
Dynamic restructuring of electrocatalysts in the activation of small molecules: challenges and opportunities
Hsiwen
Wu
a and
Jie
Zhang
 *abc
*abc
aSchool of Chemistry, Monash University, Clayton, VIC 3800, Australia
bARC Research Hub for Carbon Utilisation and Recycling, Monash University, Clayton, VIC 3800, Australia
cARC Centre of Excellence for Green Electrochemical Transformation of Carbon Dioxide, Monash University, Clayton, VIC 3800, Australia. E-mail: jie.zhang@monash.edu
First published on 3rd January 2025
Abstract
Electrochemical activation of small molecules plays an essential role in sustainable electrosynthesis, environmental technologies, energy storage and conversion. The dynamic structural changes of catalysts during the course of electrochemical reactions pose challenges in the study of reaction kinetics and the design of potent catalysts. This short review aims to provide a balanced view of in situ restructuring of electrocatalysts, including its fundamental thermodynamic origins and how these compare to those in thermal and photocatalysis, and highlighting both the positive and negative impacts of in situ restructuring on the electrocatalyst performance. To this end, examples of in situ electrocatalyst restructuring within a focused scope of reactions (i.e. electrochemical CO2 reduction, hydrogen evolution, oxygen reduction and evolution, and dinitrogen and nitrate reduction) are used to demonstrate how restructuring can benefit or adversely affect the desired process outcome. Prospects of manipulating in situ restructuring towards an energy-efficient and durable electrocatalytic process are discussed. The practicality of pulse electrolysis on an industrial scale is questioned, and the need for genius schemes, such as self-healing catalysis, is emphasized.
 Hsiwen Wu | Hsiwen Wu obtained his PhD (2021) in Chemical and Biomolecular Engineering at the Hong Kong University of Science and Technology. He is now a research fellow at the Monash University School of Chemistry. His research interests include the electrochemical reduction of CO2, electrocatalysts for oxygen reduction reactions, hydrogen fuel cells, and membrane electrode assembly design. |
 Jie Zhang | Jie Zhang is an Associate Professor at the School of Chemistry, Monash University, and a Chief Investigator, Research Program Leader and co-Theme Leader for the Australian Research Council (ARC) Centre of Excellence for Green Electrochemical Transformation of Carbon Dioxide and the ARC Research Hub for Carbon Utilisation and Recycling. His current research mainly focuses on the electrochemical activation of small molecules for energy and sensing and the development and applications of artificial intelligence in dynamic electrochemistry. |
1. Introduction
Stable performance of catalyst (i.e. maintaining its activity and selectivity over an extended duration) is a key consideration in chemical processes. In heterogeneous catalysis, the interaction between catalyst surface and adsorbed species (e.g. reactants, intermediates, products, etc.) and that with the surroundings (e.g. temperature, applied electric potential, radiation, etc.) often induce structural transformation of the catalyst during reaction. Such restructuring can be morphological and/or compositional, and the spatial and temporal scales of which can span from nano- to micrometers, and from sub-seconds to hours, respectively.1 Based on the energy input/output, heterogeneous catalysis can be categorized into thermal-, electro-, and photocatalysis, and the catalyst restructuring involved in each is highly related to the energetics near the catalyst surface.2 In thermal catalysis, energy is supplied to the catalyst as heat to produce phonons, and the change in reaction Gibbs free energy from varying reaction conditions is usually less than several tens of milli-eV. Given that reaction enthalpy is a weak function of temperature, a change in temperature by 100 °C for example only results in milli-eV level difference in reaction Gibbs free energy (the exact value depends on the molar entropy change of the reaction of interest). In electro- and photocatalysis, the Gibbs free energy involved is notably larger (e.g. an applied/output voltage of several volts is common in electrocatalysis; while photoexcitation at around 3 eV is frequently seen in semiconductors to generate charge separation in photocatalysis3,4) and therefore is accompanied by pronounced structural change and often deactivation. In addition, the flow of electrons across multiple interfaces is involved in electrocatalysis (i.e. electrolyte/catalyst, catalyst/support, support/current collector, etc.) and in photocatalysis (i.e. metal/dielectric support, etc.), where the accumulation charge tends to occur at locations of high surface curvature. Such inhomogeneity in surface charge distribution may produce extremely high local current density5 and lead to catalyst restructuring and/or corrosion of other components.6It should be noted that the restructuring of catalyst is dynamic throughout the course of reaction and that the freshly prepared catalyst functions only as a pre-catalyst, as the active sites of the former are most likely different from those during operating reaction conditions.1,2 Based on the above, promotors are often incorporated into pre-catalysts to guide the structural transformation towards enhanced catalytic performance7 (i.e. mass/volume normalized turnover frequency, selectivity towards the target product, and durability). In the Haber–Bosch process, for example, structural promoters such as Al2O3 and CaO are incorporated into iron oxides to stabilize the grain boundaries of the latter against sintering.8,9 Potassium oxide is used as an electronic promoter in the Haber–Bosch process, which in its active form facilitates the dissociative adsorption of dinitrogen.10 As another example, in electrochemical CO2 reduction (ECO2R), halide ions in the electrolyte11–16 are used as promoters to steer the restructuring of copper oxides17–22/halides23 towards a roughened Cu surface under applied potentials, which results in improved selectivity to multi-carbon products.24–26
While comprehensive reviews focusing on the in situ restructuring of electrocatalysts in one specific reaction (e.g. ECO2R,27 OER,28–32etc.) or on characterization techniques that track the species and structural evolution during electrocatalytic reactions33,34 are available, works covering the fundamental driving force of restructuring in electrocatalysis over a range of reactions35–38 and how it compares with other heterogeneous catalytic reactions (i.e. thermal and photocatalysis)2 are relatively few. In this short review, a balanced view of electrocatalyst reconstruction is provided, including its thermodynamic origins and the associated positive and negative effects on catalytic performance. While the aim is not to cover all electrocatalytic reactions involving simple molecules, we focus on the in situ restructuring of electrocatalysts during the activation of small molecules (e.g. CO2, H2, O2, N2 and NO3−) as examples to demonstrate the general applicability of the thermodynamic driving force and highlight the possibility of achieving enhanced catalytic performance through controlled restructuring. The thermodynamic origins of restructuring in electrocatalysis are first introduced and collated with those in thermal and photocatalysis. Next, examples of how in situ restructuring adversely impacts the electrocatalytic performance are presented, followed by the opposite cases where it benefits the outcome of electrocatalysis. Finally, perspectives and outlooks on regulated restructuring towards energy-efficient and durable electrocatalytic processes are presented.
2. Origins of restructuring in electrocatalysis
The restructuring of electrocatalysts occurs under reaction conditions to decrease the net free energy of the system. The fact that the state of the electrocatalytic system changes during cell operation (e.g. change in electrode potential, species concentration, exposed surface area, etc.) suggests that the driving force for catalyst restructuring also varies throughout the reaction. Origins leading to such dynamic restructuring can be classified into three categories: surface energy reduction, electrode potential, and interaction with an electrolyte and adsorbed species. The following describes how each serves as the driving force for in situ structural transformation.2.1. Surface energy reduction
In a system of particles, the thermodynamic tendency is to lower the energy of the system through the reduction of the surface free energy. This driving force is fundamental and universal, and the effects of which can be observed throughout heterogeneous catalysis (i.e. electro-, thermal-, and photocatalysis). Phenomena associated with surface energy reduction include aggregation (assembly of particles with individual boundary maintained),39 coalescence (merge of particles in which individual boundary is lost),40 sintering (merge of particles when the temperature is elevated),41 and Ostwald ripening (dissolution of high-surface-energy sites followed by redeposition onto low-surface-energy sites).40,42,43 Since surface energy reduction often results in a decreased catalytically active interface area, it is often considered as a deactivation mode in literature.44–46 To keep a focused theme of this work on the restructuring of electrocatalysts, driving forces that are more relevant to electrocatalysis are discussed in detail in the following text.2.2. Electrode potential
