
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIdentification of ligands for E3 ligases with restricted expression using fragment-based methods
Alex G.
Waterson
 ab,
Brian D.
Lehmann
c,
Zhenwei
Lu
d,
John L.
Sensintaffar
d,
Edward T.
Olejniczak
d,
Bin
Zhao
d,
Tyson
Rietz
d,
William G.
Payne
d,
Jason
Phan
d and
Stephen W.
Fesik
*abd
ab,
Brian D.
Lehmann
c,
Zhenwei
Lu
d,
John L.
Sensintaffar
d,
Edward T.
Olejniczak
d,
Bin
Zhao
d,
Tyson
Rietz
d,
William G.
Payne
d,
Jason
Phan
d and
Stephen W.
Fesik
*abd
aDepartment of Pharmacology, Vanderbilt University School of Medicine, Nashville, Tennessee, USA. E-mail: stephen.fesik@Vanderbilt.Edu
bDepartment of Chemistry, Vanderbilt University, Nashville, Tennessee, USA
cDepartment of Medicine, Vanderbilt University Medical Center, Nashville, TN, USA
dDepartment of Biochemistry, Vanderbilt University School of Medicine, Nashville, Tennessee, USA
First published on 2nd October 2025
Abstract
Heterobifunctional molecules that induce targeted degradation have emerged as powerful tools in chemical biology, target validation, and drug discovery. Despite their promise, the field is constrained by the relative paucity of ligands available for E3 ligases. Expanding the ligand repertoire for E3 ligases and other components of ubiquitin-proteasome system could significantly broaden the scope of the targeted degradation field. In this study, we report the identification of ligands for non-essential E3 ligases that are preferentially expressed in cancer tissues relative to normal tissues. Using a protein-observed NMR-based fragment screen, an ideal technique for this purpose, we identified fragment ligands and characterized their binding modes by X-ray crystallography. These ligands represent promising starting points for further optimization toward the discovery of tumor-selective degraders that may enhance the therapeutic window targeting proteins for which inhibition or degradation is associated with systemic toxicity.
Introduction
Targeted protein degradation (TPD) has emerged as a transformative paradigm in drug discovery, gaining substantial traction and impact in recent years. The approach relies on recruiting a target protein to an E3 ubiquitin ligase complex to induce ubiquitylation and degradation of that target.1 Proteolysis targeting chimeras (PROTACs), which link together ligands to the target protein and to an E3 ligase, are the most widely used strategy for inducing TPD and have been successfully applied to the degradation of a broad range of protein targets.2 Conceptually, any E3 ligase could be harnessed to induce TPD, and studies have characterized the suitability of many ligases for this purpose.3,4 By their very nature, PROTACs require availability of a suitable ligand to a degradation-competent E3 ligase. However, to date, ligands have been identified for a limited number of E3 ligases.5,6 Degradation using PROTACs derived from small molecule ligands to additional E3 ligases, such as KEAP17 and members of the DDB1 and CUL4 associated factor family (DCAF) such as DACF18 and DCAF15,9 as well as using PROTACs generated from covalently binding ligands to ligases including FEM1B10 and RNF11411 among others, has been demonstrated. But the vast majority of reported PROTACs rely on ligands to only two E3 ligases: von Hippel-Lindau tumor suppressor (VHL) and Cereblon (CRBN).Further expansion of the field would be enabled by the identification of ligands for additional E3 ligases that are capable of supporting PROTAC-induced degradation. This expansion could facilitate the targeting of previously inaccessible target proteins, overcome resistance mechanisms associated with popular E3 ligases and their ligands, and enable tissue-specific degradation.2,3,12–14 Given these potential advantages, the exploration of the ligandability of E3 ligases and validation of the suitability of these ligases for TPD is an active area of ongoing research.15
A particularly compelling, yet largely unrealized, opportunity in TPD is the ability to degrade a disease-associated protein selectively in affected tissues while sparing its function in healthy tissues. For example, one could envision degrading an oncogenic protein in tumor cells while sparing that protein in non-cancerous tissues, thereby widening the therapeutic window and minimizing associated toxicities. One strategy to achieve tumor-specific degradation would involve identification of E3 ligases that are highly expressed in tumors but minimally expressed in normal tissues. If ligands to such ligases could be identified, then PROTACs derived from them may induce selective degradation in cancer cells. This concept has been demonstrated by the PROTAC DT2216, which recruits VHL to degrade Bcl-xL.16 Since VHL is expressed at low levels in platelets, DT2216 spares Bcl-xL functions in these cells, mitigating the thrombocytopenia associated with Bcl-xL inhibitors.17
Here, we report a systematic analysis of E3 ligases expression patterns and identify ligases that are overexpressed in cancer cell lines relative to normal tissues at the protein level. Furthermore, we demonstrate the utility of using NMR-based fragment screening for identifying ligands for these ligases and employ X-ray crystallography to determine the structural basis for fragment binding to the ligases. This study provides a framework for the application of fragment-based discovery to expand the repertoire of ligands that bind to E3 ligases.
Results and discussion
Identification of E3 ligases with restricted expression profiles in cancer
To identify E3 ligases highly expressed in cancer, we first assembled a curated list of 595 candidate genes. This list comprised 339 putative E3 ligases containing E3 ligase protein domains (HECT, HECT_2, HECTc, PELI, RING, SPRY, UBOX, UBR, and zf-C2H2 domains) and 256 E3 ligase-related accessory genes containing BTB, CPSF, Cullin, F-box, OSTMP1, PHD, SOCS box, and UBA4 domains (see Table S1).18To identify E3 ligases with restricted expression patterns in cancer, we analyzed RNA-seq gene expression data from two cohorts. We merged raw count gene expression data from 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 057 tumors spanning 20 cancer types (TCGA) and 17382 normal samples from 30 tissue sites (GTEx, obtained from rapid autopsies) using Ensembl gene identifiers. This merged dataset was then normalized to read depth, scaled (e.g., by a factor of 10000 and normalized to a total count of 10000 per sample), and log transformed. Differentially expressed genes were identified using the Wilcoxon rank-sum test (Table S2).
057 tumors spanning 20 cancer types (TCGA) and 17382 normal samples from 30 tissue sites (GTEx, obtained from rapid autopsies) using Ensembl gene identifiers. This merged dataset was then normalized to read depth, scaled (e.g., by a factor of 10000 and normalized to a total count of 10000 per sample), and log transformed. Differentially expressed genes were identified using the Wilcoxon rank-sum test (Table S2).
