
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploring the immunoproteasome's substrate preferences for improved hydrolysis and selectivity†
Christine S.
Muli
a,
Cody A.
Loy
 b and
Darci J.
Trader
*abc
b and
Darci J.
Trader
*abc
aDepartment of Medicinal Chemistry and Molecular Pharmacology, Purdue University, 575 West Stadium Avenue, West Lafayette, Indiana 47907, USA
bDepartment of Pharmaceutical Sciences, University of California – Irvine, 856 Health Sciences, Irvine, California 92697, USA
cDepartment of Chemistry, University of California – Irvine, 1102 Natural Sciences 2, Irvine, California 92697, USA. E-mail: dtrader@uci.edu
First published on 7th July 2025
Abstract
The proteasome is an integral macromolecular machine responsible for regulated protein degradation, and its barrel-like core particle (CP) hydrolyzes protein substrates into peptide fragments. A proteasome isoform that is expressed under conditions of inflammation is known as the immunoproteasome (iCP), which incorporates different catalytic subunits of altered cleavage specificities from the standard proteasome (sCP). Probes and inhibitors have been generated to study iCP activity and for therapeutics, respectively; recently, the iCP has been harnessed as a prodrug enzyme to release bioactive compounds selectively into iCP-expressing cells. iCP-targeting probes, prodrugs, and inhibitors are based on peptide recognition sequences and their favorable interactions within the iCP's substrate channel. To better understand what unnatural substrates the iCP can recognize, we synthesized peptide-conjugated substrates and applied them to a liquid chromatography-mass spectrometry (LC-MS) method after incubation with purified human iCP. Structure–activity relationships of unnatural peptide-conjugated substrates revealed modifications that improved substrate selectively for the iCP by more than 3-fold compared to the original scaffold. As such, this report will be helpful to guide future iCP-targeting probes, prodrugs, and inhibitor design.
Introduction
The immunoproteasome (iCP) is a disease-specific isoform of the proteasome that is expressed upon exposure to interferon-γ, and is constitutively expressed in cells of hematopoietic origin.1,2 The barrel-like structure of the core particle (CP) is generated upon the incorporation of α and β subunits in a tightly conserved manner.3 Within the standard core particle (sCP) β subunits, three are catalytically active (β1—caspase-like, β2—trypsin-like, and β5—chymotrypsin-like) that are responsible for the cleavage of protein substrates into peptide products. When the cell is exposed to inflammatory stimuli, the CP is assembled with altered catalytic subunits (β1i—chymotrypsin-like, β2i—trypsin-like, and β5i—chymotrypsin-like), forming the newly synthesized iCP that can accept the same protein substrate as the sCP; the peptide products, however, are different.1,4 This difference in cleavage specificity allows the iCP to generate peptides that are compatible with presentation by MHC-I complexes. As such, the iCP has been found to be expressed in varying amounts across several diseases such as cancer, autoimmune diseases, and inflammation.5Several peptide-like inhibitors have been designed to fit into the β5i unprimed specificity pockets. ONX-0914 and KZR-616 are the two most utilized iCP inhibitors and have become excellent tool compounds for understanding iCP dynamics, although their clinical efficacy is not as clear.6–8 Both inhibitors contain an electrophilic epoxyketone that covalently binds to the β5i N-terminal catalytic threonine (Thr1), and alterations to their peptide-like backbone allowed for increased selectivity for the iCP over the sCP, Fig. 1A. Beyond inhibition, several probes have been generated by us and others that allow for selective monitoring of the iCP over the sCP by taking advantage of these same principles.9–12 The Ala-Thr-Met-Trp (ATMW) recognition sequence for β5i that was discovered by the Trader lab has