
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA biparatopic HER2-targeting ADC constructed via site-specific glycan conjugation exhibits superior stability, safety, and efficacy†
Qi
Xue‡
 a,
Jianjian
Peng‡
b,
Wenying
Dai‡
a,
Qingsong
Wu
a,
Jinbiao
Jiao
a,
Yudi
Hu
a,
Wanxing
Sha
a,
Yang
Yang
a,
Wenhao
Yu
a,
Siyang
Liu
a,
Ting
Xu
*b and
Jie P.
Li
*a
a,
Jianjian
Peng‡
b,
Wenying
Dai‡
a,
Qingsong
Wu
a,
Jinbiao
Jiao
a,
Yudi
Hu
a,
Wanxing
Sha
a,
Yang
Yang
a,
Wenhao
Yu
a,
Siyang
Liu
a,
Ting
Xu
*b and
Jie P.
Li
*a
aState Key Laboratory of Coordination Chemistry, School of Chemistry and Chemical Engineering, Chemistry and Biomedicine Innovation Center (ChemBIC), Nanjing University, Nanjing, China. E-mail: jieli@nju.edu.cn
bJiangsu Alphamab Biopharmaceuticals Co., Ltd., BioBay, Building C23, 218 Xinghu Street, Suzhou Industrial Park, Suzhou, Jiangsu, China. E-mail: tingxu@alphamabonc.com
First published on 25th June 2025
Abstract
HER2 is overexpressed in approximately 15–20% of cancers and is associated with aggressive disease progression. We developed JSKN003, a bispecific HER2-targeted antibody–drug conjugate (ADC), through site-specific conjugation technology based on N-glycosylation engineering. JSKN003 maintains a biantennary glycan structure and exhibits superior structural homogeneity, optimized hydrophilicity, and reduced aggregation compared to conventional thiol-maleimide chemistry. In preclinical models JSKN003 demonstrated potent antitumor efficacy, inducing tumor regression in multiple HER2-expressing tumors, such as NCI-N87, BxPC-3, and PDX tumor models. Mechanistically, JSKN003 binds specifically to HER2, undergoes efficient internalization, and traffics to the lysosome, where the payload DXd is released, leading to DNA damage and apoptosis. JSKN003 retained its cytotoxic activity against trastuzumab-resistant cells, attributed to efficient payload delivery and blockade of downstream HER2 signaling pathways, demonstrating the potential to overcome clinical trastuzumab resistance. The safety profile of JSKN003 was evaluated in cynomolgus monkeys and was found to be acceptable, with no severe toxicities observed at therapeutic doses. JSKN003 demonstrated excellent antitumor activity and a favorable safety profile in clinical trials, highlighting its potential as a promising therapeutic option for patients with HER2-positive tumors. These findings suggest that JSKN003 could be a valuable therapeutic strategy with excellent efficacy and safety for HER2-expressing tumors in the clinical setting.
Introduction
Human epidermal growth factor receptor 2 (HER2/ErbB2) is a transmembrane receptor tyrosine kinase within the HER family, which drives oncogenic signaling through heterodimerization and activation of downstream pathways. Amplification or overexpression of HER2 occurs in approximately 15–20% of breast cancers, correlating with aggressive phenotypes characterized by accelerated proliferation and metastatic potential. Consequently, HER2 has been established as a pivotal therapeutic target in oncology.1–3 In 1998, trastuzumab, a humanized monoclonal antibody targeting HER2's extracellular domain IV, received approval for the treatment of HER2-positive breast cancer, marking a significant advancement in therapy.4Beyond its direct antiproliferative effects, trastuzumab's ability to induce HER2 endocytosis has facilitated its use as a delivery vehicle for antibody–drug conjugates (ADCs). The first-generation ADC, T-DM1 (trastuzumab emtansine), conjugates the anti-microtubule agent DM1 via a non-cleavable linker, demonstrating clinical benefit in both metastatic and early-stage HER2-positive breast cancer.5,6 However, T-DM1's therapeutic window is constrained by several limitations, including off-target toxicities such as hepatotoxicity and thrombocytopenia, limited efficacy in tumors with low HER2 expression, suboptimal bystander effects, and the development of acquired resistance.7–11
To address these limitations, next-generation HER2-targeted ADCs have been developed, incorporating innovative payload-linker systems. DS-8201a (trastuzumab deruxtecan) employs a cleavable tetrapeptide linker to conjugate trastuzumab with the topoisomerase I inhibitor DXd, achieving enhanced tumor penetration and potent DNA damage response.10 Clinical trials have demonstrated its efficacy in HER2-positive metastatic breast and gastric cancers,12–14 DS-8201a exhibited superior efficacy compared to T-DM1, with significantly improved progression-free survival (median PFS: 29.0 vs. 7.2 months) and overall survival (median OS: 52.6 vs. 42.7 months).15 Nevertheless, critical challenges persist: (1) maleimide-based conjugation results in a heterogeneous drug-to-antibody ratio (DAR 7–8), where high DAR values lead to increased aggregation and accelerated hepatic clearance, ultimately compromising pharmacokinetic consistency;16–21 (2) instability of thiol-maleimide adducts causes premature DXd release, correlating with dose-limiting interstitial lung disease (ILD);22 (3) a narrow therapeutic index restricts the potential for dose escalation.23,24
To overcome these challenges, we developed JSKN003-bispecific HER2-targeting ADC that harnesses the dual epitope binding (domains II/IV) of KN026.25 The Fc glycan-directed conjugation platform allows for site-specific attachment of four DXd molecules per antibody, resulting in enhanced structural homogeneity compared to maleimide-based stochastic cysteine conjugation.26–28 This approach also improves hydrophilicity, reducing aggregation, and enhances serum stability, leading to prolonged payload retention. These properties contribute to a favorable pharmacokinetic profile and reduced off-target toxicities, further extending the therapeutic window and potentially mitigating severe adverse effects associated with DS-8201a, such as ILD. In addition to its enhanced structural stability, JSKN003 retains the superior HER2-binding and internalization properties of KN026, ensuring efficient payload delivery to trastuzumab-resistant tumor cells. This enables JSKN003 to potentially suppress the proliferation of trastuzumab-resistant tumors.
Here, we present the preclinical profile of JSKN003, including its efficacy, pharmacokinetics, and safety. JSKN003 demonstrated potent antitumor activity, favorable pharmacokinetics, enhanced serum stability, and a promising safety profile. These characteristics position JSKN003 as a promising therapeutic candidate with an expanded safety margin, particularly for HER2-driven malignancies, including those with trastuzumab resistance.
Materials and methods
General information
The solvents and reagents were obtained from commercial suppliers and used without further purification unless otherwise stated. Certain compounds were synthesized following procedures reported in the literature.Silica gel column chromatography was performed using Silica Gel 60 (200–300 mesh). Analytical thin-layer chromatography (TLC) was conducted on precoated silica gel 60 F254 plates, with shortwave UV light as the visualizing agent and KMnO4 and heat as developing agents.
NMR spectra were recorded on Bruker AVANCE III-400 or 500 spectrometers in deuterochloroform (CDCl3). 1H NMR and 13C NMR spectra were obtained at 400/500 MHz and 100/125 MHz, respectively. 19F NMR spectra were recorded at 376/470 MHz. Chemical shifts (δ) are reported relative to TMS (δ 0.00 for 1H NMR) and chloroform (δ 7.26 for 1H NMR, δ 77.00 for 13C NMR). Multiplicities are denoted as follows: s (singlet), d (doublet), dd (doublet of doublets), t (triplet), q (quartet), m (multiplet), and br (broad signal). Mass spectra were acquired using ESI-MS (LCQ Fleet, Thermo Fisher Scientific).