We analyzed the pooled data sets to identify E3 ligases that showed differential E3 ligase expression in tumor versus normal tissue samples. Fig. 1 shows log differences in the expression profiles on the X-axis, demonstrating that several E3 ligases are indeed significantly enriched in tumors compared to normal tissues.
 | ||
| Fig. 1 Differential E3 ligase transcript expression in cancer and genetic dependency. Scatterplot shows differentially expressed (Log2 FC) E3 ligases expressed in cancer (TCGA) or normal tissue (GTEx) and relative genetic dependency from CRISPR screening data compiled from 1365 cancer cell lines. Dependency values are normalized so that nonessential genes have a median score of 0 and common essential genes have a median score of −1. | ||
Some E3 ligases are likely essential for cellular function. The emergence of resistance to drugs acting on the non-essential ligase CRBN has been reported and shows no dependency based on gene expression levels or cancer type.19 However, inhibition of the function of an as-yet not validated ligase from the E3-ligase recruiting portion of a PROTAC could result in significant toxicity in multiple tissues. We evaluated E3 ligase essentiality from publicly available CRISPR knockout screens (Broad Institute's Achilles and Sanger Institute's SCORE projects). This was done by averaging gene effect scores (DepMap) across 1365 cell lines.20 Gene effect scores are normalized such that nonessential genes have a median score of 0, and common essential genes had a median score of −1. The Y-axis of Fig. 1 rank-orders the E3 ligases by their essentiality. Essential E3 ligases were highly expressed in cancer and enriched in ubiquitin/proteasome pathways in addition to cell cycle and DNA repair pathways (SKP2, CUL2, ANAPC11, BRCA1, DDB1, MDM2, PRPF19, RNF4, BARD1, RBX1, and MNAT1) (Fig. 1), similar to prior findings reported in the E3 atlas.4
We aimed to identify candidate ligases with restricted expression profiles that would enable degradation of oncogenic targets while minimizing the toxicity risk from inhibition or degradation of that target in normal tissues. An ideal tumor-specific degrader would incorporate a ligand to a ligase that is both highly expressed in tumor tissue and minimally expressed in healthy tissues, such as those in the upper right of Fig. 1. Among the ligases commonly used in the field for PROTACs are CRBN and VHL (Fig. 1, red). CRBN shows no differential expression in this analysis, lying very close to 0 on the X-axis. Thus, induction of tumor-specific degradation is not expected from CRBN-based PROTACs. VHL, while exhibiting some tumor-specific expression (positioned to the right on the X-axis), is considered essential based on CRISPR screen data (positioned near −1 on the Y-axis), and thus use of VHL-derived PROTACs may increase the chances of toxicities in normal tissues.
Our objective is to identify ligands to the E3 ligases of interest in the upper right of Fig. 1. To narrow the candidate list, we conducted a literature review to determine whether a given ligase had been previously shown to ubiquitinate and/or induce proteasomal degradation of a specific substrate. Ligases lacking such validation were excluded from consideration. We also prioritized ligases amenable to protein-observed NMR fragment screening, a technique we planned to use for the identification of hits. Thus, the availability of robust protocols for high-yield protein expression in E. coli was a final selection criterion for ligases of interest. Based on these filters, we identified two ligases for follow-up: sasitas B-lineage lymphoma c (CBL-c) and tumor necrosis factor (TNF) receptor-associated factor 4 (TRAF-4), both of which were found to be non-essential in the DepMap scoring (Fig. S1).
To demonstrate the differential expression of our candidate ligases, we plotted the distribution of mRNA expression across all normal tissues (GTEx) and all cancers (TCGA) for CBL-c and TRAF-4, alongside CRBN (Fig. 2). The similar shapes and widest portions of the density curves for CRBN in both normal and tumor tissues suggest comparable expression distributions. In contrast, both CBL-c and TRAF-4 exhibit higher overall mRNA expression in tumor samples compared to normal tissues. These results are consistent with a prior similar analysis, described in the E3 atlas.4,21 Most normal tissues do not express CBL-c, whereas a substantial proportion of cancers show detectable expression (Fig. S2). TRAF-4 is expressed at low levels across many normal tissues but shows elevated expression in various cancers (Fig. S2), suggesting a potential therapeutic window for selective targeting.
 | ||
| Fig. 2 Expression distribution of select E3 ligases across all cancer and normal tissues. Violin plot shows log-normalized transcript expression of CRBN (A), CBL-c (B), and TRAF-4 (C) in normal tissues (GTEx) or cancer (TCGA). | ||
Both ligases have been previously shown to ubiquitinate substrates. The CBL family comprises well-characterized RING E3 ligases involved in modulating receptor tyrosine kinase signaling.22 Specifically, CBL-c has been shown to ubiquitinate EGFR,23 supporting its validated ligase activity. Similarly, TRAF-4, which contains an N-terminal RING finger domain,24 has been reported to ubiquitinate Smurf2,25 CHK1,26 and IRS-1.27
Based on our mRNA expression analysis, we hypothesize that both ligases could be utilized in PROTACs to selectively degrade a target protein in cancers while sparing a given target in non-transformed tissues.
Protein expression of CBL-c
CBL-c, also known as cbl-3, is distinct from the other CBL family members (c-CBL and CBL-B) due to the absence of much of the C-terminal region.22 It also differs in its distribution, exhibiting non-uniform expression across tissues, as observed in our analysis and previously reported studies.28 CBL-c exhibits relatively high transcript expression in most tumor samples, but limited expression in normal cells, except in certain epithelial tissues (Fig. S2A). To validate CBL-c expression at the protein level, we analyzed samples from various normal tissues and a panel of cancer cell lines. Immunoblot analysis revealed low CBL-c protein expression in normal tissues (Fig. 3A, see also Fig. S3–S6), consistent with the transcriptomic data. In contrast, many cancer cell lines across multiple tumor types exhibited markedly higher CBL-c expression (Fig. 3A). | ||
| Fig. 3 Analysis of CBL-c protein expression across various normal tissues and cancer cells. (A) Immunoblot shows CBL-c protein levels across tumor specimens and cell lines. (B) Immunoblot shows CBL-c protein levels across heart tissue samples or in HT29 cells transfected with control or siRNAs targeting CBL-c or unrelated protein KLHL12. | ||
Given this differential expression, a PROTAC utilizing a CBL-c-recruiting ligand could potentially degrade oncogenic targets in tumors while sparing those in normal tissues. For instance, CBL-c is minimally expressed in cardiovascular (CV) tissues and blood vessels, suggesting utility in targeted degradation of proteins associated with CV toxicity (e.g., Bcl-xL29). To confirm these findings, we validated the specificity of the CBL-c antibody and expanded our analysis to include additional CV-related tissues. As shown in Fig. 3B (see also Fig. S7–S9), siRNA-mediated knockdown of CBL-c confirmed antibody specificity, and further western blot analysis of heart and platelet samples corroborated the low expression of CBL-c in CV tissues.