been utilized in the generation of fluorescent and luminescent probes to monitor the activity of the iCP biochemically and in cells, Fig. 1B and C.13,14 In addition, ATMW has applied to the development of prodrugs that selectively engage with the iCP for the release of Doxorubicin15 or a protein degrader cargo specifically in diseased cells, Fig. 1D.16 Due to the varied release of the Dox versus protein degrader prodrug, we hypothesized that there are favorable interactions within the primed substrate channel, which is on the other side of the Thr1, that could be taken advantage of when further developing iCP therapeutics. A previous study explored the interactions of the primed substrate channel within the sCP, however, little is known about what additional interactions could be harnessed within the iCP.17
 | ||
| Fig. 1 (A) Structures of iCP inhibitors ONX-0914 and KZR-616 that were developed based on specificity pocket (S1, S2, and S3) interactions of the β5i subunit to improve selectivity over the sCP. (B) Peptide ATMW recognition sequence (blue) appended to a cargo (pink) will interact with the iCP's β5i subunit, leading to cleavage of the cargo specifically in iCP-expressing cells. PDB: 5GJR. (C) Structures of cargo that have been appended to ATMW for activity-based probes. Top: fluorescent activity-based probe TBZ-1 (ATMW-Rh110-peptoid); bottom: luminescent activity-based probe morpholine-tagged ATMW-luciferin (m-ATMW-aLuc). (D) Structures of cargo for iCP-targeting prodrugs. Top left: m-ATMW-Doxorubicin; bottom right: m-ATMW-PABC-(ARV-771). | ||
Here, we describe the evaluation of ATMW-conjugated unnatural substrates and structure–activity relationship studies of the ATMW recognition peptide with a simplified model cargo dialanine (Ala-Ala). After synthesis of unnatural substrates, they were incubated with purified iCP and evaluated by liquid chromatography-mass spectrometry (LC-MS) to determine the substrates’ relative cleavage and isoform selectivity. With ATMW-conjugated small molecules, we observed small R groups had a lower release from the ATMW peptide than bulkier groups. In addition, groups that extended deeper into the primed channel appear to have more favorable release, potentially due to more interactions past the Thr1. While investigating the ATMW sequence with model cargo dialanine, we identified several modifications within ATMW that had improved cleavage and specificity for the iCP. These findings may be beneficial in the generation of more iCP-selective prodrugs, probes, and/or inhibitors in the future.
Results and discussion
First, we desired to understand how the iCP accepts unnatural substrates in comparison to the rhodamine–peptoid probe we had previously made, TBZ-1.13 Based on our previous studies developing probes and prodrugs for the iCP, we suspect there are favorable interactions with both sides of the scissile amide bond, Fig. 2A. Although substantial proteasomal research has been conducted for inhibitor optimization with the unprimed substrate channel, little is known on the interactions necessary in the primed substrate channel for effective hydrolysis.18 Recently, we reported that the sCP can accept different moieties in its primed substrate channel utilizing structural-activity relationships (SAR) with Leu-Leu-Val-Tyr or LLVY-conjugated substrates.17 Interestingly, polar groups within the primed substrate channel enhanced the release of the pro-moiety sequence, LLVY. To validate if the primed substrate channel of the iCP exhibited a similar trend, ATMW-conjugated substrates (including TBZ-1) were synthesized for SAR, Fig. 2B.13 These substrates were incubated with purified 5 nM iCP at 37 °C for 4 hours and analyzed by mass spectrometry, Fig. 2C. Small R groups had significantly lower ATMW released from the ATMW-R substrates, whereas substrates 6 and 7 had increased amounts proteolyzed by the iCP. These trends are similar to that of the sCP with LLVY-conjugated substrates,17 where the hydroxyl group