, S. S. Park, S. H. Park, J. Y. Jeon, H. B. Kim and B. Y. Lee, RSC Adv., 2017, 7, 5948–5956 RSC.
- C. Gutiérrez, J. F. Rodríguez, I. Gracia, A. de Lucas and M. T. García, Process Saf. Environ. Prot., 2016, 101, 144–151 CrossRef.
- A. Abdel-Hakim, T. M. El-Basheer, A. M. A. El-Aziz and M. Afifi, Polym. Test., 2021, 99, 107215 CrossRef CAS.
- S. Oh and E. E. Stache, J. Am. Chem. Soc., 2022, 144, 5745–5749 CrossRef CAS PubMed.
- C. Lu, H. Xiao and X. Chen, e-Polym., 2021, 21, 428–432 CrossRef CAS.
- P. Pfäffli, A. Zitting and H. Vainio, Scand. J. Work, Environ. Health, 1978, 4, 22–27 CrossRef.
- K. I. Dement’ev, T. A. Palankoev, O. A. Alekseeva, I. A. Babkin and A. L. Maksimova, J. Anal. Appl. Pyrolysis, 2019, 142, 104612 CrossRef.
- B. T. Ho, T. K. Roberts and S. Lucas, Crit. Rev. Biotechnol., 2018, 38, 308–320 CrossRef CAS PubMed.
- Q. Wang, H. Chen, W. Gu, S. Wang and Y. Li, Sci. Total Environ., 2024, 927, 172243 CrossRef CAS PubMed.
- M. C. D. Carter, A. Hejl, M. Janco, J. DeFelippis, P. Yang, M. Gallagher and Y. Liang, Macromolecules, 2023, 56, 5718–5729 CrossRef CAS.
- Z. Li, X. Zhang, Y. Zhao and S. Tang, Angew. Chem., Int. Ed., 2024, 63, 1–9 Search PubMed.
- V. Delplace and J. Nicolas, Nat. Chem., 2015, 7, 771–784 CrossRef CAS PubMed.
- A. Tardy, J. Nicolas, D. Gigmes, C. Lefay and Y. Guillaneuf, Chem. Rev., 2017, 117, 1319–1406 CrossRef CAS PubMed.
- D. K. Schneiderman and M. A. Hillmyer, Macromolecules, 2017, 50, 3733–3749 CrossRef CAS.
- G. Beckerab and F. R. Wurm, Chem. Soc. Rev., 2018, 47, 7739–7782 RSC.
- A. Tardy, J.-C. Honoré, J. Tran, D. Siri, V. Delplace, I. Bataille, D. Letourneur, J. Perrier, C. Nicoletti, M. Maresca, C. Lefay, D. Gigmes, J. Nicolas and Y. Guillaneuf, Angew. Chem., Int. Ed., 2017, 56, 16515–16520 CrossRef CAS PubMed.
- T. Pesenti and J. Nicolas, ACS Macro Lett., 2020, 9, 1812–1835 CrossRef CAS PubMed.
- A. W. Jackson, Polym. Chem., 2020, 11, 3525–3545 RSC.
- H. Wickel and S. Agarwal, Macromolecules, 2003, 36, 6152–6159 CrossRef CAS.
- A. W. Jackson, S. R. Mothe, L. R. Chennamaneni, A. van Herk and P. Thoniyot, Materials, 2020, 13, 2325 CrossRef CAS PubMed.
- N. Gil, B. Caron, D. Siri, J. Roche, S. Hadiouch, D. Khedaioui, S. Ranque, C. Cassagne, D. Montarnal, D. Gigmes, C. Lefay and Y. Guillaneuf, Macromolecules, 2022, 55, 6680–6694 CrossRef CAS.
- G. R. Kiel, D. J. Lundberg, E. Prince, K. E. L. Husted, A. M. Johnson, S. L. V. Lensch, P. Shieh and J. A. Johnson, J. Am. Chem. Soc., 2022, 144, 12979–12988 CrossRef CAS PubMed.
- Q. Zhang, D.-H. Qu, B. L. Feringa and H. Tian, J. Am. Chem. Soc., 2022, 144, 2022–2033 CrossRef CAS PubMed.
- Q. Zhang, Y. Deng, C.-Y. Shi, B. L. Feringa, H. Tian and D.-H. Qu, Matter, 2021, 4, 1352–1364 CrossRef CAS.
- K. R. Albanese, Y. Okayama, P. T. Morris, M. Gerst, R. Gupta, J. C. Speros, C. J. Hawker, C. Choi, J. Read de Alaniz and C. M. Bates, ACS Macro Lett., 2023, 12, 787–793 CrossRef CAS PubMed.
- C. Choi, Y. Okayama, P. T. Morris, L. L. Robinson, M. Gerst, J. C. Speros, C. J. Hawker, J. R. de Alaniz and C. M. Bates, Adv. Funct. Mater., 2022, 32, 1–6 Search PubMed.
- H. Tang and N. V. Tsarevsky, Polym. Chem., 2015, 6, 6936–6945 RSC.
- Q. Zhang, C.-Y. Shi, D.-H. Qu, Y.-T. Long, B. L. Feringa and H. Tian, Sci. Adv., 2018, 4, 8192–8199 CrossRef PubMed.
- Y. Deng, Q. Zhang, B. L. Feringa, H. Tian and D.-H. Qu, Angew. Chem., Int. Ed., 2020, 59, 5278–5283 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section including materials, synthetic procedure of tert-butyl lipoate, polymerization process, degradation process of PS-co-tBLp catalysed by DTT, degradation process of PS-co-tBLp by UV, characterization, results of polymers obtained under different conditions (Table S1), molecular weight differential distribution curves of PS-co-tBLp before and after degradation obtained under different conditions (Fig. S1), and DSC and TGA curves of PS-co-tBLp before and after degradation obtained under different conditions (Fig. S2). See DOI: https://doi.org/10.1039/d4cc05166a |
| This journal is © The Royal Society of Chemistry 2025 |