An electrocatalytic reaction involves the transfer of electrons between catalysts and reactants on an electrified surface, and the rate of which therefore depends on the energy of electrons, which is expressed as electrode potential and carries the unit of volt (or eV, as energy carried per electron). Reaction Gibbs free energy per unit charge represents the minimal voltage required to run an electrolytic cell or the maximal voltage a galvanic cell can deliver. The activation energy barrier of an electrode reaction manifests itself as overpotential, and it outlines the importance of electrocatalysts in the energy efficiency of the catalyzed reaction. Depending on the electrode reaction (i.e. half-cell reaction) of interest, the operating potential window may overlap with the potential range where the catalyst is redox active, and consequently induce changes in the oxidation state of the catalyst. Cycles of oxidation and reduction of catalysts often result in the dissolution of metal constituents, and the ensuing structural change (e.g. via Ostwald ripening) may alter the catalyst performance significantly. For example, the OER holds a high standard reduction potential of 1.23 V vs. standard hydrogen electrode, under which almost all metal catalysts become oxidized and are prone to dissolution in acidic electrolytes.31,47 As another example, in oxygen reduction reaction (ORR) in acidic media, leaching of the less noble metal in platinum alloy catalysts (PtM, M = Co,48 Ni,49,50 Cu,51 Se,52etc.) is commonly reported, and the resulting dealloyed catalysts may evolve into a smooth Pt-skin or roughened Pt-skeleton structure, depending on the initial near surface composition, (Fig. 1).53 Through in situ electrochemical leaching, PtM catalysts may also transform into solid or porous particles with Pt-rich shell and alloy core, depending on the initial particle size54,55 and electrode potential.56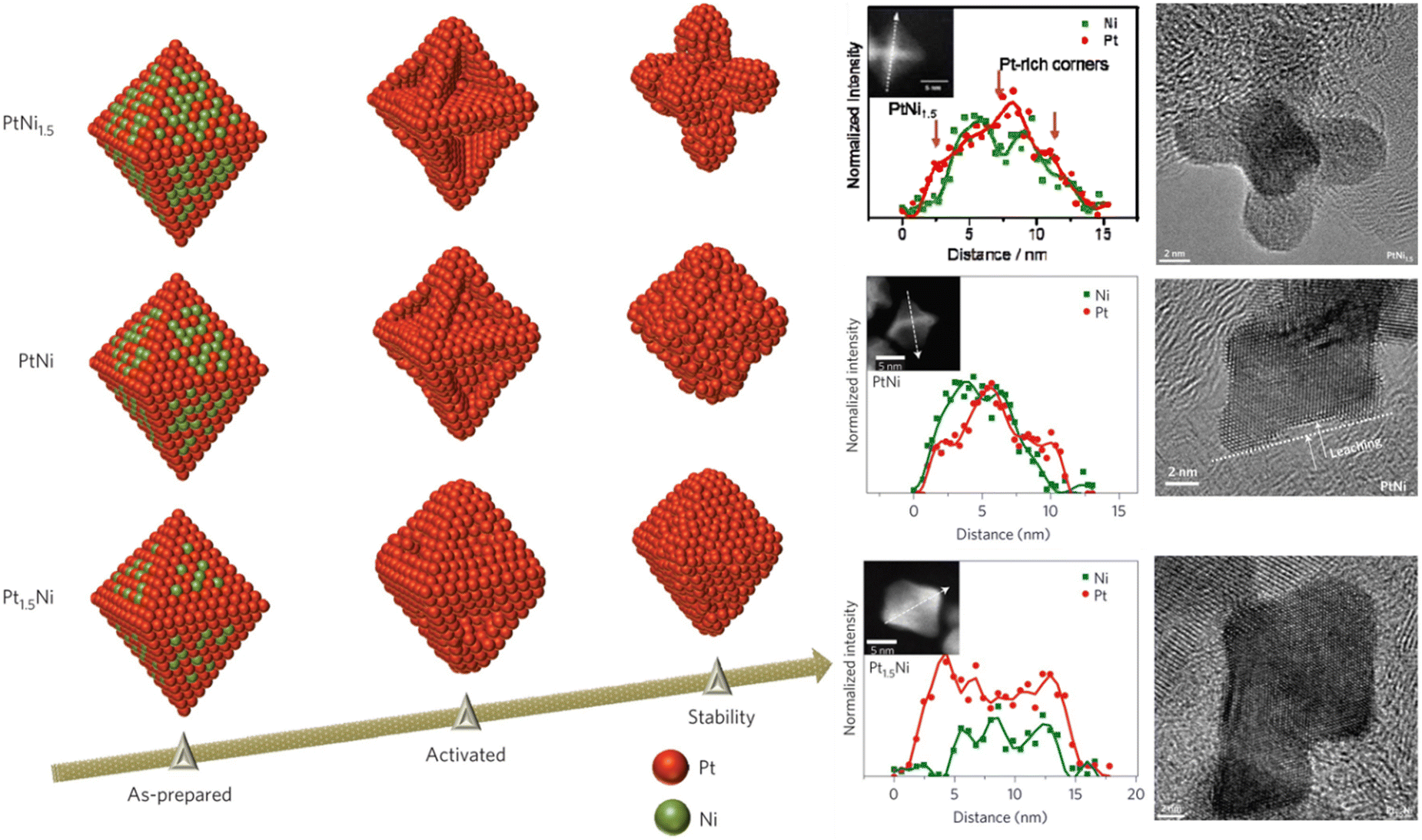 | ||
| Fig. 1 Composition-dependent structural evolution of PtNi octahedra under potential cycling in an O2-saturated 0.1 M HClO4 electrolyte. As-prepared: after 3 potential cycles (0.06–1.0 V, 100 mV s−1); Activated: after 25 potential cycles (0.06–1.0 V, 100 mV s−1); Stability: after 4000 potential cycles (0.6–1.0 V, 50 mV s−1). Figures adapted with permission from ref. 57. Copyright 2013. Springer Nature. | ||
Electrode reactions operating at strongly negative (i.e. highly reducing) potentials can also undergo restructuring resulting from the change of oxidation state(s) in electrocatalyst component(s),11,20,36,58,59 and/or due to cations present in the electrolyte.60 The latter process is termed cathodic corrosion,61,62 where cycles of cation intercalation and stripping/hydrogen evolution pulverize the crystal domain of catalysts and result in exposed high-index crystal facets.60 An exemplary case is the instability of palladium nanocrystals in acidic electrolytes, which limits its use in hydrogen evolution reaction (HER) despite its outstanding catalytic activity.63,64 The crystal structure of Pd allows facile incorporation of adsorbed hydrogen atoms into the lattice,65 a phenomenon termed H absorption, and the subsequent hydrogen gas evolution at more negative potential tends to break the crystal into smaller sizes.60 The above explains, at least partially, why restructuring of electrocatalysts is accelerated by applied electrode potential.
2.3. Interaction with the electrolyte and adsorbed species
The interaction of metal catalysts with species present in the electrolyte can lead to a considerable lowering of the reduction potential of the metal, making the dissolution of which more thermodynamically favourable. In copper-catalyzed ECO2R for example, some of the reaction products (i.e. adsorbed CO and oxalate) can form stable complexes with cuprous ions and promote dissolution-related surface reconstruction, such as Ostwald ripening.43,66 Studies on gas–solid interfaces have shown that specific adsorption (e.g. CO on Cu) lowers the formation energy of adatoms, especially on step edges and kinks.67–71 Such adsorbate-decorated adatoms have notably increased surface mobility, and tend to form nanoclusters (Fig. 2a) which function as highly active sites for thermo-catalytic reactions such CO oxidation and water–gas shift,67,68,72,73 methanol synthesis,69,74 ammonia synthesis/oxidation,75,76 and Fischer–Tropsch synthesis.76 In electrocatalytic reactions, adsorbate-promoted atom mobility on the catalyst surface is also observed in HER77,78 and ECO2R,79–81 as evidenced by the in situ formation of undercoordinated sites similar to those reported in thermos-catalytic studies. Direct observation of adsorbate-induced highly mobile Cu surface has been achieved via in situ liquid-cell transmission electron microscopy (TEM), and the former is visualized as an amorphous fluid-like layer flowing across the crystalline Cu surface (Fig. 2b–e).82 For metal alloy catalysts, the interaction with species present in the electrolyte may lead to segregation, the separation of the originally uniform phase into multiple domains of different compositions. For example, CuAg alloy catalyst during ECO2R may phase-separate into a core–shell structure with a Cu-rich surface80,83 due to stronger binding energy of CO on Cu than that on Ag.84 | ||
| Fig. 2 Adsorbate-mediated surface reconstruction. (a) Schematic of adsorbate-decorated adatom formation, diffusion and migration on the catalyst particle surface. (b) In situ liquid-cell TEM image showing the amorphous interphase between the electrolyte and crystalline Cu. Scale bar, 2 nm. (c) and (d) Time-resolved magnified TEM images of regions highlighted in (b). Yellow dots and arrows signify the disappearance of Cu atoms from the crystalline phase. Scale bar 5 Å. (e) Schematic illustration of the restructuring process at the amorphous interphase. Figures adapted with permission from ref. 67 ((a)). Copyright 2023. American Association for the Advancement of Science; and ref. 82 ((b)–(e)). Copyright 2024. Springer Nature. | ||
In many instances, the effects of the electrode potential and adsorbate/electrolyte species are intertwined, and both contribute to the restructuring of catalysts. In ECO2R, the reversible generation/disappearance of Cu nanoclusters from nitrogen-coordinated Cu single-atom catalysts with respect to applied potential has been observed via in situ X-ray absorption spectroscopy (XAS).85–87 While the formation of clusters from single-atom sites is induced by adsorbate-promoted mobility (i.e. *H–Cu88 and CO–Cu89) under applied potential, the redispersion of clusters back to nitrogen-coordinated single-atoms is facilitated by hydroxyl radicals produced from the reaction between water molecules and bicarbonate anions near cathode at open-circuit potential.88,90 In OER, the strongly oxidizing electrode potential transforms metal nanoparticle pre-catalyst into oxides, and the lattice oxygen mechanism suggests that oxygen atoms in the metal oxide crystal are constantly ejected as O2 gas, and replenished from adsorbed hydroxide anions.91–96 In this case, the effects of adsorbed hydroxide and electrode potential on OER catalyst restructuring are intertwined and both contribute to the lattice oxygen mechanism.
3. Impacts of in situ catalyst restructuring on electrocatalytic performance
In the previous section, we see that the restructuring of electrocatalysts depends on multiple (coupled) factors and that the structural change of the catalyst is dynamic throughout the course of the reaction. The impact of restructuring can be beneficial or unfavourable to the catalyzed process, depending on the targeted outcome. The following text discusses the negative and positive impacts of in situ catalyst restructuring, using reports on ORR, OER, ECO2R, HER, and nitrogen electro-redox reactions as examples.3.1. Negative impacts
In situ restructuring of electrocatalysts often results in diminished activity and/or selectivity on an industrially-relevant time scale, if not on a laboratory time scale. Therefore, control strategies towards mitigated or regulated restructuring are at the focal point of electrocatalysis research. In the following, cases where restructured electrocatalysts deliver decreased activity and/or selectivity are discussed.Electrocatalysts operating under high electrode potentials (e.g. OER and ORR catalysts) are prone to metal dissolution. In general, leaching of metal in the cell may lead to deactivation in two folds. One is the loss of electronic and geometric effects beneficial to the activity enhancement, and the other relates to the adverse effects of leached metals on other cell components (i.e. deposition on a low-potential electrode,97 deposition within the membrane electrolyte,98,99 Fenton reaction that promotes membrane degradation,100etc.). In PtM (M is a less noble metal than Pt) ORR catalysts, the outcome of in situ restructuring depends on the interplay of M dissolution and Pt surface diffusion,55 and is strongly potential dependent, as evidenced by several in situ characterization techniques on PtNi ORR catalyst.56 At the normal operating potential range of proton exchange membrane fuel cell (PEMFC) cathode, 0.6–1.0 V vs. reversible hydrogen electrode (RHE), PtNi particles maintain their structure (probed via in situ electrochemical atomic force microscopy) despite pronounced leaching of Ni content, as verified via on-line inductively coupled plasma mass spectrometry. However, at extremely high potentials of 1.3–1.5 V vs. RHE, which occur transiently during the start-up and shut down of the PEMFC, significant coalescence was observed as the in situ grazing-incidence small-angle X-ray scattering data suggested a notably increased particle diameter, standard deviation of the particle diameter, and minimum interparticle distance, while the volume fraction of Pt nanoparticles decreases.56 Upon metal leaching, the ensuing change in the catalyst particle size (i.e. wider particle size distribution) and the roughened surface with high-surface-energy sites exposed favour Ostwald ripening.101 The effects of leaching are observed in PEMFC cathode as increased catalyst particle size for Pt and PtM (M = Co, Ni) catalysts,102,103 as hollow Pt shells for Pt@Pd core–shell catalysts,104 as the appearance of a Pt-98,105 or Pd-band99 in the membrane.