Expression and purification of mutant GalT1
The mutant human GalT1 (Y289L) with an N-terminal His-tag was expressed in CHO cells via transient transfection using polyethylenimine. After 6 days of culture, the supernatant was collected, clarified, and subjected to Ni-affinity chromatography for initial purification. Eluted fractions were further purified by flow-through anion exchange chromatography using a Diamond MIX-A column to remove acidic impurities. The final protein solution was adjusted to pH 7.5, sterile-filtered (0.22 μm), and stored at −80 °C until use.LC–MS analysis of antibody conjugation and ADCs
Liquid chromatography–mass spectrometry (LC–MS) analysis was performed using a Xevo G2-S TOF mass spectrometer coupled with an Acquity UPLC system equipped with an Acquity UPLC Protein BEH C4 column (1.7 μm, 2.1 × 50 mm). The mobile phase consisted of solvent A (water with 0.1% formic acid) and solvent B (acetonitrile with 0.1% formic acid), delivered at a flow rate of 0.5 mL min−1. The gradient elution was as follows: 95% H2O (isocratic) for 2 min, 95% to 10% H2O over 4 min, 10% H2O (isocratic) for 1 min, 10% to 95% H2O over 1 min, and 95% H2O (isocratic) for 2 min. The electrospray ionization (ESI) source was operated in positive mode, with a capillary voltage of 2.0 kV and a cone voltage of 40 V. Nitrogen was used as the desolvation gas at a flow rate of 850 L h−1. Mass spectra were reconstructed using the MaxEnt algorithm in MassLynx software (v4.1, Waters). The major chromatographic peak(s) were selected for integration and further analysis, ensuring precise identification and characterization of the ion series.Synthesis of JSKN003
KN026 (5 mg) was reacted in a total volume of 500 μL containing final concentrations of 10 mg mL−1 KN026, 10 mM MnCl2, 5 mM UDP-GalNAz, 0.125 mg mL−1 mutant GalT1, and 10 mM Tris–HCl. The reaction was conducted at 30 °C for 16 h. Upon confirmation of reaction completion by mass spectrometry, the product was purified using Protein A affinity chromatography and desalted in DPBS, yielding KN026-(N3)4 (3 mg, 60% yield). KN026-(N3)4 (10 mg mL−1, 67 μM) was reacted with DBCO-DXd (15 eq., 50 mM) in the presence of DMSO (5% of total volume) in a total volume of 800 μL. The reaction was performed at 30 °C with shaking (400 rpm) for 12 h. Following confirmation of reaction completion by mass spectrometry, the product was ultrafiltered and desalted in DPBS, yielding JSKN003 (4.2 mg, 52.5% yield).Stability studies of KN026-mal and KN026-(N3)4
For stability assessment, KN026-mal (20 μL, 20 μM, in PBS) was incubated with GSH (0.8 μL, 100 mM) at 37 °C for 48 h. Similarly, KN026-(N3)4 (20 μL, 20 μM, in PBS) was incubated under identical conditions with GSH (0.8 μL, 100 mM) at 37 °C for 48 h. Stability was assessed by LC-MS to evaluate structural integrity and conjugation efficiency.N-Glycan profiling of KN026 and JSKN003
The N-glycosylation profiles of KN026 and JSKN003 were analyzed using the Waters GLYCOWORKS RAPIDFLUOR-MS N-Glycan Analysis Kit. N-linked glycans were enzymatically cleaved from the antibody backbone using GlycoWorks Rapid PNGase F. The released glycans were derivatized with RapiFluor-MS labeling reagent, enhancing ionization efficiency and MS detection sensitivity. Labeled glycans were then purified using the GlycoWorks HILIC μElution cleanup module to remove excess labeling reagent and reaction byproducts. Finally, the purified glycans were separated by hydrophilic interaction liquid chromatography (HILIC) and characterized by tandem mass spectrometry (MS/MS).HIC-HPLC analysis
KN026, DS-8201a, and JSKN003 were analyzed using hydrophobic interaction chromatography-high-performance liquid chromatography (HIC-HPLC) on an Agilent 1260 HPLC system with a TSKgel butyl-NPR column (4.6 mm × 35 mm, 2.5 μm; TOSOH). The mobile phases consisted of buffer A (20 mM sodium phosphate, 1.5 M ammonium sulfate, pH 6.9) and buffer B (75% 20 mM sodium phosphate, 25% isopropanol, pH 6.9). The separation was performed at 0.4 mL min−1 with a gradient transition from 100% buffer A to 100% buffer B over 1–13 min at 30 °C.SEC-HPLC analysis
KN026, DS-8201a, JSKN003, and Trastuzumab-(mal-DXd)8 were analyzed by size-exclusion chromatography–high-performance liquid chromatography (SEC-HPLC) using an Agilent 1260 HPLC system with an XBridge Protein BEH SEC column (7.8 mm × 150 mm, 200 Å, 3.5 μm; Waters). The mobile phase consisted of PBS (pH 7.2) with 15% isopropanol, with a flow rate of 0.6 mL min−1 at room temperature.UHPLC analysis
JSKN003 was analyzed using ultra-high performance liquid chromatography (UHPLC; Waters ACQUITY Premier system) with an Agilent PLRP-S 1000 Å column (2.1 × 100 mm, 5 μm). Mobile phases were 0.1% TFA in water (A) and 0.1% TFA in 90% acetonitrile (B). The flow rate was 0.3 mL min−1, column temperature 55 °C, and detection at 280 nm. The gradient elution transitioned from 63% A to 10% A over 12 min. Samples included blank, system suitability, test, and QC solutions. The DAR value was calculated as follows:where ADx represents the peak area of the species with x drug payloads.

Stability studies in SD rats, cynomolgus monkeys and human serum
Human serum was mixed with JSKN003 or DS-8201a to achieve a final ADC concentration of 0.2 mg mL−1. Day 0 samples were processed immediately, while remaining samples were incubated at 37 °C for 3 and 7 days. After incubation, ADCs were purified using Protein A and analyzed by LC-MS to assess structural integrity.For ELISA-based stability studies, JSKN003 and DS-8201a were diluted in serum from SD rats, cynomolgus monkeys, or humans at final concentrations of 10 μg mL−1 and 100 μg mL−1 and incubated at 37 °C for up to 21 days. Samples were collected at 0, 3, 7, 14, and 21 days, and ELISA assays were performed to measure total antibody (anti-HER2 portion) and ADC (deruxtecan-conjugated antibody) levels. HER2-His was used for total antibody detection, while anti-deruxtecan monoclonal antibody and HER2-His were used for ADC detection.
Pharmacokinetic studies
Cynomolgus monkeys were divided into three groups (n = 2 per group; one male and one female) and administered JSKN003 intravenously at 3 mg kg−1, or 30 mg kg−1. Blood samples were collected at 13 time points: pre-dose, 1 h, 2 h, 6 h, 24 h, 48 h, 72 h, 96 h, 192 h, 360 h, 528 h, and 696 h post-dose. BALB/c-nude mice (n = 3–5 per group) received JSKN003 intravenously at 1 mg kg−1, with blood samples collected at 8 time points: pre-dose, 0.03 h, 1 h, 6 h, 24 h, 48 h, 96 h, and 168 h post-dose. ELISA assays were conducted to quantify total antibody (anti-HER2 portion) and ADC (deruxtecan-conjugated antibody) levels.Binding affinity assay for HER2 protein
Serial 4-fold dilutions of the ADC or antibody (starting at 20 μg mL−1) were added to 96-well plates pre-coated with HER2-His (5 μg mL−1) and incubated at 25 °C for 2 hours. After washing, Anti-Human IgG Peroxidase Antibody (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8000 dilution) was added and incubated for an additional 2 hours. The reaction was developed using TMB substrate, stopped, and the OD450 absorbance was measured.
:8000 dilution) was added and incubated for an additional 2 hours. The reaction was developed using TMB substrate, stopped, and the OD450 absorbance was measured.
Antibodies/ADCs endocytosis assay
The endocytosis activity of Trastuzumab, DS-8201a, KN026, and JSKN003 was analyzed using the Zenon pHrodo iFL IgG Labeling Kit. A mixture of 200 nM antibody/ADC and 600 nM pHrodo-FITC was incubated in RPMI 1640 medium with 10% FBS for 15 minutes. The diluted antibody-pHrodo complex was then added to NCI-N87 cells and incubated at 37 °C for 24 hours. Following incubation, cells were washed, resuspended in FACS buffer, and analyzed by flow cytometry to measure fluorescence intensity in the FITC channel.In vitro cytotoxicity studies
Cells were seeded in a 96-well plate at 5000–10000 cells per well for 24 h at 37 °C with 5% CO2. Serial dilutions of KN026 and JSKN003 were added to the cells in complete growth medium and incubated at 37 °C with 5% CO2 for 96 h. Cell viability was evaluated using a Cell Counting-Lite 2.0 Luminescent Cell Viability Assay (Vazyme, DD1101-01). Cell viability was plotted as a percentage of untreated cells. Each measurement was taken in triplicate.
Evaluation of ADCC activity
The antibody-dependent cell-mediated cytotoxicity (ADCC) activity of KN026 and JSKN003 was assessed by measuring lactate dehydrogenase (LDH) release and luciferase activity in a co-culture system. For LDH-based ADCC assessment, pre-activated PBMCs and NCI-N87 cells were co-cultured at a 10:1 effector-to-target ratio in the presence of JSKN003, KN026, or human IgG1. After 4 hours of incubation, LDH release in the culture supernatant was measured to evaluate cytotoxicity. Additionally, ADCC activity was assessed using the Jurkat-FcγRIIIa-V158-NFAT-Luciferase reporter assay. Jurkat reporter cells and NCI-N87 cells were co-cultured at a 5:1 ratio with JSKN003 and KN026. After 6 hours of incubation, luciferase activity in Jurkat reporter cells was quantified to determine ADCC activation.