drives favorable cleavage by the CP. When purified iCP is activated with the PA28 regulator, ATMW-R substrates are hydrolyzed similarly as latent iCP, Fig. S1 (ESI†).19,20 The addition of PA28 in vitro greatly stimulates the iCP's peptidase activity without sacrificing specificity.16 In both conditions of latent or activated iCP, no ATMW-R substrate achieved greater release of ATMW than TBZ-1, indicating that the elongated structure of TBZ-1 aids with substrate engagement similar to TAS-1 with sCP.17 With the iCP's barrel-like structure, we anticipate the release of various cargo will be dependent not only on the pro-moiety in the unprimed channel but also on the its favorable interactions within the primed channel.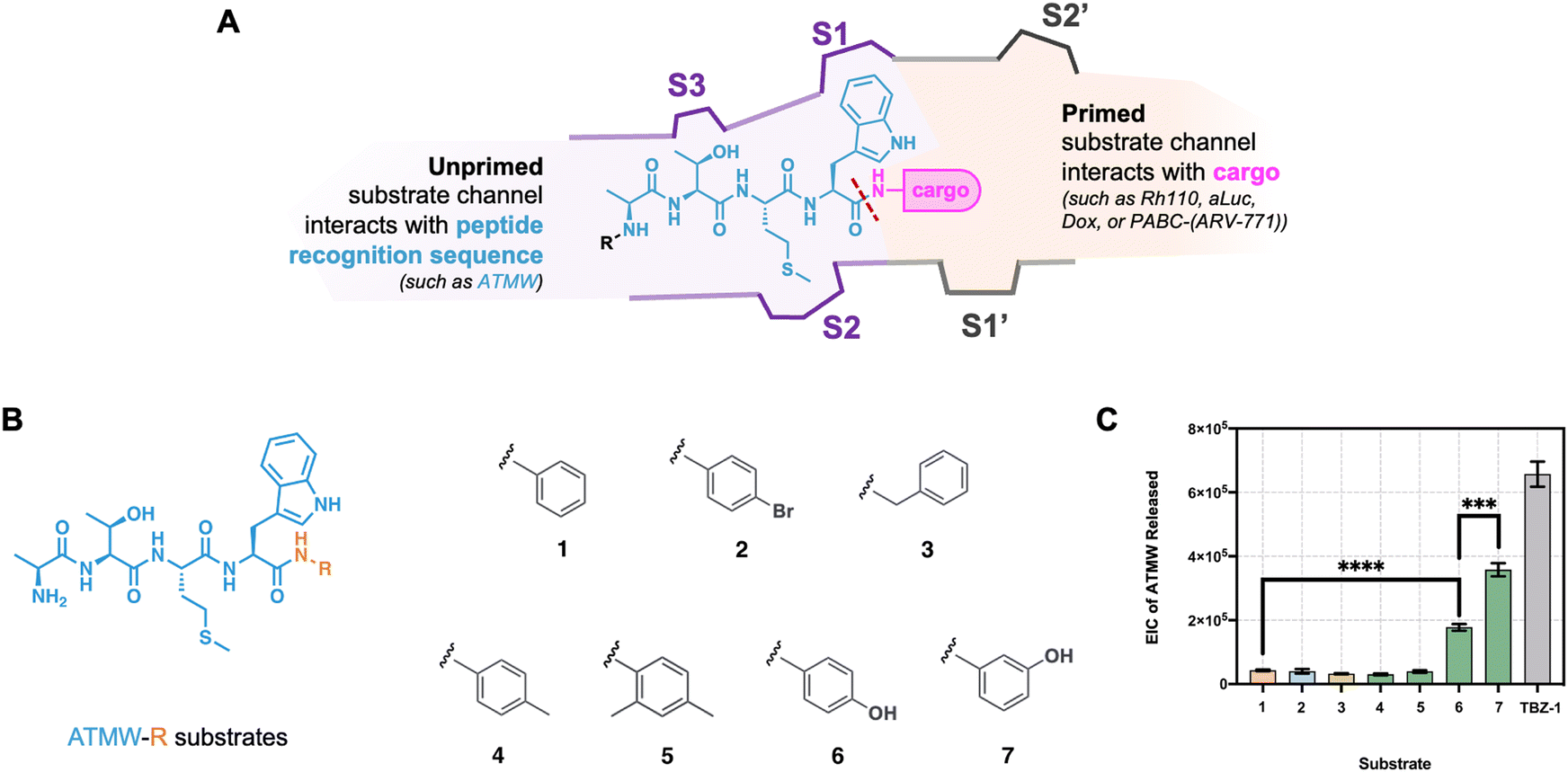 | ||
| Fig. 2 Structure–activity relationship with ATMW-R substrates to understand unnatural R groups that purified iCP can accept for hydrolysis. (A) Cartoon schematic of the iCP β5i substrate channel. While ATMW “interacts” with the unprimed specificity pockets (S1, S2, S3, and so on), cargo in ATMW-conjugated substrates will interact with the primed specificity pockets (S1’, S2’, and so on). A red dash line signifies the scissile amide bond cleaved by the iCP. (B) Structure of ATMW-R substrates that contain the iCP recognition sequence ATMW (blue) conjugated to an R group (orange). R groups for ATMW-R substrates included different substitutions on the aniline ring (substrate 1), causing an electron-withdrawing or electronic donating effect on the scissile amide bond. (C) ATMW-R substrates (including TBZ-1) were incubated with 5 nM purified iCP for 4 hours at 37 °C, and samples were subjected to mass spectrometry. EIC of ATMW Released is reported as area under the curve. Samples were performed in triplicate, represented as the mean ± standard deviation. Statistical significance by t-test is denoted by ****p ≤ 0.0001 and ***p ≤ 0.001. | ||
We previously identified the iCP as an effective target for prodrug release by demonstrating this isoform can cleave the cargo, such as Doxorubicin (Dox) and ARV-771, from the ATMW peptide. This release led to the parent drug retaining activity in higher iCP-expressing cells, while healthy cells with low iCP had no or reduced effect.16 We observed the ARV-711 cargo had improved efficacy than the Dox cargo, further supporting the hypothesis that the interactions within the primed substrate channel played a role in the ability of the iCP to release the cargos. Similar to TBZ-1 in Fig. 1B, ARV-771 has an elongated structure that could additionally aid with substrate engagement, Fig. 1D. Dox's structure potentially hinders the Thr1, leading to less of the parent drug being released in diseased cells. To understand if we could improve iCP cleavage, we were interested in performing SAR on the ATMW peptide which interacts with the unprimed substrate channel, Fig. 3A.16,18
 | ||
| Fig. 3 Structure–activity relationship with dialanine substrates to understand unnatural amino acids groups purified iCP can accept for hydrolysis. (A) In these substrates, the iCP is expected to cleave the amide bond between the recognition sequence (blue) and the cargo (orange). The cargo (dialanine) remains constant in all substrates. Then, substrates for SAR contain single-point modifications within the ATMW recognition sequence (blue) with Trp (P1), Met (P2), Thr (P3), and Ala (P4). These single point modifications within the pro-moiety sequence for dialanine substrates can later be translated to a iCP-targeting prodrug scaffold. (B) 25 μM of substrate was incubated in iCP-PA28-activated complex for 4 hours and subjected to mass spectrometry for analysis. The control substrate (ATMWAA, blue) is degraded 40% during this study. Therefore, all substrates are statistically compared to the control substrate. Statistical significance by t-test is denoted by ***p ≤ 0.001, **p ≤ 0.01, and *p ≤ 0.05. (C) To understand if the increased hydrolysis was only iCP specific, 25 μM of substrate was incubated with 9 nM sCP for 4 hours and analyzed by LC-MS. The control substrate ATMWAA achieved a 3.3X selectivity index for the activated iCP over sCP, which correlates with previously reported TBZ-1's isoform selectivity. Selectivity index was calculated by dividing the mean iCP percent degraded (purple) over sCP percent degraded (blue) of a substrate. Graphs in (B) and (C) were performed in triplicate, represented as the mean ± standard deviation. | ||