Segregation is a common issue in electrocatalysis, where the near-surface composition of a synthesized pre-catalyst is altered via nanoscale rearrangement of constituent atoms into different phases under electrochemical conditions. Aside from electrode potential and possible adsorbate that change dynamically during the reaction, the influence of ambient oxygen on catalyst segregation can be a critical factor for the stability of alloy catalysts according to density functional theory (DFT) calculations, and therefore should also be included as a design consideration.106 While catalyst segregation can be readily regulated in thermal catalysis through activation or regeneration under a selected reducing atmosphere,107–112 reliable control of segregation in electrocatalysis can be challenging due partly to the increased system complexity.106 When aiming to direct an electrocatalyst towards targeted phase(s) (e.g. solid-solution, intermetallic, core–shell, etc.) through the applied potential for example, factors such as the electrolyte pH, possible adsorbates, and the electrochemical window of the solvent are coupled to the electrode potential and impact the outcome of segregation. In ECO2R, segregation usually deactivates alloy catalysts through the removal of sites for C–C coupling,113 loss of targeted electronic interaction,80,114 or coverage of ECO2R-inactive metal on the surface.115 For example, X-ray absorption fine-structure spectroscopy (XAFS) was used to track the evolution of oxidation state and coordination environment of constituents in CuZn catalyst during ECO2R, and it was discovered that phase-segregated Cu–ZnO promotes methane production, while solid solution CuZn formed after prolonged operation suffers from pronounced HER.114 As another example, through in situ nuclear resonant inelastic X-ray scattering and XAFS, it was observed that iron atoms in CuFe alloy segregate to the surface during ECO2R due to the stronger adsorption energy to CO intermediate, resulting in an enhanced HER.115
The restructuring of the catalyst in ECO2R is specific with respect to the product–metal pair, and the combined effects of multiple processes eventually lead to deactivation of the catalyst.44–46 In Cu-catalyzed ECO2R for example, the reaction intermediate/product of *CO (adsorbed intermediate) and oxalate can interact strongly with Cu, and stabilize cuprous (Cu+) cation in the solution phase, therefore exacerbating dissolution-related structural transformation.43,66,116 The structural evolution of Cu nanoparticles in ECO2R generally follows a chronological order of cathodic corrosion, aggregation and coalescence, surface roughening and smoothening, resulting from dissolution–deposition, adatom migration, etc.117 Some of these processes are captured nicely using in situ liquid-cell TEM, such as cathodic corrosion,117–119 adsorbate-induced atomic migration82 and segregation,80 Ostwald ripening43 and coalescence (Fig. 3).117 Cu-based catalysts may initially experience a period of activation (i.e. increasing selectivity for multi-carbon products with respect to time) due to the exposure of high-index crystal facets from cathodic corrosion and adsorbate-induced formation of nanoclusters. At a time scale relevant to industrial applications, however, the activity towards the target product decreases as Ostwald ripening and coalescence gradually take effects, as shown in Fig. 3.117
 | ||
| Fig. 3 In situ liquid-cell TEM images capturing the time evolution of Cu nanoparticles at different electrode potentials. (a)–(d) at open-circuit potential at time = 0, 8, 16, and 24 seconds. (e)–(h) under −0.8 V vs. RHE at time = 0, 8, 16, and 32 seconds. Figures adapted with permission from ref. 117. Copyright 2023. Springer Nature. | ||
In electrochemical ammonia synthesis from dinitrogen120,121 or nitrate,122–125 HER is a major side-reaction similar to the case of ECO2R. In situ restructuring during ammonia synthesis may lead to increased selectivity towards HER, especially under highly cathodic overpotentials.123,126 Manipulation of the morphology of pre-catalyst has been shown to enhance the electrochemical dinitrogen reduction reaction (e-NRR) through suppressing HER127,128 and enhanced adsorption of dinitrogen on the catalyst surface.129 It should be noted that the identification of restructuring during e-NRR can be quite challenging, as the background/artefact of the dominant HER often masks the structural evolution due solely to e-NRR.130 In electrochemical dinitrogen oxidation reaction (N2OR) to nitrate, activation of dinitrogen and suppression of parasitic OER are the two major challenges in catalyst design.131 In this case, in situ restructuring of N2OR catalyst towards a defect-rich oxide/oxyhydroxide is undesirable,132–134 as such amorphous layers favor OER.135 For example, in N2OR via oxygen-vacancy-enriched tin oxide, enhanced faradaic efficiency and yield towards nitrate were attributed to decreased activation energy barrier for the activation of *N2 to *N2OH through DFT calculations.134
Aggregation and coalescence are mechanisms that apply to a system of particles, irrespective of the specific electrochemical reaction. Coalescence usually follows aggregation as the former further reduces the surface energy of the system by eliminating the surface area of the interface, and both lead to a decrease in the number of accessible active sites.136 Exemplary cases of proper anchoring of molecular catalyst (e.g. cobalt phthalocyanine) on the selected substrate (e.g. functionalized carbon nanotube, graphene) against aggregation and coalescence have been demonstrated in ORR137,138 and ECO2R,139–143 where the electronic interaction not only immobilize the catalyst molecule, but also fine-tune its catalytic property. Immobilization of single atom active sites into an extended network (i.e. metal–organic frameworks,139,144 covalent organic frameworks145) contributes to increased site density,142,143,146,147 but the stability of these frameworks under electrochemical conditions (i.e. combined effects of the applied electrode potential and electrolyte pH) remains a challenge for long-term operation.148,149 For nanoparticle catalysts, physical confinement through carbon-based support has found some success in delaying the aggregation in the HER,150 OER,151 ORR152,153 and ECO2R.154–156
3.2. Positive impacts
In instances where the active form generated from pre-catalyst demonstrates favoured catalytic performance (such as improved selectivity towards a specific product, enhanced reaction kinetics, etc.) but is rather short-lived, approaches to induce restructuring that leads to the regeneration of desired active form are often implemented. Stabilization of the active form may also be achieved through the design of pre-catalyst and/or pre-treatment methods prior to cell operation. In the following, instances where electrocatalytic activity and/or selectivity are improved through restructuring are analyzed.Regulated metal leaching can be used to prepare stable ORR catalysts prior to PEMFC assembly. Potential cycling of PtM alloy catalyst in a selected acidic electrolyte followed by thermal annealing has been shown to produce core–shell structure with a Pt-rich shell (often termed the Pt-skin catalyst) stable against metal leaching under PEMFC relevant conditions.53,57,157 For Pt@Pd catalysts, controlled Pd-leaching in the presence of a capping agent allows the rearrangement of Pt atoms in the shell, which is proposed to repair the pinhole defects. The treated Pt@Pd catalyst shows a Pt shell with increased thickness, and in PEMFC tests demonstrates notably improved durability, which is supported by the significantly lowered Pd dissolution rate.104 Non-precious metal ORR catalysts are promising alternatives to state-of-the-art Pt-based catalysts when operating under neutral or alkaline conditions.158,159 Metal leaching is also common in non-precious metal catalysts during ORR, and through rational pre-catalyst design (i.e. pairing of a metal cation and a p-block anion), a metastable amorphous layer with enhanced ORR activity can be generated via in situ restructuring.160,161 The above metal-leaching-induced restructuring of ORR catalysts was mostly studied in rotating disk electrode experiments, and caution should be given that the observed activity enhancement overlooks the possibility of ionomer/membrane poisoning in practical fuel cell test setup.
In HER, catalysts with abundant basal plane active sites have been shown to demonstrate “self-optimizing behaviour”, where stacked catalyst layers become exfoliated and expose more active sites during the course of HER (Fig. 4a–c).162 Similar to the amorphous surface layer formed through leaching of ORR catalyst component(s) introduced earlier, proper design of HER pre-catalyst leads to a metastable and highly active phase that benefits the electrocatalytic process.135,163 For example, vanadium-doped nickel sulfide is restructured to form an amorphous surface layer, which demonstrates superior HER activity relative to the control without V-doping.163 Defect engineering through the incorporation of an easily leachable component in the pre-catalyst is another way to facilitate regulated restructuring.164 In ECO2R, the stronger binding energy of *CO on defect-rich Cu sites relative to that on low-index Cu crystal facets has been shown to favour the selectivity towards multi-carbon products, through increased surface coverage of *CO and decreased energy barrier for C–C coupling.24,165 The use of Cu oxides11,18–22,24–26 or chlorides23 as pre-catalysts facilitates the generation of undercoordinated sites upon in situ reduction of Cu under ECO2R relevant potentials. The rational use of ligand promotes the in situ selective faceting of Cu nanocrystals under ECO2R conditions, and therefore increases the faradaic efficiency towards the targeted product.166,167
 | ||
| Fig. 4 In situ restructuring of electrocatalysts that lead to beneficial catalytic performance. (a) schematic illustration of the “self-optimizing behaviour” of layered TaS2 catalysts. (b), (c) TEM images of TaS2 before, and after HER potential cycling. (d) Schematic band diagrams of perovskite oxide LaCoO3 and SrCoO3, compared to the redox energy of the O2/H2O couple. (e) Galvanostatic charging of SrCoO3−δ, an oxygen-vacancy-rich oxide, in an O2- and Ar-saturated alkaline electrolyte, demonstrating the effectiveness of the lattice oxygen mechanism in lowering the reaction overpotential. Figures adapted with permission from ref. 162 ((a)–(c)). Copyright 2017. Springer Nature; and ref. 168 ((d) and (e)). Copyright 2017. Springer Nature. | ||
In a bottom-up manner, pre-catalysts consisting of atomically dispersed Cu can be directed to form reversibly nanoclusters85–88 or irreversibly nanoparticles169 based on the applied electrode potential and achieve enhanced selectivity towards multi-carbon products. It should be noted that in situ generated high-index defects are high-surface-energy sites and that these sites tend to degrade over time via modes such as (adsorbate-induced) migration, dissolution, and coalescence.44 Pulse electrolysis has been recognised as an effective approach to enhanced selectivity towards multi-carbon products in ECO2R.116,170–172 By periodically bringing the electrode potential to a less negative or even positive value, undercoordinated sites are regenerated and therefore prolong the service life of the catalyst. It should be noted that the use of pulse electrolysis to generate high-index catalyst surfaces works effectively for metals with low cohesive energy (e.g. Cu,116,170–173 Ag,171 and Bi174) because of the high surface atom mobility on these surfaces.67,175 The cohesive energy of a metal is the energy required to separate atoms in the crystal into free neutral atoms in vacuum (i.e. metal gas),175,176 and it serves as an indicator regarding the resistance of the metal against mobility-related structural change (e.g. adsorbate-induced adatom formation).76,81,177,178In situ generated high-index sites are not thermodynamically favoured, and at a longer time scale, despite the application of periodic potential pulse, the mobile metal atoms tend to migrate towards low-ohmic-resistance sites (e.g. contact planes between gas diffusion electrode and current collector)179 and other low-energy sites to stabilize the system.44
The involvement of lattice oxygen in OER on Co-based perovskite has been confirmed via in situ18O isotope labelling mass spectrometry,168 and it is expected that increasing the covalency of metal–oxygen bond makes the oxidation of lattice oxygen more thermodynamically favourable.168 On a theoretical basis, swapping the A site metal of perovskite (ABO3) to a lower-valent metal (e.g. from LaCoO3 to SrCoO3) results in a down-shift of Fermi level (Fig. 4d). When the Fermi level crosses the top of the O 2p states and moves below the O2/H2O redox energy (1.23 eV), oxidation of lattice oxygen becomes more thermodynamically favoured than that of water (Fig. 4e).168,180 The above explains why OER catalysts with low overpotential are mostly oxides with high valence metals. Such high valence metal sites are intrinsically unstable under ambient conditions, and are therefore often generated in situ under applied OER-relevant potential.