Assessment of DNA damage and apoptosis
The level of histone H2A.X phosphorylation was measured using the H2A.X Phosphorylation Assay Kit (17-344, Sigma-Aldrich, USA) to evaluate the DNA damage-inducing ability of JSKN003. Apoptosis induction was assessed using the FITC Annexin V Apoptosis Detection Kit (556547, BD Pharmingen, USA). NCI-N87 cells were incubated with serial dilutions of JSKN003 (maximum final concentration of 10 μg mL−1, 10-fold serial dilution, 5 concentrations) for 72 hours. Following incubation, cells were stained according to the manufacturer's instructions and analyzed by flow cytometry to quantify the levels of histone H2A.X phosphorylation and the proportions of apoptotic cell populations.Toxicological studies
The toxicological profile of JSKN003 was evaluated following repeated intravenous administration in cynomolgus monkeys, a species cross-reactive with JSKN003. Monkeys were randomly assigned to four groups (n = 10 per group; five males and five females) and received intravenous infusion of either JSKN003 formulation buffer (control), 10 mg kg−1, 30 mg kg−1, or 80 mg kg−1 JSKN003 once every three weeks for a total of three doses. A six-week recovery study was conducted for every group with two males and two females per group. Throughout the study, clinical signs, body weight, food consumption, and pulmonary/bronchial toxicity were monitored, including body weight, food consumption, and pulmonary/bronchial toxicity. Anti-drug antibodies (ADA) and ADA titers were analyzed prior to the third dose and at the end of the recovery phase. At the necropsy of dosing phase and recovery phase, histopathological examinations were performed on the lungs, thymus, and thyroid of surviving monkeys to assess potential long-term toxic effects.Bystander killing assay
NCI-N87 and MDA-MB-231/MDA-MB-468 cell mixtures were co-cultured at a 1:1 ratio, while MDA-MB-231 and MDA-MB-468 cells alone were seeded at the same density and incubated for 24 hours at 37 °C with 5% CO2. Subsequently, KN026 or JSKN003 (2 μg mL−1) was added, followed by an additional 96-hour incubation. MDA-MB-231 is a stable cell line overexpressing luciferase, allowing its viability to be quantified using the Dual Luciferase Reporter Assay Kit (Vazyme, DL101-01). MDA-MB-468 cell viability was assessed based on the percentage of HER2+ cells relative to untreated controls.
Establishment of acquired trastuzumab-resistant (TR) cell lines
NCI-N87 and BT474 cell lines were cultured in the presence of trastuzumab at concentrations of 2 μg mL−1 and 10 μg mL−1, respectively. Cells were passaged continuously for one year to establish acquired trastuzumab-resistant cell lines, designated as N87-RE and BT474-RE.29,30Binding affinity assay for trastuzumab-resistant cell line
BT474-RE and N87-RE cells were incubated with gradient-diluted antibody/ADC solutions at room temperature for 1 hour. After incubation, cells were washed and further incubated with PE-conjugated anti-human IgG secondary antibody (1:200 dilution) on ice for 1 hour. Following a final wash, cells were analyzed by flow cytometry for fluorescence intensity in the PE channel.
Tumor growth inhibition assay
All in vivo studies were performed in accordance with the local guidelines of the Institutional Animal Care and Use Committee.A HER2-low (IHC 2+) gastric adenocarcinoma PDX model was developed using female NOD/SCID mice. Tumor tissue was derived from an Asian patient with gastric adenocarcinoma and implanted subcutaneously at the right anterior scapula (2–3 mm tumor fragments). When the average tumor volume reached 213.40 mm3, mice were randomized into three groups, and treatment was initiated immediately. JSKN003 (10 mg kg−1) or PBS was administered intravenously once per week for two doses. Tumor volume and TGI% calculations followed the methods described previously.
Safety studies
ADCs or PBS were dosed intravenously in 6 to 8-week-old female mice at 20 mg kg−1 (3–4 mice per dose group, randomly assigned). At post-dose day 6, 3–4 serum samples were collected for hematology analysis.Results and discussions
Construction and characterization of JSKN003
JSKN003 is an ADC that combines KN026, a bispecific antibody (bsAb), with a topoisomerase I inhibitor (DXd) through site-specific modifications mediated by Fc glycosylation. KN026 targets two distinct epitopes of HER2 (ErbB2), offering a significant advantage in overcoming antibody–drug resistance compared to trastuzumab, which binds to a single epitope.25 Mass spectrometry glycoform analysis revealed that KN026 predominantly carries the FA2 N-glycan (∼82.83%), along with several minor glycoforms: A2 (2.90%), A1 (1.25%), FA1 (2.99%), and FA2G1 (4.41%) (Fig. S1A–C, ESI†). Based on these glycan structures, 85.73% of glycans (FA2 + A2) are compatible with dual small-molecule conjugation via GalT1-mediated azide installation followed by strain-promoted azide–alkyne cycloaddition (SPAAC), while 8.65% (A1 + FA1 + FA2G1) allow only single-site labeling.To generate a homogeneous ADC with stable structural properties, human galactosyltransferase1 (GalT1) were employed to introduce azide groups into the Fc region. DBCO-DXd was synthesized following the route outlined in Fig. 1A. Subsequently, strain-promoted azide–alkyne cycloaddition (SPAAC) was utilized for site-specific conjugation of DXd, resulting in a structurally uniform ADC (Fig. 1B). To further characterize the glycan modifications of JSKN003, reduced N-glycopeptide digestion followed by tandem mass spectrometry (MS/MS) analysis was performed on KN026 and JSKN003. The results indicated that KN026 predominantly contained G0F glycans, whereas JSKN003 mainly comprised G0F glycans conjugated with two DXds (Fig. 1C, D and Fig. S1D, ESI†). The molecular weight, structural uniformity, and integrity of JSKN003 were confirmed by liquid chromatography-mass spectrometry (LC-MS) analysis (Fig. 1E). The DAR of JSKN003 was determined to be 3.6, consistent with the number of G0F glycoforms present on the antibody, as measured by auto-scaled chromatographic analysis of UHPLC-MS (Fig. 1F).
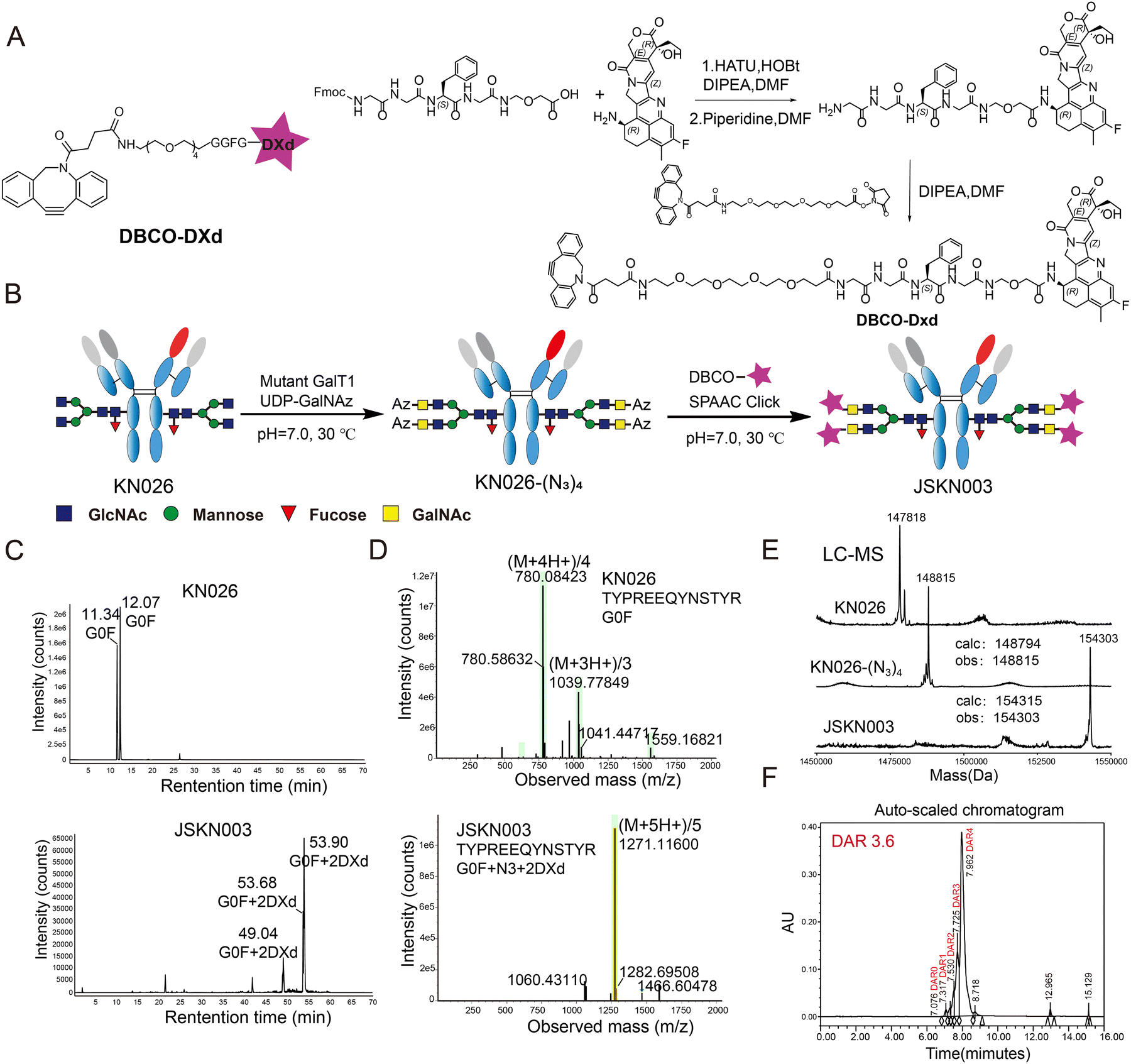 | ||
| Fig. 1 Development of JSKN003 via site-specific Fc glycosylation modification and molecular characterization. (A) Structure and synthesis route of DBCO-DXd. (B) Schematic representation of the site-specific chemoenzymatic synthesis of JSKN003 (illustrating the reaction scheme of glycans present in over 85% of the antibody population). (C) Extracted ion chromatograms (EIC) comparing the N-glycopeptide profiles of KN026 and JSKN003. (D) Electrospray ionization tandem mass spectrometry (ESI-MS/MS) fragmentation of representative N-glycopeptides from KN026 and JSKN003, highlighting structural differences in glycoforms. (E) Electrospray ionization mass spectrometry (ESI-MS) analysis of intact antibodies: KN026, KN026-(N3)4 and JSKN003. (F) Auto-scaled chromatographic analysis of JSKN003. | ||
Biophysical and serum stability comparison between JSKN003 and DS-8201a
Having established the site-specific glycan-conjugation and structural uniformity of JSKN003, we next sought to evaluate its biophysical behavior and stability in comparison to DS-8201a with conventional maleimide-conjugated ADCs.To evaluate the druggability and molecular stability of JSKN003 and compare the advantages of glycan-based conjugation with traditional thiol-based conjugation methods, hydrophobic interaction chromatography (HIC) and size exclusion chromatography (SEC) were performed on KN026, JSKN003, and DS-8201a. HIC analysis revealed that JSKN003 exhibits significantly improved hydrophilicity compared to DS-8201a, though it remains distinguishable from the parent antibody KN026 (Fig. 2B). This difference is likely due to DS-8201a's higher drug load and hydrophobic linker-payload, suggesting that the glycan-linked design of JSKN003 better preserves the antibody's native hydrophilicity. SEC showed that both JSKN003 and DS-8201a are predominantly monomeric, with main peaks accounting for >95% of the total protein (Fig. 2C). In contrast, our in-house synthesized Trastuzumab-(mal-DXd)8 (DAR ∼ 8) exhibited substantial aggregation in SEC analysis, with monomer content below 80% and a prominent high-molecular-weight (HMW) aggregate peak, accounting for approximately 24% of the total chromatographic area at 11.4 min (Fig. S2, ESI†). These findings suggest that multiple factors, including linker structure, DAR, and conjugation strategy, collectively influence the hydrophobicity and aggregation propensity of ADCs. However, the biantennary glycan-based conjugation strategy used in JSKN003 provides greater tolerance toward the inherent hydrophobicity of linkers and payloads, enabling JSKN003 to maintain good hydrophilicity despite incorporating four DBCO moieties.
 | ||
| Fig. 2 Biophysical and serum stability comparison between JSKN003 and DS-8201a. (A) Structural comparison between JSKN003 (glycoengineered ADC) and DS-8201a (commercial trastuzumab deruxtecan). (B)–(C) Hydrophobic interaction chromatography (HIC) (B) and size exclusion chromatography (SEC) (C) analysis comparing DS-8201a, KN026, and JSKN003. (D) Liquid chromatography-mass spectrometry (LC-MS) analysis demonstrating the stability of JSKN003 and DS-8201a in human and rat serum. Bar graphs show changes in drug-to-antibody ratio (DAR) values at 0 and 96 hours. Error bars are not shown as the measurements represent single technical replicates due to limited sample availability. (E) Pharmacokinetic analysis of DS-8201a and JSKN003 in tumor-bearing BALB/c mice. (F) Hematological toxicity assessment following a 20 mg kg−1 dose of ADC. Neutropenia, lymphadenopathy, and changes in monocyte levels were observed in mice. Three to four animals were administered JSKN003, DS-8201a, or PBS, and hematological markers were subsequently analyzed. Data are presented as mean ± SEM; n = 3. Student's t test. n.s, not significant; *P < 0.05. | ||
We next assessed whether glycan-based conjugation confers enhanced chemical stability under reductive and serum-based stress conditions. Maleimide-labeled antibodies and glycoengineered, site-specifically modified antibodies were first subjected to glutathione (GSH) treatment at 37 °C for 48 hours, simulating the reductive environment in vitro. LC-MS analysis revealed that the maleimide-labeled KN026-Mal reacted with GSH, forming detectable adducts after 48 hours. In contrast, the glycoengineered antibody KN026-N3 exhibited superior stability under identical GSH conditions, highlighting the enhanced stability provided by glycan modifications (Fig. S3A and B, ESI†). A significant limitation of clinical maleimide-based ADCs is their susceptibility to in vivo hydrolysis, resulting in premature payload release and associated off-target toxicities. To examine whether glycan-based conjugation could mitigate this issue, serum stability was assessed in vitro by incubating ADCs in serum at 37 °C for 96 hours. Subsequent purification of the remaining antibodies via Protein A and LC-MS analysis demonstrated that the DAR of maleimide-conjugated DS-8201a decreased substantially over 96 hours, particularly in human serum (∼50% reduction), suggesting a high propensity for off-target payload release. In marked contrast, the glycan-site conjugated JSKN003 exhibited outstanding stability in both human and rat serum, with minimal DAR loss (Fig. 2D). Extended serum stability tests performed using ELISA further confirmed the superior stability of JSKN003 compared to DS-8201a, maintaining structural integrity for up to 21 days (Fig. S3C and D, ESI†).
To corroborate these findings in vivo, BALB/c mice bearing tumors were given two doses of 10 mg kg−1 of either DS-8201a or JSKN003 on Days 0 and 7, with plasma collected on Day 11 for analysis of total antibody and ADC concentrations. JSKN003 exhibited significantly higher concentrations of both total antibody and ADC compared to DS-8201a, whereas DS-8201a showed rapid payload detachment, indicated by lower ADC concentrations relative to total antibody levels (Fig. 2E). This systemic release of DXd is a likely contributor to hematotoxicity. Conversely, the JSKN003 group displayed consistent concentrations of ADC and total antibody, confirming superior in vivo stability. To evaluate this possibility, we performed complete blood count (CBC) analysis in mice administered a single dose of 20 mg kg−1 of either DS-8201a or JSKN003. The results demonstrated fewer disturbances in blood cell populations for JSKN003-treated mice relative to those receiving DS-8201a (Fig. 2F). Collectively, these data reinforce that JSKN003 offers enhanced biosafety and stability profiles compared to DS-8201a, highlighting glycan-based conjugation as a robust strategy to overcome challenges associated with conventional maleimide-linked ADCs.
Taken together, these results demonstrate that glycan-based conjugation enables an optimal DAR (3.6) while maintaining superior colloidal stability, in contrast to thiol-based methods that often induce hydrophobic aggregation. The preserved structural integrity of JSKN003 underscores the critical influence of conjugation chemistry on ADC stability and favorable pharmacokinetic behavior. Collectively, the enhanced stability, hydrophilicity, and uniformity of JSKN003 highlight its promise as a next-generation HER2-targeted ADC with improved therapeutic potential.
Potential mechanism of action of JSKN003
ADCs exert their therapeutic effect through precise binding of the antibody to target cells, followed by internalization and intracellular release of the cytotoxic payload, ultimately leading to target cell death. To evaluate the binding capacity of JSKN003 to the target protein HER2, its binding affinity was compared with that of KN026 in HER2-positive NCI-N87 cells. The results demonstrated that JSKN003 exhibited a binding profile comparable to that of KN026, indicating that the chemical conjugation process did not compromise the antibody's ability to recognize and bind to HER2 (Fig. 3A). To confirm the effect of the drug conjugation with the anti-HER2 Antibody on internalization capability to HER2, we assess the internalization activity of JSKN003 and KN026 using flow cytometry. The results demonstrated that JSKN003 exhibited similar internalization activity to KN026 at both 5 hours and 24 hours (Fig. 3B), JSKN003 and KN026 can rapidly internalize into HER2-positive target cells after binding, indicating that drug conjugation did not affect HER2 binding and internalization.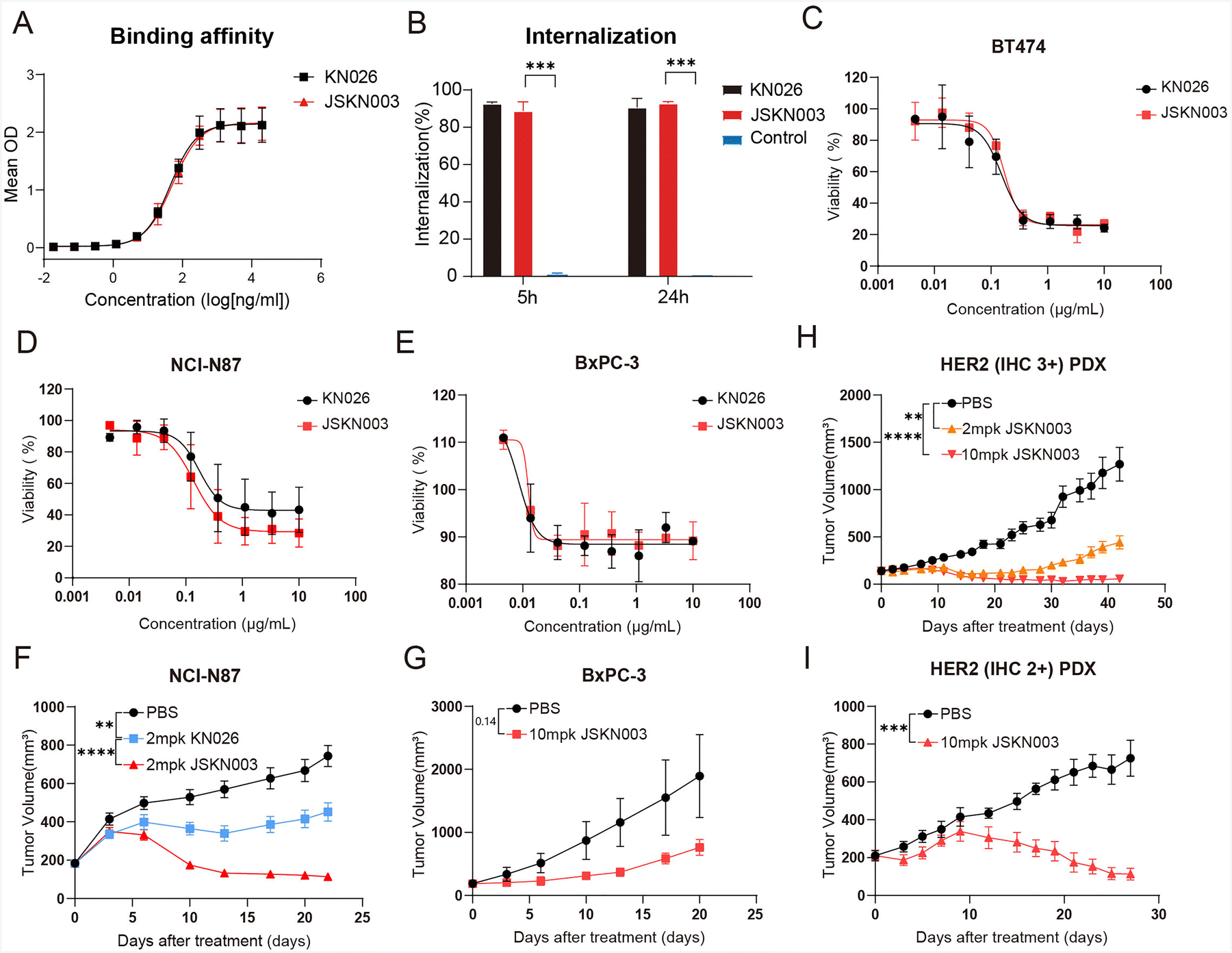 | ||
| Fig. 3 In vitro and in vivo antitumor efficacy of JSKN003 in HER2-high and HER2-low-expressing tumor models. (A) Binding affinity of KN026 and JSKN003 measured by ELISA. (B) Quantification of internalization activity of KN026 and JSKN003, presented as a bar graph. (C)–(E) In vitro cytotoxicity of KN026 and JSKN003 in HER2-high BT474 (C) and NCI-N87 (D) cells, as well as HER2-low BxPC-3 cells (E). (F)–(G) In vivo antitumor efficacy of JSKN003 in xenograft models of NCI-N87 and BxPC-3. Tumor-bearing mice were treated intraperitoneally with JSKN003 (10 mg kg−1) or vehicle control. Each treatment group included 4–6 mice. Data are presented as mean ± SEM. (H)–(I) Antitumor activity of JSKN003 in HER2-high and HER2-low patient-derived xenograft (PDX) models. (H) HER2-overexpressing (IHC 3+) adenocarcinoma PDX model; (I) HER2-low (IHC 2+) gastric adenocarcinoma PDX model. Data are presented as mean ± SEM.; n = 4–6. Student's t test. n.s, not significant; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. | ||
Having established JSKN003's capacity to bind and internalize efficiently, we next investigated whether it could induce DNA damage and apoptosis via topoisomerase I inhibition. This was assessed by measuring the phosphorylation of Histone H2A.X, a marker of DNA damage,31 and Annexin V/PI positivity, an indicator of apoptotic cell death. NCI-N87 cells were treated with JSKN003 at concentrations of 0.1, 1, and 10 μg mL−1, and analyzed by flow cytometry. A concentration-dependent increase in Histone H2A.X phosphorylation and apoptotic cell population was observed with escalating JSKN003 doses. These findings suggest that JSKN003 induces DNA damage and apoptosis through the inhibition of topoisomerase I activity (Fig. S4A, ESI†).
In addition to its direct cytotoxicity, JSKN003 may also retain immune effector functions inherited from the parental antibody. The major mechanism of action of trastuzumab is thought to involve ADCC activity mediated by its binding to FcγRIIIa on immune effector cells. To determine whether JSKN003 preserves these mechanisms of action similar to KN026, we evaluated its binding affinity to FCγRIIIa and its ADCC activity. Surface plasmon resonance (SPR) experiments demonstrated that JSKN003 maintained the strong binding affinity of KN026 to FCγRIIIa, with a dissociation constant (KD) value of 1.5 × 10−7 mol L−1 (Fig. S4B, ESI†). ADCC activity was assessed by measuring lactate dehydrogenase (LDH) release in a co-culture system of human peripheral blood mononuclear cells (PBMCs) and NCI-N87 cells. JSKN003 induced LDH release in a dose-dependent manner following a 4-hour treatment, with a maximum cytotoxicity of 45% at 1 μg mL−1, compared with 30% induced by KN026 (Fig. S4C, ESI†). Additionally, ADCC activity was evaluated using a Jurkat-FcγRIIIa-NFAT-luc reporter assay. NCI-N87 cells were co-cultured with Jurkat reporter cells in the presence of a serial dilution of JSKN003 or KN026 for 6 hours, and luciferase activity was measured as an indicator of FcγRIIIa engagement. The results showed that JSKN003 exhibited ADCC activity with an EC50 of 70 ng mL−1, which was comparable to that of KN026 (Fig. S4D, ESI†). These findings suggest that JSKN003 retains the ADCC function of KN026 even after DXd conjugation, supporting its potential immune-mediated antitumor activity.
Due to the high membrane permeability of DXd, JSKN003 demonstrated a potent “bystander” effect, enabling it to exert cytotoxicity beyond HER2-expressing cells. To validate this effect, HER2-positive NCI-N87 cells were co-cultured with HER2-negative MDA-MB-231 cells, and the cytotoxic effects of KN026 and JSKN003 on MDA-MB-231 cells were compared. The results indicated that JSKN003 effectively eliminated adjacent HER2-negative MDA-MB-231 cells following its cytotoxic action on HER2-positive NCI-N87 cells, whereas the unconjugated antibody KN026 did not induce a similar effect (Fig. S4E, ESI†). We further verified the “bystander” killing effect of JSKN003 in a co-culture system of HER2-positive NCI-N87 cells and HER2-negative MDA-MB-468 cells (Fig. S4F, ESI†). These findings suggest that JSKN003 exerts antitumor activity through a dual mechanism of action, combining the targeted pharmacologic effects of an anti-HER2 antibody with the cytotoxic potency of the topoisomerase I inhibitor DXd. This unique mechanism may contribute to its efficacy against both HER2-expressing tumor cells and surrounding HER2-negative tumor cells, further enhancing its therapeutic potential.
Anti-tumor efficacy of JSKN003
Having demonstrated that JSKN003 retains key functional properties, including HER2 binding, internalization, immune effector activity, and bystander killing, we next evaluated its antitumor efficacy in both in vitro and in vivo models. The antitumor efficacy of JSKN003 was compared directly to that of the anti-HER2 antibody KN026 across a range of human cancer cell lines, including HER2-high (NCI-N87 and BT474) and HER2-low (BxPC-3) expressing models. JSKN003 exhibited potent growth-inhibitory activity against HER2-positive cell lines, achieving IC50 values of 0.1720 μg mL−1 (BT474) and 0.1387 μg mL−1 (NCI-N87) (Fig. 3C and D). In contrast, KN026 showed moderate growth suppression in NCI-N87 cells and comparable suppression in BT474 cells under identical experimental conditions. Notably, both agents demonstrated limited cytotoxic effects against the HER2-low-expressing BxPC-3 cells, with detectable activity only at suprapharmacological concentrations (Fig. 3E). These findings collectively underscore the HER2 expression-dependent mechanism of action of JSKN003 and emphasize its enhanced potency compared to KN026 in targeting HER2-overexpressing malignancies.To further evaluate in vivo antitumor efficacy, JSKN003 was tested in both HER2-high (NCI-N87) and HER2-low (BxPC-3) xenograft models. In the HER2-high-expressing NCI-N87 xenografts, KN026 treatment at 2 mg kg−1 resulted in partial tumor growth inhibition (TGI) of 39% relative to controls at day 22 (Fig. 3F). By contrast, JSKN003 administered at the same dosage (2 mg kg−1) exhibited substantially greater antitumor activity, achieving 85% TGI, thus highlighting the enhanced therapeutic efficacy conferred by ADC conjugation. Interestingly, although JSKN003 displayed poor cytotoxicity in BxPC-3 cells in vitro, it has demonstrated notable antitumor activity in the BxPC-3 xenograft model, yielding 60% TGI (Fig. 3G) at a higher dose (10 mg kg−1).
To assess the clinical translational potential of JSKN003, additional evaluations were conducted using patient-derived xenograft (PDX) models representing both HER2-high and HER2-low tumors. In a HER2-overexpressing (IHC 3+) adenocarcinoma PDX model (18112804D09T), JSKN003 exhibited dose-dependent tumor regression; notably, a single dose of 2 mg kg−1 led to significant tumor shrinkage without observable adverse effects on body weight or overall health status (Fig. 3H and Fig. S5, ESI†). JSKN003 also demonstrated antitumor efficacy in a HER2-low (IHC 2+) gastric adenocarcinoma PDX model (Case168), further supporting its effectiveness in tumors with lower HER2 expression levels (Fig. 3I). Collectively, these findings indicate that JSKN003 possesses robust antitumor activity across HER2-high and HER2-low-expressing tumor models, highlighting its promising therapeutic potential for diverse HER2-expressing malignancies.
JSKN003 has the potential to overcome resistance to trastuzumab
Given its demonstrated efficacy in HER2-low models, we further explored whether JSKN003 could also overcome therapeutic resistance in tumors previously exposed to HER2-targeted therapy. Trastuzumab has revolutionized the treatment of HER2-positive breast and gastric cancers; however, its therapeutic efficacy is often limited by the development of drug resistance. KN026, a bispecific anti-HER2 antibody that targets two distinct epitopes, exhibits superior activity in blocking ErbB2 heterodimerization and effectively abrogates ErbB2 signaling in trastuzumab-resistant cancer cell lines.32 Consequently, JSKN003 has potential in overcoming trastuzumab resistance, offering promising therapeutic prospects. To model trastuzumab resistance, NCI-N87 and BT474 cells were continuously exposed to trastuzumab for one year, generating trastuzumab-resistant N87-RE and BT474-RE cell lines. To determine whether JSKN003 could effectively bind to trastuzumab-resistant cells, we evaluated the binding affinity of DS-8201a and JSKN003 in these resistant cell lines. The results showed that JSKN003 exhibited superior binding affinity to N87-RE and BT474-RE cells compared with DS-8201a (Fig. 4B). These findings highlight the ability of bispecific antibodies to maintain strong target engagement in trastuzumab-resistant cells, a property that is preserved in the JSKN003 ADC. To assess whether ADCs could undergo efficient internalization in resistant cells, we incubated DS-8201a, and JSKN003 with pHrodo iFL IgG labeling reagent for 15–20 minutes. The labeled ADCs were then added to N87-RE and BT474-RE cells. Upon lysosomal entry and acid hydrolysis, the pHrodo Green-labeled antibodies emitted green fluorescence, allowing quantification of internalization via FITC fluorescence intensity. After 5 hours, JSKN003 exhibited enhanced internalization in trastuzumab-resistant cells compared to DS-8201a, further supporting the advantage of bispecific antibody-based ADCs in resistant tumors (Fig. 4C and D). Finally, we evaluated the cytotoxicity of DS-8201a and JSKN003 in trastuzumab-resistant cells. Although the addition of Dxd has improved the cytotoxicity of DS-8201a, the reduced binding affinity and internalization capacity resulted in modestly enhancement of tumor cell killing. As expected, DS-8201a exhibited moderate cytotoxicity in resistant cell lines. In contrast, JSKN003, displayed significantly stronger cytotoxicity against N87-RE and BT474-RE cell lines (Fig. 4C and D). The ability of JSKN003 to maintain tumor cell binding, facilitate efficient internalization and exhibit potent cytotoxicity underscores the clinical potential of JSKN003 in overcoming trastuzumab resistance.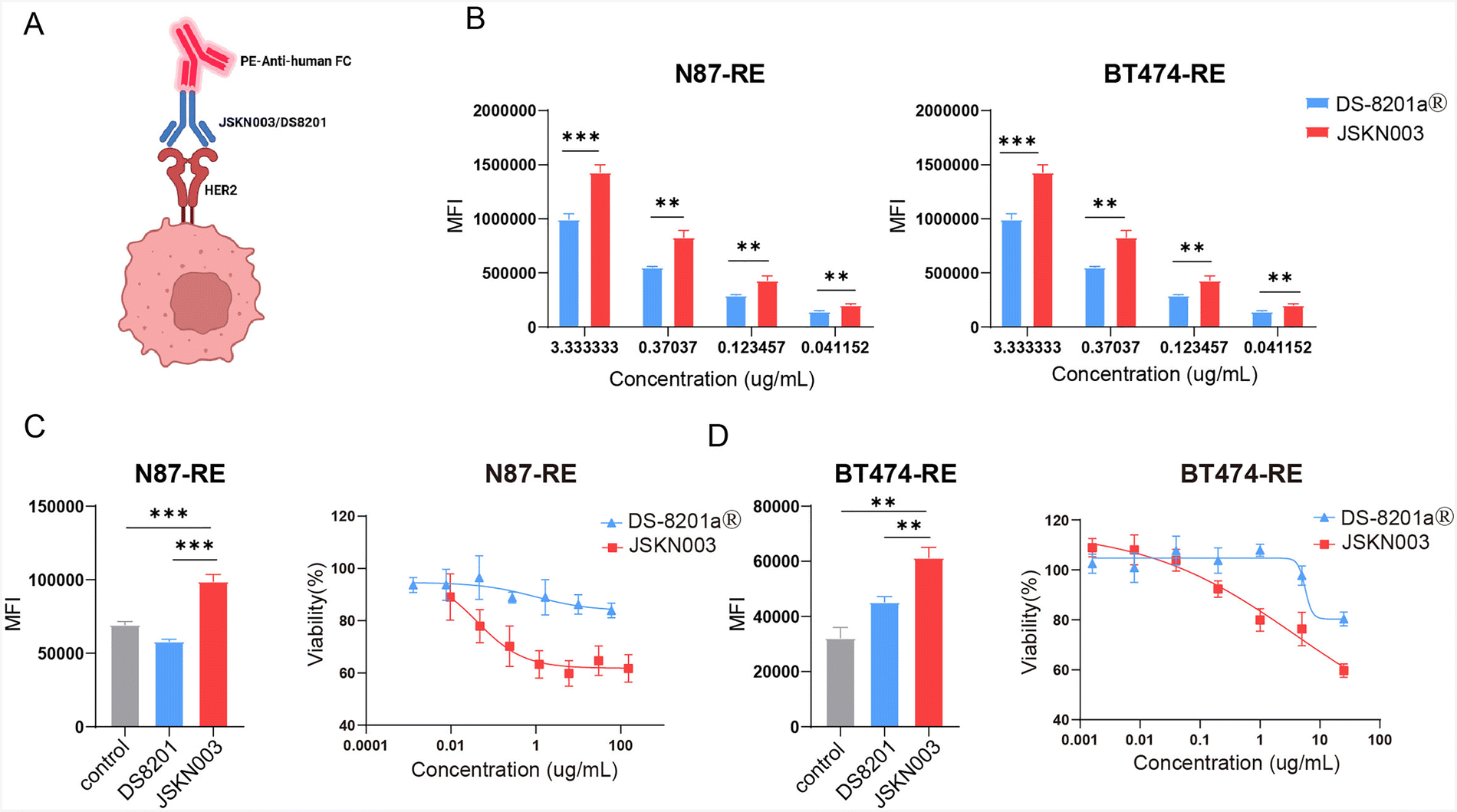 | ||
| Fig. 4 JSKN003 overcomes trastuzumab resistance in N87-RE and BT474-RE cells. (A) Schematic illustration of ADCs binding mechanisms. (B) Binding affinity of DS-8201a and JSKN003 to trastuzumab-resistant N87-RE and BT474-RE cells. (C) Internalization activity of DS-8201a and JSKN003 in N87-RE cells (left); cytotoxic activity of DS-8201a and JSKN003 in N87-RE cells (right). (D) Internalization activity of DS-8201a and JSKN003 in BT474-RE cells (left); cytotoxic activity of DS-8201a and JSKN003 in BT474-RE cells (right). Data are presented as mean ± SEM.; n = 4–6. Student's t test. n.s, not significant; *P < 0.05; **P < 0.01; ***P < 0.001. | ||
Pharmacokinetics of JSKN003 in BALB/c-nude mice and cynomolgus monkeys
To further characterize translational potential of JSKN003, we next evaluated its pharmacokinetic stability in murine and non-human primate models. To evaluate the in vivo stability and systemic persistence of JSKN003, pharmacokinetic profile was assessed in BALB/c-nude mice and cynomolgus monkeys. Plasma concentrations of JSKN003 declined in an exponential manner following a single administration. In cynomolgus monkeys, JSKN003 at 3 mg mL−1 persisted for more than 20 days, whereas the 30 mg mL−1 formulation demonstrated extended stability, remaining detectable for over 30 days (Fig. 5A and B). Pharmacokinetic analysis revealed that JSKN003 exhibited comparable systemic exposure (AUC) and half-life to total antibody. No significant differences were observed in the pharmacokinetic profiles between JSKN003 and the total antibody, indicating that JSKN003, with its payload conjugated via Fc glycan linkage, remains stable in plasma. These findings support the favorable pharmacokinetic properties of JSKN003, which may contribute to its sustained antitumor activity in clinical trials.33–36 Given JSKN003's prolonged systemic exposure and structural stability, we further investigated its safety profile through repeated-dose toxicology studies in cynomolgus monkeys. | ||
| Fig. 5 Pharmacokinetics of JSKN003 in BALB/c-nude mice and cynomolgus monkeys. (A) and (B) Pharmacokinetic profile of JSKN003 in BALB/c-nude mice (A) and cynomolgus monkeys (B). Each treatment group included 5 mice. | ||
Safety profile of JSKN003
To assess systemic toxicity and determine a tolerable dosing range, a repeated intravenous dosing study (every 3 weeks for a total of 3 doses) was conducted in cynomolgus monkeys, a species cross-reactive with JSKN003, and to assess its toxicological profile. No mortality or life-threatening toxicities were observed at dose levels up to 80 mg kg−1. However, at the highest dose of 80 mg kg−1, animals exhibited deteriorated clinical conditions, including reduced body weight, decreased food consumption, and lung and bronchial toxicity (Table 1). Histopathological findings in the lungs, thymus, and thyroid of surviving monkeys are summarized in ESI,† Table S1. Pulmonary and bronchial toxicity, characterized by inflammation and edema, was also noted at this dose. In the thymus, minimal to mild decrease of cellularity in cortex and/or medulla was detected in monkeys receiving 10 mg kg−1 and 30 mg kg−1. Additionally, repeated administration of JSKN003 induced minimal to mild thyroid toxicity at 30 and 80 mg kg−1, with findings with severity graded as minimal following a 6-week recovery period at doses of 30 and 80 mg kg−1. Based on the severity of observed toxicities, the highest non-severely toxic dose (HNSTD) in cynomolgus monkeys was determined to be 30 mg kg−1.| Species | Cynomolgus monkeys |
|---|---|
| Doses | 0, 10, 30, and 80 mg kg−1 |
| Regimens | Intravenous, every 3 weeks |
| Days 1, 22, 43 (3 times in total) | |
| No. of animals | 5/sex/group (main): all dose groups |
| 2/sex/group (recovery): all dose groups | |
| Lethal dose | Not reached |
| Body weight | ≤30 mg kg−1: normal |
| 80 mg kg−1: decreased by 5–15% between day 1–14, recovery observed by later days | |
| ADA (anti-drug antibodies) | Day 43 (pre-dose): 0/5 (10 mg kg−1), 1/5 (30 mg kg−1), 0/5 (80 mg kg−1) |
| Day 85 (recovery period): 1/2 (10 mg kg−1), 0/2 (30 mg kg−1), 0/2 (80 mg kg−1) | |
| ADA titers | 312.95 (30 mg kg−1); 1407.43 (10 mg kg−1) |
| Clinical observations | Mild erythema and reduced food intake in some animals at higher doses (≥30 mg kg−1) |
| Target organs and tissues | ≥30 mg kg−1: thymus, thyroid gland |
| 80 mg kg−1: thymus, thyroid gland, lung and bronchi | |
| STD10/HNSTD | HNSTD: 30 mg kg−1 |
Conclusions
Most ADCs rely on amino- or thiol-based conjugation methods,37 resulting in heterogeneous DAR, compromised pharmacokinetics, and increased off-target toxicities.16–19 Maleimide linkers further contribute to instability, leading to premature payload release and severe side effects.20–22 To overcome these limitations, we developed JSKN003, a novel bispecific HER2-targeted ADC employing site-specific Fc glycan conjugation. JSKN003 features a consistent DAR (3.6), enhanced structural stability, improved hydrophilicity, and significantly better serum stability.To determine whether these molecular design advantages translate into therapeutic benefits, JSKN003 was evaluated in multiple in vitro and in vivo tumor models. JSKN003 exhibited robust antitumor efficacy across HER2-high (NCI-N87), HER2-low (BxPC-3), and PDX tumor models, outperforming the unconjugated antibody KN026. Notably, JSKN003 demonstrated a strong “bystander effect”, effectively eliminating adjacent HER2-negative cells, beneficial for treating heterogeneous tumors. Across multiple phase I studies, JSKN003 also demonstrated robust clinical efficacy in heavily pretreated patients with advanced solid tumors. In a pooled analysis of HER2-positive (IHC 3+) gastric, colorectal, and biliary tract tumors, the objective response rate (ORR) reached 75.0%, and the disease control rate (DCR) reached 89.3% while in solid tumors with variable HER2 expression, JSKN003 achieved an ORR of 51.1% and a DCR of 93.3%, indicating activity beyond HER2-amplified tumors.34,36
Encouraged by these therapeutic results, we next investigated JSKN003's safety and tolerability. Preclinical safety studies demonstrated that JSKN003 possesses a favorable toxicological profile compared to DS-8201a, characterized by minimal dose-dependent toxicity and superior linker stability. Consistent with these findings, clinical data from pooled phase I trials (JSKN003-101 [NCT05494918], JSKN003-102 [NCT05744427]) revealed a markedly improved safety profile. Only 6.3% of patients experienced grade ≥3 treatment-related adverse events (TRAEs), in contrast to approximately 40–50% reported for DS-8201a.33–36,38,39 Importantly, the incidence of ILD with JSKN003 was low (with no grade ≥3 events), whereas ILD occurred in 10.9% of patients receiving DS-8201a, including 1.8% grade ≥3 events and fatal cases. Hematologic toxicities were also significantly milder with JSKN003-only a single case of grade 3 anemia was observed, with no reported severe neutropenia-compared to DS-8201a, which is associated with ∼21% grade ≥3 neutropenia. Notably, no patients treated with JSKN003 required dose reductions or treatment discontinuation due to adverse events. These findings underscore the potential of glycan-based site-specific conjugation to enhance the safety and clinical manageability of antibody–drug conjugates.
Beyond its broad-spectrum efficacy and tolerability, JSKN003 also addresses a critical clinical challenge-trastuzumab resistance. With the widespread clinical use of trastuzumab, its therapeutic efficacy is limited in tumors where downstream HER2 signaling is impaired or bypassed. KN026, a bispecific antibody targeting the epitopes of both trastuzumab and pertuzumab, exhibits enhanced activity by more effectively blocking ErbB2 heterodimerization and abrogating HER2-driven signaling, even in trastuzumab-resistant cancer cell lines. Moreover, through efficient intracellular payload delivery, JSKN003 retains potent cytotoxic activity against trastuzumab-resistant tumor cells, offering a promising strategy to overcome resistance mechanisms associated with conventional HER2-targeted therapies.
Together, these findings strongly support the continued clinical development of JSKN003 as an innovative HER2-targeted ADC. Building on encouraging results from Phase I trials conducted in Australia and China, JSKN003 is currently undergoing evaluation in three pivotal Phase III studies across diverse HER2-expressing malignancies. JSKN003-302 (NCT06079983) aims to enroll 400 participants to examine efficacy and safety in HER2-low metastatic breast cancer patients who have failed previous chemotherapy treatments. JSKN003-301 (NCT06846437) is comparing JSKN003 with T-DM1 in patients with advanced HER2-positive breast cancer previously treated with trastuzumab and taxanes. Additionally, JSKN003-306 (NCT06751485) will evaluate JSKN003's effectiveness and safety against standard chemotherapy options—(doxorubicin, paclitaxel, and topotecan—in patients with platinum-resistant epithelial ovarian, primary peritoneal, or fallopian tube cancers. Collectively, these clinical advancements underscore JSKN003's potential as a transformative treatment option for diverse HER2-expressing malignancies.
Author contributions
Conceptualization: J. P. Li, T. Xu. Supervision: J. P. Li, T. Xu. Investigation: Q. Xue, J. J. Peng, W. Y. Dai, Q. S. Wu, J. B. Jiao, Y. D. Hu, W. X. Sha. Methodology: Q. S. Wu, J. B. Jiao, Y. D. Hu, W. X. Sha. Formal analysis: Q. Xue, J. J. Peng, W. Y. Dai. Data curation: Q. Xue, J. J. Peng, W. Y. Dai. Validation: W. H. Yu, S. Y. Liu, Y. Yang. Visualization: W. H. Yu, S. Y. Liu. Writing – original draft: Q. Xue, J. J. Peng. Writing – review & editing: J. P. Li, T. Xu. Funding acquisition: J. P. Li, T. Xu. All authors have read and approved the final version of the manuscript.Ethics statement for animal research
All procedures involving mice were conducted in accordance with the Guidelines for the Care and Use of Laboratory Animals of Nanjing University and were approved by the Animal Ethics Committee of Nanjing University (IACUC No. 2410008). All procedures involving cynomolgus monkeys were conducted in accordance with the Guidelines for the Care and Use of Laboratory Animals of Medicilon and were approved by the Animal Ethics Committee of Medicilon (IACUC No. 19046-23001).Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting the findings of this study are available within the main article and its ESI.† Additional supporting data may be made available from the corresponding author upon reasonable request.Acknowledgements
This work was supported by the Natural Science Foundation of Jiangsu Province, China (BK20232020, BK20240007), and the Yachen Funding for Innovation. We also gratefully acknowledge financial support from Alphamab Biopharmaceuticals Co., Ltd for experimental studies including hydrophobic interaction chromatography (HIC), size-exclusion chromatography (SEC), UHPLC analysis, toxicological studies, pharmacokinetic assessments in cynomolgus monkeys, and tumor growth inhibition assays. Additional support from Nanjing University is also acknowledged. The authors thank all team members for their contributions to experimental execution, data analysis, and manuscript preparation.References
- Z. Yi, G. Rong and Y. Guan, et al., Molecular landscape and efficacy of her2-targeted therapy in patients with her2-mutated metastatic breast cancer, npj Breast Cancer, 2020, 6, 59 CrossRef CAS.
- Y. N. Kim, Y. S. Chung and E. Park, et al., Human epidermal growth factor receptor-2 expression and subsequent dynamic changes in patients with ovarian cancer, Sci. Rep., 2024, 14, 7992 CrossRef CAS.
- A. Ooi, T. Takehana and X. Li, et al., Protein overexpression and gene amplification of her-2 and egfr in colorectal cancers: An immunohistochemical and fluorescent in situ hybridization study, Mod. Pathol., 2004, 17, 895–904 CrossRef CAS PubMed.
