
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA state-of-the-art view: G-quadruplex-targeting for platinum complexes’ treatment of tumors
Jinrong
Yang
a,
Yu
Chen
 *a and
Hui
Chao
*ab
*a and
Hui
Chao
*ab
aMOE Key Laboratory of Bioinorganic and Synthetic Chemistry, State Key Laboratory of Anti-Infective Drug Discovery and Development, Guangdong Basic Research Center of Excellence for Functional Molecular Engineering, School of Chemistry, Sun Yat-Sen University, Guangzhou, 510006, P. R. China. E-mail: chenyu63@mail.sysu.edu.cn; ceschh@mail.sysu.edu.cn
bMOE Key Laboratory of Theoretical Organic Chemistry and Functional Molecule, School of Chemistry and Chemical Engineering, Hunan University of Science and Technology, Xiangtan, 400201, P. R. China
First published on 29th April 2025
Abstract
Cisplatin and its analogs are extensively utilized as metal-based anticancer agents in clinical settings due to their mechanism of action, which involves targeting genomic double-stranded DNA to induce cytotoxicity in cancer cells. However, the associated severe side effects and DNA damage repair-inducing drug resistance present significant challenges. In recent years, G-quadruplex nucleic acids, formed through the self-assembly of guanine-rich nucleic acid sequences, have emerged as a compelling target for the design of novel anticancer therapeutics. The strategic design of platinum complexes that selectively interact with, stabilize, or cleave G-quadruplex structures represents a promising approach for developing effective anticancer agents to overcome cisplatin resistance. This review will emphasize the advancements made over the past decade in interacting G-quadruplexes with platinum complexes as potential anticancer therapeutics. The ongoing development of platinum complexes spans from targeting nuclear DNA G-quadruplexes to mitochondrial DNA and cytoplasmic RNA G-quadruplexes, evolving from monotherapy approaches, such as chemotherapy and photodynamic therapy, to a combination of radiotherapy, immunotherapy, and more, highlighting the dynamic progress of platinum complexes. At the end, we have summarized 4 points of pending issues in this fast-growing field, which we hope can provide some help to the development of this field.
Introduction
The coordination chemistry of metallodrugs has emerged as a well-established interdisciplinary area of research.1 As investigations in this field progress, the potential to develop effective metal-based chemotherapeutic agents for cancer treatment becomes increasingly feasible.2,3 Currently, the most commonly utilized chemotherapeutic agents for managing human tumors that have received regulatory approval include cisplatin, carboplatin, and oxaliplatin.2 Cisplatin, the first metal antitumor drug approved for clinical use, works by targeting DNA and results in the dysfunction of transcription, translation, and other processes, ultimately causing tumor cell death.4,5The antitumor mechanism of cisplatin involves entering the cells and replacing chloride ligands with water, forming aquated species that react with nucleophilic sites in the cellular macromolecules.6 It has been widely used to treat testicular, ovarian, head and neck, cervical, and non-small cell lung cancer.7 Unfortunately, platinum-based anticancer drugs have some clinical problems and need to be improved in design. They are administered indiscriminately to all rapidly dividing cells, including bone marrow. They are known to cause hematopoiesis and exert pressure on the kidneys, resulting in nephrotoxicity, neurotoxicity, myelosuppression, and ototoxicity during removal from the body.8 Cisplatin has also shown poor distribution through the body and poor tumor penetration.9 More importantly, the cytotoxicity of cisplatin results from its binding to DNA, resulting in extensive DNA damage. However, there are multi-pathway DNA damage repair mechanisms in cells, including nucleotide excision, base excision, and mismatch repair, which significantly increase the resistance of tumor cells to cisplatin.10,11 Therefore, researchers have shifted their focus from double-stranded DNA to DNA of other configurations, such as G-quadruplexes DNA.
G-tetrad is formed by stacking four guanines connected by Hoogsteen hydrogen bonds, and G-quadruplex DNA is formed by self-assembling a series of planar G-tetrad that stack and are connected by the intervening sequences, which can be further stabilized by the presence of mono- and divalent cations (Fig. 1a and b).12–15 G-quadruplex structures can be formed intramolecularly within single-stranded nucleic acid sequences and inter-molecularly from two or more individual strands.16 Depending on the distant ways the exterior loops connect the G-quarters and the relative orientation of the tetra-stranded helices, G-quadruplexes display a wide range of topologies. They contain one, two, or four separate stands, referred to as unimolecular, bimolecular, and tetramolecular, respectively. Common examples of G-quadruplex topologies are illustrated in Fig. 1c: parallel, anti-parallel, and hybrid of parallel and anti-parallel.17,18 The variety of configurations makes it possible to perform unique functions regulating life processes selectively, but it also makes it difficult to design specific ligands.
 | ||
| Fig. 1 (a) Schematic representation of the G-quartet formed by Hoogsteen hydrogen bonds between four guanines; (b) view of the X-ray crystal structure of an intramolecular G-quadruplex DNA formed from a single oligonucleotide strand (PDB 1KF1); (c) structural topologies of commonly observed G-quadruplexes.18 Copyright©2024, Tayler D. Prieto Otoya. | ||
Due to the enrichment of guanines in the DNA sequence of telomeres, nuclease-sensitive promoter regions, or promoters of proto-oncogenes in eukaryotes,19,20 when it is induced to form G-quadruplex structures, it will prevent the binding of related enzymes to DNA, thereby inhibiting enzyme function. Take telomerase as an example. Telomerase binds to telomere sequence DNA, participates in maintaining chromosome stability, and has been found to immortalize tumor cells through overexpression.21,22 However, telomerase can only specifically recognize linear telomere sequence DNA rather than G-quadruplex. Therefore, inducing the formation of G-quadruplex structures in telomere sequence DNA can inhibit telomerase function. Similar situations are also found in the promoter region of c-myc, c-kit, KRAS, PDGF-A, hTERT, HIF, and so on, which are considered molecular switches in transcriptional regulation.23,24 G-quadruplexes inducers and/or stabilizers prevent enzymatic DNA elongation or the expression of oncogenic proteins.25 As a result, the design of G-quadruplex stabilizers is regarded as a promising antitumor strategy.26
Although there are reports on G-quadruplex ligands based on organic molecules,27 metal complexes have significant advantages over organic molecules in this field. Metal complexes display unique metal-centered structures and chemical properties, such as reversible coordination bonds and more flexible atomic interaction than strictly covalent bonds.28 Metal complexes also provide access to a much broader chemical space than their purely organic counterparts due to coordination geometry around the metal center, for example, planar, tetrahedral, pyramidal, and octahedral.29,30 This change in geometry leads to larger potential bonding patterns, which is more conducive to the design and synthesis of chemical ligands with strong specificity for G-quadruplex systems with various structures.31 In addition, metal complexes are usually positively charged, which is beneficial to approaching DNA's negatively charged phosphate backbone. It is worth mentioning that an increasing number of metal complexes, such as platinum, ruthenium, iridium, iron, copper complexes, and so on, have been reported as G-quadruplex DNA binding agents.32–36 Due to layout restrictions, this review will focus on platinum complexes as G-quadruplex DNA ligands during the decades.