The existence of high valence metal sites (i.e. Ni4+) during the OER has been confirmed using in situ soft XAS,181 and the generation of such sites usually results from dissolution of other constituent metals therefore forming a defect-rich surface layer.96,181–187 For example, perovskite hydroxide CoSn(OH)6 was used as a pre-catalyst, and in situ transformed into a highly active OER catalyst via electrochemical etching (galvanostatic activation in the reference) of Sn component.182 As another example, the pre-catalyst of La- and Mn-codoped cobalt spinel was transformed into a surface-defect-rich OER catalyst upon leaching of lanthanum content as La3+.183 It is also proposed that the M–O–M motifs (M = Fe, Co, Ni) within the in situ formed amorphous layers of metal oxyhydroxide function as the active sites for enhanced OER activity.188 Under the harsh reaction environment of acidic OER, even iridium suffers from notable dissolution from the pre-catalyst. Through manipulation of the composition of Ir-based perovskite pre-catalysts and dissolution/electrochemical test conditions, it is proposed that the IrOx amorphous layer formed in situ through continuous dissolution/precipitation functions as the true catalyst determining the OER performance.189
A self-healing electrocatalyst spontaneously regenerates its active sites under the operating conditions (i.e. dynamic equilibrium of multiple reactions).190,191 In such a system, therefore, both processes of loss and regeneration of active sites are continuous and dynamic throughout the course of reaction.190,191 Distinction should be made between self-healing and self-repairing (Fig. 5a), where in the latter, the regeneration of active sites only occurs when the cell operation is stopped (e.g. at open-circuit).190,191 Based on the above, the reported on–off electrolysis192 and the self-assembled metal–organic macrocycle catalyst193 in ECO2R are self-repairing at best. Compared to pulse electrolysis, self-repairing catalysts offer notable merits by removing sophisticated control and continuous power supply. The more desirable self-healing catalysts, however, are yet to be reported for ECO2R. Self-healing catalysts in OER operate based on parallel dissolution and precipitation of metal oxide clusters (Fig. 5b).190
 | ||
| Fig. 5 Comparison between conventional oxygen evolution catalysts (OECs), self-repairing OECs, and self-healing OECs. (a) Summary and schematic illustration of the operating principle of self-healing OECs. (b) Mechanistic schemes of the OER on conventional catalysts (left) versus that on self-healing OECs (Co-OEC assembly, right). Figures adapted with permission from ref. 190. Copyright 2022. Springer Nature. | ||
Selection of a pre-catalyst metal (e.g. Mn, Co, and Ni) cation and solution phase oxyanion (e.g. phosphate, methylphosphonate, and borate) pair is crucial to ensure the self-healing feature of the electrocatalytic system. An appropriate cation-oxyanion pair allows dissolved cations to precipitate as oxides/oxyhydroxides, and the size of the latter is regulated via metallate capping formed from the reaction between oxyanions and dissolved metal cations.190,194 The pre-catalysts of such self-healing OER catalysts usually contain a metal component that upon restructuring serves as a porous conductive support (e.g. indium tin oxide,194 fluorine-doped tin oxide,195 lead oxide,196etc.) to ensure facile electronic and ionic conducting pathways. In HER, although not specifically stated as a self-healing system, certain metal alloys also display self-healing properties, where the dissolved molybdenum cations from pre-catalysts polymerize to form polymolybdate which stabilizes the in situ generated catalyst with enhanced HER activity.197 It has been demonstrated that reversible in situ restructuring can be achieved, which allows the catalyst to convert between two active forms depending on the operating condition (i.e. a bifunctional electrocatalyst).198,199 The pre-catalysts of such bifunctional electrocatalysts are usually prepared via electrodeposition in pH-buffered oxyanion solution, where the incorporated phosphorus198 or boron199 content in the near surface region facilitates the ensuing reversible restructuring. In electrochemical nitrate reduction reaction, amorphous surface layers resulting from in situ restructuring are often observed, which deliver improved faradaic efficiencies towards ammonia via stabilization of *NOH intermediates200,201 and/or suppression of HER,202,203 or through improved adsorption to nitrate anions.204
The above two sections have discussed the positive and negative impacts of in situ restructuring of catalysts on the outcome of electrochemical process. A tabular comparison summarizing the discussion and examples presented is shown in Table 1.
| Electrocatalytic reaction | Example catalyst | Restructuring event | Negative impact | Positive impact |
|---|---|---|---|---|
| ORR | PtM (M = Co, Ni, Cu, Se, etc.) | Metal leaching | Dissolved metal cations poison the ionomer and membrane in fuel cells.98,99 Fenton-like reaction leading to membrane degradation100 | Under the right conditions, the resulting Pt-rich shell offers higher specific activity53,57,157 |
| OER | Metal oxides | Metal leaching | Loss of active sites. Corrosion and amorphous layer formation compromise the mechanical integrity of the electrode29,30,32 | Under the right combination of electrolyte and (mixed) metal oxide catalyst, self-healing can be achieved with high activity and stability190,194,196 |
| ECO2R | Cu-based particles | Formation of an amorphous surface layer | At long time scales, undercoordinated sites are consumed through Ostwald ripening and other processes, eventually leading to decreased activity and selectivity43,66,117 | At short time scales, the generated undercoordinated sites improve the selectivity towards multi-carbon products20,205 |
| HER | Transition metal chalcogenides | Formation of the restructured surface layer | Prolonged operation favors the formation of highly crystalline MoS2 domains, or inactive oxides/oxyhydroxides on the surface, which deactivates the catalyst206 | In situ formation of an amorphous layer exposes highly active sites162,206 |
4. Perspectives and outlook
The fundamental origins and impacts of in situ restructuring of catalysts on electrochemical processes have been reviewed. Designs of pre-catalysts and pre-treatment/activation methods have found notable success, but the long-term operational stability of some of these strategies developed in a laboratory scale is questionable when advancing to industrial scales. Design strategies of pre-catalysts (e.g. alloying, doping, molecular tuning, size and facet engineering, oxidation state, defect engineering, etc.) have been covered in excellent reviews,27,28,31,36 and the effectiveness of these approaches rely on controlled/guided restructuring towards the most active and/or selective form of the electrocatalyst. In terms of manipulating restructuring through pre-treatment/activation methods, some of the proposed approaches, such as pulse electrolysis179,207 and in situ electrochemical dealloying,52,55 still face critical challenges in industrially-relevant setups, due to the impracticality of implementing three-electrode systems and the stability issue related to the leached metals, respectively. We’d like to present four perspectives and outlooks regarding the application of in situ restructuring of electrocatalysts in practical industrial settings.(1) Pulse electrolysis has found success in ECO2R in regulating active sites to generate targeted products with improved stability and/or selectivity when the restructuring process is reversible under electrochemical conditions. However, it is worth noting that such a technique faces challenges in scaling up towards commercialization. A process that relies on the precise control of the potential of an electrode requires the implementation of a reference electrode (i.e. a three-electrode cell). Such a half-cell approach has completely disregarded possible impacts of pulse on the efficiency and stability of counter electrode catalyst, which are equally important in an electrochemical process. Therefore, in large-scale industrial electrochemical processes, the long-term use of reference electrodes throughout the operation is very rare if not non-existent. Instead, constant cell voltage or current is the most practiced mode of operation due to its simplicity and practicality. In the Hall–Héroult electrochemical process for aluminium production for example, constant voltage electrolysis is maintained by adjusting the electrolyte resistance (i.e. distance between the anode and cathode).208 In chlor-alkali electrolysis, a constant current is applied and the cell voltage is monitored, which serves as an indicator for reactant (i.e. brine solution) flowrate adjustment to keep the operation at the highest energy efficiency.209,210
(2) Approaches to inducing in situ restructuring of electrocatalysts towards a more active/selective form and stabilizing them for a prolonged period of time under practical conditions (including transient operation such as start-up and shut-down) are essential to the development of energy-efficient and durable electrochemical processes. Revisiting the concept of promoter discussed at the beginning of this article, the introduction of appropriate species into the electrolyte together with the pre-catalyst to form a catalytic system has shown substantial promise in the OER (e.g. phosphate–Co194), HER (polymolybdate–Ni197), ECO2R (imidazolium-based-ionic-liquid-Ag,211 functionalized-iron-porphine-Cu212), and e-NRR (LiClO4–Cu213). The fact that even the state-of-the-art Haber–Bosch process relies on the addition of a promoter suggests that this approach towards regulated restructuring deserves more research attention in electrocatalysis.
(3) The concept of self-healing catalysts, where highly active/selective sites are generated, consumed upon reaction, and renewed constantly, provides prospects for an energy-efficient and durable process. Considering that efficient and stable OER has been achieved under harsh conditions (e.g. 80 °C and pH = 1) through a self-healing catalyst,196 the next step towards commercialization should be the improvement in impurity tolerance of the catalyst system, and insights can be obtained from seawater electrolysis.214,215 How self-healing can be realized for reactions other than OER and HER, which will require appropriate selection and pairing of solution phase species and catalyst metal components, is still an area rarely explored.
(4) In order to develop more reliable control of in situ restructuring of electrocatalysts to our advantage, advances in characterization techniques on electrochemical interfaces and a rigorous understanding of electrokinetics and other potential activity/selectivity descriptors beyond adsorption energy216 should propel the field greatly. Advances in the above areas will allow us greater access to the in situ restructuring of more complex electrocatalytic reactions, such as electrosynthesis of ammonia through co-reduction of dinitrogen217–219 or nitrate220,221 with CO2. The formation of the C–N bond requires the atomic-scale proximity of intrinsically different sites (i.e. strong *N2 or *NOH binding vs. strong *CO2 binding), which poses challenges in catalyst synthesis. The greatly increased number of possible intermediates and side reactions further complicates the in situ restructuring scenario. Considering the fact that the catalyst restructuring in the e-NRR is still masked by the dominant competing HER, a breakthrough in characterization techniques is required to elucidate the structural evolution attributed to the electrochemical C–N coupling from CO2 and N2.
In summary, the dynamic nature of electrocatalyst surface has introduced both challenges and opportunities to the industrial applications of electrochemical processes. In situ restructuring impacts negatively the electrocatalytic outcome when the evolved stable form of the catalyst during operation is of low activity and/or selectivity, or when other device components become poisoned or failed in the course of restructuring. Positive impacts may also be observed on restructured electrocatalysts if the stable form delivers improved activity and/or selectivity than the pre-catalysts. The authors hope that this review article will inspire researchers to advance electrochemical technologies and to ensure access to affordable, reliable and sustainable energy for all.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Australian Research Council (ARC) Centre of Excellence for Green Electrochemical Transformation of Carbon Dioxide (CE230100017), the ARC Research Hub for Carbon Utilisation and Recycling (IH220100012), an ARC Discovery Project grant (DP220100316), and a Monash-Woodside Energy Partnership grant. Open access publishing was facilitated by Monash University, as part of the Royal Society of Chemistry – Monash University agreement via the Council of Australian University Librarians.References
- C. Vogt and B. M. Weckhuysen, Nat. Rev. Chem., 2022, 6, 89–111 CrossRef.
- L. Liu and A. Corma, Nat. Rev. Chem., 2021, 5, 256–276 CrossRef PubMed.
- T. Luttrell, S. Halpegamage, J. Tao, A. Kramer, E. Sutter and M. Batzill, Sci. Rep., 2014, 4, 4043 CrossRef PubMed.
- M. A. Henderson, Surf. Sci. Rep., 2011, 66, 185–297 CrossRef.
- M. Liu, Y. Pang, B. Zhang, P. De Luna, O. Voznyy, J. Xu, X. Zheng, C. T. Dinh, F. Fan and C. Cao, Nature, 2016, 537, 382–386 CrossRef.
- F. Li, X. V. Medvedeva, J. J. Medvedev, E. Khairullina, H. Engelhardt, S. Chandrasekar, Y. Guo, J. Jin, A. Lee and H. Thérien-Aubin, Nat. Catal., 2021, 4, 479–487 CrossRef.
- G. J. Hutchings, Catal. Lett., 2001, 75, 1–12 CrossRef.
- H. Liu, Chin. J. Catal., 2014, 35, 1619–1640 CrossRef.