Identification and characterization of ligands for CBL-c
To realize the potential of inducing selective degradation of a specific protein target using an E3 ligase with a restricted expression profile, ligands suitable for use in PROTACs must first be identified. We employed a protein-observed NMR-based fragment screening protocol30 to identify compounds that bind to CBL-c, due to the unparalleled levels of information provided by this technique and its established applicability to challenging protein targets.31 To facilitate NMR-based screening for CBL-c binders, we cloned, expressed, and purified uniformly 15N labeled protein, comprising the tyrosine kinase binding (TKB) domain of CBL-c (residues 1–323). The protein was obtained in a good yield of 5 mg L−1 and displayed a suitably resolved 1H–15N SOFAST HMQC spectrum (see Fig. 4A). CBL-c, like other members of the CBL family, interacts with protein tyrosine kinases and helps to regulate their activity. Notably, CBL-c has been shown to bind to and downregulate the epidermal growth factor receptor (EGFR).22,32 To elucidate the NMR shift pattern corresponding to binding to CBL-c in its recognition domain, we obtained a peptide fragment of the Src Homology 3 domain of EGFR and assessed its binding to CBL-c using NMR (see Fig. 4B). | ||
| Fig. 4 NMR spectral analysis of the CBL-c TKB domain. (A) The 1H–15N SOFAST HMQC spectrum of the TKB domain of CBL-c. (B) Shifts in CBL-c are induced by interaction with an EGFR peptide fragment EDSFLQRpYSSDPT. Shifts induced by a fragment mixture (C) and fragment 1 (D) binding to CBL-c. | ||
Using 1H–15N SOFAST HMQC NMR, we screened our in-house custom fragment library comprising 13824 compounds. Screening for fragment binding was first performed using pooled mixtures of 12 fragments, each at a concentration of 800 μM. Individual compounds from mixtures that showed shifts were subsequently analyzed separately to determine the specific fragment responsible for inducing the observed shifts. Several compounds were found to induce significant shifts in the CBL-c HMQC spectrum. As shown in Fig. 4C and D, different shift patterns were observed with the fragment hits. In some cases, the shift patterns matched to portions of that induced by the peptide fragment, but other shifts were more distinct, suggesting a binding site outside the peptide-binding location.
In total, 15 fragment hits showing strong chemical shifts were obtained. This relatively modest number of hits establishes the ligandability of the TKB domain of CBL-c but also indicates that this is a relatively challenging protein domain for small molecule binding. We acquired structurally related analogs of the chemical templates represented in the hit set by mining a combination of internal and commercial sources. These analogs were also evaluated by NMR to assess their binding to CBL-c, helping to clarify structure–activity relationships (SAR) and expanding the pool of validated binders. Fragment hits and analogs displaying strong shifts were titrated against the protein by NMR, and binding affinities were determined by analyzing the dose-responsive shift magnitudes.
The hit compounds clustered into three chemical series, all containing a carboxylic acid. Compound 1 exemplifies an anthranilic acid scaffold (Fig. 5). While the acid moiety was conserved across all compounds, the extension from the aniline nitrogen tolerated additional variation, with lipophilic alkyl groups and aromatic moieties generally demonstrating enhanced affinity. Compound 5 exhibited the highest affinity in this series, with a dissociation constant of 430 μM. Sub-millimolar affinities were also observed from a narrow set of quinoline acid-based hits, with some tolerance for aromatic ring substitutions at the 6-position (7a–d, Table 1).
 | ||
| Fig. 5 Anthranilic acid fragments that bind to CBL-c. KD values assessed by titration of induced protein shifts over at least 5 doses of compound. | ||
|
|
||
|---|---|---|
| Compound | R | K D (μM) |
| 7a | H | 620 |
| 7b | 6-OMe | 720 |
| 7c | 6-F | 840 |
| 7d | 6-CH3 | 1200 |

Additionally, a series of indole acids demonstrated binding to CBL-c with affinities as low as 400 μM (Table 2). In this series, substitutions at multiple positions of the indole ring system retained binding to the protein, with 6-position showing the best affinity values (e.g., 8c).
|
|
||||
|---|---|---|---|---|
| Compound | R1 | R2 | R3 | K D (μM) |
| 8a |

|
H | H | 610 |
| 8b |
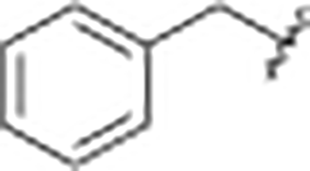
|
–CH3 | –OCH3 | 600 |
| 8c | –CH3 | –CH3 | –OCH2CH3 | 400 |
| 8d |

|
–CH3 | –OCH3 | 800 |
| 8e |
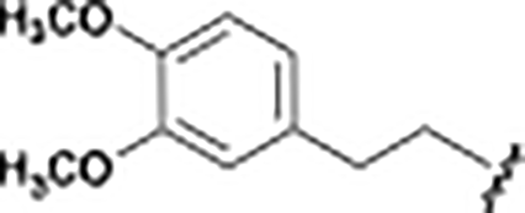
|
–CH3 | –OCH3 | 1100 |

We used X-ray crystallography to determine the binding location and orientation of the hit compounds. We successfully obtained a high resolution (1.8 Å) structure of compound 1 bound to the TKB domain of CBL-c. Consistent with the NMR shift pattern observed (Fig. 4C), this compound binds at an allosteric site distinct from the EGFR peptide binding site22 (Fig. 6A). Within this allosteric pocket, 1 is anchored by interactions between the carboxylic acid group and the backbone NH of Ser76 and two bound waters, one bridging to the backbone NH of Ser 76 and the other interacting with Arg 62 (Fig. 6B). The anthranilic acid aromatic ring is stabilized by a weak π-stacking interaction with His 147 and van der Waals contacts with Pro 70. The amide NH of 1 forms an internal hydrogen bond with the carboxylic acid of the compound, orienting the thiophene moiety into a hydrophobic subpocket. This subpocket likely accommodates the side chains of other active compounds such as 5 and 6. Notably, a charged subpocket above Arg 62 (Fig. 6B and C) remains unoccupied by 1 and represents a promising avenue for the identification of higher affinity future analogs. It is plausible that the substitutions on the quinoline acids 7b–c or the indole acids 8b–e may extend into this region, offering potential templates for optimization. It is noteworthy that the sequence homology within this region of the CBL family is low, suggesting that the compounds binding to the allosteric site, and derivatives, may selectively bind to CBL-c.
 | ||
| Fig. 6 X-ray structure of compound 1 bound to CBL-c. (A) Overlay of the structure of 1 with the structure of phospho-EGFR peptide (PDB ID: 3VRP). 1 is shown in blue sticks, while the peptide substrate from 3VRP is shown in orange. (B) Close-up view of 1 bound to the allosteric site on CBL-c, with surface representation. (C) Binding interactions of 1 in the allosteric site. Likely hydrogen bonding inactions shown as dashed blue lines. Bound waters are represented as red spheres. | ||
Protein expression of TRAF-4
The tumor necrosis factor (TNF) receptor-associated factor (TRAF) family comprises a small group of signaling molecules involved in various signal transduction events downstream of receptors such as TNF-R, the interleukin 1 receptor/toll-like receptor (TLR), the nucleotide-binding oligomerization domain-like receptor (NLR), the RIG-I like receptor (RLR), and certain cytokine receptors.24 A RING domain with E3 ligase activity is located at the N-terminal end of most family members, and a TRAF-C domain, which is responsible for mediating interactions with the receptors at the cell membrane, is located at the C-terminus in all family members except TRAF-7. These domains are separated by a long alpha-helical coiled-coil section (TRAF-N). TRAF proteins typically forms trimers in solution, resulting in an overall structure that resembles a mushroom.24TRAF-4 is unique among TRAF family members in that it does not interact with TNF receptors.33 Its expression profile is also distinct, as it has been reported to be overexpressed in several cancers, including breast, lung, ovary, prostate, and colon.33 This expression patten is reflected in observed transcriptomic differences (Fig. S2B). TRAF-4 exhibits high mRNA expression in most tumor samples compared to heart, brain, skin, and several other critical normal tissues. To validate TRAF-4 expression at the protein level, we used Western blotting on protein lysates from cancer cells and tissue samples. Our results demonstrate generally high expression of TRAF-4 in representative cancer cell lines compared to samples of normal tissues (Fig. 7A, see also Fig. S10–S12). As with CBL-c, extended our analysis to a panel of cardiovascular tissue samples and confirmed antibody specificity with siRNA-mediated knockdown of TRAF-4 (Fig. 7B, see also Fig. S13–S15). Notably, protein-level expression differences between tumor cells and normal tissues were more pronounced than those observed at the mRNA level. These findings suggest that TRAF-4 may also be viable for selective protein degradation. For example, the difference in TRAF-4 expression between lung cancer and heart tissue suggests a potential therapeutic window for TPD by minimizing the potential for cardiac toxicity.