Platinum complexes for nuclear G-quadruplexes
G-quadruplexes have a stacked planar structure; therefore, molecules with a planar structure can bind to and stabilize the G-quadruplex through π–π stacking. Platinum(II) complexes usually form planar squares with tetra coordination and have an exemplary planar configuration. In addition, the positive charge of most platinum(II) compounds also plays a relevant role, which enhances G-quadruplex binding through electrostatic interactions with the negative charges of the backbone, making them excellent candidates for G-quadruplex stabilizers.37Mononuclear platinum complexes
 | ||
| Fig. 2 (a) and (b) The chemical structures of representative mono-chelated platinum complexes 1–6 reported in the literature. (c) The G-quadruplex-binding platinum complexes 4 and 5 can damage DNA, increase micronucleus formation, activate the RIG-I pathway, and induce ICD in vivo.39 Copyright©2023, John Wiley and Sons. | ||
Pyridostatin (PDS) is a well-known G-quadruplex inducer and stabilizer.22 However, its target gene is still unknown.40 Betzer and co-workers assembled PDS and an N-heterocyclic carbene-platinum into complex 7 (Fig. 3a), which possesses antitumor activity and can induce the loss or disruption of intracellular TRF2 from telomerase.41 With a similar strategy, another interesting work was published by Ma et al. in 2024.42 The integration of PDS with cisplatin and its analogs 8–10 (Fig. 3b) has led to identifying PDS as the most frequently utilized small molecule for binding to G-quadruplex DNA structures.43 Experimental findings indicate that after incubation with 8 and 9, the expression levels of AIM2, ASC, cleaved caspase-1, and GSDMD-N, a protein that oligomerizes to form pores in the plasma membrane and increases membrane permeability during pyroptosis, were significantly increased in the cells. Flow cytometry results showed that the intracellular fluorescence of HMGB1 decreased and CRT increased, respectively, in a concentration-dependent manner after 8 and 9 treatments (Fig. 3c). However, complex 10 had a weaker effect on immunogenic cell death. This activation subsequently triggers an immune response and leads to potent antitumor effects.
 | ||
| Fig. 3 (a) The chemical structure of complex 7 and its schematic diagram of the antitumor mechanism.41 Copyright©2016, American Chemical Society. (b) The structures of PDS–platinum complexes 8–10; (c) a schematic diagram of the antitumor mechanism of complexes 8–10. Complexes 8–10 effectively inhibit the expressions of c-myc and VEGF to inhibit tumor proliferation and further damage G-quadruplex structures to activate the immune-related cGAS-STING pathway and AIM2-ASC-related pyroptosis, triggering a strong immune response and significantly enhancing the therapeutic effect.42 Copyright©2024, Royal Society of Chemistry. | ||
Bidentate ligand-coordinated platinum complexes
Sleiman et al. presented the initial bidentate coordinated Pt(II) G-quadruplex selective binders, complexes 11–13 (Fig. 4a). By increasing the π system and expanding the aromatic ligand plane, complexes 12 and 13 exhibit improved affinity and selectivity with the G-quadruplex.37 Yang and coworkers reported two simple platinum(II) complexes coordinated with 1,10-phenanthroline (14, Fig. 4b) or 2,2′-bipyridine (15, Fig. 4b) for efficient G-quadruplex stabilization. According to the results of circular dichroism (CD) and fluorescence resonance energy transfer (FRET) experiments, the ability of complex 14 (ΔTm = 21 °C) to stabilize the G-quadruplex is better than that of complex 15 (ΔTm = 7 °C).44 Subsequently, compound 14 was employed to explore further the solution structure and effects of the c-myc G-quadruplex, which can potentially target G-quadruplexes in living cells and inhibit myc gene expression in cancerous cells.45 | ||
| Fig. 4 (a) The chemical structures of bidentate ligand-coordinated platinum complexes 11–13. (b) The chemical structures of complexes 14–15 and the schematic diagram of complex 14 binding with c-myc G-quadruplexes. Copyright©2024 Royal Society of Chemistry.45 (c) The chemical structures of bidentate ligand-coordinated platinum complexes 16–22. | ||
The results above do not indicate that a larger conjugate plane yields a better effect. In 2008, Yan et al. synthesized a series of platinum-dipyridophenazine (Pt-dppz) derivatives 16–22 (Fig. 4c). Among them, the introduction of the carboxyl group has, to some extent, disrupted the terminal planar structure of complex 16, which exhibited the highest telomerase inhibitor effect in vitro, with an IC50 value of 760 nM.46
Traditionally, it is believed that the electrostatic attraction between the positive charges (complexes) and negative charges (DNA phosphate backbone) enhances the interaction between the complex and G-quadruplex structures. However, since the amount of double helix DNA in cells is significantly higher than that of G-quadruplexes, the selectivity of these positively charged platinum complexes for G-quadruplexes presents a considerable challenge. To address this issue, the research group led by Zhu et al. developed a neutral platinum(II) complex 23 (Fig. 5a). In addition to the non-charged characteristic, complex 23 also has the characteristics of a non-planar structure. Upon the solution containing G-quadruplex DNA, complex 23 can rapidly transform into a positive monovalent planar structure accompanied by Cl− dissociation and then bind G-quadruplexes intelligently. Complex 23 preferentially interacts with the vascular endothelial growth factor (VEGF) G-quadruplex. ESI-MS and NMR observed a the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complex of the dechlorinated 23 and VEGF-G4. The authors demonstrated that complex 23 can bind G-quadruplexes but nearly does not bind dsDNA. With the G-quadruplex fluorescence lifetime probe, (4,4′,4′′-(nitrilotris(ben-zene-4,1-diyl))tris(1-ethylpyridin-1-ium)iodide) (NBTE), the G4s peak area decreased from 41 ± 2% to 21 ± 2% when adding complex 23 into the FLIM images. Complex 23 effectively diminished the expression of VEGF and inhibited the growth of blood vessels (Fig. 5c and e), thereby contributing to its antitumor efficacy.47
:1 complex of the dechlorinated 23 and VEGF-G4. The authors demonstrated that complex 23 can bind G-quadruplexes but nearly does not bind dsDNA. With the G-quadruplex fluorescence lifetime probe, (4,4′,4′′-(nitrilotris(ben-zene-4,1-diyl))tris(1-ethylpyridin-1-ium)iodide) (NBTE), the G4s peak area decreased from 41 ± 2% to 21 ± 2% when adding complex 23 into the FLIM images. Complex 23 effectively diminished the expression of VEGF and inhibited the growth of blood vessels (Fig. 5c and e), thereby contributing to its antitumor efficacy.47
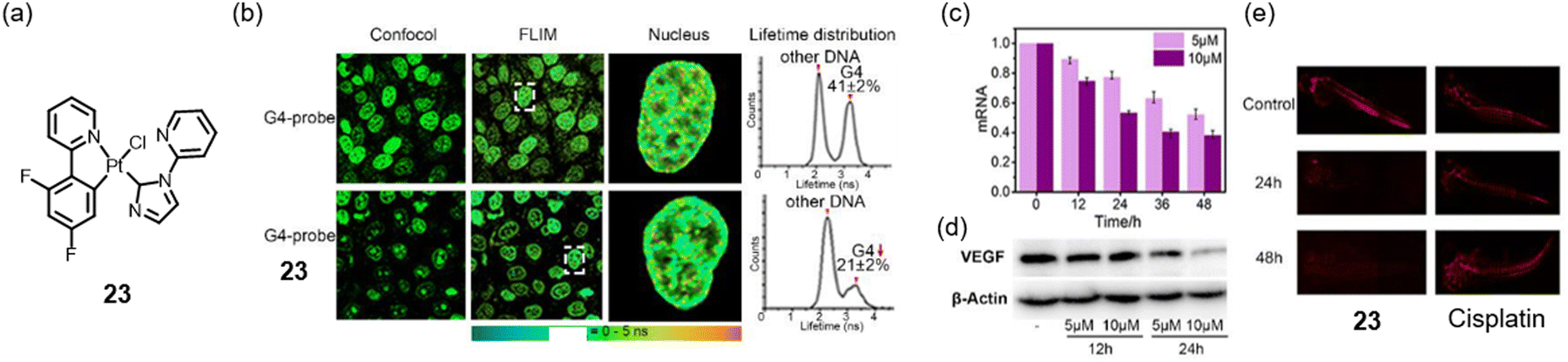 | ||
| Fig. 5 (a) The chemical structures of the neutral complex 23. (b) FLIM experiments of HeLa cells with 10 μM G-quadruplex-probe NBTE and 23 for 24 h. The descent of the G4-peak proved that 23 enters the nucleus and binds G4. (c) and (d) qRT-PCR and western blot results showed that 23 decreases VEGF mRNA transcriptions and VEGF protein levels in HeLa cells. (e) Complex 23 can effectively inhibit blood vessel growth in zebrafish, as red fluorescence changes show.47 Copyright©2021, Wiley-VCH GmbH. | ||
Subsequently, Liang et al. employed the organic compounds 1-azabenzanthrone or 6-hydroxyloxoisoaporphine alkaloid scaffolds that coordinate with platinum to form a π broader conjugate with G-quadruplexes. They synthesized organo-platinum(II) complexes with oxoisoaporphine, 24–25 (Fig. 6a), and improved the π–π interactions with G-quadruplexes. In addition, both inhibited telomerase activity, but 25 was more potent (Fig. 6b). As shown in Fig. 6c, these complexes exhibited antitumor effects on SK-OV-3/DDP cells in vitro, operating through mechanisms associated with cellular senescence and apoptosis.48 The in vivo antitumor result is consistent with the in vitro one.