- J. Folke, K. Dembélé, F. Girgsdies, H. Song, R. Eckert, S. Reitmeier, A. Reitzmann, R. Schlögl, T. Lunkenbein and H. Ruland, Catal. Today, 2022, 387, 12–22 CrossRef.
- D. R. Strongin and G. A. Somorjai, J. Catal., 1988, 109, 51–60 CrossRef.
- Y. Kwon, Y. Lum, E. L. Clark, J. W. Ager and A. T. Bell, ChemElectroChem, 2016, 3, 1012–1019 CrossRef.
- D. Gao, F. Scholten and B. Roldan Cuenya, ACS Catal., 2017, 7, 5112–5120 CrossRef.
- A. S. Varela, W. Ju, T. Reier and P. Strasser, ACS Catal., 2016, 6, 2136–2144 CrossRef.
- P.-P. Yang, X.-L. Zhang, P. Liu, D. J. Kelly, Z.-Z. Niu, Y. Kong, L. Shi, Y.-R. Zheng, M.-H. Fan and H.-J. Wang, J. Am. Chem. Soc., 2023, 145, 8714–8725 CrossRef PubMed.
- P. Liu, H. Liu, S. Zhang, J. Wang and C. Wang, Electrochim. Acta, 2020, 354, 136753 CrossRef.
- S. Lee, D. Kim and J. Lee, Angew. Chem., 2015, 127, 14914–14918 CrossRef.
- C. W. Li and M. W. Kanan, J. Am. Chem. Soc., 2012, 134, 7231–7234 CrossRef.
- C. W. Li, J. Ciston and M. W. Kanan, Nature, 2014, 508, 504–507 CrossRef PubMed.
- D. Ren, Y. Deng, A. D. Handoko, C. S. Chen, S. Malkhandi and B. S. Yeo, ACS Catal., 2015, 5, 2814–2821 CrossRef.
- X. Wang, K. Klingan, M. Klingenhof, T. Möller, J. Ferreira de Araújo, I. Martens, A. Bagger, S. Jiang, J. Rossmeisl and H. Dau, Nat. Commun., 2021, 12, 794 CrossRef.
- T. Möller, F. Scholten, T. N. Thanh, I. Sinev, J. Timoshenko, X. Wang, Z. Jovanov, M. Gliech, B. Roldan Cuenya and A. S. Varela, Angew. Chem., 2020, 132, 18130–18139 CrossRef.
- W. Fu, Z. Liu, T. Wang, J. Liang, S. Duan, L. Xie, J. Han and Q. Li, ACS Sustainable Chem. Eng., 2020, 8, 15223–15229 CrossRef.
- C. S. Chen, A. D. Handoko, J. H. Wan, L. Ma, D. Ren and B. S. Yeo, Catal. Sci. Technol., 2015, 5, 161–168 RSC.
- A. S. Malkani, M. Dunwell and B. Xu, ACS Catal., 2018, 9, 474–478 CrossRef.
- K. Jiang, Y. Huang, G. Zeng, F. M. Toma, W. A. Goddard III and A. T. Bell, ACS Energy Lett., 2020, 5, 1206–1214 CrossRef.
- J. E. Pander III, D. Ren, Y. Huang, N. W. X. Loo, S. H. L. Hong and B. S. Yeo, ChemElectroChem, 2018, 5, 219–237 CrossRef.
- J. Zhang, S. Xia, Y. Wang, J. Wu and Y. Wu, iScience, 2024, 27, 110005 CrossRef.
- L. Wang, Q. Meng, M. Xiao, C. Liu, W. Xing and J. Zhu, Renewables, 2024, 2, 272–296 CrossRef.
- J. Wang, Chem, 2023, 9, 1645–1657 Search PubMed.
- J. Wang, Curr. Opin. Electrochem., 2023, 39, 101304 CrossRef.
- L. Gao, X. Cui, C. D. Sewell, J. Li and Z. Lin, Chem. Soc. Rev., 2021, 50, 8428–8469 RSC.
- J. Chen, H. Chen, T. Yu, R. Li, Y. Wang, Z. Shao and S. Song, Electrochem. Energy Rev., 2021, 4, 566–600 CrossRef.
- Y. Zhu, T.-R. Kuo, Y.-H. Li, M.-Y. Qi, G. Chen, J. Wang, Y.-J. Xu and H. M. Chen, Energy Environ. Sci., 2021, 14, 1928–1958 RSC.
- B. Hasa, Y. Zhao and F. Jiao, Annu. Rev. Chem. Biomol. Eng., 2023, 14, 165–185 CrossRef PubMed.
- H. Jiang, Q. He, Y. Zhang and L. Song, Acc. Chem. Res., 2018, 51, 2968–2977 CrossRef PubMed.
- W. Wang, J. Duan, Y. Liu and T. Zhai, Adv. Mater., 2022, 34, 2110699 CrossRef PubMed.
- C. Xia, F.-M. Li, C. He, S. Zaman, W. Guo and B. Y. Xia, Fundam. Res., 2024 DOI:10.1016/j.fmre.2024.04.017.
- Y. Huang, W. Quan, H. Yao, R. Yang, Z. Hong and Y. Lin, Inorg. Chem. Front., 2023, 10, 352–369 RSC.
- G. Nichols, S. Byard, M. J. Bloxham, J. Botterill, N. J. Dawson, A. Dennis, V. Diart, N. C. North and J. D. Sherwood, J. Pharm. Sci., 2002, 91, 2103–2109 CrossRef PubMed.
- A. D. McNaught and A. Wilkinson, IUPAC compendium of chemical terminology, Royal Society of Chemistry, 1997 Search PubMed.
- E. Goudeli and S. E. Pratsinis, AIChE J., 2016, 62, 589–598 CrossRef.
- Z. Zhang, Z. Wang, S. He, C. Wang, M. Jin and Y. Yin, Chem. Sci., 2015, 6, 5197–5203 RSC.
- J. Vavra, T. H. Shen, D. Stoian, V. Tileli and R. Buonsanti, Angew. Chem., 2021, 133, 1367–1374 CrossRef.
- H. Wu, H. Yu, Y.-L. Chow, P. A. Webley and J. Zhang, Adv. Mater., 2024, 36, 2403217 CrossRef PubMed.
- W. Lai, Y. Qiao, Y. Wang and H. Huang, Adv. Mater., 2023, 35, 2306288 CrossRef PubMed.
- J. Chen and L. Wang, Adv. Mater., 2022, 34, 2103900 CrossRef PubMed.
- S. Cherevko, S. Geiger, O. Kasian, N. Kulyk, J.-P. Grote, A. Savan, B. R. Shrestha, S. Merzlikin, B. Breitbach, A. Ludwig and K. J. J. Mayrhofer, Catal. Today, 2016, 262, 170–180 CrossRef.
- J. Choi, J. Cho, C.-W. Roh, B.-S. Kim, M. S. Choi, H. Jeong, H. C. Ham and H. Lee, Appl. Catal., B, 2019, 247, 142–149 CrossRef.
- T.-Y. Jeon, S. K. Kim, N. Pinna, A. Sharma, J. Park, S. Y. Lee, H. C. Lee, S.-W. Kang, H.-K. Lee and H. H. Lee, Chem. Mater., 2016, 28, 1879–1887 CrossRef.
- C. Roiron, V. Martin, K. Kumar, L. Dubau and F. Maillard, Electrochim. Acta, 2024, 477, 143760 CrossRef.
- K. Jayasayee, J. A. R. V. Veen, T. G. Manivasagam, S. Celebi, E. J. M. Hensen and F. A. de Bruijn, Appl. Catal., B, 2012, 111–112, 515–526 CrossRef.
- W. Niu, S. Pakhira, G. Cheng, F. Zhao, N. Yao, J. L. Mendoza-Cortes and B. E. Koel, Nat. Mater., 2024, 23, 1704–1711 CrossRef.
- V. R. Stamenkovic, B. S. Mun, K. J. J. Mayrhofer, P. N. Ross and N. M. Markovic, J. Am. Chem. Soc., 2006, 128, 8813–8819 CrossRef.
- L. Gan, M. Heggen, R. O’Malley, B. Theobald and P. Strasser, Nano Lett., 2013, 13, 1131–1138 CrossRef.
- P. Strasser and S. Kühl, Nano Energy, 2016, 29, 166–177 CrossRef.
- I. Khalakhan, M. Bogar, M. Vorokhta, P. Kúš, Y. Yakovlev, M. Dopita, D. J. S. Sandbeck, S. Cherevko, I. Matolínová and H. Amenitsch, ACS Appl. Mater. Interfaces, 2020, 12, 17602–17610 CrossRef.
- C. Cui, L. Gan, M. Heggen, S. Rudi and P. Strasser, Nat. Mater., 2013, 12, 765–771 CrossRef PubMed.
- Y. Zhao, X. Chang, A. S. Malkani, X. Yang, L. Thompson, F. Jiao and B. Xu, J. Am. Chem. Soc., 2020, 142, 9735–9743 Search PubMed.
- S. H. Lee, J. C. Lin, M. Farmand, A. T. Landers, J. T. Feaster, J. E. Avilés Acosta, J. W. Beeman, Y. Ye, J. Yano and A. Mehta, J. Am. Chem. Soc., 2020, 143, 588–592 CrossRef PubMed.
- T. Wirtanen, T. Prenzel, J.-P. Tessonnier and S. R. Waldvogel, Chem. Rev., 2021, 121, 10241–10270 CrossRef PubMed.
- A. I. Yanson, P. Rodriguez, N. Garcia-Araez, R. V. Mom, F. D. Tichelaar and M. T. Koper, Angew. Chem., 2011, 123, 6470–6474 CrossRef.
- B. Vanrenterghem, M. Bele, F. Zepeda, M. Šala, N. Hodnik and T. Breugelmans, Appl. Catal., B, 2018, 226, 396–402 CrossRef.
- S. Sarkar and S. C. Peter, Inorg. Chem. Front., 2018, 5, 2060–2080 RSC.
- C. Liu, F. Chen, B.-H. Zhao, Y. Wu and B. Zhang, Nat. Rev. Chem., 2024, 8, 277–293 CrossRef PubMed.
- A. Zalineeva, S. Baranton, C. Coutanceau and G. Jerkiewicz, Sci. Adv., 2017, 3, e1600542 CrossRef.
- J. Vavra, G. P. L. Ramona, F. Dattila, A. Kormányos, T. Priamushko, P. P. Albertini, A. Loiudice, S. Cherevko, N. Lopéz and R. Buonsanti, Nat. Catal., 2024, 7, 89–97 CrossRef.
- L. Xu, K. G. Papanikolaou, B. A. Lechner, L. Je, G. A. Somorjai, M. Salmeron and M. Mavrikakis, Science, 2023, 380, 70–76 CrossRef PubMed.
- B. Eren, D. Zherebetskyy, L. L. Patera, C. H. Wu, H. Bluhm, C. Africh, L.-W. Wang, G. A. Somorjai and M. Salmeron, Science, 2016, 351, 475–478 CrossRef.
- M. Roiaz, L. Falivene, C. Rameshan, L. Cavallo, S. M. Kozlov and G. N. Rupprechter, J. Phys. Chem. C, 2018, 123, 8112–8121 CrossRef PubMed.
