
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEngineering regiospecific methylation of the pladienolides†
Emily R.
Smith‡
 a,
Dua H.
Al-Smadi‡
bc,
Yitao
Dai
b,
Manead
Khin
b,
Joanna E.
Burdette
b,
Brendan M.
Duggan
d,
James J.
La Clair
a,
Alessandra S.
Eustáquio
*b and
Michael D.
Burkart
*a
a,
Dua H.
Al-Smadi‡
bc,
Yitao
Dai
b,
Manead
Khin
b,
Joanna E.
Burdette
b,
Brendan M.
Duggan
d,
James J.
La Clair
a,
Alessandra S.
Eustáquio
*b and
Michael D.
Burkart
*a
aDepartment of Chemistry and Biochemistry, University of California, 9500 Gilman Drive, La Jolla, San Diego, California 92093, USA. E-mail: mburkart@ucsd.edu
bDepartment of Pharmaceutical Sciences, Retzky College of Pharmacy, University of Illinois Chicago, Chicago, IL 60607, USA. E-mail: ase@uic.edu
cDepartment of Medicinal Chemistry and Pharmacognosy, College of Pharmacy, Jordan University of Science and Technology, Irbid, 22110, Jordan
dSkaggs School of Pharmacy and Pharmaceutical Sciences, 9500 Gilman Drive, La Jolla, San Diego, California 92093, USA
First published on 8th May 2025
Abstract
Well-recognized for the ability to modulate spliceosome activity, the pladienolide family of polyketide natural products has been the recent subject of intense synthetic and bioactivity studies. However, our understanding of their biosynthesis remains incomplete. Here, we report the biosynthetic gene cluster of FD-895 from Streptomyces hygroscopicus A-9561 and explore the installation of a key methylation important for metabolite stability. We demonstrate the in vitro and in vivo application of an O-methyltransferase for regioselective methylation of pladienolide B at the C21 position, a post-synthase modification critical for compound stability. These findings provide a crucial next step in developing systems to engineer this important family of splicing modulatory anti-tumor agents.
Introduction
Since the first report of FD-895 (1) in 1994,1 considerable attention has focused on a family of 12-membered ring macrolides commonly known as the pladienolides (1–4, Fig. 1).2 These compounds belong to a select group of polyketides capable of modulating eukaryotic RNA splicing3,4 through binding within a pocket of the SF3b spliceosome complex.5 Potent in vitro activity and efficacy in animal models have led to clinical trial entry of two pladienolide analogues, E7107 (5)6 and H3B-8800 (6).7 A combination of off-target effects and bioavailability concerns with these analogues has been linked to metabolic stability challenges,8,9 a feature observed in independent comprehensive structure–activity relationship (SAR) studies.10 Our recent efforts with rebecsinib (17S-FD-895, 4)11 highlight the critical importance of methylation at the C21 hydroxyl group for aqueous stability. These findings suggest that the increased metabolic lability of some pladienolides (2 and 3, Fig. 1) compared to others (1 and 4, Fig. 1) can be attributed to the absence of methylation at the C21 hydroxyl group. | ||
| Fig. 1 Structures of the pladienolide family of natural products, including FD-895 (1), pladienolide B (2), and D (3) along with structures of clinically evaluated analogues E7107 (5) and H3B-8800 (6) and preclinical lead rebecsinib (17S-FD-895, 4). While FD-895 was reported first,1 the pladienolides were coined upon the isolation of 2 and 3.2 Blue shading denotes regions of SAR studies. | ||
First reported with synthetic precursors12 and later with the intact natural product,13 pladienolide B (2) undergoes an intramolecular cyclization resulting in C18 epoxide ring opening from attack by the C21 hydroxyl group, forming inactive cyclic ether (7, Fig. 2). Blocking this side reaction by methylation of the C21 hydroxyl group, as achieved naturally in 1, provides analogues like 4 with increased stability.14 The absence of the C21 hydroxyl group in 1 and 4 improves the stability of these compounds relative to 2.
 | ||
| Fig. 2 Facile decomposition of 2 arises through an intramolecular ring opening between the hydroxyl group at C21 and epoxide at C18, forming tetrahydrofuran 7. Methylation at C21 in 2 by conversion to 21-methoxypladienolide B (8). | ||
Herein we utilize a previously unknown enzyme, FddK, to elucidate an important step in FD-895 biosynthesis and prepare the first pathway engineered pladienolide analogue with potential clinical relevance.
Results and discussion
Since the FD-895 (1) producing strain was initially unavailable, we first attempted to engineer the pladienolide B (2) producer, Streptomyces platensis Mer-11107, by expressing the O-methyltransferase (O-MT)15 gene herF from the herboxidiene producer, S. chromofuscus ATCC 49982 (Fig. S1–S3 and Table S1, ESI†). However, incorporating herF into S. platensis Mer-11107 proved HerF unsuitable for O-methylation at the C21 hydroxyl group of the pladienolides; no product was detected by HPLC (Fig. S4, ESI†). Since herF is translationally coupled with the upstream herE gene that encodes an epoxidase (Fig. S1, ESI†), we also evaluated the expression of herEF. Although epoxidation to 2 was observed, we did not observe methylation (Fig. S5 and Table S2, ESI†). We turned our attention to the FD-895 (1) producer S. hygroscopicus A-9561, as it is known to methylate at C21.A high-fidelity genome sequence of S. hygroscopicus A-9561 was obtained by Nanopore sequencing (ESI). After biosynthetic gene cluster (BGC) analysis by antiSMASH,16 we compared the biosynthetic pathways of pladienolide B (Fig. 3a and b) to FD-895 (Fig. 3c and d).17 Both routes contain type I polyketide synthase (PKS) pathways, each composed of four megasynthases collectively organized into eleven modules (M): loading module (LM) to M10 (Fig. 3a and c). Both synthases bear the same domain organization and share a high sequence similarity (Fig. 3 and Fig. S6, ESI†).