With peptide sequence-dependent release of Dox by the iCP, we then embarked on an SAR study to optimize the ATMW sequence. Modifications to the ATMW sequence could not only improve cellular stability of iCP-targeting prodrugs but also increase iCP hydrolysis of challenging or unfavorable cargo, such as Dox. For this SAR study, unnatural amino acids were chosen to build upon our original OBOC library screen in TBZ-1's design,13 which was based upon the crystal structure of the sCP/iCP.21 To expedite synthesis and purification, dialanine cargo was utilized on the C-terminus instead of Dox. Dialanine cargo has been previously used with the recognition sequence LLVY for the sCP in the optimization of sCP activity-based probe, TAS-1 into TAS-2/TAS-3.22 After four hours of incubation with iCP-PA28-activated complex, substrates were analyzed by mass spectrometry, Fig. 3B. The control ATMWAA was hydrolyzed approximately 40% during this time frame, and all samples that contained one single modification from the ATMW recognition sequence were statistically compared to the ATMWAA control. All P1 modified ATMWAA analogs, except for Trp(N-Me), were more degraded than the control ATMWAA. Interestingly, the Ala(3-benzothienyl) (Abt) substrate was hydrolyzed the most, which only substitutes the Trp's nitrogen atom with a sulfur. It appears that having the hydrogen bonding from the indole or the benzothiophene are important for hydrolysis. For P2 substitutions, no substantial increase with the substrate analogs was observed. The hydrogen bonding amino acids in P3 all performed better than the original Thr, suggesting that a longer group in the S3 pocket would be favorable.
Higher hydrolysis by the iCP may also increase non-specific hydrolysis by the sCP. To confirm how iCP isoform selectivity is maintained or changed, dialanine substrates were incubated with purified 9 nM of sCP for 4 hours at 37 °C and their hydrolysis products were analyzed by mass spectrometry, Fig. 3C. In this study, sCP was not activated with PA28 like the iCP since the latent sCP appears to readily accept smaller peptides compared to latent iCP. Latent sCP also mimics previous TBZ-1 isoform selectivity studies, where there is an approximate 3-fold selectivity index between the iCP and sCP.13 While this mimics previous experiments, this limitation could introduce bias in interpretation of the iCP/sCP selectivity index in the dialanine series; and therefore, nonpeptidic cargo should be further validated. In addition, this more accurately resembles the complexes within the cell as PA28 is also an IFN-γ inducible subunit known to associate with iCP.19,23
With ATMWAA under these conditions, we measured a 3.3-fold selectivity index for the iCP over the sCP. Most substrates either maintained or increased selectivity for the iCP; in several cases, the substrate is barely degraded by the sCP (like with AT(Met![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O2)WAA and A(Orn)MWAA), so the selectivity index appears larger. ATM(2-Nal)AA was the most favored substrate by the sCP, so even though the ATM(2-Nal)AA substrate is hydrolyzed well by the iCP, this substrate is not isoform selective. Although no substrates were found with better hydrolysis in P2 group substitutions, the MetO2 group could increase the substrate selectivity for the iCP more than 2-fold. Further, we investigated if any cleavage outside of the P1–P1′ site was performed by the iCP and sCP, and none was observed (with an examples shown in Fig. S2 and S3, ESI†).