- P. L. Hansen, J. B. Wagner, S. Helveg, J. R. Rostrup-Nielsen, B. S. Clausen and H. Topsøe, Science, 2002, 295, 2053–2055 CrossRef PubMed.
- B. Eren and M. Salmeron, J. Phys. Chem. C, 2018, 123, 8171–8176 CrossRef.
- A. A. Gokhale, J. A. Dumesic and M. Mavrikakis, J. Am. Chem. Soc., 2008, 130, 1402–1414 CrossRef PubMed.
- W. D. Williams, M. Shekhar, W.-S. Lee, V. Kispersky, W. N. Delgass, F. H. Ribeiro, S. M. Kim, E. A. Stach, J. T. Miller and L. F. Allard, J. Am. Chem. Soc., 2010, 132, 14018–14020 CrossRef PubMed.
- S. Kuld, M. Thorhauge, H. Falsig, C. F. Elkjær, S. Helveg, I. Chorkendorff and J. Sehested, Science, 2016, 352, 969–974 CrossRef PubMed.
- F. Wang, J. Ma, G. He, M. Chen, C. Zhang and H. He, ACS Catal., 2018, 8, 2670–2682 CrossRef.
- L. Xu and M. Mavrikakis, JACS Au, 2023, 3, 2216–2225 CrossRef PubMed.
- H. Matsushima, C. Haak, A. Taranovskyy, Y. Gründer and O. M. Magnussen, Phys. Chem. Chem. Phys., 2010, 12, 13992–13998 RSC.
- H. Matsushima, A. Taranovskyy, C. Haak, Y. Gründer and O. M. Magnussen, J. Am. Chem. Soc., 2009, 131, 10362–10363 CrossRef CAS PubMed.
- R. Amirbeigiarab, J. Tian, A. Herzog, C. Qiu, A. Bergmann, B. Roldan Cuenya and O. M. Magnussen, Nat. Catal., 2023, 6, 837–846 CrossRef CAS.
- P. Wilde, P. B. O'Mara, J. R. Junqueira, T. Tarnev, T. M. Benedetti, C. Andronescu, Y.-T. Chen, R. D. Tilley, W. Schuhmann and J. J. Gooding, Chem. Sci., 2021, 12, 4028–4033 RSC.
- L. Xu, M. Rebarchik, S. Bhandari and M. Mavrikakis, Surf. Sci., 2024, 122613, DOI:10.1016/j.susc.2024.122613.
- Q. Zhang, Z. Song, X. Sun, Y. Liu, J. Wan, S. B. Betzler, Q. Zheng, J. Shangguan, K. C. Bustillo, P. Ercius, P. Narang, Y. Huang and H. Zheng, Nature, 2024, 630, 643–647 CrossRef CAS PubMed.
- E. L. Clark, C. Hahn, T. F. Jaramillo and A. T. Bell, J. Am. Chem. Soc., 2017, 139, 15848–15857 CrossRef CAS PubMed.
- A. Bagger, W. Ju, A. S. Varela, P. Strasser and J. Rossmeisl, ChemPhysChem, 2017, 18, 3266–3273 CrossRef CAS PubMed.
- D. Karapinar, N. T. Huan, N. Ranjbar Sahraie, J. Li, D. Wakerley, N. Touati, S. Zanna, D. Taverna, L. H. Galvão Tizei and A. Zitolo, Angew. Chem., Int. Ed., 2019, 58, 15098–15103 CrossRef CAS PubMed.
- H. Xu, D. Rebollar, H. He, L. Chong, Y. Liu, C. Liu, C.-J. Sun, T. Li, J. V. Muntean and R. E. Winans, Nat. Energy, 2020, 5, 623–632 CrossRef CAS.
- Z. Weng, Y. Wu, M. Wang, J. Jiang, K. Yang, S. Huo, X.-F. Wang, Q. Ma, G. W. Brudvig and V. S. Batista, Nat. Commun., 2018, 9, 415 CrossRef PubMed.
- X. Bai, X. Zhao, Y. Zhang, C. Ling, Y. Zhou, J. Wang and Y. Liu, J. Am. Chem. Soc., 2022, 144, 17140–17148 CrossRef CAS PubMed.
- C. M. Gunathunge, X. Li, J. Li, R. P. Hicks, V. J. Ovalle and M. M. Waegele, J. Phys. Chem. C, 2017, 121, 12337–12344 CrossRef CAS.
- S. Mu, H. Lu, Q. Wu, L. Li, R. Zhao, C. Long and C. Cui, Nat. Commun., 2022, 13, 3694 CrossRef CAS PubMed.
- S. Fierro, T. Nagel, H. Baltruschat and C. Comninellis, Electrochem. Commun., 2007, 9, 1969–1974 CrossRef CAS.
- O. Diaz-Morales, F. Calle-Vallejo, C. de Munck and M. T. M. Koper, Chem. Sci., 2013, 4, 2334–2343 RSC.
- R. Kötz, S. Stucki, D. Scherson and D. M. Kolb, J. Electroanal. Chem. Interfacial Electrochem., 1984, 172, 211–219 CrossRef.
- N. Danilovic, R. Subbaraman, K.-C. Chang, S. H. Chang, Y. J. Kang, J. Snyder, A. P. Paulikas, D. Strmcnik, Y.-T. Kim, D. Myers, V. R. Stamenkovic and N. M. Markovic, J. Phys. Chem. Lett., 2014, 5, 2474–2478 CrossRef CAS PubMed.
- S. Cherevko, A. R. Zeradjanin, A. A. Topalov, N. Kulyk, I. Katsounaros and K. J. J. Mayrhofer, ChemCatChem, 2014, 6, 2219–2223 CrossRef CAS.
- E. Fabbri, M. Nachtegaal, T. Binninger, X. Cheng, B.-J. Kim, J. Durst, F. Bozza, T. Graule, R. Schäublin, L. Wiles, M. Pertoso, N. Danilovic, K. E. Ayers and T. J. Schmidt, Nat. Mater., 2017, 16, 925–931 CrossRef CAS PubMed.
- M. Dunwell, Q. Lu, J. M. Heyes, J. Rosen, J. G. Chen, Y. Yan, F. Jiao and B. Xu, J. Am. Chem. Soc., 2017, 139, 3774–3783 CrossRef CAS PubMed.
- S. Cherevko, N. Kulyk and K. J. J. Mayrhofer, Nano Energy, 2016, 29, 275–298 CrossRef CAS.
- A. Kongkanand, N. P. Subramanian, Y. Yu, Z. Liu, H. Igarashi and D. A. Muller, ACS Catal., 2016, 6, 1578–1583 CrossRef CAS.
- P. C. Okonkwo, I. Ben Belgacem, W. Emori and P. C. Uzoma, Int. J. Hydrogen Energy, 2021, 46, 27956–27973 CrossRef CAS.
- D. K. Pattadar and F. P. Zamborini, Langmuir, 2019, 35, 16416–16426 CrossRef PubMed.
- R. L. Borup, A. Kusoglu, K. C. Neyerlin, R. Mukundan, R. K. Ahluwalia, D. A. Cullen, K. L. More, A. Z. Weber and D. J. Myers, Curr. Opin. Electrochem., 2020, 21, 192–200 Search PubMed.
- Z. Xu, H. Zhang, H. Zhong, Q. Lu, Y. Wang and D. Su, Appl. Catal., B, 2012, 111–112, 264–270 CrossRef.
- H. Wu, F. Xiao, J. Wang, M. Gu and M. Shao, Nano Res., 2024, 17, 8772–8784 CrossRef.
- R. Sgarbi, W. Ait Idir, Q. Labarde, M. Mermoux, P. Wu, J. Mainka, J. Dillet, C. Marty, F. Micoud, O. Lottin and M. Chatenet, Ind. Chem. Mater., 2023, 1, 501–515 RSC.
- E. L. Clark, R. Nielsen, J. E. Sørensen, J. L. Needham, B. Seger and I. Chorkendorff, ACS Energy Lett., 2023, 8, 4414–4420 CrossRef PubMed.
- Y. Guo, S. Mei, K. Yuan, D.-J. Wang, H.-C. Liu, C.-H. Yan and Y.-W. Zhang, ACS Catal., 2018, 8, 6203–6215 CrossRef.
- J. C. Matsubu, V. N. Yang and P. Christopher, J. Am. Chem. Soc., 2015, 137, 3076–3084 CrossRef PubMed.
- F. Wang, C. Li, X. Zhang, M. Wei, D. G. Evans and X. Duan, J. Catal., 2015, 329, 177–186 CrossRef.
- E. Gioria, L. Duarte-Correa, N. Bashiri, W. Hetaba, R. Schomaecker and A. Thomas, Nanoscale Adv., 2021, 3, 3454–3459 RSC.
- K. Larmier, W. C. Liao, S. Tada, E. Lam, R. Verel, A. Bansode, A. Urakawa, A. Comas-Vives and C. Copéret, Angew. Chem., Int. Ed., 2017, 56, 2318–2323 CrossRef PubMed.
- J. Li, Y. He, L. Tan, P. Zhang, X. Peng, A. Oruganti, G. Yang, H. Abe, Y. Wang and N. Tsubaki, Nat. Catal., 2018, 1, 787–793 CrossRef.
- Y. Qiao, G. Kastlunger, R. C. Davis, C. A. G. Rodriguez, A. Vishart, W. Deng, Q. Xu, S. Li, P. Benedek and J. Chen, ACS Catal., 2023, 13, 9379–9391 CrossRef.
- H. S. Jeon, J. Timoshenko, F. Scholten, I. Sinev, A. Herzog, F. T. Haase and B. Roldan Cuenya, J. Am. Chem. Soc., 2019, 141, 19879–19887 CrossRef CAS PubMed.
- S. Kunze, P. Grosse, M. Bernal Lopez, I. Sinev, I. Zegkinoglou, H. Mistry, J. Timoshenko, M. Y. Hu, J. Zhao and E. E. Alp, Angew. Chem., 2020, 132, 22856–22863 CrossRef.
- R. M. Arán-Ais, F. Scholten, S. Kunze, R. Rizo and B. Roldan Cuenya, Nat. Energy, 2020, 5, 317–325 CrossRef.
- Y. Yang, S. Louisia, S. Yu, J. Jin, I. Roh, C. Chen, M. V. Fonseca Guzman, J. Feijóo, P.-C. Chen and H. Wang, Nature, 2023, 614, 262–269 CrossRef CAS.
- R. M. Arán-Ais, R. Rizo, P. Grosse, G. Algara-Siller, K. Dembélé, M. Plodinec, T. Lunkenbein, S. W. Chee and B. R. Cuenya, Nat. Commun., 2020, 11, 3489 CrossRef PubMed.
- P. Grosse, A. Yoon, C. Rettenmaier, A. Herzog, S. W. Chee and B. Roldan Cuenya, Nat. Commun., 2021, 12, 6736 CrossRef CAS PubMed.
- G. Qing, R. Ghazfar, S. T. Jackowski, F. Habibzadeh, M. M. Ashtiani, C.-P. Chen, M. R. Smith, III and T. W. Hamann, Chem. Rev., 2020, 120, 5437–5516 CrossRef CAS PubMed.