 | ||
| Fig. 6 (a) The chemical structures of complex 24 (Pt1) and 25 (Pt2). (b) The induction of apoptosis by 24 (Pt1) and 25 (Pt2) was examined by FACS analysis with PI and Annexin V-FITC staining. (c) SK-OV-3/DDP cells were treated with 24 (Pt1) and 25 (Pt2) for 12 h, respectively, and then were processed for 53BP1 (green) and TRF2 (red) immunofluorescence. The nuclei were stained (DAPI, blue) in the merged images.48 Copyright©2015, American Chemical Society. | ||
Tridentate ligand-coordinated platinum complexes
In addition to the bidentate-coordinated Pt(II) complexes, ones with tridentate ligands also exhibit suitable planar structures. Vilar et al. subsequently investigated a series of mono- and bimetallic terpyridine complexes 26–28 (Fig. 7a). Their findings, derived from fluorescent indicator displacement assays (FID) and CD experiments, indicated that the dinuclear platinum complex 28 exhibited a greater affinity for the human telomere (HTelo) and c-myc G-quadruplex DNA than the mono-nuclear metal complexes. Furthermore, complex 28 displayed a preferential selectivity for G-quadruplex DNA over double-stranded DNA.49 Based on the results of complexes 26–28, to improve the selection of c-myc G-quadruplex DNA, Georgiades et al. have documented a family of organoplatinum complexes 29–33 (Fig. 7b) that exhibit similar structures but replace terpyridine with the cyclometallic ligand phenyl-bipyridine. Among them, complex 30 emerged as the most effective binder for c-myc, exhibiting a binding constant (Ka) of 5.2 × 105 M−1, with a ΔTm value for the optimal binder exceeding 30 °C. Additionally, complexes 31–32 are enantiomers, with the L-isomer displaying greater potency against c-myc.50 | ||
| Fig. 7 The chemical structures of reported tridentate ligand-coordinated platinum complexes (26–44) as G-quadruplexes ligands. | ||
Unlike the bidentate-coordinated platinum complex, the tridentate-coordinated platinum complex retains a dissociative CL ligand and can conveniently introduce functional ligands. At the same time, tridentate ligands, such as terpyridine, are relatively easy to be functionalized. For example, based on complexes 26–28, complexes 34–36 were prepared by replacing Cl with 2-(piperidin-1-yl) ethan-1-thiol (Fig. 7c). With this strategy, their group also synthesized complexes 37–42 (Fig. 7d). Overall, compared with complexes that retain Cl coordination, the ability of complexes to interact with G-quadruplexes is improved after introducing substituent groups, mainly due to the introduction of flexible chains that can fit better into the loops and grooves of G-quadruplex DNA.51
This may be a more universal strategy for designing G-quadruplex stabilizers. On the other hand, the mono-metallic complex 37, which contains an uncoordinated cyclen moiety, shows similar affinities for G-quadruplex as the Pt–Cu (38) and Pt–Zn complexes (39). Coupled with the cyclometallic coordinated complex results, the positive charge number's effect on the complex's binding ability with G-quadruplex is not as significant as expected.
Wei and co-workers reported another example.52 They designed a novel platinum(II) complex (43, Fig. 7e) with a berberine derivative serving as a bioactive ligand. Their results demonstrated that complex 43 interacted favorably with G-quadruplex DNA over double-stranded DNA, inhibited tumor cell proliferation, and induced apoptosis. Specifically, complex 43 had a binding affinity of 17.9 μM for the bcl-2 G-quadruplex and did not cause any conformational changes in the structure of the bcl-2 G-quadruplex. In addition to this work, Wei and co-workers have reported several examples of G-quadruplex stabilizers based on Pt complexes. Still, due to space constraints, we only list the literature sources here.53,54 These studies offer novel insights for developing promising platinum(II) antitumor agents based on G-quadruplex structures.
The above studies focus on the nuclear G-quadruplex. Mitochondrial DNA also contains many regions that can form G-quadruplex structures, but this field is often ignored. Recently, Jia and colleagues designed and synthesized complex 44 (Fig. 7f) as the first mitochondrial and nuclear G-quadruplex dual-targeting stabilizer. Complex 44 binds to mitochondrial G-quadruplexes and directly disrupts mitochondrial DNA's replication and translation processes. Moreover, they provided the first evidence that most mitochondrial ribosome genes are highly enriched in G4 structures in their promoter regions, and thus, complex 44 indirectly inhibits their expression by dampening the recruitment of TAF1 and NELFB to the G-quadruplex in nuclear DNA.55
Tetradentate ligand-coordinated platinum complexes
Few reports on platinum complexes bearing tetradentate ligands focus on Salphen ligands. As a molecule with large planar structures and strong coordination ability, Salphen and its derivatives have been introduced to construct metal Salphen complexes, mainly the first-row transition metal.56,57 In 2024, a series of Pt(II)–Salphen complexes 45–56 (Fig. 8a) were investigated by Vilar to assess the influence of various substituents on their binding properties to G-quadruplex DNA, their ability to generate photo-induced reactive oxygen species (ROS), and their cellular behavior.58 They found that when the R group is the same, the nature and position of the substituents on the lower rings have an important effect on the position of the emission maxima. The general pattern is that the 4-position substitution leads to a blue shift in the absorption/emission spectra, while the 5-position substitution is red-shifted compared to the unsubstituted complexes. The enhancement of the size of the aromatic system (e.g., Ph of complex 45vs. Naph of complex 54) improves their effective π–π interactions with the G-tetrad. The R substituents influence the interactions between the complexes and the DNA phosphate backbone. The –NMe3+ group favors electrostatic interactions with the negatively charged DNA. In contrast, the morpholine and piperidine complexes contain larger and cyclic amines with a protonation state that depends on the local pH conditions. In addition, the R substituents modulate the reactive oxygen species yield of the complexes. Complexes bearing the –NMe3+ group generally yield higher oxygen species than the other two substituents. Complex 54 exhibits the highest reactive oxygen species yield and, thus, the most phototoxic ability.58 | ||
| Fig. 8 (a)–(c) The chemical structures of Pt(II)–Salphen complexes 45–63. For complexes 59–63, R = arginine, K = lysine. (d) The chemical structure of Pt(IV)–Salphen complexes 64. (e) Upon addition of bio-reductants such as glutathione, complex 65 is readily reduced to the corresponding square planar Pt(II) complex 66, which binds to G-quadruplex DNA with high affinity. (f) Emission spectra of Pt(II) complex 66, Pt(IV) complex 65, and 65 + glutathione.59 Copyright©2018 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. (g) The chemical structures of Pt(II)–Salphen complexes 67. (h) Schematic representation of the synthetic route of Pt(IV) complexes 68 and 69. | ||