O2)WAA and A(Orn)MWAA), so the selectivity index appears larger. ATM(2-Nal)AA was the most favored substrate by the sCP, so even though the ATM(2-Nal)AA substrate is hydrolyzed well by the iCP, this substrate is not isoform selective. Although no substrates were found with better hydrolysis in P2 group substitutions, the MetO2 group could increase the substrate selectivity for the iCP more than 2-fold. Further, we investigated if any cleavage outside of the P1–P1′ site was performed by the iCP and sCP, and none was observed (with an examples shown in Fig. S2 and S3, ESI†).
These results all relied on the pro-moiety sequence incorporating only one of the changes to its scaffold. Since modifying one amino acid appeared to give different selectivity and cleavage results, we then synthesized a recognition sequence that changes each position, Fig. 4A. morph-Ala-Orn-(MetO2)-Trp(7,Me) or mAO(MetO2)W(7,Me) incorporated changes at the P1, P2, and P3 positions that should have more favorable interactions than the original ATMW sequence to increase cleavage and improve selectivity for the iCP. Once the new recognition sequence with the dialanine cargo was prepared, its cleavage was assessed with iCP and sCP to see if there was improved properties compared to the parent ATMW scaffold. By mass spectrometry, the new recognition sequence with unnatural amino acids had slightly improved hydrolysis of dialanine and increased selectivity about 3-fold, Fig. 4B. Although the improved hydrolysis was minimal, the increase in selectivity of the iCP recognition sequence can have a positive impact when translating the cargo to different therapeutic agents.
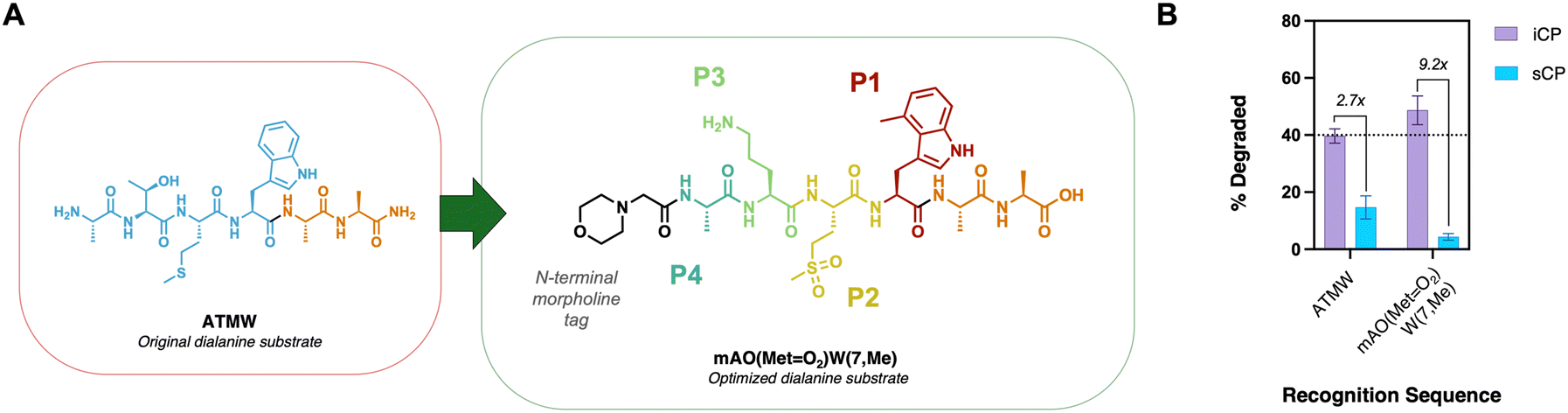 | ||
| Fig. 4 (A) ATMW recognition sequence (blue) is altered to incorporate positive hits from the SAR screen. The cargo (dialanine, orange) remained constant in both substrates. (B) 25 μM of substrate was incubated in iCP or sCP for 4 hours and subjected to mass spectrometry for analysis. The control substrate ATMWAA achieved a 2.7X selectivity index for the iCP over sCP, while the optimized scaffold increased selectivity by >3-fold. Selectivity index was calculated by dividing the mean iCP percent degraded (purple) over sCP percent degraded (blue) of a substrate. Graph (B) was performed in triplicate, represented as the mean ± standard deviation. | ||
To explore the affect these changes have on translating to nonpeptidic cargo, we swapped the dialaline for the fluorescent reporter 7-amino, 4-methyl coumarin (AMC). AMC-based fluorescent probes have been vital in characterizing proteasome activity biochemically and in cell lysates.24,25 Application of our iCP-based peptide sequences to AMC allows monitoring of iCP cleavage as a function of fluorescence as opposed to mass spectrometry. To do this, we synthesized an N-terminal morpholine tagged original scaffold ATMW coupled to AMC (mATMW-AMC), as well as newly discovered sequence (mAO(MetO2)W(7,Me)-AMC). The two AMC probes were tested biochemically with purified iCP with or without iCP selective inhibitor ONX-0914, as a kinetic read scanning every minute for one hour. Upon completion of the time points, the slopes were compared. Excitingly, our optimized peptide sequence hydrolyzed the AMC cargo more efficiently than the original ATMW, Fig. 5. In addition, upon co-treatment with iCP selective inhibitor ONX-0914, both probes had signal drastically reduced, further confirming the selectivity of our probes. The optimized peptide had its signal reduced slightly more than the original ATMW peptide, combined with its improved hydrolysis provides further validation that this new sequence is more selective for the iCP.
 | ||
| Fig. 5 Original ATMW scaffold or newly optimized AO(MetO2)W(7,Me) sequence were conjugated to nonpeptidic cargo, 7-amino, 4-methyl coumarin (AMC). (Left) Structure of morph-peptide-AMC either with ATMW or AO(MetO2)W(7,Me). (Right) 10 μM of probe with or without 10 μM ONX-0914 was incubated in 9 nM iCP and kinetically read by fluorescent plate reader over 1 hour. Slopes of lines were determined by linear regression analysis and plotted in GraphPad prism and bar graphs. Experiment was performed in triplicate, represented as the mean ± standard deviation. | ||
Conclusion
Taking the structure of activity-based probe TBZ-1, we first evaluated the capacity of purified iCP to hydrolyze ATMW-conjugated unnatural substrates. ATMW-R substrates were degraded by the iCP in a similar trend as LLVY-R substrates with the sCP, displaying that the iCP can hydrolyze cargo from ATMW at varying efficiencies. Since hydroxyl substitution in the P1’ position was favorable in both sCP17 LLVY-R and iCP ATMW-R substrates, we expect that polar cargo will contribute to increased cleavage by both proteasome isoforms but not affect isoform selectivity. Instead, specificity between the iCP and sCP can be improved in the unprimed peptide recognition sequence, so we evaluated the iCP ATMW pro-moiety sequence to optimize for future iCP prodrugs. We performed an SAR study with unnatural amino acids to determine modifications suitable for increased substrate hydrolysis and selectivity. With the use of dialanine cargo for simplified and expedited synthesis, the exchange of the ATMW sequence with unnatural amino acids and the P1–P3 individual substitutions were found to be superior to the original sequence. Additionally, P1–P3 individual substitutions were observed to have maintained or improved iCP isoform selectivity over the sCP. While multiple substitutions within the recognition sequence only minimally increased iCP hydrolysis, there was a significant increase in isoform selectivity. To validate the translatability of our findings, we then prepared fluorescent containing probes using the original scaffold and the newly identified optimized sequence. Upon testing the probes with iCP, we were able to confirm that the new sequence outperformed the original, both in hydrolysis of cargo and in selectivity, providing an improved recognition sequence for the iCP that can be used in further studies.Conflicts of interest
D. J. T. is a shareholder and consultant for Booster Therapeutics, GmbH. Other authors declare no conflict of interest.Acknowledgements
This work was supported through a start-up package from Purdue University School of Pharmacy, the Purdue Center of Cancer Research, a grant from NIH NIAID (1R01AI50847). C. S. M. was supported through an NIH National Cancer Institute Ruth L. Kirschstein Predoctoral Individual National Research Service Award to Promote Diversity in Health-Related Research F31 Fellowship (5F31CA247327).References
- M. Aki, N. Shimbara, M. Takashina, K. Akiyama, S. Kagawa, T. Tamura, N. Tanahashi, T. Yoshimura, K. Tanaka and A. Ichihara, Interferon-γ Induces Different Subunit Organizations and Functional Diversity of Proteasomes1, J. Biochem., 1994, 115(2), 257–269, DOI:10.1093/oxfordjournals.jbchem.a124327.
- B. Boes, H. Hengel, T. Ruppert, G. Multhaup, U. H. Koszinowski and P. M. Kloetzel, Interferon Gamma Stimulation Modulates the Proteolytic Activity and Cleavage Site Preference of 20S Mouse Proteasomes, J. Exp. Med., 1994, 179(3), 901–909, DOI:10.1084/jem.179.3.901.
- M. Groll, L. Ditzel, J. Löwe, D. Stock, M. Bochtler, H. D. Bartunik and R. Huber, Structure of 20S Proteasome from Yeast at 2.4Å Resolution, Nature, 1997, 386(6624), 463–471, DOI:10.1038/386463a0.
- Y. Yang, J. B. Waters, K. Früh and P. A. Peterson, Proteasomes Are Regulated by Interferon Gamma: Implications for Antigen Processing, Proc. Natl. Acad. Sci. U. S. A., 1992, 89(11), 4928–4932, DOI:10.1073/pnas.89.11.4928.
- B. L. Zerfas, M. E. Maresh and D. J. Trader, The Immunoproteasome: An Emerging Target in Cancer and Autoimmune and Neurological Disorders, J. Med. Chem., 2020, 63(5), 1841–1858, DOI:10.1021/acs.jmedchem.9b01226.
- T. W. Jenkins, S. L. Downey-Kopyscinski, J. L. Fields, G. J. Rahme, W. C. Colley, M. A. Israel, A. V. Maksimenko, S. N. Fiering and A. F. Kisselev, Activity of Immunoproteasome Inhibitor ONX-0914 in Acute Lymphoblastic Leukemia Expressing MLL–AF4 Fusion Protein, Sci. Rep., 2021, 11(1), 10883, DOI:10.1038/s41598-021-90451-9.
- R. Furie, S. Parikh, J. Wang, D. Bomba, R. Leff, C. Kirk and N. Henig, Pos0695 Kzr-616, A Selective Immunoproteasome Inhibitor For The Treatment Of Systemic Lupus Erythematosus: Results From The Completed Dose Escalation Phase 1b Portion Of The Mission Study, Ann. Rheum. Dis., 2021, 80(1), 595–596, DOI:10.1136/annrheumdis-2021-eular.2158.
- J. Lickliter, D. Bomba, J. Anderl, A. Fan, C. J. Kirk and J. Wang, AB0509 Kzr-616, a Selective Inhibitor of the Immunoproteasome, Shows a Promising Safety and Target Inhibition Profile in a Phase i, Double-Blind, Single (SAD) and Multiple Ascending Dose (MAD) Study in Healthy Volunteers. In SLE, Sjögren's and APS – treatment; BMJ Publishing Group Ltd and European League Against, Rheumatism, 2018, 1413–1414, DOI:10.1136/annrheumdis-2018-eular.3344.
- B. L. Zerfas and D. J. Trader, Monitoring the Immunoproteasome in Live Cells Using an Activity-Based Peptide–Peptoid Hybrid Probe, J. Am. Chem. Soc., 2019, 141(13), 5252–5260, DOI:10.1021/jacs.8b12873.
- C. A. Loy and D. J. Trader, Caged Aminoluciferin Probe for Bioluminescent Immunoproteasome Activity Analysis, RSC Chem. Biol., 2024, 5(9), 877–883, 10.1039/D4CB00148F.
- G. de Bruin, B. T. Xin, M. Kraus, M. Van Der Stelt, G. A. Van Der Marel, A. F. Kisselev, C. Driessen, B. I. Florea and H. S. Overkleeft, A Set of Activity-Based Probes to Visualize Human (Immuno)Proteasome Activities, Angew. Chem., Int. Ed., 2016, 55(13), 4199–4203, DOI:10.1002/anie.201509092.
- M. B. Winter, F. La Greca, S. Arastu-Kapur, F. Caiazza, P. Cimermancic, T. J. Buchholz, J. L. Anderl, M. Ravalin, M. F. Bohn, A. Sali, A. J. O’Donoghue and C. S. Craik, Immunoproteasome Functions Explained by Divergence in Cleavage Specificity and Regulation, eLife, 2017, 6, e27364, DOI:10.7554/eLife.27364.
- B. L. Zerfas and D. J. Trader, Monitoring the Immunoproteasome in Live Cells Using an Activity-Based Peptide–Peptoid Hybrid Probe, J. Am. Chem. Soc., 2019, 141(13), 5252–5260, DOI:10.1021/jacs.8b12873.