- D. R. MacFarlane, P. V. Cherepanov, J. Choi, B. H. R. Suryanto, R. Y. Hodgetts, J. M. Bakker, F. M. Ferrero Vallana and A. N. Simonov, Joule, 2020, 4, 1186–1205 CrossRef CAS.
- Y. Wang, C. Wang, M. Li, Y. Yu and B. Zhang, Chem. Soc. Rev., 2021, 50, 6720–6733 RSC.
- D. Liu, L. Qiao, S. Peng, H. Bai, C. Liu, W. F. Ip, K. H. Lo, H. Liu, K. W. Ng, S. Wang, X. Yang and H. Pan, Adv. Funct. Mater., 2023, 33, 2303480 CrossRef CAS.
- S. Han, H. Li, T. Li, F. Chen, R. Yang, Y. Yu and B. Zhang, Nat. Catal., 2023, 6, 402–414 CrossRef CAS.
- K. Fan, W. Xie, J. Li, Y. Sun, P. Xu, Y. Tang, Z. Li and M. Shao, Nat. Commun., 2022, 13, 7958 CrossRef CAS PubMed.
- A. Biswas, S. Nandi, N. Kamboj, J. Pan, A. Bhowmik and R. S. Dey, ACS Nano, 2021, 15, 20364–20376 CrossRef CAS PubMed.
- D. Gupta, A. Kafle, S. Kaur, P. P. Mohanty, T. Das, S. Chakraborty, R. Ahuja and T. C. Nagaiah, J. Mater. Chem. A, 2022, 10, 20616–20625 RSC.
- U. K. Ghorai, S. Paul, B. Ghorai, A. Adalder, S. Kapse, R. Thapa, A. Nagendra and A. Gain, ACS Nano, 2021, 15, 5230–5239 CrossRef CAS PubMed.
- J. Wang, B. Huang, Y. Ji, M. Sun, T. Wu, R. Yin, X. Zhu, Y. Li, Q. Shao and X. Huang, Adv. Mater., 2020, 32, 1907112 CrossRef CAS PubMed.
- B. H. R. Suryanto, H.-L. Du, D. Wang, J. Chen, A. N. Simonov and D. R. MacFarlane, Nat. Catal., 2019, 2, 290–296 CrossRef CAS.
- Y. Wang, T. Li, Y. Yu and B. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202115409 CrossRef CAS PubMed.
- S. Paul, A. Adalder, N. Barman, R. Thapa, A. Bera, K. Mitra and U. K. Ghorai, Adv. Funct. Mater., 2024, 34, 2408314 CrossRef CAS.
- A. Adalder, S. Paul, B. Ghorai, S. Kapse, R. Thapa, A. Nagendra and U. K. Ghorai, ACS Appl. Mater. Interfaces, 2023, 15, 34642–34650 CrossRef CAS PubMed.
- R. Singh, A. Biswas, N. Barman, M. Iqbal, R. Thapa and R. S. Dey, Small, 2024, 20, 2406718 CrossRef CAS PubMed.
- J. Liu and L. Guo, Matter, 2021, 4, 2850–2873 CrossRef CAS.
- F. Lv, N. Han, Y. Qiu, X. Liu, J. Luo and Y. Li, Coord. Chem. Rev., 2020, 422, 213435 CrossRef CAS.
- W. Fan, Z. Duan, W. Liu, R. Mehmood, J. Qu, Y. Cao, X. Guo, J. Zhong and F. Zhang, Nat. Commun., 2023, 14, 1426 CrossRef CAS PubMed.
- S. Aralekallu, S. Hadimane, Shantharaja, M. Nemakal and L. Koodlur Sannegowda, Fuel, 2024, 361, 130736 CrossRef CAS.
- R. Matheu, E. Gutierrez-Puebla, M. Á. Monge, C. S. Diercks, J. Kang, M. S. Prévot, X. Pei, N. Hanikel, B. Zhang and P. Yang, J. Am. Chem. Soc., 2019, 141, 17081–17085 CrossRef CAS PubMed.
- N. Han, Y. Wang, L. Ma, J. Wen, J. Li, H. Zheng, K. Nie, X. Wang, F. Zhao and Y. Li, Chem, 2017, 3, 652–664 CAS.
- J. Choi, P. Wagner, S. Gambhir, R. Jalili, D. R. MacFarlane, G. G. Wallace and D. L. Officer, ACS Energy Lett., 2019, 4, 666–672 CrossRef CAS.
- N. Corbin, J. Zeng, K. Williams and K. Manthiram, Nano Res., 2019, 12, 2093–2125 CrossRef CAS.
- L. Sun, V. Reddu, A. C. Fisher and X. Wang, Energy Environ. Sci., 2020, 13, 374–403 RSC.
- N. Kornienko, Y. Zhao, C. S. Kley, C. Zhu, D. Kim, S. Lin, C. J. Chang, O. M. Yaghi and P. Yang, J. Am. Chem. Soc., 2015, 137, 14129–14135 CrossRef CAS PubMed.
- S. Lin, C. S. Diercks, Y.-B. Zhang, N. Kornienko, E. M. Nichols, Y. Zhao, A. R. Paris, D. Kim, P. Yang and O. M. Yaghi, Science, 2015, 349, 1208–1213 CrossRef CAS PubMed.
- P. Shao, L. Yi, S. Chen, T. Zhou and J. Zhang, J. Energy Chem., 2020, 40, 156–170 CrossRef.
- C. S. Diercks, Y. Liu, K. E. Cordova and O. M. Yaghi, Nat. Mater., 2018, 17, 301–307 CrossRef PubMed.
- Y. An, X. Lv, W. Jiang, L. Wang, Y. Shi, X. Hang and H. Pang, Green Chem. Eng., 2024, 5, 187–204 CrossRef.
- W. Zheng, M. Liu and L. Y. S. Lee, ACS Catal., 2020, 10, 81–92 CrossRef.
- H. Jiang, Y. Lin, B. Chen, Y. Zhang, H. Liu, X. Duan, D. Chen and L. Song, Mater. Today, 2018, 21, 602–610 CrossRef.
- X. Cui, P. Ren, C. Ma, J. Zhao, R. Chen, S. Chen, N. P. Rajan, H. Li, L. Yu, Z. Tian and D. Deng, Adv. Mater., 2020, 32, 1908126 CrossRef PubMed.
- C. Galeano, J. C. Meier, V. Peinecke, H. Bongard, I. Katsounaros, A. A. Topalov, A. Lu, K. J. Mayrhofer and F. Schüth, J. Am. Chem. Soc., 2012, 134, 20457–20465 CrossRef PubMed.
- E. Padgett, V. Yarlagadda, M. E. Holtz, M. Ko, B. D. Levin, R. S. Kukreja, J. M. Ziegelbauer, R. N. Andrews, J. Ilavsky and A. Kongkanand, J. Electrochem. Soc., 2019, 166, F198 CrossRef.
- J.-Y. Kim, D. Hong, J.-C. Lee, H. G. Kim, S. Lee, S. Shin, B. Kim, H. Lee, M. Kim and J. Oh, Nat. Commun., 2021, 12, 3765 CrossRef PubMed.
- Y. Li, F. Cui, M. B. Ross, D. Kim, Y. Sun and P. Yang, Nano Lett., 2017, 17, 1312–1317 CrossRef PubMed.
- Z. Zhang, G. Wen, D. Luo, B. Ren, Y. Zhu, R. Gao, H. Dou, G. Sun, M. Feng and Z. Bai, J. Am. Chem. Soc., 2021, 143, 6855–6864 CrossRef PubMed.
- B. Han, C. E. Carlton, A. Kongkanand, R. S. Kukreja, B. R. Theobald, L. Gan, R. O’Malley, P. Strasser, F. T. Wagner and Y. Shao-Horn, Energy Environ. Sci., 2015, 8, 258–266 RSC.
- A. A. Gewirth, J. A. Varnell and A. M. Diascro, Chem. Rev., 2018, 118, 2313–2339 CrossRef PubMed.
- S. D. Bhoyate, J. Kim, F. M. de Souza, J. Lin, E. Lee, A. Kumar and R. K. Gupta, Coord. Chem. Rev., 2023, 474, 214854 CrossRef CAS.
- B. Yan, D. Krishnamurthy, C. H. Hendon, S. Deshpande, Y. Surendranath and V. Viswanathan, Joule, 2017, 1, 600–612 CrossRef CAS.
- M. B. Stevens, M. E. Kreider, A. M. Patel, Z. Wang, Y. Liu, B. M. Gibbons, M. J. Statt, A. V. Ievlev, R. Sinclair, A. Mehta, R. C. Davis, J. K. Nørskov, A. Gallo, L. A. King and T. F. Jaramillo, ACS Appl. Energy Mater., 2020, 3, 12433–12446 CrossRef CAS.
- Y. Liu, J. Wu, K. P. Hackenberg, J. Zhang, Y. M. Wang, Y. Yang, K. Keyshar, J. Gu, T. Ogitsu, R. Vajtai, J. Lou, P. M. Ajayan, B. C. Wood and B. I. Yakobson, Nat. Energy, 2017, 2, 17127 CrossRef CAS.
- X. Shang, K.-L. Yan, Y. Rao, B. Dong, J.-Q. Chi, Y.-R. Liu, X. Li, Y.-M. Chai and C.-G. Liu, Nanoscale, 2017, 9, 12353–12363 RSC.
- Y. Zhu, H.-C. Chen, C.-S. Hsu, T.-S. Lin, C.-J. Chang, S.-C. Chang, L.-D. Tsai and H. M. Chen, ACS Energy Lett., 2019, 4, 987–994 CrossRef CAS.
- A. Verdaguer-Casadevall, C. W. Li, T. P. Johansson, S. B. Scott, J. T. McKeown, M. Kumar, I. E. Stephens, M. W. Kanan and I. Chorkendorff, J. Am. Chem. Soc., 2015, 137, 9808–9811 CrossRef CAS PubMed.
- K. Yao, J. Li, A. Ozden, H. Wang, N. Sun, P. Liu, W. Zhong, W. Zhou, J. Zhou, X. Wang, H. Liu, Y. Liu, S. Chen, Y. Hu, Z. Wang, D. Sinton and H. Liang, Nat. Commun., 1749, 2024, 15 Search PubMed.
- S. Yu, S. Louisia and P. Yang, JACS Au, 2022, 2, 562–572 CrossRef CAS PubMed.
- A. Grimaud, O. Diaz-Morales, B. Han, W. T. Hong, Y.-L. Lee, L. Giordano, K. A. Stoerzinger, M. T. M. Koper and Y. Shao-Horn, Nat. Chem., 2017, 9, 457–465 CrossRef CAS PubMed.
- H. Sun, L. Chen, L. Xiong, K. Feng, Y. Chen, X. Zhang, X. Yuan, B. Yang, Z. Deng and Y. Liu, Nat. Commun., 2021, 12, 6823 CrossRef CAS PubMed.
- Z. Li, L. Wang, T. Wang, L. Sun and W. Yang, J. Am. Chem. Soc., 2023, 145, 20655–20664 CrossRef CAS PubMed.
- Y. Xu, J. P. Edwards, S. Liu, R. K. Miao, J. E. Huang, C. M. Gabardo, C. P. O’Brien, J. Li, E. H. Sargent and D. Sinton, ACS Energy Lett., 2021, 6, 809–815 CrossRef CAS.