With the synthesis of an increasing number of ligands that could selectively target the G-quadruplex, their group proposed a second generation of dimeric metal–Salphen complexes, 58–63 (Fig. 8b). The monomeric Salphen complex 57 (Fig. 8c) displayed excellent stability towards G-quadruplex. Still, the stability of the dimer G-quadruplex was higher than its monomeric counterpart. Both dinuclear complexes 58–63 showed minimal thermal stability compared to monomeric G-quadruplex DNA, with ΔTm values ranging from 1.1 to 1.5 °C. When the polyether joint length (complex 50) is increased, the dimeric G-quadruplex DNA can exhibit enhanced stabilization. Furthermore, the stability of the dimeric G-quadruplex G2T6 improved progressively following the transition from a polyether to a peptide junction, indicating that the affinity, selectivity, and stability of the complex to the dimeric G-quadruplex are influenced by multiple factors.60 Several Pt(IV) coordination complexes have been proposed as inert prodrugs to address the inherent limitations associated with Pt(II) anticancer agents. These prodrugs are designed to release the corresponding tetragonal Pt(II) active species upon reduction by cellular reducing agents or through photoactivation.61,62 Abiding by this thought, a platinum(IV) Salphen complex, as for complex 64 (Fig. 8d), was developed. It is characterized by a symmetrical metal-binding ligand incorporating two phenolic groups and two imines as metal-binding moieties.30,56 In the same year, Vilar's group also designed and synthesized a Pt(IV) Salphen complex 65 (Fig. 8e), making an instance of a reduction-activated G-quadruplex DNA binder.59 Their findings show that the octahedral complex 65 has a poor affinity for G-quadruplexes. However, adding bio-reductant agents such as glutathione or ascorbic acid as a simulant for the tumor microenvironment can result in in situ redox-triggered conversion to generate Pt(II) complex 66 that effectively binds to G-quadruplex DNA. As illustrated in Fig. 8f, no significant emission was recorded for 65, while the emission was restored upon adding glutathione. As a result, adding glutathione to the solution containing 65 and c-myc DNA resulted in a substantial increase in the thermal stability of c-myc DNA, with a ΔTm = 22°C.59 To gain further insight into the interaction between Pt(IV) Salphen and G-quadruplexes, Sicilia and co-workers performed computational analysis. Their results proved that H-bonds drive complex 67, a protonated form of complex 65, binding G-quadruplex with π–π stacking interactions. Furthermore, strong electrostatic attraction between the positively charged piperidine groups and the negatively charged phosphate backbone of the G-quadruplex allows the metal complex to approach the biomolecule binding site easily.61
In contrast to the above studies focusing on modifying the Salphen ligand, Shao altered the Pt(IV) complex axial ligand by replacing Cl with an HDAC inhibitor. This axial lipophilic ligand assists Pt(IV) pro-probe 68 in rapidly entering live cells and reaching the nucleus. In situ reduction of complex 68 restores parental Pt(II) complex 69 and simultaneously lights up RNA and DNA G-quadruplexes in live cancerous cells. Moreover, complex 68 shows potent cytotoxicity after a long-time incubation as a dual-functional theranostic agent.63
Multinuclear platinum complexes
As mentioned above, molecules with large conjugated π planes are, traditionally, more likely to stabilize G-quadruplexes through π–π interactions. Compared to the single metal complex, bimetallic or polymetallic centers are more straightforward in expanding the plane, which has become a promising strategy for developing metal complexes with high G-quadruplex selectivity and affinity.64 In 2005, Bombard's group developed dinuclear Pt(II) complexes 70–71 (Fig. 9a) with varying alkyl chain lengths, which demonstrated the ability to cross-link the two quadruplex structures of the human telomeric sequence AG3(T2AG3)3.65 Aldrich-Wright's team reported the synthesis of four dinuclear platinum-terpyridine complexes, designated as 72–75 (Fig. 9b). Upon examining various linkers, it was observed that complex 72, which features the shortest linker, is characterized by insufficient length and flexibility. In contrast, complex 73, which is linked via sulfonamide (SOS) and exhibits a melting temperature change of (ΔTm) 11.2 °C for Q1 and 12.7 °C for Q2 (Fig. 9c and d), along with complex 74 (ΔTm = 17 °C), demonstrates an intermediate linker length and the potential for hydrogen bonding interactions with the G-quadruplex. Additionally, complex 75 (ΔTm = 17 °C), with the aromatic linker, exhibits high selectivity toward QDNA in the presence of up to a 600-fold concentration of dsDNA.66 | ||
| Fig. 9 (a), (b) and (e) Chemical structures of dinuclear platinum complexes; (c) SRCD melt spectra of HTelo (inset: PDB ID:2HY9); (d) melt spectra of HTelo with 74.66 Copyright© 2016, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. Two docking structures of the dual cross-linked 80-G-quadruplex complex (f: conf1; g: conf2). 80 and G2/G19 were shown in the Licorice model (Pt: brown; N: blue; C: cyan; Cl: red; G2: orange; G19: purple), and the rest of the G-quadruplex was shown in the ribbon model (T: pink; A: gray; G: green; G2: orange; G19: purple). (h) The hydrogen bond formed between 3′-A22A21T20 trinucleotides and the top G-tetrad (G triad in cyan(C)/blue(N)/red(O); 3′-trinucelotides in green(C)/violet(N)/pink(O)).67 Copyright©2018, Springer Nature & Lei He et al. | ||
Based on the mononuclear complex 76, Shao and co-workers reported four dinuclear Pt(II) complexes 77–80 (Fig. 9e) with different lengths of alkyl chain linkage. These complexes are expected to bind c-myc G-quadruplex via a dual functional clamp: (1) non-covalently π-chelated to guanines at both 3′- and 5′-ends (Fig. 9f–h).67 Among them, complex 79 shows the highest combination ability, indicating that a suitably designed bridging length is vital for dinuclear platinum complexes for optimal stabilization and selectivity towards the G-quadruplex.
Adding more metal Pt centers can construct supramolecules with the configuration of supramolecular structures, bringing more unexpected effects.68 For example, Liu and coworkers broke the traditional idea that G-quadruplex-targeting platinum(II) complexes require a planar configuration. They developed two fan-shaped trinuclear Pt(II) complexes 81–82 (Fig. 10a).69 Out of those expected, 81 and 82 are practical and selective telomeric G-quadruplex binders, exhibiting strong telomerase inhibitions and antitumor ability. They obtained IC50 values of 6.41 ± 0.042 μM and 2.67 ± 0.035 μM for 81 and 82, respectively. Encouraged by this result, they developed another fan-shaped trinuclear Pt(II) complex 83 (Fig. 10b).70 Similar to complexes 81–82, complex 83 is capable of specifically binding to telomeric G-quadruplex (Tel26, Fig. 10c and d). Further research shows that 83 interacts with Tel26 through different proportions and modalities, including π–π stacking, hydrogen bonding, and electronic interactions. NMR spectroscopy revealed that complex 83 initially binds to the 5′-end of Tel26 (Fig. 10e). With the additional amounts of complex 83 introduced, it subsequently binds to the 3′ terminal, forming a distinctive dimeric 4:2 structure. The TRAP-LIG assay demonstrated the excellent telomerase inhibition properties of 83 (Fig. 10f).