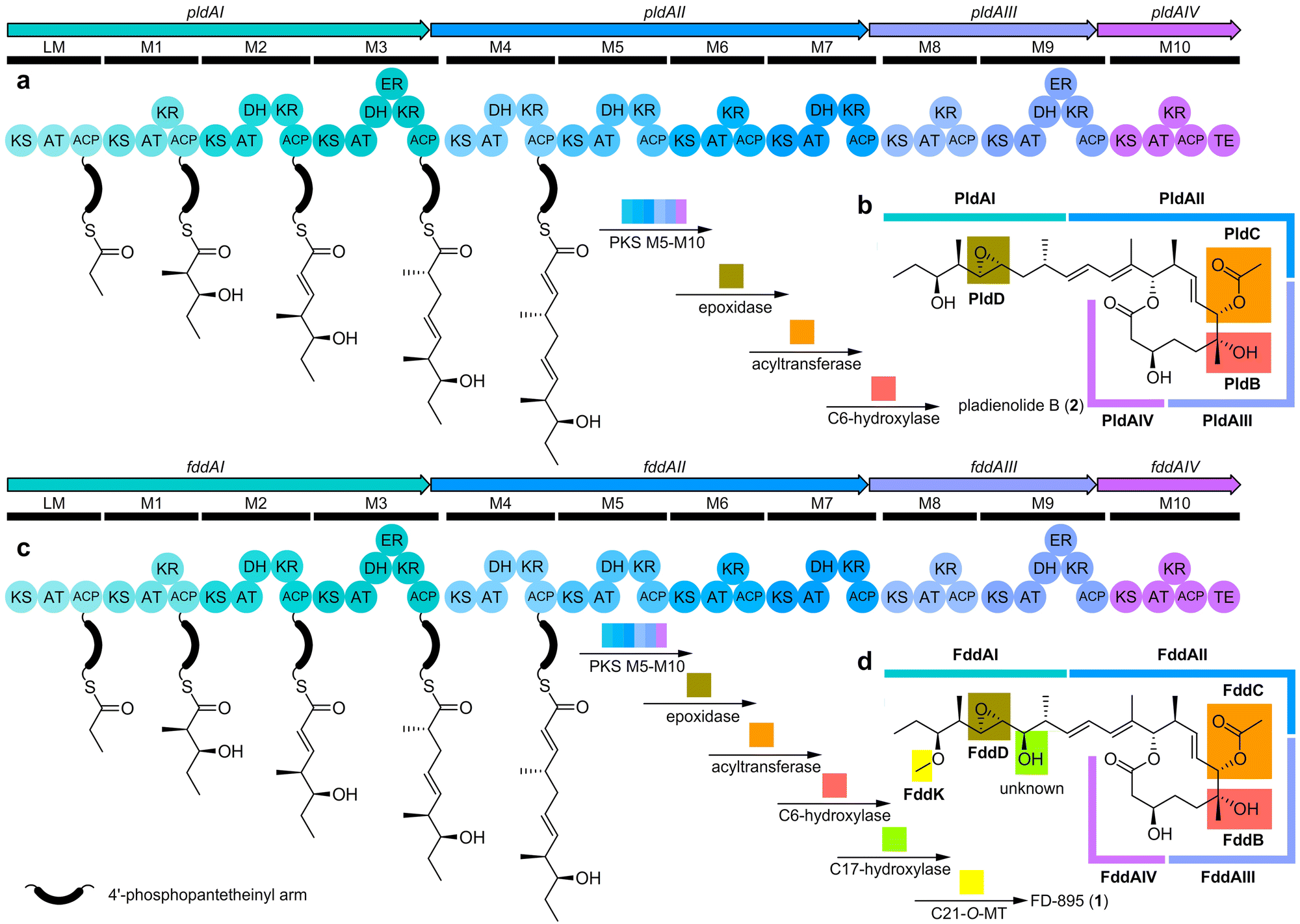 | ||
| Fig. 3 Biosyntheses of pladienolide B (2) and FD-895 (1). (a) Pladienolide B BGC from S. platensis Mer-11107 showing domain architecture for the 11 PKS modules (LM-M10). (b) Schematic depiction of the tailoring enzyme genes (pldBCD) and role of each biosynthetic enzyme in the production of 2. (c) FD-895 BGC from S. hygroscopicus A-9561 showing the domain architecture of the 11 PKS modules (LM-M10). (d) A schematic overview of the role of each biosynthetic enzyme including putative tailoring enzyme genes (fddBCD, fddK). The order of steps for the post-PKS pathways has not been established. An expanded comparison of these BGCs, including the position of each post-PKS tailoring enzyme as well as neighboring genes, is provided in Fig. S6 (ESI†). | ||
Through pld and fdd gene cluster comparison, we identified a putative O-MT, FddK, in fdd. We predicted FddK to catalyze S-adenosyl-L-methionine (SAM)-dependent O-methylation at C21, given that a fddK homologue is absent from the pladienolide pathway. The importance of studying FddK arises from its anticipated ability to engineer regiospecific methylation of pladienolides at C21, a position crucial for aqueous stability. Blast-P searches of FddK identified a close relationship to GfsG (67% amino acid sequence identity, Fig. S7, ESI†), an O-MT associated with FD-891 (10) biosynthesis (Fig. S8, ESI†) in S. graminofaciens A-8890. Kudo et al. demonstrated that recombinant GfsG catalyzes SAM-dependent O-methylation of FD-892 (Fig. S8, ESI†).18
Establishing in vitro methylation with FddK
With this data in hand, we prepared FddK by cloning the fddk gene (Twist Biosciences) into a pET28a-TEV vector containing an N-terminal His6-tag and expressing and purifying the recombinant FddK by immobilized metal affinity chromatography with Ni-NTA resin (ESI†). This procedure provided effective access to FddK, which was stored at 1–4 mg mL−1 in 50 mM K3PO4, pH 7.5, 250 mM NaCl, 10% glycerol v/v.To confirm the function of FddK, pladienolide B (2) was used as a starting material for in vitro methylation. Via combined NMR and LC–MS spectral data, we could rapidly identify methylation at C21 by detecting conversion of 2 to 21-methoxypladienolide B (8, Fig. 4b–d and Fig. S9–S11, ESI†). Enzyme activity was further screened for its dependence on temperature, pH, ionic salts, SAM concentration, and reaction time optimal to enhance in vitro production of 8. Using initial reaction conditions from Darsandhari et al.,19 we screened 50 μL aliquots containing 1 mg mL−1 FddK, 2 mM SAM, 2 mM 2, 20 mM MgCl2, 100 mM Tris pH 7.5 for 2 h at 37 °C. While NMR (Fig. S12, S13 and Tables S3, S4, ESI†) provided unequivocal validation, we found that conventional TLC analyses allowed for the most rapid evaluation. Screening from 20 °C to 50 °C at 5 °C intervals, we found that the reaction was optimal at 40 °C (Fig. S12 and Table S3, ESI†). Parallel screening returned pH 7.5 as the optimal pH, with reductions at pH 7.0 and 8.0, respectively (Fig. S13 and Table S4, ESI†). Among divalent ion salts tested (MgCl2, CaCl2, and ZnCl2), MgCl2 provided the highest yield.