- C. A. Loy and D. J. Trader, Caged Aminoluciferin Probe for Bioluminescent Immunoproteasome Activity Analysis, RSC Chem. Biol., 2024, 5, 877–883, 10.1039/D4CB00148F.
- E. Maurits, M. J. Van De Graaff, S. Maiorana, D. P. A. Wander, P. M. Dekker, S. Y. Van Der Zanden, B. I. Florea, J. J. C. Neefjes, H. S. Overkleeft and S. I. Van Kasteren, Immunoproteasome Inhibitor–Doxorubicin Conjugates Target Multiple Myeloma Cells and Release Doxorubicin upon Low-Dose Photon Irradiation, J. Am. Chem. Soc., 2020, 142(16), 7250–7253, DOI:10.1021/jacs.9b11969.
- C. S. Muli, C. A. Loy and D. J. Trader, Immunoproteasome as a Target for Prodrugs, J. Med. Chem., 2025, 68(6), 6507–6517, DOI:10.1021/acs.jmedchem.4c03017.
- C. S. Muli and D. J. Trader, 20S Proteasome Hydrolysis of LLVY Substrates to Determine Preferences for Moieties in Its Primed Substrate Channel, Bioorg. Med. Chem. Lett., 2023, 85, 129233, DOI:10.1016/j.bmcl.2023.129233.
- C. A. Loy and D. J. Trader, Primed for Interactions: Investigating the Primed Substrate Channel of the Proteasome for Improved Molecular Engagement, Molecules, 2024, 29(14), 3356, DOI:10.3390/molecules29143356.
- J. Chen, Y. Wang, C. Xu, K. Chen, Q. Zhao, S. Wang, Y. Yin, C. Peng, Z. Ding and Y. Cong, Cryo-EM of Mammalian PA28αβ-iCP Immunoproteasome Reveals a Distinct Mechanism of Proteasome Activation by PA28αβ, Nat. Commun., 2021, 12(1), 739, DOI:10.1038/s41467-021-21028-3.
- K. Schwarz, M. Eggers, A. Soza, U. H. Koszinowski, P.-M. Kloetzel and M. Groettrup, The Proteasome Regulator PA28α/β Can Enhance Antigen Presentation without Affecting 20S Proteasome Subunit Composition, Eur. J. Immunol., 2000, 30(12), 3672–3679 CrossRef.
- E. M. Huber, M. Basler, R. Schwab, W. Heinemeyer, C. J. Kirk, M. Groettrup and M. Groll, Immuno- and Constitutive Proteasome Crystal Structures Reveal Differences in Substrate and Inhibitor Specificity, Cell, 2012, 148(4), 727–738, DOI:10.1016/j.cell.2011.12.030.
- B. L. Zerfas, R. A. Coleman, A. F. Salazar-Chaparro, N. J. Macatangay and D. J. Trader, Fluorescent Probes with Unnatural Amino Acids to Monitor Proteasome Activity in Real-Time, ACS Chem. Biol., 2020, 15(9), 2588–2596, DOI:10.1021/acschembio.0c00634.
- T. Preckel, W.-P. Fung-Leung, Z. Cai, A. Vitiello, L. Salter-Cid, O. Winqvist, T. G. Wolfe, M. V. Herrath, A. Angulo, P. Ghazal, J.-D. Lee, A. M. Fourie, Y. Wu, J. Pang, K. Ngo, P. A. Peterson, K. Früh and Y. Yang, Impaired Immunoproteasome Assembly and Immune Responses in PA28−/− Mice, Science, 1999, 286(5447), 2162–2165, DOI:10.1126/science.286.5447.2162.
- A. F. Kisselev and A. L. Goldberg. Monitoring Activity and Inhibition of 26S Proteasomes with Fluorogenic Peptide Substrates, Methods in Enzymology; Ubiquitin and Protein Degradation, Academic Press, 2005; Vol. 398, pp 364–378. DOI:10.1016/S0076-6879(05)98030-0.
- K. J. Rodgers and R. T. Dean, Assessment of Proteasome Activity in Cell Lysates and Tissue Homogenates Using Peptide Substrates, Int. J. Biochem. Cell Biol., 2003, 35(5), 716–727, DOI:10.1016/S1357-2725(02)00391-6.
Footnote |
| † Electronic supplementary information (ESI) available: Methods, supporting figures, LC-MS and plate reader methods, and LC-MS traces of purified substrates/peptides/peptidomimetics. See DOI: https://doi.org/10.1039/d5cb00114e |
| This journal is © The Royal Society of Chemistry 2025 |