 | ||
| Fig. 10 (a) and (b) Dinuclear and multinuclear platinum complexes are reported as G-quadruplex ligands. (c) The folding topology of the hybrid-1 human telomeric G-quadruplex adopted by Tel26 in K+ solution. (d) CD spectra of the Tel26 titrated by the Pt-tripod in 100 mM K+ solution. Ratios of 83/Tel26 are shown in the spectra. (e) Imino proton regions of the 1D 1H NMR titration spectra of Tel26 interacting with 83. (f) 83 exhibited excellent telomerase inhibition properties by the TRAP-LIG assay.70 Copyright© 2018, Springer Nature & Liu et al. | ||
Mao's group developed square-shaped tetranuclear Pt(II) complexes 84–85 with quinoxaline bridges. Complexes 84–85 have effective telomerase inhibition and excellent anticancer efficacy.71 The binding stoichiometric ratio of Pt(II) square/G-quadruplexes is 6:1, the first case in the reported G-quadruplex binders. Zhang designed a similarly structured square-shaped tetranuclear Pt(II) 86 (Fig. 11a) as a TPK2 G-quadruplex stabilizer. The 86-treatment group had a significantly increased number of undetectable telomeres, implying telomere-free ends in both HeLa and MG63 cells (Fig. 11b). Indeed, the percentage of cells with telomere dysfunction-induced foci (TIFs; 53BP1 foci colocalized with telomere) increased gradually after 2.0 μM of complex 86 treatment for 90 days (MG63) or 60 days (HeLa). (Fig. 11c) Consequently, 81.7% of MG63 or 83.9% of HeLa cells were senescent after 106 days or 93 days of complex 86 treatment, respectively (Fig. 11d and e). This mechanism results in a dual anticancer effect through the induction of telomere dysfunction and the inhibition of focal adhesion kinase (FAK)-mediated adhesion and migration.72
 | ||
| Fig. 11 (a) Multinuclear platinum complexes reported as G-quadruplex ligands. (b) A Q-FISH assay was performed to detect the telomere-free ends in untreated or 86-treated HeLa (2.0 μM, 90 days) or MG63 cells (2.0 μM, 60 days). (c) Immunofluorescence and FISH assays were used to detect telomere dysfunction-induced foci (TIFs; 53BP1 foci colocalized with telomere) in 0.01% DMSO (Control) or 2.0 μM 86-treated HeLa cells (106 days). (d) and (e) β-Galactosidase staining to detect senescent cells of untreated or 86-treated HeLa and MG63 cells after long-term proliferation of 106 days for HeLa cells and 86 days for MG63 cells.72 Copyright © 2022, Elsevier B.V. (f) Substitution-inert polynuclear platinum complex structures of 87–92. | ||
The design of the above polynuclear Pt(II) supramolecules still refers to the characteristics of mononuclear complexes, i.e., containing a particular planar configuration to improve the interaction with G-quadruplexes through π–π interactions. Brabec et al. jumped out of this framework restriction and synthesized a series of chained polynuclear Pt(II) complexes 87–92 (Fig. 11f).73 These complexes were demonstrated to stabilize G-quadruplexes and terminate DNA polymerization on templates containing G-quadruplex-forming sequences. Interestingly, the stabilization ability increased with the overall charge of the metal complex. Among them, tetra-Pt(II) complex 92 and tri-Pt(II) complex 91 carrying the highest charges of +10 and +8, respectively, were the most potent G4 binders and DNA synthesis inhibitors.
Platinum complexes for mitochondrial G-quadruplexes
Research on platinum complex-based G-quadruplex stabilizers has focused on nuclear G-quadruplexes, possibly due to the targeting ability of traditional platinum-based antitumor agents like cisplatin (Table 1). In contrast, mitochondrial G-quadruplexes are often neglected. Mitochondria are the powerhouse of cells. Their abnormal functions are highly related to cancer, aging, and various other diseases. Since mitochondrial DNA regulates mitochondria function, while the formation of mitochondrial G-quadruplexes is related to mitochondrial genome stability and the regulation of mitochondrial DNA replication and transcription,74,75 mitochondrial G-quadruplexes have emerged as a promising anticancer target and have attracted significant interest.76| Target | Complex | Area of the tumor | Application |
|---|---|---|---|
| a Only solution-level experiments were performed. | |||
| Human telomere | 1–3 | Lung cancer | Chemotherapy |
| 7 | Ovarian cancer | Chemotherapy | |
| 16 | Cervical cancer | Chemotherapy | |
| 24–25 | Ovarian cancer | Chemotherapy | |
| 26–28 | |||
| 45–56 | Cervical cancer | Photodynamic therapy | |
| 58–63 | Bone osteosarcoma | Chemotherapy | |
| 65–66 | |||
| 70–71 | |||
| 72–75 | |||
| 81–82 | |||
| 83 | |||
| 84–85 | |||
| 86 | Bone osteosarcoma, cervical cancer | Chemotherapy | |
| c-myc | 8 | Breast cancer | Chemoimmunotherapy |
| 14–15 | Cervical cancer | Chemotherapy | |
| 26–28 | |||
| 29–33 | |||
| 34–42 | Bone osteosarcoma | Chemotherapy | |
| 45–46 | Cervical cancer | Photodynamic therapy | |
| 67 | |||
| 72–75 | |||
| 77–80 | |||
| 87–92 | Breast cancer | Chemotherapy | |
| c-kit | 45–56 | Cervical cancer | Photodynamic therapy |
| 87–92 | Breast cancer | Chemotherapy | |
| VEGF | 8 | Breast cancer | Chemoimmunotherapy |
| 23 | Cervical cancer | Chemotherapy | |
| bcl-2 | 43 | Cervical cancer | Chemotherapy |
| MYT1L | 4–5 | Breast cancer | Immunotherapy |
| Nuclear and mitochondrial G-quadruplex | 44 | Ovarian cancer | Chemotherapy |
| Nuclear DNA and cytoplasmic RNA G-quadruplex | 68–69 | Cervical cancer, lung cancer | Chemotherapy |
Reported mitochondrial G-quadruplexes are major in organic compounds. For example, Zhang et al. designed a series of new naphthalimide derivatives, benzothiophenonaphthalimides, to selectively induce a G-rich HRCC DNA sequence in the mitochondria to form a G-quadruplex structure and stabilized it.77 The molecular engineering of a fluorescent G4 ligand, 3,6-bis(1-methyl-4-vinylpyridinium) carbazole diiodide (BMVC), can verify the existence of mtDNA G4s in live cells and eventually causes cell death.78 Platinum complexes are in their infancy in this area. As mentioned above, complex 44 (Fig. 7f) is the first mitochondrial and nuclear G-quadruplex dual-targeting stabilizer. The authors demonstrated that complex 44 localizes in mitochondria and plays a significant role in the mechanisms underlying mitochondrial toxicity.55 Aside from this sole report, the area remains underexplored.
Platinum complexes for cytoplasm G-quadruplexes (RNA G-quadruplexes)
RNA is also capable of forming G-quadruplex structures,79 similar to the DNA-based G-quadruplexes mentioned earlier. However, studies on RNA G-quadruplexes are lagging behind those of cytosolic DNA G-quadruplexes, much like those on mitochondrial DNA G-quadruplexes. To the best of our knowledge, complex 68 (Fig. 8h), developed by Shao and co-workers, is the first and the only example of a platinum-based RNA G-quadruplex binder.63 Due to the rich functions of RNA in several fields such as epigenetics, developing more platinum complex-based RNA G-quadruplex ligands will be meaningful and vital research.Concluding remarks
G-quadruplexes have emerged as promising targets for antitumor therapies. The significant potential of metal complexes to bind to and stabilize G-quadruplex structures, thereby inhibiting enzyme activity or modulating the expression of specific oncogenes, has garnered considerable interest. Among various metal complexes, platinum complexes are excellent candidates for G-quadruplex ligands due to their widespread use in clinical drugs like cisplatin and their unique planar configuration and positive charges. The targets and the corresponding numbers of complexes are listed in Table 1 for ease of reading and searching.Despite numerous reports and successful cases, G-quadruplex ligands based on platinum complexes still face many issues that need to be addressed.