 | ||
| Fig. 7 Analysis of TRAF-4 protein expression across various normal tissues and cancer cells. (A) Immunoblot shows TRAF-4 protein levels across tumor specimens and cell lines. (B) Immunoblot shows TRAF-4 protein levels across heart tissue samples and in HT29 cells transfected with control or TRAF-4-targeting siRNA. | ||
Identification and characterization of ligands for TRAF-4
To identify fragments that bind to TRAF-4, we cloned, expressed, and purified a uniformly 15N-labeled version of the TRAF-C domain of TRAF-4. The protein was obtained in a good yield (5–10 mg L−1) and its NMR spectrum was very well resolved. Binding-induced chemical shifts were readily observed upon addition of a peptide derived from a known TRAF-4 binding partner GPIbβ34 (Fig. 8A). Similar to our findings with CBL-c, a relatively modest number of hits was obtained, with 17 discrete fragments out of the 13824-membered library inducing strong shifts in the NMR spectrum. These hits produce shift patterns that overlap with those observed with the GPIbβ-derived peptide (compare Fig. 8A and B), suggesting that they bind within the narrow substrate recognition groove of TRAF-4.26
 | ||
| Fig. 8 NMR spectral analysis of the TRAF-C domain of TRAF-4. 1H–15N SOFAST HMQC NMR spectrum of the TRAF-C domain of TRAF-4 (A), showing shifts resulting from the binding of (B) a peptide fragment of GPIbβ and (C) fragment 9. | ||
To expand upon the initially identified fragment hits, we conducted a limited SAR expansion mining internally available compounds and acquiring additional analogs from commercial sources. In total, this effort yielded two primary hit series, along with fragments lacking close structural analogs. While many of the compounds displayed relatively weak binding to TRAF-4, compounds with affinities below 500 μM were identified in both series. Representative compounds and their NMR-derived affinities are shown in Fig. 9 and 10 as well as Table 3. For compounds without titration data, binding strength was inferred from the magnitude of the chemical shift perturbations induced by compounds at 800 μM.
 | ||
| Fig. 9 Quinoxaline-like fragment hits that bind to TRAF-4. KD values assessed by titration of induced protein shifts over at least 5 doses of compound. | ||
 | ||
| Fig. 10 Singleton fragment hits for TRAF-4. KD values assessed by titration of induced protein shifts over at least 5 doses of compound. | ||
|
|
||||
|---|---|---|---|---|
| RNA-seq analysis compound | R1 | R2 | R3 | K D (μM) |
| 14a | H | –CH3 |

|
140 |
| 14b | –H | –CH3 |
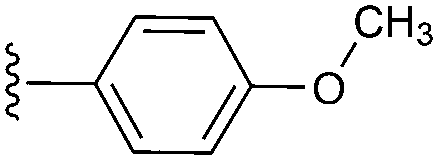
|
Weak shifts |
| 14c | –CH3 | –CH2CH3 |

|
Weak shifts |
| 14d | H | –CH3 |
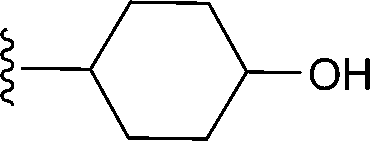
|
Weak shifts |
| 14e | H | –CH3 |

|
Weak shifts |
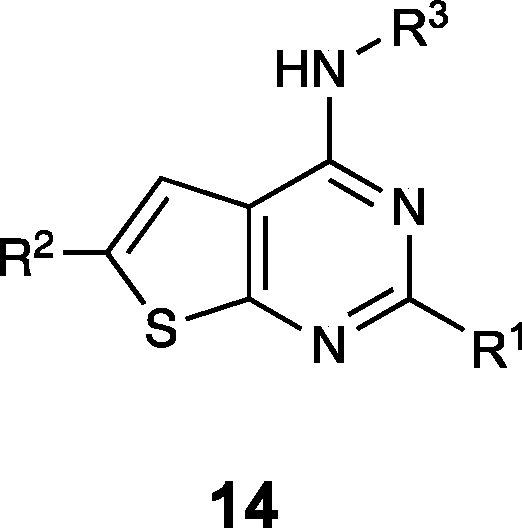
The thienopyrimidine-based compounds (Table 3) demonstrated the strongest binding to TRAF-4 and appear highly chemically tractable for future medicinal chemistry optimization. However, the template closely resembles known kinase inhibitors, raising concerns about potential off-target effects and selectivity. As a result, we focused our structural studies on elucidating the binding mode of the quinoxaline template.
We successfully obtained the X-ray structure of both compound 9 and a peptide fragment derived from EGFR which has also been shown to associate with TRAF-435 (residues 1198 to 1207, LRVAPQSSEF) in complex with the TRAF-C domain. As predicted from the NMR shift pattern, 9 occupies the peptide recognition groove of TRAF-4 (Fig. 11A). The compound binds in close proximity to Tyr 366 (Fig. 11B and C) and is anchored by hydrogen bonds to the backbone carbonyl and NH of Gly 433 and Gly 435, respectively. Additionally, the thiazole nitrogen likely participates in an interaction with the side chain of Asn 355. Based on the smaller shifts induced by compounds with changes to the thiazole moiety (shown in Fig. 9), we infer that both the interaction with Asn 355 and van der Waals contacts with adjacent residues contribute significantly to the binding affinity. These findings suggest that the affinity could be improved further by structure-guided modifications of this thiazole as well as extensions from the quinoxaline.
 | ||
| Fig. 11 Structural elucidation of binding to TRAF-4. (A) Overlay of the X-ray costructure of 9 bound to TRAF-4 overlain with the costructure of a bound EGFR-derived peptide. (B) View of 9 in the substrate groove, with surface representation on. (C) Interactions of 9 in the binding pocket. Likely hydrogen bonding interactions are shown with blue dashed lines. | ||
Experimental
The E3 ligase and accessory genes were compiled from several existing resources (Cell Signaling, hUbiquitome database, UbiProt Database, and DUDE v. 1.0, available at (https://esbl.nhlbi.nih.gov/Databases/KSBP2/Targets/Lists/E3-ligases/)).12Raw count reads for 11057 tumors spanning 20 cancer types from TCGA (GDC-PANCAN.htseq_counts.tsv) were downloaded (https://xenabrowser.net/). Details of alignment parameters can be found at https://docs.gdc.cancer.gov/Data/Bioinformatics_Pipelines/Expression_mRNA_Pipeline/. Normal RNA-seq raw counts (GTEx_Analysis_2017-06-05_v8) for 17382 tissues from rapid autopsies were obtained from the GTEx portal (https://gtexportal.org/). Raw counts from TCGA and GTEx were merged by ensemble gene id, normalized to read depth, scaled (10000) and log transformed. Differentially expressed genes were identified with the Van Elteren test,36 a stratified version of non-parametric Wilcoxon rank-sum test. All statistical tests were performed in R (version 4.4).