 | ||
Fig. 4 Selective C21 O-methylation of pladienolide B (2). (a) Structures of 2, 8, and 7-desacetoxy-21-methoxypladienolide B (9). (b) The 1H NMR spectrum of the optimized, crude in vitro reaction depicting the C25 methyl group protons (Fig. S22, ESI†) for staring material 2 (purple) and product 8 (orange). An internal standard, 3-chloro-5-cholestene (green dot denotes the C18 methyl of 3-chloro-5-cholestene, Fig. S22, ESI†), was added at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio with 2 at the start of the reaction. (c) The 1H,13C-HSQC spectrum of purified 8 depicts the one bond correlations between H31-C31 (blue) and H21-C21 (red). (d) The 1H,13C-HMBC spectrum depicts the three bond correlations between the H31-C21 and H21-C31, demonstrating regioselective methylation of the C21 hydroxyl group by FddK. Additional LC–MS spectra of 8 along with HRMS and NMR spectra on 8 and 9 are provided in the ESI.† :1 molar ratio with 2 at the start of the reaction. (c) The 1H,13C-HSQC spectrum of purified 8 depicts the one bond correlations between H31-C31 (blue) and H21-C21 (red). (d) The 1H,13C-HMBC spectrum depicts the three bond correlations between the H31-C21 and H21-C31, demonstrating regioselective methylation of the C21 hydroxyl group by FddK. Additional LC–MS spectra of 8 along with HRMS and NMR spectra on 8 and 9 are provided in the ESI.† | ||
With reaction conditions identified, we turned to optimizing the SAM concentration. Screening from 0.2–7.0 equivalents of the starting condition concentration, we found that the optimal yield was obtained at 4.0 equivalents (TLC analysis). The fact that the reaction could not be driven to completion by the addition of increased SAM at the start or during the reaction suggested that the products may feedback inhibit FddK.20 Further screening also determined that the reaction produced the highest yield within 6 h, although TLC analysis could identify traces of product just minutes after the reaction start time.
With optimal conditions determined, we scaled the reaction up 50-fold to provide an accurate yield and validate the structure of 8. Over multiple repetitions, we observed consistent conversion reaching a 1:1 mixture of 2 and 8. The structure of 8 was validated by HRMS (Fig. S14–S20 (ESI†), m/z 568.3833 NH4+ adduct and 573.3391 Na+ adduct observed, with m/z 568.3844 and m/z 573.3398 calculated, respectively) in addition to identifying the correlation between the proton on C21 and the carbon on the methoxy group (C31), and vice versa, by 2D NMR analyses including 1H,13C-HSQC ASAP (Fig. 4c and d) and additional 1H,13C-HSQC, 1H,1H-gCOSY, and 1H,13C-HMBC ASAP data (ESI†). Quantification using an internal standard (3-chloro-5-cholestene), as shown in Fig. 4b and Fig. S22 (ESI†), allowed us to validate the in vitro production of 8 at 44.1% yield (79% based on recovery of starting material (BRSM)).
Engineering of FddK methylation in vivo
In parallel with in vitro methods, we explored the development of microbial production of 8 by incorporating fddK from S. hygroscopicus A-9561 into the pladienolide B producer S. platensis Mer-11107. Here, the fddK gene was cloned from S. hygroscopicus A-9561 into an integrative vector (pSET152) under the control of the constitutive promoter ermE* to generate pOMET011 (Table S1, ESI†). Plasmid pOMET011 was then introduced into S. platensis Mer-11107 via conjugation from E. coli S17-1. The obtained exconjugants were confirmed by PCR (Fig. S23, ESI†). HPLC (Fig. S24, ESI†) and LC–MS (Fig. S25, ESI†) analyses of culture extracts agreed with the conversion of pladienolide B (2) into a methylated (+14 Da) pladienolide B derivative in vivo. Analysis of the purified product by 1D and 2D NMR (spectral data provided in the ESI†) confirmed the production of 21-methoxypladienolide B (8). Using three replicates, we obtained titers of 2.2 ± 0.4 mg L−1 of 8 from cultures of the pOMET011 engineered strain, whose NMR and HRMS spectral data matched those obtained by the recombinantly produced enzyme (Fig. 4b and spectral data provided in the ESI†). Interestingly, complete conversion of 2 to 8 was consistently observed in vivo from the pOMET011 engineered strain (Fig. S24 and S25, ESI†). In addition to 8, we obtained trace amounts of 7-desacetoxy-21-methoxypladienolide B (9) (Fig. S21 (ESI†), validated by HRMS with m/z of 508.3400, Na+ and NH4+ adducts of 531.3292 and 526.3738, respectively) from these cultures, suggesting that the C21 methylation step can also occur prior to acetylation (Fig. 4d).Activity analyses validate 8 to possess a cytotoxicity profile comparable to existing splice modulators
Finally, we performed activity studies on the new natural product analogue 8. Screening against ovarian cancer cell lines (OVCAR3 and OV81.2), in which 2 had previously shown activity,21 indicated that 21-methoxypladienolide B (8) displays nanomolar activity comparable to both pladienolide B (2) and 17S-FD-895 (4), with 4 showing the most potent activity (Table S6, ESI†). All three compounds (2, 4 and 8) were found to be more active than Taxol (positive control), with 2–3-fold increase in ability to inhibit cell proliferation in ovarian cancer lines associated with tumors with a very low 5-year survival rate (<45%).22 Although 4 demonstrates increased potent activity compared to 8 in both cell lines tested, 2 is more potent than 4 in OV81.2 (Table S6, ESI†). This is not surprising as pladienolides demonstrate varying levels of comparable activity depending on the cell line.10 Thus, it is likely that when evaluated in other cells lines, 8 may exhibit increased cytotoxicity compared to 2 and 4. While most pladienolide activity studies to date have focused on bloodborne cancers,7,11 the fact that many other tumor types are susceptible to spliceosome modulators suggests their broad fundamental potentials.Conclusions
Here, we elucidated important steps in the biosynthesis and translation of potent spliceosome modulating natural products. We demonstrated activity of FddK as a post-PKS O-MT in FD-895 biosynthesis. Additionally, the isolation of a previously unreported intermediate 9 revealed that O-acetylation of the polyketide can occur as the last tailoring step proceeded by hydroxylation, epoxidation, and O-methylation (Fig. 4d). This finding refutes the need of O-acetylation as a critical first step for downstream enzymes recognition, contrary to previous understanding of these post-PKS systems.13 Through genome sequencing and pathway elucidation, we detailed post-PKS methylation through both strain engineering in vivo and in vitro enzymatic transformations. Enzymatic semi-synthetic methylation, currently conducted by chemical synthesis,4,7,8,23 represents a potential tool to enable access to analogues with increased stability and potency. A pathway engineering approach, that included heterologous expression of FddK, led to maximal conversion of 2in vivo, providing efficient production of 8. Engineered compound 8 displays antitumor activity in vitro, validating a new splice-modulating analogue and offering a biological platform to access novel bioactive polyketides. Efforts are now underway to optimize the engineered strains as well as develop methods to design producer organisms that can accommodate oxidation at the C17 position of pladienolides.Experimental
General experimental methods and reagents
Chemical solvents and reagents were obtained from Acros Organics, Alfa Aesar, Chem-Impex Int., Fischer Scientific, Fluka, Oakwood Chemical, Sigma-Aldrich, Spectrum Chemical Mfg. Corp., or TCI Chemicals. Solvents used for extraction were ACS grade. Restriction enzymes were acquired from New England Biolabs and Thermo Fisher Scientific. Gene inserts were purchased from Twist Bioscience. Oligonucleotide primers were synthesized by Sigma-Aldrich. S-Adenosyl-L-methionine was purchased from New England Biolabs and Sigma-Aldrich. Deuterated NMR solvents were obtained from Cambridge Isotope Laboratories. Unless otherwise noted, all reactions were conducted in ½ dram vials equipped with a Teflon lined cap and a 3 mm stir bar. Analytical Thin Layer Chromatography (TLC) was performed on Silica Gel 60 F254 precoated glass plates (EM Sciences). Visualization was achieved with UV light and/or an appropriate stain (ceric ammonium molybdate). NMR spectra were recorded on a Bruker Avance 300, a JEOL400, a Varian VX500 (equipped with an XSens Cold probe), a Bruker Avance III 600, or a Bruker Avance 800 (equipped with a triple resonance TXO cryoprobe) spectrometer. Chemical shift δ values for 1H and 13C spectra are reported in parts per million (ppm) and multiplicities are abbreviated as s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br = broad. 13C-NMR spectra were recorded with proton decoupling. FID files were processed using MestraNova 12.0.3 (MestreLab Research) and TopSpin 4.3 (Bruker). Electrospray (ESI) mass spectrometric analyses were performed using a ThermoFinnigan LCQ Deca spectrometer. A Thermo Fisher Scientific LTQ Orbitrap XL mass spectrometer was used for high-resolution electrospray ionization mass spectrometry analysis (HR-ESI-MS). Spectral data and procedures are provided for all new compounds and copies of select spectra have been provided in the ESI.†Expression of herF and herEF in S. platensis
Streptomyces chromofuscus ATCC 49982 genomic DNA (gDNA) was isolated using GenElute Bacterial Genomic DNA Kit (Sigma-Aldrich) and used as template for amplification of a 1077-bp herF gene fragment or a 2558-bp fragment containing herE and herF genes. Q5 high-fidelity DNA polymerase (NEB) with Q5 high GC enhancer was used for PCR amplification. Primer sequences are given in Table S1 (ESI†).Construction of pYD001 used for herF expression
Primer pair P1_NdeI_herF_F/P2_XbaI_herF_R was used to amplify the herF gene from S. chromofuscus gDNA. The PCR mixture (50 μL) consisted of ∼100 ng of gDNA, 0.2 mM of each dNTP, 250 nM of each primer, and 0.02 U μL−1 Q5 high-fidelity DNA polymerase (NEB) in 1× Q5 reaction buffer and 1× Q5 high GC enhancer supplied with the enzyme and using the following thermal cycling parameters: 60 s at 98 °C; 30 cycles of 98 °C for 10 s, 69 °C for 30 s and 72 °C for 30 s; with a final extension at 72 °C for 120 s; and hold at 10 °C. Primers P1_NdeI_herF_F and P2_XbaI_herF_R introduced the restrictions sites XbaI and NdeI into the final PCR product. The PCR product was co-digested with XbaI and NdeI (NEB) in 1× CutSmart buffer supplied with the enzymes and ligated into the same sites of pTA004 (a pSET152-based vector containing the constitutive ermE* promoter)24–26 using T4 DNA ligase (Thermo Fisher Scientific) to yield pYD001 after transformation into E. coli DH5α. The pYD001 construct was confirmed by restriction digest and Sanger sequencing.Construction of pYD005 used for herEF expression
pYD005 was constructed as described for pYD001 with minor modifications. The primer pair P1_herEF_F and P2_herEF_R was used to amplify a DNA fragment containing both herE and herF genes. The PCR (50 μL) was as for pYD001, except that the annealing temperature was 66 °C and the extension step (72 °C) was done for 90s. Primers P1_herEF_F and P2_herEF_R introduced the restrictions sites XbaI and NdeI into the final PCR product. The PCR product was co-digested with XbaI and NdeI enzymes and was ligated into the same sites of pTA004. The ligation product was transformed into E. coli DH5α to yield pYD005. The pYD005 construct was confirmed by restriction digest and Sanger sequencing.Transfer of herF or herEF into S. platensis
Plasmids pYD001 and pYD005 containing herF and herEF, respectively, were transferred into S. platensis FERM BP-7812 via conjugation from E. coli S17 as previously reported with some modifications.17E. coli S17 with either pYD001 or pYD005 was cultured in 10 mL LB containing apramycin (50 μg mL−1) at 37 °C, 200 rpm, to an OD600 of 0.4–0.6 before harvesting cells by centrifugation at 4000 × g, 4 °C. Cells were washed twice with fresh LB to remove antibiotics prior to resuspension in 2 mL LB. Around 108S. platensis spores were harvested by centrifugation at 8000 × g, 4 °C and the supernatant was discarded. The spore pellet was carefully resuspended in 0.5 mL of E. coli S17 cell suspension and plated onto four ISP4 plates without antibiotics (2 μL, 10 μL, 100 μL, and the rest; when platting samples < 50 μL, 100 μL LB was used to help spread the sample on the plate). Negative control plates were prepared by plating only E. coli S17 cell suspension or only S. platensis wild-type spore suspension onto ISP4 plates. After plates were incubated at 30 °C for 16–20 h, each plate was overlaid with 1 mL water containing nalidixic acid at 25 μg mL−1 (selectively kills E. coli) and apramycin at 50 μg mL−1 (for plasmid selection). Conjugation plates with evenly spread antibiotics were incubated at 30 °C for about a week. Single colonies were streaked onto ISP4 plates containing nalidixic acid (25 μg mL−1) and apramycin (50 μg mL−1) to confirm apramycin resistance (apraR) and to isolate exconjugants. Single apraR clones with black spores formed, were picked from the ISP4 plates, and inoculated into 250 mL Erlenmeyer flasks containing stainless-steel springs (to increase aeration and separation of cell aggregates) and 25 mL TSB medium containing nalidixic acid (25 μg mL−1) and apramycin (50 μg mL−1). After flasks were incubated at 220 rpm, 30 °C for two days, cells from 2 mL aliquots were harvested and used to isolate gDNA with the GenElute Bacterial Genomic DNA Kit (Sigma-Aldrich) and to streak on ISP2 plates containing no antibiotics to check for contamination and morphology. The gDNA was used as template in PCR below:PCR was used to confirm the presence of the herF gene in exconjugants using primers P1_herF_noext_F and P2_herF_noext_R in reactions (20 μL) containing 0.2 mM each dNTP, 3% DMSO, 250 nM (each) primer, 100 ng gDNA, and 1.25 U DreamTaq DNA polymerase (Thermo Fisher Scientific) in the buffer supplied with enzyme. Thermocycling conditions were initial denaturation for 3 min at 95 °C; amplification for 30 cycles (95 °C for 30 s, 53 °C annealing for 30 s, 72 °C for 30 s); a final extension for 12 min at 72 °C, and an infinite hold at 10 °C. The PCR results were visualized via gel electrophoresis at 110 V for 60 min (Fig. S2, ESI†).
PCR was used to confirm the presence of herEF genes in exconjugants using primers P1_herEF_noext_F and P2_herEF_noext_R in reactions (20 μL) containing 0.2 mM each dNTP, 3% DMSO, 250 nM (each) primer, 100 ng gDNA, and 1.25 U DreamTaq DNA polymerase (Thermo Fisher Scientific) in the buffer supplied with enzyme. Thermocycling conditions were identical to that of the herF PCR with the annealing temperature at 53 °C. The PCR results were visualized via running gel electrophoresis at 110 V for 60 min (Fig. S3, ESI†).
S. platensis Mer-11107 cultivation
The production protocol was conducted according to Machida et al.17 The S. platensis Mer-11107 seed culture was obtained by inoculating either 50 μL spore suspension into a 250 mL Erlenmeyer flask containing 25 mL TSB medium and a stainless-steel spring (no antibiotics added). After flasks were incubated at 220 rpm, 30 °C for two days, 1 mL of the seed culture was transferred into a 250 mL Erlenmeyer flask containing 60 mL SPCGB medium (5% soluble starch, 3% Pharmamedia, 2% β-cyclodextrin, 0.1% CaCO3, 1% glucose, pH 7.5) and without a spring. After each flask was incubated at 200 rpm, 25 °C for five days, the culture (50 mL left due to evaporation) was collected.Compound extraction
Production culture (25 mL) was mixed with 1:1 ratio DI water and transferred to a separatory funnel. Hexanes (60 mL) was added to the funnel and mixed thoroughly. The organic layer was removed, and the aqueous layer was extracted with EtOAc (3 × 60 mL). The combined organic phases were dried by rotary evaporation. Acetonitrile was used to dissolve the crude extract, which was transferred to a 20 mL glass vial and dried under reduced pressure. Dried extracts were kept at −20 °C until analysis (3 × 6 mL acetonitrile and dried by rotary evaporation).
Genome sequencing of S. hygroscopicus A-9561
S. hygroscopicus A-9561 mycelia were cultured in liquid suspension by inoculation of spore suspension into streptomyces ISP2 liquid media and grown at 30 °C on a rotary shaker (200 rpm) for 7 days. The culture was centrifuged in sterile test tubes at 2000 rpm and provided to SeqCenter (Pittsburgh, PA) for DNA extraction and sequencing by Nanopore trimmed (long) reads, Illumina read pairs, genome assembly, and annotation. Quality control and adapter trimming was performed with bcl-convert (https://support-docs.illumina.com/SW/BCL_Convert/Content/SW/FrontPages/BCL_Convert.html) and porechop (https://github.com/rrwick/Porechop) for Illumina and ONT sequencing respectively. Sequencing metrics were as follows: total length 10158679, 34 contigs, largest contig 3766290, and 71.52% GC. Hybrid assembly with Illumina and ONT reads was performed with Unicycler.27 Assembly statistics were recorded with QUAST.28 Assembly annotation was performed with Prokka.29 The following versions were used with default parameters: bcl-convert 3.9.3, porechop 0.2.3_sequan2.1.1, unicycler 0.4.8, quast 5.0.2, prokka 1.14.5 with rfam, and guppy 5.0.16 with effbat8. The assembled genome was deposited into the NCBI genome database.
Cloning, expression, and purification of FddK
The codon-optimized insert containing the fddK gene was flanked by NdeI and XhoI restriction sites. Following the NdeI/XhoI double digestion of the fddK insert and pET28a-TEV plasmid (using the New England Biolabs (NEB) double digest protocol), fddK was cloned into pET28a-TEV then transformed into DH5α E. coli. Cloning was confirmed using Sanger Sequencing through Azenta Life Sciences. The pET28a-TEV-fddK plasmid was then transformed into BL21 E. coli and cells were grown at 150 rpm in Luria Bertani (LB) media supplemented with a final concentration of 0.1 M kanamycin at 37 °C for heterologous expression of fddK. Once an OD600 value of 0.7–0.8 was reached, cells were chilled on ice then induced with isopropyl β-D-1-thiogalactopyranoside (IPTG) to a final concentration of 0.5 mM and finally grown at 16 °C for 18 h. The cells were collected using centrifugation (45 min, 2000 rpm, 4 °C), resuspended in phosphate lysis buffer (50 mM K3PO4, 250 mM NaCl, 10% glycerol v/v, pH 7.5), and finally disrupted by sonication. Cell lysate was isolated by two rounds of centrifugation (20 min, 5000 rpm, 4 °C) in which lysate collected from the first round was re-centrifuged. The target FddK protein was purified using metal-affinity chromatography with Ni-NTA resin for polyhistidine (His6)-tag purification. FddK was eluted with subsequent buffer washes (5 mL, 7 column volumes) of increasing imidazole concentrations (0 mM, 10 mM, 50 mM, 100 mM, and 250 mM). For sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) analysis, 12% (w/v) polyacrylamide gel was used, and protein bands were visualized using Coomassie Brilliant Blue dye. Imidazole was removed via dialysis in the lysis buffer (16 h, 4 °C) using 10 K MWCO SnakeSkin dialysis tubing. FddK was concentrated (3.36 mg mL−1) in the lysis buffer using centrifugation (3000 rpm, 4 min, 4 °C).Screening and optimization of FddK methylation of pladienolide B (2)
Initially, reaction conditions were replicated from Darsandhari et al.19 as follows: 0.11 mM FddK, 2 mM S-adenosyl-L-methionine (SAM) and pladienolide B (2, substrate), 20 mM MgCl2, in 100 mM Tris-base buffer pH 7.5 with 100 μL reaction volume. Most reactions were run at 37 °C for 2 h and then quenched with a 2× volume of cold methanol. Reactions were monitored via thin layer chromatography (TLC) with TLC solvent conditions of 11:9 acetone/hexanes. The “standard” reaction temperature was increased to 40 °C based on proton nuclear magnetic resonance (1H NMR) spectroscopy data indicating in vitro reactions run at 40 °C produce a slightly higher yield than those run at 37 °C (Fig. S3, ESI†). Data from control reactions lacking enzyme and lacking SAM are shown in Fig. S26 and S27 (ESI†), respectively.