Firstly, the rationale behind the design strategy for complexes remains unclear. While a few reports mention non-planar configurations, such as fan-shaped trinuclear Pt(II) complexes 81–83, current research primarily focuses on the π–π stacking between planar configurations and G-quadruplexes. Nevertheless, planar configurations will likely interact with double-stranded DNA, leading to poor selectivity. Moreover, there are only a few reports on strategies such as introducing flexible chains to enhance the binding of loops and grooves (complexes 35–39 and 41–42) and constructing dinuclear complexes through flexible chain bridging to achieve “sandwich” binding (complexes 72–75 and 77–80), making it challenging to summarize the structure–activity relationship.
Secondly, research has focused on nuclear G-quadruplexes, precise sequences such as telomeres, c-myc, c-kit, and KRAS. As can be seen in Table 1, telomeric G-quadruplexes occupy half of the studies, with the next most commonly studied being c-myc. RNA G-quadruplexes, mitochondrial G-quadruplex DNA, and other targets are rarely explored. However, in recent years, the function of mitochondrial G-quadruplexes has gained increasing importance and may become a significant research focus in the future. It is going to be a vast field of study.
Thirdly, most reported G-quadruplex ligands have been evaluated under non-physiological and dilute conditions. Cellular environments, especially nuclear environments, are molecular crowding conditions, and previous studies have shown that these conditions can distort the G-quadruplex configuration, significantly reducing the binding ability of ligands.80,81 Our group reported the first example of a Ru(II)–Pt(II) dinuclear complex as a G-quadruplex stabilizer under molecular crowding conditions.82 Despite this preliminary insight, there remains a lack of understanding of how molecular crowding environments influence the binding affinity.
Finally, as listed in Table 1, existing studies have mainly utilized these platinum-based G-quadruplex stabilizers to achieve chemotherapy. However, the efficacy of chemotherapy is often limited by toxic side effects and susceptibility to drug resistance. Combination strategies, particularly in chemotherapy combined with radiotherapy, are a trend. Raghavan's results show that regions of DNA enriched in G-quadruplex structures are less sensitive to ionizing radiation compared with B-DNA in vitro and inside cells. The planar G-quartet of G4-DNA is shielded from ionizing radiation-induced free radicals, which provides an avenue for reducing the toxic side effects of radiotherapy.83 On the other hand, it has also been shown that terpyridine-platinum as a G-quadruplex ligand is able to enhance the sensitivity to ionizing radiation of human glioblastoma and non-small cell lung cancer cells.84 These two opposite results suggest that the G-quadruplex, as a complex system, has many mechanisms yet to be explored. In addition, the combination of chemotherapy and immunotherapy, known as chemoimmunotherapy, is an approach that is receiving attention today. A commercial G-quadruplex ligand, TMPyP4, was found to enhance the antitumor immune response by triggering DNA damage and activating the cGAS-STING pathway, which fosters CD8+ T cell activation and dendritic cell maturation.85 Research has shown platinum compounds, such as compounds 4, 5, 8, and 9, target G-quadruplex to activate immunogenic cell death and increase cytotoxic T cells in tumors by disrupting the G-quadruplex. The combination of G-quadruplex-directed cisplatin chemotherapy and immunotherapy has led to a broader and more effective anti-tumor therapy. It has also opened up more therapeutic avenues for antitumor treatment with platinum compounds.
Overall, we hope this overview will increase the understanding of platinum complex-based G-quadruplex ligands and lay the foundation for designing new G-quadruple-targeting antitumor agents. We believe that this research field has a bright future.
Author contributions
Jinrong Yang: gathered data and wrote the drafts; Yu Chen: wrote the drafts, contributed to the editing and funding acquisition; Hui Chao: project administration, editing, and funding acquisition.Data availability
There is no additional data to share.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science Foundation of China (No. 22120102002, 92353301, and 22477148), the Natural Science Foundation of Guangdong Province for Distinguished Young Scholars (No. 2021B1515020102), and the Science and Technology Innovation Program of Hunan Province of China (No. 2021RC5028).Notes and references
- K. D. Mjos and C. Orvig, Chem. Rev., 2014, 114, 4540–4563 CrossRef CAS PubMed.
- S. Ghosh, Bioorg. Chem., 2019, 88, 202925 CrossRef.
- F. Trudu, F. Amato, P. Vanhara, T. Pivetta, E. M. Peña-Méndez and J. Havel, J. Appl. Biomed., 2015, 13, 79–103 CrossRef.
- M. Crul, R. C. A. M. van Waardenburg, J. H. Beijnen and J. H. M. Schellens, Cancer Treat. Rev., 2002, 28, 291–303 CrossRef CAS PubMed.
- Y. P. Ho, S. C. F. Au-Yeung and K. K. W. To, Med. Res. Rev., 2003, 23, 633–655 CrossRef CAS PubMed.
- M. Kartalou and J. M. Essigmann, Mutat. Res., Fundam. Mol. Mech. Mutagen., 2001, 478, 23–43 CrossRef CAS PubMed.
- E. R. Jamieson and S. J. Lippard, Chem. Rev., 1999, 99, 2467–2498 CrossRef CAS PubMed.
- G. Süss-Fink, Dalton Trans., 2010, 39, 1673–1688 RSC.
- C. Carlier, B. Laforce, S. J. M. Van Malderen, F. Gremonprez, R. Tucoulou, J. Villanova, O. De Wever, L. Vincze, F. Vanhaecke and W. Ceelen, J. Pharm. Biomed., 2016, 131, 256–262 CrossRef CAS PubMed.
- H. R. Li, H. J. Dai, T. Y. Shi, X. Cheng, M. H. Sun, K. X. Chen, M. Y. Wang and Q. Y. Wei, Carcinogenesis, 2020, 41, 1229–1237 CrossRef CAS PubMed.
- C. Q. Liu, S. Li and Y. L. Tang, Cancer Chemother. Pharmacol., 2023, 92, 329–340 CrossRef CAS PubMed.
- J. J. Xia, J. L. Chen, J. H. Zhou, M. P. Cheng, X. Z. Zhuang, C. F. Cai, H. X. Ju, J. L. Mergny and J. Zhou, J. Phys. Chem. B, 2024, 128, 11077–11087 CrossRef CAS PubMed.
- G. Qin, Z. Q. Liu, J. Yang, X. F. Liao, C. Q. Zhao, J. S. Ren and X. G. Qu, Nat. Cell Biol., 2024, 26, 1212–1224 CrossRef CAS PubMed.
- J. L. Xu, H. Y. Huang and X. Zhou, JACS Au, 2021, 1, 2146–2161 CrossRef CAS PubMed.
- J. Q. Chen, Y. Y. Chen, L. J. Zhao, L. Y. Feng, F. F. Xing, C. Q. Zhao, L. Z. Hu, J. S. Ren and X. G. Qu, J. Mater. Chem. C, 2019, 7, 13947–13952 RSC.
- J. Spiegel, S. Adhikari and S. Balasubramanian, Trends Chem., 2020, 2, 123–136 CrossRef CAS PubMed.
- H. G. Miserachs, D. Donghi, R. Börner, S. Johannsen and R. K. O. Sigel, J. Biol. Inorg. Chem., 2016, 21, 975–986 CrossRef PubMed.