Genetic dependency
Gene effect scores derived from CRISPR knockout screens published by Broad's Achilles and Sanger's SCORE projects (DepMap 22Q2 Public) were downloaded from the DepMap portal (https://depmap.org/).20 Scores are normalized such that nonessential genes have a median score of 0 and independently identified common essentials have a median score of −1.Protein expression analysis
Protein expression and purification
The gene encoding CBL-c (residues 5–322) was synthesized with codon optimization and subcloned into pET28a(+) vector containing an N-terminal TEV-cleavable His6-tag. The genes encoding the TRAF domain of TRAF-4 (residues 292–466) and the corresponding truncate that lacks residues involved in trimerization (residues 300–466) were synthesized with codon optimization for bacterial expression and cloned into pET22b(+) and pET28a(+) vectors, respectively, by GenScript Biotech Corporation. Both DNA constructs contain a C-terminal His6-tag for IMAC purification with the latter thrombin-cleavable.The recombinant plasmids were transformed into electrocompetent BL21-Gold (DE3) E. coli strain and the bacterial cells were cultured in either Luria-Bertani broth or M9 minimal media containing 15NH4Cl for isotopic labeling. The media was supplemented with 50 μg mL−1 kanamycin or 100 μg mL−1 ampicillin and growth was carried out with shaking at 37 °C until the optical density at 600 nm reached 0.8. Protein expression was induced by the addition of 0.5 mM IPTG and incubated for 20 h at 16 °C and 18 h at 18 °C for TRAF-4 and CBL-c, respectively. The cell pellet was harvested by centrifugation at 5000g for 15 minutes, re-suspended in lysis buffer (TRAF-4: 50 mM Na2HPO4, 10 mM KH2PO4 pH 7.4, 300 mM NaCl, 5 mM BME, and 1 mM PMSF, CBL-c: 25 mM Tris, pH 8.0, 500 mM NaCl, 5 mM BME, 10 mM imidazole, 100 μM PMSF), and lysed using the APV-2000 homogenizer (SPX flow) in 3 cycles of 400–900 bar. Cell lysate was clarified by centrifugation at 15000g for 45 minutes, filtered, and loaded onto a HisTrap FF column (Cytiva). The column was washed with 10 column volumes of buffer A (TRAF-4: 50 mM Na2HPO4, 10 mM KH2PO4 pH 7.4, 300 mM NaCl, 20 mM imidazole, 5 mM BME, CBL-c: lysis buffer minus PMSF) and His6-tagged TRAF-4 protein was eluted from the column using a linear gradient from 0 to 100% of buffer B (TRAF-4: 50 mM Na2HPO4, 10 mM KH2PO4 pH 7.4, 300 mM NaCl, 500 mM imidazole, 5 mM BME, CBL-c: buffer A plus 500 mM imidazole) over 10 column volumes. His6-tagged CBL-c protein was eluted in 3 steps of linear gradients from 0 to 6% B in 6 column volumes (CV), washing for 3 CV's, then to 50% B in 6 CV's, washing for 3 CV's, then to 100% B and washing for 4 CV's. The fractions containing TRAF-4 (residues 292–466) and CBL-c were incubated with Thrombin and TEV protease, respectively, overnight to remove the His6-tag and dialyzed against Buffer A without imidazole. For CBL-c, NaCl concentration was reduced to 300 mM in the dialysis buffer. The tag-free protein was purified from the reaction mix by reverse Ni-NTA affinity and concentrated with an Amicon Stirred Cell (MilliporeSigma). Truncated TRAF-4 and CBL-c proteins were exchanged into an optimized buffer with low ionic strength (25 mM sodium phosphate pH 7.4, 25 mM NaCl, 1 mM DTT) and (25 mM HEPES pH 7.0, 100 mM NaCl, 5 mM BME), respectively for NMR-based fragment screen. For X-ray crystallography, TRAF-4 (residues 292–466) was further purified by size-exclusion chromatography (HiLoad 26/600, Superdex 75 pg, Cytiva) using Buffer C (20 mM Tris-HCl, pH 7.4, 300 mM NaCl, 3 mM DTT, 0.01% NaN3). CBL-c was further purified by SEC on the same column using NMR buffer and concentrated to 1 mg mL−1 for NMR and 2 mg mL−1 for crystallization. Protein concentration was quantified by Pierce 660 nm assay (ThermoFisher).
NMR experiments
All NMR experiments were performed at 25 °C using a 600 MHz Bruker Avance III spectrometer equipped with a 5 mm single-axis x-gradient cryoprobe and a Bruker SampleJet. Gradient-enhanced, two-dimensional 1H–15N heteronuclear multiple-quantum coherence (SOFAST-HMQC)37 spectra of TRAF-4 and CBL-c were recorded using 32 scans of 16-minute and 100 scans of 50-minute acquisition time respectively, and analyzed using Topspin 4.1.0 (Bruker). A fragment library comprising 13824 compounds was screened as mixtures of 12 fragments prepared in twelve 96-well plates. Each NMR sample was made of 25 μM of 15N-labeled truncated TRAF-4 or 40 μM of 15N-labeled CBL-c, 800 μM of each fragment, and 5% DMSO-d6 for spectrometer locking in 5 mm-diameter NMR tubes. Screening hits from the fragment mixtures were identified by comparing the chemical shifts of backbone resonances to a ligand-free protein spectrum and then deconvoluted by screening individual fragments to determine which component of the mixture was responsible for inducing the shift perturbations.
Protein crystallization and structure determination
Fresh batches of TRAF-4 and CBL-c proteins were concentrated to 7.5 mg mL−1 and 2 mg mL−1, respectively, and screened for crystallization conditions in the presence of 2-fold or 10-fold molar excess of ligand. Crystals were obtained by mixing 300 nL protein with 300 nL reservoir solution (TRAF-4: 10–15% PEG 3350, 0.1 M Bis-TRIS pH 6.5, CBL-c: 10–20% PEG 3350, 0.1 M ammonium formate, pH 6.5–7.5) using the sitting drop vapor diffusion method at 18 °C. Crystals appeared within a few weeks and were flash frozen in liquid nitrogen cryo-protected with 20% glycol for TRAF-4 or 20% glycerol for CBL-c. X-ray experiments were performed at 100K on the Life Sciences Collaborative Access Team (LS-CAT) Sector-21 beamlines at the advanced photon source (APS), Argonne National Laboratory. Diffraction data were indexed, integrated, and scaled with HKL2000.38 Phasing was accomplished by molecular replacement with Phaser39 using the structure of CBL-c (PDB:3VRN) and TRAF-4 (PDB:4M4E) as starting models. Ligand structures were built by AceDRG and manually placed into their corresponding electron densities. Final protein–ligand complex structures were obtained by several cycles of refinement in Phenix40 and manual model fitting in COOT.41Chemical compounds
All molecules reported in this work are commercially available, with stated purities generally >95%. These molecules were used as obtained.Conclusions
In this study, we applied an NMR-based fragment screening platform to two E3 ligases (CBL-c and TRAF-4) for which no small molecule ligands have been previously identified. In both cases, the screening approach successfully identified highly tractable small molecule hits that represent the first known small molecule binders to each protein. This further reinforces and extends the applicability of fragment-based methods for discovering molecules that bind to challenging protein targets. Furthermore, we characterized the binding modes of these hits to their respective target proteins, enabling future structure-guided optimization toward higher affinity molecules that could be used in a PROTAC approach to recruit CBL-c or TRAF4 to a target protein.The ligases selected for this study were prioritized based on expression profiling using a filtering and selection strategy applied to publicly available datasets. Both CBL-c and TRAF-4 exhibit elevated protein expression levels in many cancer cell lines relative to normal tissues. This differential expression suggests that PROTACs built from optimized ligands to the proteins could achieve more potent and selective degradation of target proteins in cancerous tissues while minimizing effects in normal tissues.
To validate this concept, several follow-up activities would be required. Most prominently, higher affinity ligands are needed for incorporation into PROTACs that can subsequently be used to confirm the ability of these ligases to participate in selective TPD. The discovery of higher affinity ligands to each ligase to enable PROTAC construction would require a medicinal chemistry optimization campaign that can be guided by the X-ray crystal structures of the hit fragments bound to respective proteins. Vectors to grow into additional unused space in the binding pockets are already evident with the structures obtained. Furthermore, since the hits share common shift patterns that indicate a high probability of a shared binding location, application of features of one hit template to another would produce improved compounds whose tighter binding would be revealed by examination with NMR or other biophysical or biochemical assays as appropriate. Given the ligandability demonstrated in our work, we anticipate that ligands based on the identified fragment hits and with an affinity appropriate for use in PROTACs could be obtained for both ligases.
The crystal structure of fragment hit 1 bound to CBL-c reveals solvent-exposed vectors extending from the anthranilic acid ring, which would also be required for developing linker attachment points for a PROTAC. The allosteric binding site occupied by this fragment poses an intriguing question for the use of future optimized versions of these compounds for PROTACs. As most PROTACs used in TPD bind to their respective E3 ligase substrate recognition pockets, the impact of allosteric binding to this domain on inducing, but not inhibiting, protein ubiquitylation mediated by CBL-c remains an unanswered question.
TRAF-4 presents a different set of challenges compared to CBL-c. TRAF-4 has previously been shown to mediate protein degradation in a biological context.3 The structure of compound 9 bound to TRAF-4 also reveals solvent-exposed vectors, particularly from the quinoxaline ring system, that may serve as a viable site for PROTAC linker attachment in optimized analogs. While these compounds bind to the substrate recognition groove, the unique architecture of the TRAF-4 family, specifically the spatial separation between the TRAF-C domain and the RING domain enforced by the TRAF-N domain, raises questions about the influence of this structural arrangement on the ability to achieve efficient degradation using a PROTAC bound to the TRAF-C domain of TRAF-4.
Taken together, our findings highlight a useful application of fragment-based screening and open new avenues for expanding the scope of PROTACs and TPD strategies.
Author contributions
AGW: conceptualization, data curation, project administration, supervision, visualization, writing – original draft, writing – review & editing, BDL: conceptualization, methodology, investigation, software, visualization, writing – original draft, writing – review & editing, JLS: investigation, visualization, writing – review & editing, ETO: methodology, investigation, writing – review & editing, BZ: investigation, writing – review & editing, TR: investigation, writing – review & editing, WGP: investigation, writing – review & editing, JP: investigation, data curation, supervision, writing – review & editing, SWF: conceptualization, funding acquisition, project administration, supervision, writing – original draftConflicts of interest
There are no conflicts to declare.Data availability
Supplementary information (SI): Tables showing the full list of E3 ligases evaluated and the differential expression statistics for those ligases. Figures showing DepMap results and an expanded transcriptomics profile for CBLC and TRAF4, and raw western blots. See DOI: https://doi.org/10.1039/d5cb00198f.
Expression data: the E3 ligase and accessory genes were compiled from several existing resources (Cell Signaling, hUbiquitome database, UbiProt Database, and DUDE v. 1.0, available at (https://esbl.nhlbi.nih.gov/Databases/KSBP2/Targets/Lists/E3-ligases)). Raw count reads for 11057 tumors spanning 20 cancer types from TCGA (GDC-PANCAN.htseq_counts.tsv) were downloaded (https://xenabrowser.net). Details of alignment parameters can be found at https://docs.gdc.cancer.gov/Data/Bioinformatics_Pipelines/Expression_mRNA_Pipeline. Normal RNA-seq raw counts (GTEx_Analysis_2017-06-05_v8) for 17382 tissues from rapid autopsies were obtained from the GTEx portal (https://gtexportal.org). Genetic dependency: gene effect scores derived from CRISPR knockout screens published by Broad's Achilles and Sanger's SCORE projects (DepMap 22Q2 Public) were downloaded from the DepMap portal (https://depmap.org). Crystallography: atomic coordinates and structure factors for CBL-c and TRAF-4 complexed with fragments or peptides can be accessed in the Protein Data Bank via the following accession codes: 9OGW, compound 1, 9OGV, compound 9, 9OLB, EGFR peptide respectively. The authors will release the atomic coordinates upon article publication.
Acknowledgements
We thank the Vanderbilt High-Throughput Screening Core facility for compound management and the Vanderbilt University Biomolecular NMR Facility for use of Bruker NMR spectrometers. We also thank beamline scientists and staff at the Advanced Photon Source (APS) for facilitating our synchrotron access. The use of the Advanced Photon Source, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the U.S. DOE under Contract No. DE-AC02-06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (Grant 085P1000817) The use of Vanderbilt NMR facility was supported in part by NIH SIG Grant 1S-10RR025677-01 and Vanderbilt University matching funds. B. D. L was supported by National Cancer Institute grant CA098131.Notes and references
- J. M. Tsai, R. P. Nowak, B. L. Ebert and E. S. Fischer, Targeted protein degradation: from mechanisms to clinic, Nat. Rev. Mol. Cell Biol., 2024, 25, 740–757, DOI:10.1038/s41580-024-00729-9.
- M. Békés, D. R. Langley and C. M. Crews, PROTAC targeted protein degraders: the past is prologue, Nat. Rev. Drug Discovery, 2022, 21, 181–200, DOI:10.1038/s41573-021-00371-6.
- J. Poirson, H. Cho, A. Dhillon, S. Haider, A. Z. Imrit, M. H. Y. Lam, N. Alerasool, J. Lacoste, L. Mizan, C. Wong, A.-C. Gingras, D. Schramek and M. Taipale, Proteome-scale discovery of protein degradation and stabilization effectors, Nature, 2024, 628, 878–886, DOI:10.1038/s41586-024-07224-3.
- Y. Liu, J. Yang, T. Wang, M. Luo, Y. Chen, C. Chen, Z. Ronai, Y. Zhou, E. Ruppin and L. Han, Expanding PROTACtable genome universe of E3 ligases, Nat. Commun., 2023, 14, 6509, DOI:10.1038/s41467-023-42233-2.
- P. Jevtic, D. L. Haakonsen and M. Rapé, An E3 ligase guide to the galaxy of small-molecule-induced protein degradation, Cell Chem. Biol., 2021, 28, 1000–1013, DOI:10.1016/j.chembiol.2021.04.002.
- A. Rodríguez-Gimeno and C. Galdeano, Drug discovery approaches to target E3 ligases, ChemBioChem, 2025, 26, e202400656, DOI:10.1002/cbic.202400656.
- G. Du, J. Jiang, N. J. Henning, N. Safaee, E. Koide, R. P. Nowak, K. A. Donovan, H. Yoon, I. You, H. Yue, N. A. Eleuteri, Z. He, Z. Li, H. T. Huang, J. Che, B. Nabet, T. Zhang, E. S. Fischer and N. S. Gray, Exploring the target scope of KEAP1 E3 ligase-based PROTACs, Cell Chem. Biol., 2022, 29(10), 1470–1481, DOI:10.1016/j.chembiol.2022.08.003.
- M. Schröder, M. Renatus, X. Liang, F. Meili, T. Zoller, S. Ferrand, F. Gauter, X. Li, F. Sigoillot, S. Gleim, T.-M. Stachyra, J. R. Thomas, D. Begue, M. Khoshouei, P. Lefeuvre, R. Andraos-Rey, B. Chung, R. Ma, B. Pinch, A. Hofmann, M. Schirle, N. Schmiedeberg, P. Imbach, D. Gorses, K. Calkins, B. Bauer-Probst, M. Maschlej, M. Niederst, R. Maher, M. Henault, J. Alford, E. Ahrne, L. Tordella, G. Hollingworth, N. H. Thomä, A. Vulpetti, T. Radimerski, P. Holzer, S. Carbonneau and C. R. Thoma, DCAF1-based PROTACs with activity against clinically validated targets overcoming intrinsic- and acquired-degrader resistance, Nat. Commun., 2024, 15, 275, DOI:10.1038/s41467-023-44237-4.
- S. C. C. Lucas, A. Ahmed, S. N. Ashraf, A. Argyrou, M. R. Bauer, G. M. De Donatis, S. Demanze, F. Eisele, L. Fusani, A. Hock, G. Kadamur, S. Li, A. Macmillan-Jones, I. N. Michaelides, C. Phillips, M. Rehnström, M. Richter, M. C. Rodrigo-Brenni, F. Shilliday, P. Wang and R. I. Storer, Optimization of Potent Ligands for the E3 Ligase DCAF15 and Evaluation of Their Use in Heterobifunctional Degraders, J. Med. Chem., 2024, 67(7), 5538–5566, DOI:10.1021/acs.jmedchem.3c02136.
- N. J. Henning, A. G. Manford, J. N. Spradlin, S. M. Brittain, E. Zhang, J. M. McKenna, J. A. Tallarico, M. Schirle, M. Rape and D. K. Nomura, Discovery of a Covalent FEM1B Recruiter for Targeted Protein Degradation Applications, J. Am. Chem. Soc., 2022, 144(2), 701–708, DOI:10.1021/jacs.1c03980.
- M. Luo, J. N. Spradlin, L. Boike, B. Tong, S. M. Brittain, J. M. McKenna, J. A. Tallarico, M. Schirle, T. J. Maimone and D. K. Nomura, Chemoproteomics-enabled discovery of covalent RNF114-based degraders that mimic natural product function, Cell Chem. Biol., 2021, 28, 559–566, DOI:10.1016/j.chembiol.2021.01.005.
- M. Cieślak and M. Slowianek, Cereblon-Recruiting PROTACs: Will New Drugs Have to Face Old Challenges, Pharmaceutics, 2023, 15(3), 812, DOI:10.3390/pharmaceutics15030812.
- L. Zhang, B. Riley-Gillis, P. Vijay and Y. Shen, Acquired Resistance to BET-PROTACs (Proteolysis-Targeting Chimeras) Caused by Genomic Alterations in Core Components of E3 Ligase Complexes, Mol. Cancer Ther., 2019, 18(7), 1302–1311, DOI:10.1158/1535-7163.MCT-18-1129.
- R. S. Z. Saleem, M. P. Schwalm and S. Knapp, Expanding the ligand spaces for E3 ligases for the design of protein degraders, Biog. Med. Chem., 2024, 105, 117718, DOI:10.1016/j.bmc.2024.117718.
- A. Rodríguez-Gimeno and C. Galdeano, Drug discovery approaches to target E3 ligases, ChemBioChem, 2025, 26(1), e202400656, DOI:10.1002/cbic.202400656.
- S. Khan, X. Zhang, D. Lv, Q. Zhang, Y. He, P. Zhang, X. Liu, D. Thummuri, Y. Yuan, J. S. Wiegand, J. Pei, W. Zhang, A. Sharma, C. R. McCurdy, V. M. Kuruvilla, N. Baran, A. A. Ferrando, Y.-M. Kim, A. Rogojina, P. J. Houghton, G. Huang, R. Hromas, M. Konopleva, G. Zheng and D. Zhou, A selective BCL-XL PROTAC degrader achieves safe and potent antitumor activity, Nat. Med., 2019, 25, 1938–1947, DOI:10.1038/s41591-019-0668-z.
- A. Kaefer, J. Yang, P. Noertersheuser, S. Mensing, R. Humerickhouse, W. Awni and H. Xiong, Mechanism-based pharmacokinetic/pharmacodynamic meta-analysis of navitoclax (ABT-263) induced thrombocytopenia, Cancer Chemother. Pharmacol., 2014, 74, 593–602, DOI:10.1007/s00280-014-2530-9.
- B. Medvar, V. Raghuram, T. Pisitkun, A. Sarkar and M. A. Knepper, Comprehensive database of human E3 ubiquitin ligases: application to aquaporin-2 regulation, Physiol. Genomics, 2016, 48, 502–512, DOI:10.1152/physiolgenomics.00031.2016.
- S. Gooding, N. Ansari-Pour, F. Towfic, M. O. Estévez, P. P. Chamberlain, K.-T. Tsai, E. Flynt, M. Hirst, D. Rozelle, P. Dhiman, P. Neri, K. Ramasamy, N. Bahlis, P. Vyas and A. Thakurta, Multiple cereblon genetic changes are associated with acquired resistance to lenalidomide or pomalidomide in multiple myeloma, Blood, 2021, 137(2), 232–237, DOI:10.1182/blood.2020007081.
- C. Pacini, J. M. Dempster, I. Boyle, E. Goncalves, H. Najgebauer, E. Karakoc, D. van der Meer, A. Barthorpe, H. Lightfoot, P. Jaaks, J. M. McFarland, M. J. Garnett, A. Tsherniak and F. Iorio, Integrated cross-study datasets of genetic dependencies in cancer, Nat. Commun., 2021, 12, 1661, DOI:10.1038/s41467-021-21898-7.
- E3 atlas results for CBLC and TRAF4 can be found at the following links: https://hanlaboratory.com/E3Atlas/E3_detail?E3=CBLC and https://hanlaboratory.com/E3Atlas/E3_detail? E3=TRAF4.
- B. Mohapatra, G. Ahmad, S. Nadeau, N. Zutshi, W. An, S. Scheffe, L. Dong, D. Feng, B. Goetz, P. Arya, T. A. Bailey, N. Palermo, G. E. O. Borgstahl, A. M. Natarajan, S. M. Raja, M. Naramura, V. Band and H. Band, Protein tyrosine kinase regulation by ubiquitination: Critical roles of Cbl-family ubiquitin ligases, Biochim. Biophys. Acta, 2013, 1833(1), 122–139, DOI:10.1016/j.bbamcr.2012.10.010.
- P. E. Ryan, S. C. Kales, R. Yadavalli, M. M. Nau, H. Zhang and S. Lipowitz, Cbl-c Ubiquitin Ligase Activity Is Increased via the Interaction of Its RING Finger Domain with a LIM Domain of the Paxillin Homolog, Hic 5, PLoS One, 2012, 7(11), e49428, DOI:10.1371/journal.pone.0049428.
- H. H. Park, Structure of TRAF Family: Current Understanding of Receptor Recognition, Front. Immunol., 2018, 9, 1999, DOI:10.3389/fimmu.2018.01999.
- J. Li, P. Wang, Z. Xie, S. Wang, S. Cen, M. Li, W. Liu, S. Tang, G. Ye, G. Zheng, H. Su, M. Ma, X. Wu, Y. Wu and H. Shen, TRAF-4 positively regulates the osteogenic differentiation of mesenchymal stem cells by acting as an E3 ubiquitin ligase to degrade Smurf2, Cell Death Differ., 2019, 26, 2652–2666, DOI:10.1038/s41418-019-0328-3.
- X. Yu, W. Li, H. Liu, Q. Deng, X. Wang, H. Hu, Z. Y. Xu-Monette, W. Xiong, Z. Lu, K. H. Young, W. Wang and Y. Li, Ubiquitination of the DNA-damage checkpoint kinase CHK1 by TRAF-4 is required for CHK1 activation, J. Hematol. Oncol., 2020, 13, 40, DOI:10.1186/s13045-020-00869-3.
- W. Yu, R. Singh, Z. Wang, B. W. O’Malley and P. Yi, The E3 ligase TRAF-4 promotes IGF signaling by mediating atypical ubiquitination of IRS-1, J. Biol. Chem., 2021, 296, 100739, DOI:10.1016/j.jbc.2021.100739.
- M. M. Keane, S. A. Ettenberg, M. M. Nau, P. Banerjee, M. Cuello, J. Penninger and S. Lipkowitz, cbl-3: a new mammalian cbl family protein, Oncogene, 1999, 18, 3365–3375, DOI:10.1038/sj.onc.1202753.
- A. S. Judd, B. Bawa, W. R. Buck, Z.-F. Tao, Y. Li, M. J. Mitten, M. Bruncko, N. Catron, G. Doherty, K. R. Durbin, B. Enright, R. Frey, D. Haasch, S. Haman, A. R. Haight, T. A. Henriques, J. Holms, K. Izeradjene, R. A. Judge, G. J. Jenkins, A. Kunzer, J. D. Leverson, R. L. Martin, D. Mitra, S. Mittelstadt, L. Nelson, P. Nimmer, J. Palma, R. Peterson, D. C. Phillips, S. L. Ralston, S. H. Rosenberg, X. Shen, X. Song, K. S. Vaidya, X. Wang, J. Wang, Y. Xiao, H. Zhang, X. Zhang, E. A. Blomme, E. R. Boghaert, J. C. Kalvass, A. Phillips and A. J. Souers, BCL-XL targeting antibody-drug conjugates are active in preclinical models and mitigate on-mechanism toxicity of small-molecule inhibitors, Sci. Adv., 2024, 10, eado7120, DOI:10.1126/sciadv.ado7120.
- S. A. Shuker, P. J. Hajduk, R. P. Meadows and S. W. Fesik, Discovering High-Affinity Ligands for Proteins: SAR by NMR, Science, 1996, 274, 1531–1534, DOI:10.1126/science.274.5292.153.
- M. J. Harner, A. O. Frank and S. W. Fesik, Fragment-based drug discovery using NMR spectroscopy, J. Biomol. NMR, 2013, 56(2), 65–75, DOI:10.1007/s10858-013-9740-z.
- K. Takeshita, T. Tezuka, Y. Isozaki, E. Yamashita, M. Suzuki, M. Kim, Y. Yamanashi, T. Yamamoto and A. Nakagawa, Structural flexibility regulates phosphopeptide-binding activity of the tyrosine kinase binding domain of Cbl-c, J. Biochem., 2012, 152(5), 487–495, DOI:10.1093/jb/mvs085.
- A. Rousseau, M.-C. Rio and F. Alpy, TRAF-4, at the Crossroad between Morphogenesis and Cancer, Cancers, 2011, 3, 2734–2749, DOI:10.3390/cancers3022734.
- C. M. Kim, Y.-J. Son, S. Kim, S. Y. Kim and H. H. Park, Molecular basis for unique specificity of human TRAF-4 for platelets GPIbβ and GPVI, Proc. Natl. Acad. Sci. U. S. A., 2017, 114(43), 11422–11427, DOI:10.1073/pnas.1708688114.
- G. Cai, L. Zhu, X. Chen, K. Sun, C. Lui, G. C. Sen, G. C. Stark, J. Qin and X. Li, TRAF4 binds to the juxtamembrane region of EGFR directly and promotes kinase activation, Proc. Natl. Acad. Sci. U. S. A., 2018, 115(45), 11531–11536, DOI:10.1073/pnas.1809599115.
- S. Liang, Q. Liang, R. Chen and K. Chen, Stratified Test Alleviates Batch Effects in Single-Cell Data, Algorithms for Computational Biology, 2020, vol. 1209, ISBN: 978-3-030-42265-3 Search PubMed.
- P. Schanda, Ē. Kupče and B. Brutscher, SOFAST-HMQC experiments for recording two-dimensional deteronuclear correlation spectra of proteins within a few seconds, J. Biomol. NMR, 2005, 33, 199–211, DOI:10.1007/s10858-005-4425-x.
- Z. Otwinowski and W. Minor, Processing of X-Ray Diffraction Data Collected in Oscillation Mode, Methods Enzymol., 1997, 276, 307–326, DOI:10.1016/S0076-6879(97)76066-X.
- A. J. McCoy, R. W. Grosse-Kunstleve, P. D. Adams, M. D. Winn, L. C. Storoni and R. J. Read, Phaser Crystallographic Software, J. Appl. Crystallogr., 2007, 40(4), 658–674, DOI:10.1107/S0021889807021206.
- P. V. Afonine, R. W. Grosse-Kunstleve, N. Echols, J. J. Headd, N. W. Moriarty, M. Mustyakimov, T. C. Terwilliger, A. Urzhumtsev, P. H. Zwart and P. D. Adams, Towards Automated Crystallographic Structure Refinement with phenix.refine, Acta Crystallogr. D: Biol. Crystallogr., 2012, 68(4), 352–367, DOI:10.1107/S0907444912001308.
- P. Emsley, B. Lohkamp, W. G. Scott and K. Cowtan, Features and Development of Coot, Acta Crystallogr., D:Biol. Crystallogr., 2010, 66(4), 486–501, DOI:10.1107/S0907444910007493.
| This journal is © The Royal Society of Chemistry 2025 |