- C. A. Hudis, Trastuzumab-mechanism of action and use in clinical practice, N. Engl. J. Med., 2007, 357, 39–51 CrossRef CAS.
- T. T. Junttila, G. Li and K. Parsons, et al., Trastuzumab-dm1 (t-dm1) retains all the mechanisms of action of trastuzumab and efficiently inhibits growth of lapatinib insensitive breast cancer, Breast Cancer Res. Treat., 2011, 128, 347–356 CrossRef CAS PubMed.
- G. D. Lewis Phillips, G. Li and D. L. Dugger, et al., Targeting her2-positive breast cancer with trastuzumab-dm1, an antibody-cytotoxic drug conjugate, Cancer Res., 2008, 68, 9280–9290 CrossRef CAS PubMed.
- V. Diéras, D. Miles and S. Verma, et al., Trastuzumab emtansine versus capecitabine plus lapatinib in patients with previously treated her2-positive advanced breast cancer (emilia): A descriptive analysis of final overall survival results from a randomised, open-label, phase 3 trial, Lancet Oncol., 2017, 18, 732–742 CrossRef.
- Y. Endo, N. Mohan and M. Dokmanovic, et al., Mechanisms contributing to ado-trastuzumab emtansine-induced toxicities: A gateway to better understanding of adc-associated toxicities, Antibiot. Ther., 2021, 4, 55–59 CAS.
- E. A. Perez, C. Barrios and W. Eiermann, et al., Trastuzumab emtansine with or without pertuzumab versus trastuzumab with taxane for human epidermal growth factor receptor 2-positive advanced breast cancer: Final results from marianne, Cancer, 2019, 125, 3974–3984 CrossRef CAS.
- Y. Ogitani, T. Aida and K. Hagihara, et al., Ds-8201a, a novel her2-targeting adc with a novel DNA topoisomerase i inhibitor, demonstrates a promising antitumor efficacy with differentiation from t-dm1, Clin. Cancer Res., 2016, 22, 5097–5108 CrossRef CAS.
- R. Colombo and J. R. Rich, The therapeutic window of antibody drug conjugates: A dogma in need of revision, Cancer Cell, 2022, 40, 1255–1263 CrossRef CAS.
- S. Modi, C. Saura and T. Yamashita, et al., Trastuzumab deruxtecan in previously treated her2-positive breast cancer, N. Engl. J. Med., 2020, 382, 610–621 CrossRef CAS PubMed.
- J. Cortés, S.-B. Kim and W.-P. Chung, et al., Trastuzumab deruxtecan versus trastuzumab emtansine for breast cancer, N. Engl. J. Med., 2022, 386, 1143–1154 CrossRef.
- K. Shitara, Y.-J. Bang and S. Iwasa, et al., Trastuzumab deruxtecan in previously treated her2-positive gastric cancer, N. Engl. J. Med., 2020, 382, 2419–2430 CrossRef CAS PubMed.
- J. Cortés, S. A. Hurvitz and S. A. Im, et al., Trastuzumab deruxtecan versus trastuzumab emtansine in her2-positive metastatic breast cancer: Long-term survival analysis of the destiny-breast03 trial, Nat. Med., 2024, 30, 2208–2215 CrossRef.
- K. J. Hamblett, P. D. Senter and D. F. Chace, et al., Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate, Clin. Cancer Res., 2004, 10, 7063–7070 CrossRef CAS.
- Y. T. Adem, K. A. Schwarz and E. Duenas, et al., Auristatin antibody drug conjugate physical instability and the role of drug payload, Bioconjugate Chem., 2014, 25, 656–664 CrossRef CAS PubMed.
- N. S. Beckley, K. P. Lazzareschi and H. W. Chih, et al., Investigation into temperature-induced aggregation of an antibody drug conjugate, Bioconjugate Chem., 2013, 24, 1674–1683 CrossRef CAS.
- M. A. Kasper, A. Stengl and P. Ochtrop, et al., Ethynylphosphonamidates for the rapid and cysteine-selective generation of efficacious antibody-drug conjugates, Angew. Chem., Int. Ed., 2019, 58, 11631–11636 CrossRef CAS PubMed.
- N. Toda, S. Asano and C. F. Barbas 3rd, Rapid, stable, chemoselective labeling of thiols with Julia-Kocieński-like reagents: A serum-stable alternative to maleimide-based protein conjugation, Angew. Chem., Int. Ed., 2013, 52, 12592–12596 CrossRef CAS PubMed.
- Q. Wu, Q. Xue and J. Li, et al., Identification of styryl sulfonyl fluoride as a near-perfect Michael acceptor for diversified protein bioconjugations, CCS Chem., 2023, 5, 2251–2263 CrossRef CAS.
- Z. Abuhelwa, A. Alloghbi and A. Alqahtani, et al., Trastuzumab deruxtecan-induced interstitial lung disease/pneumonitis in erbb2-positive advanced solid malignancies: A systematic review, Drugs, 2022, 82, 979–987 CrossRef CAS PubMed.
- R. P. Lyon, T. D. Bovee and S. O. Doronina, et al., Reducing hydrophobicity of homogeneous antibody-drug conjugates improves pharmacokinetics and therapeutic index, Nat. Biotechnol., 2015, 33, 733–735 CrossRef CAS.
- J. R. Junutula, H. Raab and S. Clark, et al., Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index, Nat. Biotechnol., 2008, 26, 925–932 CrossRef CAS PubMed.
- H. Wei, H. Cai and Y. Jin, et al., Structural basis of a novel heterodimeric fc for bispecific antibody production, Oncotarget, 2017, 8, 51037–51049 CrossRef PubMed.
- R. van Geel, M. A. Wijdeven and R. Heesbeen, et al., Chemoenzymatic conjugation of toxic payloads to the globally conserved N-glycan of native mabs provides homogeneous and highly efficacious antibody-drug conjugates, Bioconjugate Chem., 2015, 26, 2233–2242 CrossRef CAS PubMed.
- W. Huang, J. Giddens and S. Q. Fan, et al., Chemoenzymatic glycoengineering of intact igg antibodies for gain of functions, J. Am. Chem. Soc., 2012, 134, 12308–12318 CrossRef CAS PubMed.
- Q. Zhou, J. E. Stefano and C. Manning, et al., Site-specific antibody-drug conjugation through glycoengineering, Bioconjugate Chem., 2014, 25, 510–520 CrossRef CAS PubMed.
- J. Park, S. K. Kang and W. S. Kwon, et al., Novel her2-targeted therapy to overcome trastuzumab resistance in her2-amplified gastric cancer, Sci. Rep., 2023, 13, 22648 CrossRef CAS.
- S. Zhang, W. C. Huang and P. Li, et al., Combating trastuzumab resistance by targeting src, a common node downstream of multiple resistance pathways, Nat. Med., 2011, 17, 461–469 CrossRef CAS.
- E. P. Rogakou, D. R. Pilch and A. H. Orr, et al., DNA double-stranded breaks induce histone h2ax phosphorylation on serine 139, J. Biol. Chem., 1998, 273, 5858–5868 CrossRef CAS.
- B. Li, Y. Meng and L. Zheng, et al., Bispecific antibody to erbb2 overcomes trastuzumab resistance through comprehensive blockade of erbb2 heterodimerization, Cancer Res., 2013, 73, 6471–6483 CrossRef CAS PubMed.
- C. Beecroft, B. Gao and J. Park, et al., Abstract ct179: Safety and efficacy of jskn003 in patients with advanced/metastatic solid tumors: A first-in-human, dose-escalation, multicenter, open-label, phase i study, Cancer Res., 2024, 84, CT179–CT179 CrossRef.
- X. Liu, J. Zhang and L. Shen, et al., Evaluation of the safety, pharmacokinetics, and efficacy of jskn003 in patients with advanced solid tumors: A phase i/ii clinical study, J. Clin. Oncol., 2024, 42, 3031 CrossRef.
- Q. Rao, Y. Chen and B. Gao, et al., Jskn003, a her2-targeting antibody-drug conjugate, in patients with platinum-resistant ovarian cancer: A pooled analysis of two studies, Ann. Oncol., 2024, 35, S575–S576 CrossRef.
- L. Shen, D. Liu and J. J. W. Park, et al., Evaluation of the safety and efficacy of jskn003 in patients with advanced her2-positive (ihc 3 +) solid tumors (excluding breast cancer), Ann. Oncol., 2024, 35, S527–S528 CrossRef.
- C. Dumontet, J. M. Reichert and P. D. Senter, et al., Antibody-drug conjugates come of age in oncology, Nat. Rev. Drug Discovery, 2023, 22, 641–661 CrossRef CAS.
- H. S. Rugo, G. Bianchini and J. Cortes, et al., Optimizing treatment management of trastuzumab deruxtecan in clinical practice of breast cancer, ESMO Open, 2022, 7, 100553 CrossRef CAS PubMed.
- M. Yagisawa, H. Taniguchi and T. Satoh, et al., Trastuzumab deruxtecan in advanced solid tumors with human epidermal growth factor receptor 2 amplification identified by plasma cell-free DNA testing: A multicenter, single-arm, phase ii basket trial, J. Clin. Oncol., 2024, 42, 3817–3825 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cb00096c |
| ‡ These authors contributed equally: Qi Xue, Jianjian Peng, Wenying Dai. |
| This journal is © The Royal Society of Chemistry 2025 |