- T. D. P. Otoya, K. T. Mcquaid and C. J. Cardin, Med. Chem. Res., 2024, 33, 2001–2019 CrossRef.
- D. Rhodes and H. J. Lipps, Nucleic Acids Res., 2015, 43, 8627–8637 CrossRef CAS PubMed.
- H. Fernando, A. P. Reszka, J. Huppert, S. Ladame, S. Rankin, A. R. Venkitaraman, S. Neidle and S. Balasubramanian, Biochemistry, 2006, 45, 7854–7860 CrossRef CAS.
- N. H. Campbell, N. H. Abd Karim, G. N. Parkinson, M. Gunaratnam, V. Petrucci, A. K. Todd, R. Vilar and S. Neidle, J. Med. Chem., 2012, 55, 209–222 CrossRef CAS.
- H. M. Su, J. L. Xu, Y. Q. Chen, Q. Wang, Z. Lu, Y. G. Chen, K. Chen, S. Q. Han, Z. T. Fang, P. Wang, B. F. Yuan and X. Zhou, J. Am. Chem. Soc., 2021, 143, 1917–1923 CrossRef CAS.
- S. Balasubramanian, L. H. Hurley and S. Neidle, Nat. Rev. Drug Discovery, 2011, 10, 261–275 CrossRef CAS PubMed.
- T. A. Brooks, S. Kendrick and L. Hurley, FEBS J., 2010, 277, 3459–3469 CrossRef CAS PubMed.
- R. Chaudhuri, S. Bhattacharya, J. Dash and S. Bhattacharya, J. Med. Chem., 2021, 64, 42–70 CrossRef CAS.
- S. Neidle, Nat. Rev. Chem., 2017, 1, 0041 CrossRef CAS.
- A. Awadasseid, X. D. Ma, Y. L. Wu and W. Zhang, Biomed. Pharmacother., 2022, 148, 111550 CrossRef PubMed.
- C. Metcalfe and J. A. Thomas, Chem. Soc. Rev., 2003, 32, 215–224 RSC.
- M. R. Gill, J. Garcia-Lara, S. J. Foster, C. Smythe, G. Battaglia and J. A. Thomas, Nat. Chem., 2009, 1, 662–667 CrossRef CAS PubMed.
- J. Zegers, M. Peters and B. Albada, J. Biol. Inorg. Chem., 2023, 28, 117–138 CrossRef CAS PubMed.
- Q. Cao, Y. Li, E. Freisinger, P. Z. Qin, R. K. O. Sigel and Z. W. Mao, Inorg. Chem. Front., 2017, 4, 10–32 RSC.
- Z. Yu, M. L. Han and J. A. Cowan, Angew. Chem., Int. Ed., 2015, 54, 1901–1905 CrossRef CAS PubMed.
- S. N. Georgiades, N. H. Abd Karim, K. Suntharalingam and R. Vilar, Angew. Chem., Int. Ed., 2010, 49, 4020–4034 CrossRef CAS PubMed.
- S. K. Jia, W. J. Wang, S. S. Qin, S. J. Xie, L. S. Zhan, Q. Wei, Z. Lu, X. L. Zhou, C. Chen, K. Chen, S. Yan, C. P. Tan, Z. W. Mao and X. Zhou, Chin. Chem. Lett., 2023, 34, 107517 CrossRef CAS.
- W. Tuntiwechapikul and M. Salazar, Biochemistry, 2001, 40, 13652–13658 CrossRef CAS PubMed.
- W. Tuntiwechapikul, T. L. Jeong and M. Salazar, J. Am. Chem. Soc., 2001, 123, 5606–5607 CrossRef CAS PubMed.
- R. Kieltyka, J. Fakhoury, N. Moitessier and H. F. Sleiman, Chem. – Eur. J., 2008, 14, 1145–1154 CrossRef CAS PubMed.
- A. J. Pickard, F. Liu, T. F. Bartenstein, L. G. Haines, K. E. Levine, G. L. Kucera and U. Bierbach, Chem. – Eur. J., 2014, 20, 16174–16187 CrossRef CAS PubMed.
- L. Y. Liu, T. Z. Ma, Y. L. Zeng, W. T. Liu, H. Zhang and Z. W. Mao, Angew. Chem., Int. Ed., 2023, 62, e202305645 CrossRef CAS PubMed.
- Y. Z. Hou, T. L. Gan, T. T. Fang, Y. Zhao, Q. Luo, X. K. Liu, L. Y. Qi, Y. Y. Zhang, F. F. Jia, J. J. Han, S. M. Li, S. J. Wang and F. Y. Wang, Nucleic Acids Res., 2022, 50, 3070–3082 CrossRef CAS PubMed.
- J. F. Betzer, F. Nuter, M. Chtchigrovsky, F. Hamon, G. Kellermann, S. Ali, M. A. Calméjane, S. Roque, J. Poupon, T. Cresteil, M. P. Teulade-Fichou, A. Marinetti and S. Bombard, Bioconjugate Chem., 2016, 27, 1456–1470 CrossRef CAS PubMed.
- T. Z. Ma, L. Y. Liu, Y. L. Zeng, K. Ding, H. Zhang, W. T. Liu, Q. Cao, W. Xia, X. S. Xiong, C. Wu and Z. W. Mao, Chem. Sci., 2024, 15, 9756–9774 RSC.
- L. Y. Liu, T. Z. Ma, Y. L. Zeng, W. T. Liu and Z. W. Mao, J. Am. Chem. Soc., 2022, 144, 11878–11887 CrossRef CAS PubMed.
- J. T. Wang, X. H. Zheng, Q. Xia, Z. W. Mao, L. N. Ji and K. Wang, Dalton Trans., 2010, 39, 7214–7216 RSC.
- W. T. Liu, B. C. Zhu, L. Y. Liu, X. Y. Xia, J. Jang, J. Dickerhoff, D. Z. Yang and Z. W. Mao, Nucleic Acids Res., 2024, 52, 9397–9406 CrossRef CAS PubMed.
- D. L. Ma, C. M. Che and S. C. Yan, J. Am. Chem. Soc., 2009, 131, 1835–1846 CrossRef CAS PubMed.
- B. C. Zhu, J. He, W. T. Liu, X. Y. Xia, L. Y. Liu, B. B. Liang, H. G. Yao, B. Liu, L. N. Ji and Z. W. Mao, Angew. Chem., Int. Ed., 2021, 60, 15340–15343 CrossRef CAS PubMed.
- Z. F. Chen, Q. P. Qin, J. L. Qin, Y. C. Liu, K. B. Huang, Y. L. Li, T. Meng, G. H. Zhang, Y. Peng, X. J. Luo and H. Liang, J. Med. Chem., 2015, 58, 2159–2179 CrossRef CAS PubMed.
- K. Suntharalingam, A. J. P. White and R. Vilar, Inorg. Chem., 2010, 49, 8371–8380 CrossRef CAS PubMed.
- L. Savva, M. Fossépré, O. Keramidas, A. Themistokleous, N. Rizeq, N. Panagiotou, M. Leclercq, E. Nicolaidou, M. Surin, S. C. Hayes and S. N. Georgiades, Chem. – Eur. J., 2022, 28, e202201497 CrossRef CAS PubMed.
- V. S. Stafford, K. Suntharalingam, A. Shivalingam, A. J. P. White, D. J. Mann and R. Vilar, Dalton Trans., 2015, 44, 3686–3700 RSC.
- S. L. Zhang, H. M. Fu, Y. X. Ma, Q. F. Lin, Y. L. Xu, Q. Y. Yang, P. He and Z. Z. Wei, Org. Biomol. Chem., 2025, 23, 1219 RSC.