Scaled up enzymatic production of 21-methoxypladienolide B (8)
We scaled up our enzymatic reaction by fifty-fold to produce sufficient material for structure elucidation. Using multiple repeats at a 5 mL scale, we were able to obtain 2.429 mg of 8 by treating 5.3671 mg 2 with FddK by incubating 0.1041 mM FddK, 2 mM S-adenosyl-L-methionine (SAM), 20 mM MgCl2, in 100 mM Tris-base buffer pH 7.5 for 40 °C for 2 h and then quenching with a 2× volume of cold methanol. The crude product was obtained by extraction with EtOAc (3 × 5 mL), drying with Na2SO4, and solvent removal by rotary evaporation. Pure 8 (2.4 mg, 44%) was obtained after preparative TLC (20 × 20 plate, 25 mm) eluting with 11:9 acetone/hexanes. We also recovered (2.3 mg, 44%) 2via preparative TLC.
Cultivation conditions for S. platensis Mer-11107 engineered with FddK
Escherichia coli DH5α was used for cloning pSET152-based vectors.30 while E. coli S17 was used for conjugation. Both E. coli strains were routinely cultured in Difco Luria-Bertani (LB) medium or agar at 37 °C. Apramycin at 50 μg mL−1 was used for selection of E. coli harboring pSET152-based vectors. Solid cultures of Streptomyces platensis were prepared on Difco International Streptomyces Project-4 (ISP-4) agar and incubated at 30 °C until sporulation was observed, generally after 14 days. Liquid cultures of S. platensis were prepared in Bacto Tryptic Soy Broth (TSB) and cultivated at 30 °C and 220 rpm for two days. For selection of S. platensis exconjugants containing pSET152-based vectors, the medium was supplemented with apramycin and nalidixic acid at 50 μg mL−1 and 25 μg mL−1, respectively.17,30S. platensis production cultures of both the wild-type (FERM BP-7812) and the exconjugants were done according to protocols mentioned before.17 All strains were cryo-preserved in 20% glycerol at −80 °C.Plasmid construction for S. platensis Mer-11107 engineered with FddK
S. hygroscopicus A-9561 genomic DNA (gDNA) was used as a template for the amplification of gene fddK, an 813-bp fragment putatively encoding an O-methyltransferase. The gDNA was isolated using the GenElute Bacterial Genomic DNA Kit (Sigma-Aldrich). A Bio-Rad T100 thermal cycler was used for PCR. PCR conditions were (50 μL): 100 ng gDNA, 0.5 μM of each primer P3 P_O-metase_F and P4 P_O-metase_R, 0.2 mM dNTP mix (Thermo Scientific), 1× Q5 High GC Enhancer (New England Biolabs), 0.02 U μL−1 Q5 High Fidelity DNA polymerase with 1× Q5 reaction buffer (New England Biolabs). The PCR cycling conditions were: 60 s at 98 °C; 30 cycles of 98 °C for 10 s, 70 °C for 30 s, and 72 °C for 30 s; final extension for 2 min at 72 °C; and an indefinite hold at 10 °C. The replicative vector pUWL20125 was used as a template for the amplification of the ermE* constitutive promoter.25,26 PCR conditions were the same as above but using primer pair P1 P_ermE*_F and P2 P_ermE*_R and an annealing temperature of 69 °C instead of 70 °C. Splicing by overlap extension (SOE)-PCR was used to fuse the promoter and gene fragments together with the following reaction (50 μL): purified PCR products 1 and 2 (∼100 ng), 0.2 mM dNTP mix, 1× Q5 High GC Enhancer, 0.02 U μL−1 Q5 polymerase with 1× Q5 reaction buffer (New England Biolabs). Extension reactions were performed using the following parameters: 60 s at 98 °C; 5 cycles of 98 °C for 10 s, 69 °C for 30 s, and 72 °C for 30 s; and a final hold at 10 °C. The primer pair P1 P_ermE*_F and P4 P_O-metase_R was added to the extension reaction at 0.1 μM concentration before starting amplification: 60 s at 98 °C; 30 cycles of 98 °C for 10 s, 68 °C for 30 s, and 72 °C for 30 s; and a final extension for 10 min at 72 °C. The SOE-PCR product was cloned into the pSET152 vector using restriction-ligation. Primers P1 P_ermE*_F and P4 P_O-metase_R introduced the restrictions sites BglII and XbaI into the final product of the SOE-PCR. Primer sequences are given in Table S1 (ESI†). The product was co-digested with BglII and XbaI (New England Biolabs) enzymes and then ligated using T4 DNA ligase (Thermo Fisher Scientific) into the same sites of pSET152 vector. The ligation mixture was chemically transformed into E. coli DH5α yielding pOMET011. The pOMET011 plasmid was confirmed by restriction digest and whole plasmid sequencing (Primordium).Transfer of O-methyltransferase fddK into S. platensis Mer-11107
Plasmid pOMET011 containing the fddK gene was subsequently transferred into E. coli S17.1 and S. platensis Mer-11107 via electroporation and conjugation, respectively. Nalidixic acid (25 μg mL−1) and apramycin (50 μg mL−1) were used to isolate exconjugants. PCR was used to confirm the presence of the ermE* promoter and fddK gene in exconjugants using primers P_ermE*_F and P_O-metase_R (Fig. S1, ESI†).S. platensis engineered with FddK cultivation and extraction
The production protocol was conducted according to ref. 4. The S. platensis seed culture was obtained by inoculating either 50 μL spore suspension (108 spores) or 100 μL mycelium cryostock into a 250 mL Erlenmeyer flask containing 25 mL TSB medium without antibiotics and a stainless-steel spring. After flasks were incubated at 220 rpm, 30 °C for two days, 1 mL of the seed culture was transferred into a 250 mL Erlenmeyer flask (without a spring) containing 60 mL SPCGB medium. Production cultures were incubated at 200 rpm, 25 °C for five days. A whole culture extraction method was used. The production culture was mixed with 1:1 ratio DI water and transferred to a separatory funnel. The culture was extracted three times with 1:1 ratio of EtOAc. The combined EtOAc extract was dried in a round-bottom flask using a rotary evaporator. To defat the sample, 50 mL hexane was added to the round-bottom flask, the hexane was swirled a couple of times and then removed from the flask using a pipette and the extract dried using the rotary evaporator. Acetonitrile was used to dissolve the crude extract, which was transferred to a 20 mL glass vial and dried under reduced pressure. Dried extracts were kept at −20 °C until analysis.
HPLC and LC/MS analyses
The crude extract was dissolved in methanol to reach 1 mg mL−1 concentration before being analyzed by HPLC and LC–MS. HPLC analyses were performed on an Agilent 1260 Infinity system equipped with a Kinetex C18 reverse-phase column (150 × 4.6 mm, 5 μm particle size, 100 Å pore size, Phenomenex) and a SecurityGuard Ultra Column Guard with a C18 stationary phase (4.6 mm ID). Elution was performed in methanol (solvent A) and water (solvent B) at a flow rate of 1 mL min−1 with a solvent gradient as follows: initial hold at 40% A for 5 min (equilibration), linear gradient from 40 to 100% A for 26 min, hold at 100% A for 4 min, linear gradient from 100 to 40% A for 1 min, and terminal hold at 40% A for 4 min. The detection wavelength range was 200–600 nm, and chromatograms were extracted at λ at 240 nm. The pladienolide B retention time was 15 min according to an authentic standard (Cayman Chemical) (Fig. S4 and S5, ESI†). The FD-895 retention time was 13.6 min according to the synthetic standard. Further analysis of the extract was performed using liquid chromatography–mass spectrometry (LC–MS) to confirm the production of 21-methoxypladienolide B (8). At UIC, we used Bruker Compact Quadrupole Time of Flight (qToF) which couples an ultra-high-performance liquid chromatography (UHPLC) to a qToF mass spectrometer (MS) with electrospray ionization (ESI). Additionally, a Waters Acquity LC–MS with a single SQ detector run with MassLynx software was used for data acquisition and analysis at UC San Diego.Cytotoxicity analyses
Human ovarian cancer cells OVCAR3 and OV81.2 were used in this cytotoxicity assay. Both cell lines were propagated at 37 °C in 5% CO2, with OVCAR3 and OV81.2 in RPMI 1640 medium or DMEM respectively, both supplemented with fetal bovine serum (10%), penicillin (100 units per mL), and streptomycin (100 μg mL−1). Cells in log phase growth were harvested by trypsinization followed by two washes to remove all traces of enzyme. A total of 5000 cells were seeded per well of a 96-well clear, flat-bottom plate (Microtest 96, Falcon) and incubated overnight (37 °C in 5% CO2). Samples dissolved in DMSO were then diluted and added to the appropriate wells. The cells were incubated in the presence of test substance for 72 h at 37 °C and evaluated for viability with a commercial absorbance assay (CellTiter 96 Aqueous One Solution Cell Proliferation Assay, Promega Corp, Madison, WI) that measured viable cells.Author contributions
E. R. S., D. H. A., J. J. L., A. S. E. and M. D. B. conceived the project. E. R. S., J. J. L and M. D. B. conducted the in vitro studies. D. H. A., Y. D. and A. S. E. conducted the in vivo engineering studies. M. K. and J. B. conducted the activity assays. J. J. L. and B. M. D. collected the NMR data. E. R. S., B. M. D. and J. J. L. processed and assigned the NMR spectra. E. R. S. and D. H. A. conducted the LC–MS analyses. E. R. S. and J. J. L. collected and processed the HRMS data. All authors analyzed the data. The manuscript was written by E. R. S., D. H. A., B. M. D., J. J. L, A. S. E and M. D. B. with contributions and proofreading from all authors.Data availability
Copies of NMR and MS spectra have been provided in the ESI.† Raw data files are openly free upon request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the American Type Culture collection for Streptomyces chromofuscus ATCC 49982, the International Patent Organism Depositary of the National Institute of Technology and Evaluation, Japan, for S. platensis Mer-11107 (FERM BP-7812), and Prof. Kazuo Shin-ya (AIST, Japan) for S. hygroscopicus A-9561. Financial support for this work was provided by NIH R21CA280558 to M. D. B. and A. S. E., NIH P01CA125066 to J. E. B., and startup funds from the Department of Pharmaceutical Science, College of Pharmacy, University of Illinois at Chicago to A.S.E. Emily R. Smith was supported in part by the Chemistry-Biology Interfaces Training Grant, NIH Grant T32GM146648. We thank Y. Su for assistance with acquisition of mass spectral data.Notes and references
- M. Seki-Asano, T. Okazaki, M. Yamagishi, N. Sakai, Y. Takayama Y, K. Hanada, S. Morimoto, A. Takatsuki and K. Mizoue, J. Antibiot., 1994, 47, 1395–1401 CrossRef CAS PubMed.
- T. Sakai, N. Asai, A. Okuda, N. Kawamura and Y. Mizui, J. Antibiot., 2004, 74, 180–187 CrossRef PubMed.
- T. Schneider-Poetsch, J. K. Chhipi-Shrestha and M. Yoshida, J. Antibiot., 2021, 74, 603–616 CrossRef CAS PubMed.
- B. León, M. K. Kashyap, W. C. Chan, K. A. Krug, J. E. Castro, J. J. La Clair and M. D. Burkart, Angew. Chem., Int. Ed., 2017, 56, 12052–12063 CrossRef CAS PubMed; J. Sim, E. Jang, H. J. Kim and H. Jeon, Molecules, 2021, 26, 5938 CrossRef PubMed; N. A. Larsen, Subcell. Biochem., 2021, 96, 409–432 Search PubMed; J. C. Lin, Int. J. Mol. Sci., 2017, 19, 75 CrossRef PubMed; D. Pham and K. Koide, Nat. Prod. Rep., 2016, 33, 637–647 RSC.
- C. Cretu, A. A. Agrawal, A. Cook, C. L. Will, P. Fekkes, P. G. Smith, R. Lührmann, N. Larsen, S. Buonamici and V. Pena, Mol. Cell, 2018, 70, 265–273 CrossRef CAS PubMed.
- D. S. Hong, R. Kurzrock, A. Naing, J. J. Wheler, G. S. Falchook, J. S. Schiffman, N. Faulkner, M. J. Pilat, J. O’Brien, P. LoRusso and I. Aphase, Invest. New Drugs, 2014, 32, 436–444 CrossRef CAS PubMed.
- D. P. Steensma, M. Wermke, V. K. Klimek, P. L. Greenberg, P. Font, R. S. Komrokji, J. Yang, A. M. Brunner, H. E. Carraway, L. Ades, A. Al-Kali, J. M. Alonso-Dominguez, A. Alfonso-Piérola, C. C. Coombs, H. J. Deeg, I. Flinn, J. M. Foran, G. Garcia-Manero, M. B. Maris, M. McMasters, J.-B. Micol, J. P. De Oteyza, F. Thol, E. S. Wang, J. M. Watts, J. Taylor, R. Stone, V. Gourineni, A. Marino, A. J. Yao, B. Destenaves, X. Yuan, K. Yu, S. Dar, L. Ohanjanian, K. Kuida, J. Xiao, C. Scholz, A. Gualberto and U. Platzbecker, Leukemia, 2021, 35, 3542–3550 CrossRef CAS PubMed.
- W. C. Chan, B. León, K. A. Krug, A. Patel, J. J. La Clair and M. D. Burkart, ACS Med. Chem. Lett., 2018, 9, 1070–1072 CrossRef CAS PubMed.
- D. Zhang and F. Meng, ChemMedChem, 2020, 15, 2098–2120 CrossRef CAS PubMed.
- W. C. Chan, K. A. Trieger, J. J. La Clair, C. H. M. Jamieson and M. D. Burkart, J. Med. Chem., 2023, 66, 6577–6590 CrossRef CAS PubMed.
- L. A. Crews, W. Ma, L. Ladel, J. Pham, L. Balaian, S. K. Steel, P. K. Mondala, R. H. Diep, C. N. Wu, C. N. Mason, I. van der Werf, I. Oliver, E. Reynoso, G. Pineda, T. C. Whisenant, P. Wentworth, J. J. La Clair, Q. Jiang, M. D. Burkart and C. H. M. Jamieson, Cell Stem Cell, 2023, 30, 250–263 CrossRef CAS PubMed.
- A. L. Mandel, B. D. Jones, J. J. La Clair and M. D. Burkart, Bioorg. Med. Chem. Lett., 2007, 17, 5159–5164 CrossRef CAS PubMed.
- T. J. Booth, J. A. Kalaitzis, D. Vuong, A. Crombie, E. Lacey, A. M. Piggott and B. Wilkinson, Chem. Sci., 2020, 11, 8249–8255 RSC.
- V. Villa, M. K. Kashyap, D. Kumar, T. J. Kipps, J. E. Castro, J. J. La Clair and M. D. Burkart, J. Med. Chem., 2013, 56, 6576–6582 CrossRef CAS PubMed; M. A. Skiba, M. M. Bivins, J. R. Schultz, S. M. Bernard, W. D. Fiers, Q. Dan, S. Kulkarni, P. Wipf, W. H. Gerwick, D. H. Sherman, C. C. Aldrich and J. L. Smith, ACS Chem. Biol., 2018, 13, 3221–3228 CrossRef PubMed.
- D. Yu, F. Xu, S. Zhang, L. Shao, S. Wang and J. Zhan, Bioorg. Med. Chem. Lett., 2013, 23, 5667–5670 CrossRef CAS PubMed.
- K. Blin, S. Shaw, A. M. Kloosterman, Z. Charlop-Powers, G. P. van Wezel, M. H. Medema and T. Weber, Nucleic Acids Res., 2021, 49(W1), W29–W35 CrossRef CAS PubMed.
- K. Machida, A. A. Kazuhiro, T. Susumu, T. Toshio, A. Yasuhide, M. Yoshida and H. Ikeda, Biosci., Biotechnol., Biochem., 2008, 72, 2946–2952 CrossRef CAS PubMed.
- K. Kudo, M. Atsushi, M. Kazutoshi and T. Eguchi, ChemBioChem, 2010, 11, 1574–1582 CrossRef PubMed; K. Kudo, K. Koichi, F. Takashi, Y. Hiroto, M. Atsushi, K. Akiko, N. Mario, M. Akimasa and T. Eguchi, ChemBioChem, 2016, 17, 233–238 CrossRef PubMed.
- S. Darsandhari, D. Dhakal, B. Shrestha, P. Parajuli, J.-H. Seo, T.-S. Kim and J. K. Sohng, Enzyme Microb. Technol., 2018, 113, 29–36 CrossRef CAS PubMed.
- D. Popadić, D. Mhaindarkar, M. H. N. Dang Thai, H. C. Hailes, S. Mordhorst and J. N. Andexer, RSC Chem. Biol., 2021, 2, 883–891 RSC.
- Y. Mizui, T. Sakai, M. Iwata, T. Uenaka, K. Okamoto, H. Shimizu, T. Yamori, K. Yoshimatsu and M. Asada, J. Antibiot., 1994, 47, 188–196 Search PubMed.
- M. M. Cavalluzzi, M. Viale, N. P. Rotondo, V. Ferraro and G. Lentini, Anticancer Agents Med. Chem., 2024, 24, 637–647 CrossRef CAS PubMed.
- W. C. Chan, J. J. La Clair, B. León, K. A. Trieger, M. Q. Slagt, M. T. Verhaar, D. U. Bachera, M. T. Rispens, R. M. Hofman, V. L. de Boer, R. van der Hulst, R. Bus, P. Hiemstra, M. L. Neville, K. A. Mandla, J. S. Figueroa, C. M. H. Jamieson and M. D. Burkart, Cell Rep. Phys. Sci., 2020, 1, 100277 CrossRef CAS.
- M. Bierman, R. Logan, K. O’Brien, E. T. Seno, R. Nagaraja Rao and B. E. Schoner, Gene, 1992, 116, 43–49 CrossRef CAS PubMed.
- Practical Streptomyces Genetics, ed. Kieser, T., Innes, Norwich, 2000 Search PubMed.
- M. J. Bibb, G. R. Janssen and J. M. Ward, Gene, 1985, 38, 215–226 CrossRef CAS PubMed.
- R. R. Wick, J. M. Judd, C. L. Gorrie and K. E. Holt Unicycler, PLoS Comput. Biol., 2018, 13, e1005595 CrossRef PubMed.
- A. Gurevich, V. Saveliev, N. Vyahhi and G. Tesler, Bioinformatics, 2013, 29, 1072–1075 CrossRef CAS PubMed.
- T. Seemann, Bioinformatics, 2014, 30, 2068–2069 CrossRef CAS PubMed.
- M. J. Bibb, J. White, J. M. Ward and G. R. Janssen, Mol. Microbiol., 1994, 14, 533–545 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures and copies of NMR spectral data. See DOI: https://doi.org/10.1039/d5cb00068h |
| ‡ Denotes co-first authors. |
| This journal is © The Royal Society of Chemistry 2025 |