- H. S. Jeon, J. Timoshenko, C. Rettenmaier, A. Herzog, A. Yoon, S. W. Chee, S. Oener, U. Hejral, F. T. Haase and B. Roldan Cuenya, J. Am. Chem. Soc., 2021, 143, 7578–7587 CrossRef CAS PubMed.
- C. Kim, L.-C. Weng and A. T. Bell, ACS Catal., 2020, 10, 12403–12413 CrossRef CAS.
- H. Liu, T. Yan, S. Tan, L. Sun, Z. Zhang, S. Hu, S.-H. Li, X. Kang, Y. Lei, L. Jiang, T. Hou, L. Liu, Q. Yu and B. Liu, J. Am. Chem. Soc., 2024, 146, 5333–5342 CrossRef CAS PubMed.
- C. Kittel, Introduction to solid state physics, Wiley, Hoboken, NJ, 8th edn, 2005 Search PubMed.
- U. Mizutani, M. Inukai, H. Sato and E. S. Zijlstra, Physical Metallurgy, ed. D. E. Laughlin and K. Hono, Elsevier, Oxford, 2014, 5th edn, pp. 103–202 DOI:10.1016/B978-0-444-53770-6.00002-2.
- L. Xu and M. Mavrikakis, J. Catal., 2024, 431, 115373 CrossRef CAS.
- B. Hammer and J. K. Norskov, Nature, 1995, 376, 238–240 CrossRef CAS.
- J. Kok, J. de Ruiter, W. van der Stam and T. Burdyny, J. Am. Chem. Soc., 2024, 146, 19509–19520 CrossRef CAS PubMed.
- J. B. Goodenough, Chem. Mater., 2014, 26, 820–829 CrossRef CAS.
- X. Zheng, B. Zhang, P. De Luna, Y. Liang, R. Comin, O. Voznyy, L. Han, F. P. García de Arquer, M. Liu, C. T. Dinh, T. Regier, J. J. Dynes, S. He, H. L. Xin, H. Peng, D. Prendergast, X. Du and E. H. Sargent, Nat. Chem., 2018, 10, 149–154 CrossRef CAS PubMed.
- F. Song, K. Schenk and X. Hu, Energy Environ. Sci., 2016, 9, 473–477 RSC.
- L. Chong, G. Gao, J. Wen, H. Li, H. Xu, Z. Green, J. D. Sugar, A. J. Kropf, W. Xu, X.-M. Lin, H. Xu, L.-W. Wang and D.-J. Liu, Science, 2023, 380, 609–616 CrossRef CAS PubMed.
- J. Huang, J. Chen, T. Yao, J. He, S. Jiang, Z. Sun, Q. Liu, W. Cheng, F. Hu, Y. Jiang, Z. Pan and S. Wei, Angew. Chem., Int. Ed., 2015, 54, 8722–8727 CrossRef CAS PubMed.
- Q. He, H. Xie, Z. U. Rehman, C. Wang, P. Wan, H. Jiang, W. Chu and L. Song, ACS Energy Lett., 2018, 3, 861–868 CrossRef CAS.
- R. Ram, L. Xia, H. Benzidi, A. Guha, V. Golovanova, A. Garzón Manjón, D. Llorens Rauret, P. Sanz Berman, M. Dimitropoulos, B. Mundet, E. Pastor, V. Celorrio, C. A. Mesa, A. M. Das, A. Pinilla-Sánchez, S. Giménez, J. Arbiol, N. López and F. P. García de Arquer, Science, 2024, 384, 1373–1380 CrossRef CAS PubMed.
- B.-Q. Li, Z.-J. Xia, B. Zhang, C. Tang, H.-F. Wang and Q. Zhang, Nat. Commun., 2017, 8, 934 CrossRef PubMed.
- S. Ye, Y. Lei, T. Xu, L. Zheng, Z. Chen, X. Yang, X. Ren, Y. Li, Q. Zhang and J. Liu, Appl. Catal., B, 2022, 304, 120986 CrossRef CAS.
- R. Zhang, N. Dubouis, M. Ben Osman, W. Yin, M. T. Sougrati, D. A. D. Corte, D. Giaume and A. Grimaud, Angew. Chem., Int. Ed., 2019, 58, 4571–4575 CrossRef PubMed.
- A. E. Thorarinsdottir, S. S. Veroneau and D. G. Nocera, Nat. Commun., 2022, 13, 1243 CrossRef PubMed.
- M. R. Mohammadi, S. Loos, P. Chernev, C. Pasquini, I. Zaharieva, D. González-Flores, P. Kubella, K. Klingan, R. D. L. Smith and H. Dau, ACS Catal., 2020, 10, 7990–7999 CrossRef.
- T. N. Nguyen, Z. Chen, A. S. Zeraati, H. S. Shiran, S. M. Sadaf, M. G. Kibria, E. H. Sargent and C.-T. Dinh, J. Am. Chem. Soc., 2022, 144, 13254–13265 CrossRef PubMed.
- J. Wang, X. Jing, Y. Yang, B. Xu, R. Jia and C. Duan, J. Am. Chem. Soc., 2024, 146, 19951–19961 CrossRef PubMed.
- M. W. Kanan and D. G. Nocera, Science, 2008, 321, 1072–1075 CrossRef PubMed.
- C. Feng, F. Wang, Z. Liu, M. Nakabayashi, Y. Xiao, Q. Zeng, J. Fu, Q. Wu, C. Cui, Y. Han, N. Shibata, K. Domen, I. D. Sharp and Y. Li, Nat. Commun., 2021, 12, 5980 CrossRef PubMed.
- M. Chatti, J. L. Gardiner, M. Fournier, B. Johannessen, T. Williams, T. R. Gengenbach, N. Pai, C. Nguyen, D. R. MacFarlane, R. K. Hocking and A. N. Simonov, Nat. Catal., 2019, 2, 457–465 CrossRef.
- W. Du, Y. Shi, W. Zhou, Y. Yu and B. Zhang, Angew. Chem., Int. Ed., 2021, 60, 7051–7055 CrossRef PubMed.
- S. Cobo, J. Heidkamp, P.-A. Jacques, J. Fize, V. Fourmond, L. Guetaz, B. Jousselme, V. Ivanova, H. Dau, S. Palacin, M. Fontecave and V. Artero, Nat. Mater., 2012, 11, 802–807 CrossRef PubMed.
- C. He, X. Wu and Z. He, J. Phys. Chem. C, 2014, 118, 4578–4584 CrossRef.
- Y. Wang, W. Zhou, R. Jia, Y. Yu and B. Zhang, Angew. Chem., Int. Ed., 2020, 59, 5350–5354 CrossRef PubMed.
- L. Qiao, D. Liu, A. Zhu, J. Feng, P. Zhou, C. Liu, K. W. Ng and H. Pan, Appl. Catal., B, 2024, 340, 123219 CrossRef.
- S. Li, P. Ma, C. Gao, L. Liu, X. Wang, M. Shakouri, R. Chernikov, K. Wang, D. Liu, R. Ma and J. Wang, Energy Environ. Sci., 2022, 15, 3004–3014 RSC.
- J. Zhang, T. Quast, B. Eid, Y.-T. Chen, R. Zerdoumi, S. Dieckhöfer, J. R. C. Junqueira, S. Seisel and W. Schuhmann, Nat. Commun., 2024, 15, 8583 CrossRef PubMed.
- K. Wu, C. Sun, Z. Wang, Q. Song, X. Bai, X. Yu, Q. Li, Z. Wang, H. Zhang, J. Zhang, X. Tong, Y. Liang, A. Khosla and Z. Zhao, ACS Mater. Lett., 2022, 4, 650–656 CrossRef.
- H. Jung, S. Y. Lee, C. W. Lee, M. K. Cho, D. H. Won, C. Kim, H.-S. Oh, B. K. Min and Y. J. Hwang, J. Am. Chem. Soc., 2019, 141, 4624–4633 CrossRef PubMed.
- S. C. Lee, J. D. Benck, C. Tsai, J. Park, A. L. Koh, F. Abild-Pedersen, T. F. Jaramillo and R. Sinclair, ACS Nano, 2016, 10, 624–632 CrossRef PubMed.
- T. Ito, J. Raj, T. Zhang, S. Roy and J. Wu, EES Catal., 2024, 2, 997–1005 RSC.
- A. T. Tabereaux and R. D. Peterson, Treatise on Process Metallurgy, ed. S. Seetharaman, R. Guthrie, A. McLean, S. Seetharaman and H. Y. Sohn, Elsevier, 2nd edn, 2024, pp. 625–676 DOI:10.1016/B978-0-323-85373-6.00004-1.
- U. DOE-OIT, Prepared by Energetics Incorporated, Columbia, Maryland, USA, 2000.
- H. Ito and A. Manabe, in Electrochemical Power Sources: Fundamentals, Systems, and Applications, ed. T. Smolinka and J. Garche, Elsevier, 2022, pp. 281–304 DOI:10.1016/B978-0-12-819424-9.00011-2.
- S.-F. Zhao, M. Horne, A. M. Bond and J. Zhang, J. Phys. Chem. C, 2016, 120, 23989–24001 CrossRef.
- F. Li, Y. C. Li, Z. Wang, J. Li, D.-H. Nam, Y. Lum, M. Luo, X. Wang, A. Ozden and S.-F. Hung, Nat. Catal., 2020, 3, 75–82 CrossRef.
- A. Guha, S. Narayanaru, N. M. Kaley, D. Krishna Rao, J. Mondal and T. N. Narayanan, Mater. Today Commun., 2019, 21, 100700 CrossRef.
- J. Guo, Y. Zheng, Z. Hu, C. Zheng, J. Mao, K. Du, M. Jaroniec, S.-Z. Qiao and T. Ling, Nat. Energy, 2023, 8, 264–272 Search PubMed.
- M. A. Khan, T. Al-Attas, S. Roy, M. M. Rahman, N. Ghaffour, V. Thangadurai, S. Larter, J. Hu, P. M. Ajayan and M. G. Kibria, Energy Environ. Sci., 2021, 14, 4831–4839 RSC.
- L. Rebollar, S. Intikhab, N. J. Oliveira, Y. Yan, B. Xu, I. T. McCrum, J. D. Snyder and M. H. Tang, ACS Catal., 2020, 10, 14747–14762 CrossRef.
- S. Paul, S. Sarkar, A. Adalder, A. Banerjee and U. K. Ghorai, J. Mater. Chem. A, 2023, 11, 13249–13254 RSC.
- J. Mukherjee, S. Paul, A. Adalder, S. Kapse, R. Thapa, S. Mandal, B. Ghorai, S. Sarkar and U. K. Ghorai, Adv. Funct. Mater., 2022, 32, 2200882 CrossRef.
- L. Niu, L. An, X. Wang and Z. Sun, J. Energy Chem., 2021, 61, 304–318 CrossRef.
- H. Cai, Z. Wang, G. Meng, T. Wei, Y. Liu, J. Luo, Q. Liu, G. Hu and X. Liu, Inorg. Chem., 2024, 63, 20935–20939 CrossRef PubMed.
- Y. Xiong, Y. Wang, J. Zhou, F. Liu, F. Hao and Z. Fan, Adv. Mater., 2024, 36, 2304021 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2025 |