- Z. Z. Wei, Q. P. Qin, T. Meng, C. X. Deng, H. Liang and Z. F. Chen, Eur. J. Med. Chem., 2018, 145, 360–369 CrossRef CAS PubMed.
- J. L. Qin, Q. P. Qin, Z. Z. Wei, Y. C. Yu, T. Meng, C. X. Wu, Y. L. Liang, H. Liang and Z. F. Chen, Eur. J. Med. Chem., 2016, 124, 417–427 CrossRef CAS PubMed.
- K. L. Kuang, C. Y. Li, F. Maksut, D. Ghosh, R. Vinck, M. L. Wang, J. Poupon, R. Xiang, W. Li, F. Li, Z. Wang, J. R. Du, M. P. Teulade-Fichou, G. Gasser, S. Bombard and T. Jia, J. Biomed. Sci., 2024, 31, 104 CrossRef PubMed.
- A. Łęczkowska, J. Gonzalez-Garcia, C. Perez-Arnaiz, B. Garcia, A. J. P. White and R. Vilar, Chem. – Eur. J., 2018, 24, 11785–11794 CrossRef PubMed.
- R. Vilar, Adv. Inorg. Chem., 2020, 75, 425–445 CrossRef CAS.
- M. Bartlett, J. Burke, P. Sherin, M. K. Kuimova, M. Barahona and R. Vilar, Chem. – Eur. J., 2024, 30, e202402465 CrossRef CAS PubMed.
- S. Bandeira, J. Gonzalez-Garcia, E. Pensa, T. Albrecht and R. Vilar, Angew. Chem., Int. Ed., 2018, 57, 310–313 CrossRef CAS PubMed.
- T. Kench, V. Rakers, D. Bouzada, J. Gomez-González, J. Robinson, M. K. Kuimova, M. V. López, M. E. Vázquez and R. Vilar, Bioconjugate Chem., 2023, 34, 911–921 CrossRef CAS PubMed.
- V. Vigna, S. Scoditti, A. Spinello, G. Mazzone and E. Sicilia, Int. J. Mol. Sci., 2022, 23, 15579 CrossRef CAS PubMed.
- E. M. Bolitho, C. Sanchez-Cano, H. Shi, P. D. Quinn, M. Harkiolaki, C. Imberti and P. J. Sadler, J. Am. Chem. Soc., 2021, 143, 20224–20240 CrossRef CAS PubMed.
- Q. Q. Guo, M. J. Huang, C. C. Wang and F. W. Shao, J. Inorg. Biochem., 2022, 237, 111988 CrossRef CAS PubMed.
- M. G. Mendoza-Ferri, C. G. Hartinger, M. A. Mendoza, M. Groessl, A. E. Egger, R. E. Eichinger, J. B. Mangrum, N. P. Farrell, M. Maruszak, P. J. Bednarski, F. Klein, M. A. Jakupec, A. A. Nazarov, K. Severin and B. K. Keppler, J. Med. Chem., 2009, 52, 916–925 CrossRef CAS PubMed.
- I. Ourliac-Garnier, M. A. Elizondo-Riojas, S. Redon, N. P. Farrell and S. Bombard, Biochemistry, 2005, 44, 10620–10634 CrossRef CAS PubMed.
- D. L. Ang, B. W. J. Harper, L. Cubo, O. Mendoza, R. Vilar and J. Aldrich-Wright, Chem. – Eur. J., 2016, 22, 2317–2325 CrossRef CAS PubMed.
- L. He, Z. Y. Meng, D. C. Xu and F. W. Shao, Sci. Rep., 2018, 8, 767 CrossRef PubMed.
- N. P. Farrell, Chem. Soc. Rev., 2015, 44, 8773–8785 RSC.
- C. X. Xu, L. Y. Liu, B. Lv, H. Y. Zhao, Q. Cao, T. Zhai and Z. W. Mao, Dalton Trans., 2020, 49, 9322–9329 RSC.
- W. T. Liu, Y. F. Zhong, L. Y. Liu, C. T. Shen, W. J. Zeng, F. Y. Wang, D. Z. Yang and Z. W. Mao, Nat. Commun., 2018, 9, 3496 CrossRef PubMed.
- X. H. Zheng, H. Y. Chen, M. L. Tong, L. N. Ji and Z. W. Mao, Chem. Commun., 2012, 48, 7607–7609 RSC.
- Z. Shen, R. L. Zheng, H. M. Yang, S. H. Xing, X. X. Jin, H. Yan, J. F. Zhu, Y. A. Mei, F. Lin and X. H. Zheng, Int. J. Biol. Macromol., 2022, 213, 858–870 CrossRef CAS PubMed.
- J. Malina, H. Kostrhunova, N. P. Farrell and V. Brabec, Inorg. Chem. Front., 2021, 8, 3371–3381 RSC.
- G. Battogtokh, Y. Y. Cho, J. Y. Lee, H. S. Lee and H. C. Kang, Front. Pharmacol., 2018, 9, 922 CrossRef PubMed.
- B. X. Zheng, W. Long, W. D. Zheng, Y. X. Zeng, X. C. Guo, K. H. Chan, M. T. She, A. S. L. Leung, Y. J. Lu and W. L. Wong, J. Med. Chem., 2024, 67, 6292–6312 CrossRef CAS PubMed.
- L. Holden, R. C. Curley, G. Avella, C. N. Long and T. E. Keyes, Angew. Chem., Int. Ed., 2024, 63, e202408581 CrossRef CAS PubMed.
- Q. Huang, X. Wang, A. Chen, H. Zhang, Q. M. Yu, C. F. Shen, A. Awadasseid, X. Y. Zhao, X. Q. Xiong, Y. L. Wu and W. Zhang, Biochem. Pharmacol., 2022, 201, 115062 CrossRef CAS PubMed.
- W. C. Huang, T. Y. Tseng, Y. T. Chen, C. C. Chang, Z. F. Wang, C. L. Wang, T. N. Hsu, P. T. Li, C. T. Chen, J. J. Lin, P. J. Lou and T. C. Chang, Nucleic Acids Res., 2015, 43, 10102–10113 CAS.
- P. Agarwala, S. Pandey and S. Maiti, Org. Biomol. Chem., 2015, 13, 5570–5585 RSC.
- D. Miyoshi, H. Karimata and N. Sugimoto, J. Am. Chem. Soc., 2006, 128, 7957–7963 CrossRef CAS PubMed.
- B. Heddi and A. T. Phan, J. Am. Chem. Soc., 2011, 133, 9824–9833 CrossRef CAS PubMed.
- K. Xiong, C. Ouyang, J. Q. Liu, J. Karges, X. L. Lin, X. Chen, Y. Chen, J. Wan, L. N. Ji and H. Chao, Angew. Chem., Int. Ed., 2022, 61, e202204866 CrossRef CAS PubMed.
- N. Kumari, S. V. Vartak, S. Dahal, S. Kumari, S. S. Desai, V. Gopalakrishnan, B. Choudhary and S. C. Raghavan, Iscience, 2019, 21, 288–307 CrossRef CAS PubMed.
- P. Merle, M. Gueugneau, M. P. Teulade-Fichou, M. Müller-Barthélémy, S. Amiard, E. Chautard, C. Guetta, V. Dedieu, Y. Communal, J. L. Mergny, M. Gallego, C. White, P. Verrelle and A. Tchirkov, Sci. Rep., 2015, 5, 16255 CrossRef CAS PubMed.
- P. S. Li, D. W. Zhou, Y. M. Xie, Z. Yuan, M. Z. Huang, G. P. Xu, J. F. Huang, Z. K. Zhuang, Y. X. Luo, H. C. Yu and X. L. Wang, Cell Death Dis., 2024, 15, 816 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |