DOI:
10.1039/D4CB00316K
(Paper)
RSC Chem. Biol., 2025,
6, 1148-1155
An iridium(III) 3-chloro-6-thio-1,2,4,5-tetrazine complex for cysteine conjugation, bioimaging and photoactivated therapy†
Received
20th December 2024
, Accepted 6th May 2025
First published on 7th May 2025
Abstract
Photoactivatable systems have received considerable attention in the development of diagnostics and therapeutics due to their noninvasive nature and precise spatiotemporal control. Of particular interest is the 3,6-dithio-1,2,4,5-tetrazine (S,S-tetrazine) unit, which can not only act as a photolabile protecting group for constructing photoactivatable systems but also as a bioorthogonal scaffold that enables the inverse electron-demand Diels–Alder (IEDDA) cycloaddition reaction with strained alkynes. In this study, we designed and synthesised a cyclometallated iridium(III) complex modified with a 3-chloro-6-thio-1,2,4,5-tetrazine moiety (1) for cysteine conjugation. The complex was conjugated with an integrin-targeting peptide c(RGDfC) to afford a tumour-targeting conjugate (1-RGD) for bioimaging and photoactivated therapy. An RGD-free analogue (2) was also prepared for comparison studies. Unlike common iridium(III) complexes, excitation of conjugate 1-RGD and complex 2 resulted in weak emission and negligible singlet oxygen (1O2) generation due to the quenching effect of the tetrazine unit. Upon continuous light irradiation, the S,S-tetrazine moiety in conjugate 1-RGD and complex 2 underwent efficient photodissociation, yielding thiocyanate (3) and amide (4) complexes as photoproducts with increased emission intensities and enhanced 1O2 generation efficiencies. Interestingly, the IEDDA cycloaddition reaction of the S,S-tetrazine-containing conjugate 1-RGD and complex 2 with (1R,8S,9s)-bicyclo[6.1.0]non-4-yn-9-ylmethanol (BCN-OH) led to significant emission enhancement. Notably, conjugate 1-RGD showed higher cellular uptake and (photo)cytotoxicity (IC50,dark = 26 μM, IC50,light = 0.08 μM) in U87-MG cells, which overexpress integrin, compared to MCF-7 (IC50,dark = 52 μM, IC50,light = 0.22 μM) and HEK293 cells (IC50,dark > 50 μM, IC50,light = 1.3 μM) with lower integrin levels. This work will contribute to the development of photoactivatable transition metal complexes for cancer-targeted imaging and therapy.
Introduction
In the past few decades, significant advancements have been made in the development of therapeutic agents for the effective treatment of various human diseases.1–3 However, challenges remain in achieving effective therapy at specific disease sites due to the limited selectivity of existing therapeutics, which can lead to potential side effects on healthy tissues.4,5 To overcome this hurdle, different therapeutic agents have been developed that are responsive to endogenous (e.g., pH, enzymes and redox reactions) and exogenous stimuli (e.g., light, ionizing irradiation and magnetic fields).6,7 Photoactivatable systems that undergo photochemical reactions and generate radical species or release molecules of interest upon light irradiation have gained significant attention.8,9 By adjusting parameters such as irradiation wavelength, intensity and duration, these light-based approaches offer precise spatiotemporal control, enabling efficient accumulation of therapeutic agents at the target site and potent therapeutic effects.10
With their rich photophysical and photochemical properties, transition metal complexes have been widely utilised for the construction of photoactivatable therapeutics.11–14 In particular, transition metal complexes that can undergo photoactivated chemotherapy (PACT) are of significant interest because they are noncytotoxic in the dark but become highly (photo)cytotoxic after light irradiation due to photoinduced ligand dissociation.15,16 For example, ruthenium(II) polypyridine complexes with distorted octahedral geometry can undergo phototriggered ligand loss with significantly enhanced cytotoxicity upon light irradiation due to the generation of solvent-substituted photoproducts that can covalently bind to DNA.17 Another example used a sterically hindered ruthenium(II) complex conjugated to a nicotinamide phosphoribosyltransferase inhibitor, which undergoes photodissociation upon light irradiation to induce cytotoxic effects.18 Apart from the release of ligands from the coordination sphere of the metal centre, the attachment of organic photolabile protecting groups (PPGs) to ligands is another effective strategy to induce controllable cytotoxicity. Previously, we have developed iridium(III) complexes modified with a poly(ethylene glycol) (PEG) chain using a 2-nitrobenzyl moiety as a photosensitive linker.19 Upon light irradiation, the departure of the biocompatible PEG moiety leads to enhanced cytotoxicity of the resultant complex.
1,2,4,5-Tetrazine, known for its exceptional ability to quench various luminophores through Förster resonance energy transfer20,21 or photoinduced electron transfer,22 can serve as a versatile bioorthogonal scaffold that enables rapid and selective inverse electron-demand Diels–Alder (IEDDA) cycloaddition reactions with strained alkynes.23 Interestingly, studies on 3,6-dithio-1,2,4,5-tetrazine (S,S-tetrazine) derivatives have shown their potential as useful phototriggers, allowing the investigation of early events in peptide/protein-folding.24,25 These S,S-tetrazines display an additional absorption band centred at 410 nm compared to traditional tetrazines, which can be attributed to an n → π* transition or charge-transfer involving the sulfur atoms, leading to photodissociation upon visible-light activation.26 The photodissociation process occurs rapidly in the picosecond timescale, yielding inert photoproducts (thiocyanates and dinitrogen) with a high yield (Scheme 1).25
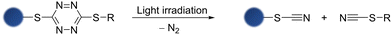 |
| | Scheme 1 Photodissociation reaction of an S,S-tetrazine into thiocyanates and dinitrogen. | |
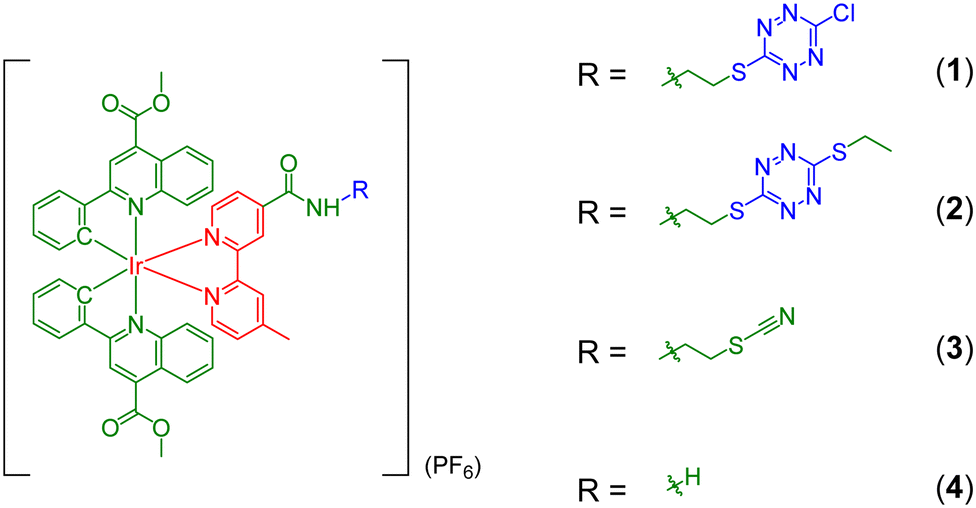
A previous study has revealed that 3-chloro-6-thio-1,2,4,5-tetrazine (Cl,S-tetrazine) can selectively label cysteine residues in proteins to yield conjugates bearing an S,S-tetrazine moiety.27 It is anticipated that the integration of a Cl,S-tetrazine moiety into luminescent iridium(III) polypyridine complexes can afford a new class of labelling reagents for cysteine residues, leading to the construction of photoactivatable bioconjugates. In this study, we designed and synthesised an iridium(III) Cl,S-tetrazine complex, [Ir(pqe)2(bpy-CONH-S-Tz-Cl)](PF6) (Hpqe = 2-phenylquinoline-4-carboxylic acid methyl ester; bpy-CONH-S-Tz-Cl = 4-(S-(6-chloro-1,2,4,5-tetrazin-3-yl)-N-mercaptoethylaminocarbonyl)-4′-methyl-2,2′-bipyridine) (1) (Scheme 2). Its S,S-tetrazine-containing counterpart [Ir(pqe)2(bpy-CONH-S-Tz-S-Et)](PF6) (bpy-CONH-S-Tz-S-Et = 4-(S-(6-ethylthio-1,2,4,5-tetrazin-3-yl)-N-mercaptoethylaminocarbonyl)-4′-methyl-2,2′-bipyridine) (2) and the S,S-tetrazine-free analogues, [Ir(pqe)2(bpy-CONH-SCN)](PF6) (bpy-CONH-SCN = 4-(N-(thiocyanatoethyl)aminocarbonyl)-4′-methyl-2,2′-bipyridine) (3) and [Ir(pqe)2(bpy-CONH2)](PF6) (bpy-CONH2 = 4-aminocarbonyl-4′-methyl-2,2′-bipyridine) (4) were also prepared for comparison studies. All the complexes were characterised by high-resolution ESI-MS, NMR and IR spectroscopy. Detailed synthetic procedures and characterisation data are included in the ESI.† Upon photoexcitation, the tetrazine complexes 1 and 2 exhibited substantially weaker emission intensities and lower singlet oxygen (1O2) generation efficiencies compared to complexes 3 and 4 due to the quenching effect of the tetrazine unit. Notably, upon continuous light irradiation, the S,S-tetrazine moiety in complex 2 underwent efficient dissociation, yielding the thiocyanate and amide complexes 3 and 4 as the photoproducts with increased emission intensities and enhanced 1O2 photogeneration efficiencies. Additionally, complex 2 can undergo IEDDA cycloaddition reaction with (1R,8S,9s)-bicyclo[6.1.0]non-4-yn-9-ylmethanol (BCN-OH), leading to significant emission enhancement. The photosensitivity of S,S-tetrazine motivated us to utilise complex 1 to modify the cysteine residue of an integrin-binding peptide c(RGDfC), affording an S,S-tetrazine-containing peptide conjugate [Ir(pqe)2(bpy-CONH-S-Tz-S-RGD)](CF3COO) (1-RGD) that showed intriguing photoactivatable characteristics and tumour-targeting capabilities. Remarkably, the conjugate displayed efficient cellular uptake and potent photocytotoxicity towards integrin-overexpressing human glioblastoma U87-MG cells.
 |
| | Scheme 2 Structures of complexes 1–4. | |
Results and discussion
Photophysical properties
The electronic absorption data and spectra of complexes 1–4 are presented in Table S1 and Fig. S1 (ESI†). The complexes displayed intense spin-allowed intraligand (1IL) (π → π*) (N^N/pqe) absorption (ca. 255–385 nm) and weaker spin-allowed metal-to-ligand charge-transfer (1MLCT) (dπ(Ir) → π*(N^N/pqe)) absorption bands/shoulders (ca. 390–475 nm).28,29 The weak absorption tail beyond ca. 550 nm is assigned to spin-forbidden 3MLCT (dπ(Ir) → π*(N^N/pqe)) transitions. Additionally, a weak absorption band at around 510–530 nm was observed, which is ascribed to the n → π* transition of the tetrazine unit (Fig. S1, ESI†). Upon photoexcitation, complexes 1–4 exhibited a structureless emission band with positive solvatochromism in fluid solutions at 298 K (Table 1 and Fig. S2, ESI†). The emission maxima of the complexes showed significant blueshifts when the samples were cooled to 77 K, suggesting a 3MLCT (dπ(Ir) → π*(N^N/pqe)) excited state (Fig. S2, ESI†). Due to the efficient emission quenching property of the tetrazine unit, complexes 1 and 2 showed much lower emission quantum yields (Φem ≤ 0.004) than complexes 3 and 4 (Φem ≥ 0.14) (Table 1). The 1O2 quantum yields (ΦΔ) of complexes 1–4 were evaluated in aerated CH3CN using 1,3-diphenylisobenzofuran as the 1O2 scavenger (Table S2, ESI†). The ΦΔ values of the tetrazine complexes 1 and 2 were determined to be 0.55 and 0.60, respectively, which are lower than those of the tetrazine-free complexes 3 and 4 (0.81 and 0.79, respectively). These results indicate that the tetrazine unit can not only quench the emission of the complexes but also suppress their 1O2 photogeneration efficiencies.
Table 1 Photophysical data of complexes 1–4 and conjugate 1-RGD
| Complex/conjugate |
Medium (T/K) |
λ
em
/nm |
Φ
em
|
τ
o
/μs |
λ
ex = 350 nm.
[Ru(bpy)3]Cl2 was used as a reference (Φem = 0.040 in aerated H2O, λex = 455 nm).30
The lifetimes were measured at the emission maxima (λex = 375 nm).
Potassium phosphate buffer (50 mM, pH 7.4)/CH3CN (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v).
EtOH/MeOH (4:1, v/v). sh: shoulder :1, v/v).
EtOH/MeOH (4:1, v/v). sh: shoulder |
|
1
|
CH3CN (298) |
632 |
0.003 |
0.68 |
| Bufferd (298) |
650 |
0.002 |
0.23 |
| Glasse (77) |
596, 646 sh |
|
4.25 |
|
2
|
CH3CN (298) |
631 |
0.004 |
0.60 |
| Bufferd (298) |
650 |
0.002 |
0.18 |
| Glasse (77) |
594, 644 sh |
|
4.38 |
|
3
|
CH3CN (298) |
634 |
0.14 |
0.69 |
| Bufferd (298) |
648 |
0.02 |
0.35 |
| Glasse (77) |
598, 645 sh |
|
4.79 |
|
4
|
CH3CN (298) |
631 |
0.15 |
0.67 |
| Bufferd (298) |
648 |
0.02 |
0.34 |
| Glasse (77) |
597, 647 sh |
|
4.23 |
|
1-RGD
|
CH3CN (298) |
631 |
0.003 |
0.58 |
| Bufferd (298) |
648 |
0.001 |
0.19 |
| Glasse (77) |
596, 644 sh |
|
4.14 |
Photoactivatable properties and phosphorogenic response
The photodissociation of the S,S-tetrazine moiety in complex 2 in phosphate-buffered saline (PBS)/DMSO (9:1, v/v) upon photoirradiation at 450 nm (10 mW cm−2) for 20 min was monitored by reverse-phase high-performance liquid chromatography (RP-HPLC). Notably, two new peaks at tR = 16.53 and 16.05 min in the HPLC spectra were detected (Fig. 1a) after the photoreaction. ESI-MS analyses revealed that the photoreleased products were the thiocyanate complex 3 and amide complex 4 (which appears to be a photodecomposed/hydrolysed product), respectively (Scheme 3 and Fig. 1b and c). Impressively, the photodissociation of the S,S-tetrazine unit led to a significant increase in the emission intensity (I/Io = 10.7) and lifetime (τ = 0.29 μs) of the solution (Table 2 and Fig. 1d). Due to the presence of a tetrazine moiety, complex 2 was also expected to undergo IEDDA cycloaddition reaction with BCN-OH to afford 2-BCN (Scheme 4). The phosphorogenic response of complex 2 towards BCN-OH was investigated. The IEDDA reaction of complex 2 (10 μM) with excess BCN-OH (500 μM) in PBS/DMSO (9:1, v/v) at 298 K produced a non-quenching pyridazine product (Fig. S3, ESI†), leading to significant emission enhancement and lifetime extension (I/Io = 38.9; τ = 0.37 μs; Table 2).
 |
| | Fig. 1 (a) HPLC chromatograms of complex 2 (10 μM) in aerated PBS/DMSO (9:1, v/v) treated before (black) or after (red) photoirradiation. ESI-mass spectra of the photodissociation products collected at (b) tR = 16.53 min and (c) tR = 16.05 min of complex 2 (10 μM) in aerated PBS/DMSO (9:1, v/v) upon photoirradiation. (d) Emission spectra of complex 2 (10 μM) in aerated PBS/DMSO (9:1, v/v) treated before (black) or after (red) photoirradiation. Photoirradiation conditions for all experiments were carried out at 450 nm (10 mW cm−2) for 20 min at 298 K. | |
 |
| | Scheme 3 Photodissociation reaction of the S,S-tetrazine moiety in complex 2. | |
Table 2 Emission enhancement factors (I/Io) and lifetimes (τ) of complex 2 and conjugate 1-RGD (10 μM) upon photoirradiation at 450 nm (10 mW cm−2) in aerated PBS/DMSO (9:1, v/v) for 20 min or incubation with BCN-OH (500 μM) in aerated PBS/DMSO (9:1, v/v) in the dark for 16 h. The emission lifetimes of complex 2 and conjugate 1-RGD alone under the same conditions could not be determined with accuracy due to very weak emission
|
|
Complex/conjugate + hv |
Complex/conjugate + BCN-OH |
| Complex/conjugate |
λ
em
/nm |
I/Iob |
τ
/μs |
λ
em
/nm |
I/Iod |
τ
/μs |
|
λ
ex = 350 nm.
I
o and I are the emission intensities of complex 2 or conjugate 1-RGD (10 μM) before and after photoirradiation at 450 nm (10 mW cm−2) for 20 min, respectively.
The lifetimes were measured at the emission maxima (λex = 375 nm).
I
o and I are the emission intensities of complex 2 or conjugate 1-RGD (10 μM) in the absence and presence of BCN-OH (500 μM, 16 h), respectively.
|
|
2
|
648 |
10.7 |
0.29 |
631 |
38.9 |
0.37 |
|
1-RGD
|
648 |
9.9 |
0.26 |
632 |
31.9 |
0.33 |
 |
| | Scheme 4 The IEDDA cycloaddition reaction of complex 2 with BCN-OH. | |
Construction of an integrin-targeting peptide conjugate bearing an S,S-tetrazine moiety
Having examined that the iridium(III) S,S-tetrazine complex displayed remarkable phosphorogenic response upon both photodissociation and bioorthogonal reactions, we used complex 1 as a labelling reagent for the construction of S,S-tetrazine-containing peptide conjugates that can be applied for tumour-targeted phototherapy. Integrins, consisting of α- and β-subunits, play an important role in cell adhesion mediation and signalling.31 The RGD sequence, as the minimal integrin-binding motif, has been widely applied in imaging and tumour-targeted therapy due to its high binding affinity with integrins.32,33 A cyclic RGD peptide containing a cysteine c(RGDfC) was selected for conjugation with complex 1 to afford the peptide conjugate 1-RGD (Scheme 5) that is expected to exhibit enhanced accumulation and photoactivatable therapeutic effects in cancer cells overexpressing integrin αvβ3. The reactivity of complex 1 (25 μM) towards c(RGDfC) (25 μM) was examined in acetate buffer (50 mM, pH 6.0)/DMF (9:1, v/v, 1 mL) containing tris(2-carboxyethyl)phosphine (TCEP) (100 μM) at 37 °C. After incubation for 4 h, HPLC analyses revealed that almost all the complex was transformed into the conjugation product 1-RGD, with a conversion yield of >95% (Fig. S4, ESI†). Conjugate 1-RGD was purified by semi-preparative RP-HPLC and the purified product was characterised by HPLC and ESI-MS (Fig. S5, ESI†). Upon excitation, it showed weak emission and low 1O2 generation efficiency due to the presence of the S,S-tetrazine unit (Table 1 and Table S2, ESI†). The photodissociation reaction of the S,S-tetrazine moiety in conjugate 1-RGD was examined by steady-state irradiation at 450 nm at 298 K; the reaction was found to complete in 20 min, yielding the thiocyanate (complex 3) and amide (complex 4) photoproducts, as indicated by HPLC and ESI-MS analyses (Fig. 2a–c). Interestingly, the sample solution displayed emission enhancement of 9.9 fold after photoirradiation (Fig. 2d). Additionally, conjugate 1-RGD exhibited substantial emission enhancement and lifetime extension (I/Io = 31.9, τ = 0.33 μs; Table 2 and Fig. S6, ESI†) upon reacting with BCN-OH. These results indicate that complex 2 and conjugate 1-RGD bearing an S,S-tetrazine moiety can serve as efficient photoactivatable reagents and phosphorogenic bioorthogonal probes.
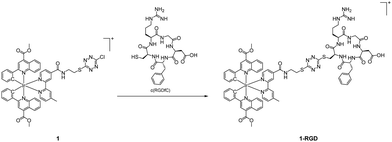 |
| | Scheme 5 Conjugation of complex 1 with peptide c(RGDfC). | |
 |
| | Fig. 2 (a) HPLC chromatograms of conjugate 1-RGD (10 μM) in PBS/DMSO (9:1, v/v) treated before (black) or after (red) photoirradiation. ESI-mass spectra of the photodissociation products collected at (b) tR = 16.45 min and (c) tR = 15.99 min of conjugate 1-RGD (10 μM) in PBS/DMSO (9:1, v/v) upon photoirradiation. (d) Emission spectra of conjugate 1-RGD (10 μM) in PBS/DMSO (9:1, v/v) treated before (black) or after (red) photoirradiation. Photoirradiation conditions for all experiments were carried out at 450 nm (10 mW cm−2) for 20 min at 298 K. | |
Cellular studies
The higher expression of integrins in cancer cells contributes to tumour progression and metastasis by promoting tumour cell proliferation, migration and invasion.34 To examine the targeting efficiency of conjugate 1-RGD towards integrin-overexpressing cancer cell lines, cancerous brain (U87-MG; high integrin αvβ3 expression), breast (MCF-7; low integrin αvβ3 expression) and normal kidney (HEK293; low integrin αvβ3 expression) cells were used as model cell lines.35–38 The cellular uptake of conjugate 1-RGD and complexes 2–4 was determined by ICP-MS and the results are listed in Table S3 (ESI†). Conjugate 1-RGD showed higher accumulation in U87-MG cells (0.70 fmol) than in MCF-7 and HEK293 cells (0.32 and 0.032 fmol, respectively). Interestingly, pretreatment of U87-MG cells with free RGD peptide (50 μM, 30 min) substantially inhibited the uptake of conjugate 1-RGD (0.34 fmol) while only exerting a slight effect in MCF-7 and HEK293 cells (0.25 and 0.030 fmol, respectively). The reduced uptake for conjugate 1-RGD is attributed to the competitive binding with free RGD peptide to the integrin binding sites, indicative of the specificity of the conjugate towards integrin. Noteworthily, complexes 2–4 also displayed higher uptake towards U87-MG and MCF-7 cells (1.1–2.5 fmol) and much lower accumulation in HEK293 cells (0.25–0.77 fmol). However, the cellular internalisation of these complexes was not influenced by the preincubation of the cells with free RGD, indicative of no integrin binding specificity.
Based on the results, it appears that conjugate 1-RGD was internalised by cells through an integrin-mediated mechanism, particularly in cell lines that overexpress integrins, such as U87-MG cells. The cellular uptake mechanism was further investigated using various inhibitors. Upon treating U87-MG cells with the conjugate at low temperature (4 °C), the amount of intracellular iridium significantly decreased compared to cells incubated at 37 °C (Fig. S7, ESI†). However, the internalisation of the conjugate remained unaffected by pretreatment with a cation transporter inhibitor (tetraethylammonium chloride). Preincubation of the cells with metabolic inhibitors (2-deoxy-D-glucose and oligomycin) or endocytosis inhibitors (ammonium chloride or chloroquine) also substantially reduced the cellular uptake efficiency of the conjugate. These results collectively indicate that conjugate 1-RGD was taken up by cells via an energy-dependent endocytic pathway.
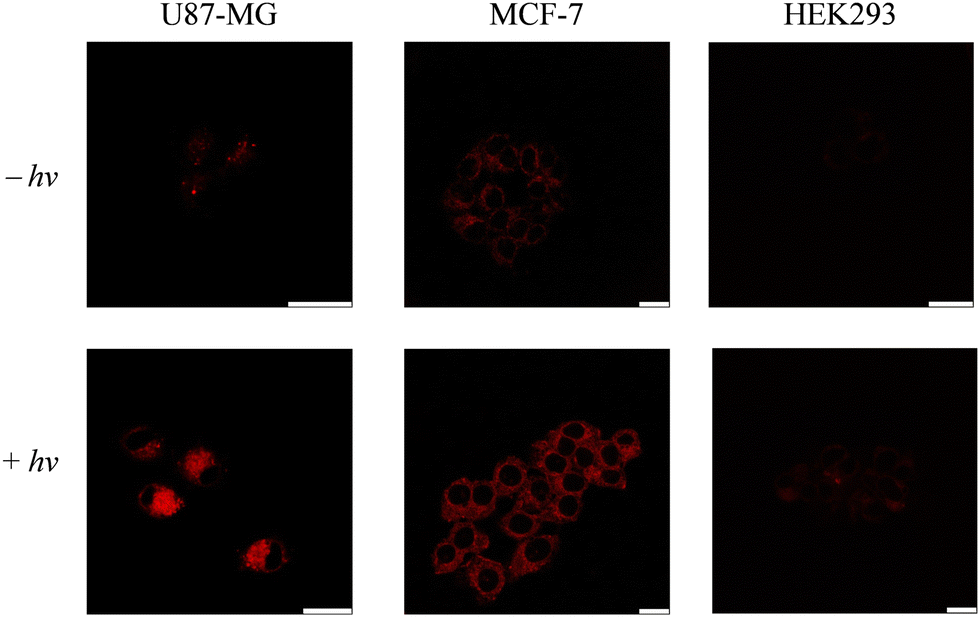
To further examine the specificity of conjugate 1-RGD towards integrin αvβ3, cells were preincubated without or with free RGD, treated with the conjugate and exposed to steady-state irradiation. As revealed by laser-scanning confocal microscopy (LSCM), the intracellular emission intensity significantly decreased in U87-MG and MCF-7 cells pretreated with the free RGD peptide, while no noticeable changes were observed for HEK293 cells pretreated with or without the RGD peptide (Fig. 3). Notably, preincubation with the RGD peptide did not reduce the emission intensity of complex 2 towards any of the three cell lines (Fig. S8, ESI†), which further supports the integrin-mediated cellular uptake of conjugate 1-RGD. The phosphorogenic property of conjugate 1-RGD in live cells was also investigated by LSCM. Significant emission enhancement was observed for U87-MG and MCF-7 cells that were treated with the conjugate and subsequently photoirradiated, compared to the cells treated with the conjugate under dark conditions (Fig. 4). The emission enhancement is attributed to the photodissociation of the S,S-tetrazine moiety in conjugate 1-RGD within the cells. In contrast, only minimal emission changes were observed for HEK293 cells treated with conjugate 1-RGD under both dark and light conditions, probably due to limited cellular uptake of the conjugate as a result of the lower integrin expression levels of the cells. This is further supported by flow cytometric measurements, which showed higher intracellular emission enhancement for U87-MG and MCF-7 cells treated with conjugate 1-RGD after light irradiation compared to HEK293 cells (Fig. S9, ESI†). Additionally, ESI-MS analyses of U87-MG cell lysates indicate that the conjugate was transformed into the thiocyanate and amide products after photoirradiation (Fig. S10, ESI†). Similar emission enhancement was observed for U87-MG and MCF-7 cells treated with the RGD-free complex 2 while substantially lower emission enhancement was observed in HEK293 cells (Fig. S11, ESI†). The intracellular localisation of conjugate 1-RGD and complexes 2–4 was investigated by LSCM. The phosphorogenic IEDDA reaction was utilised to study the intracellular localisation of conjugate 1-RGD and complex 2 due to the intrinsically weak emission. The U87-MG cells were first treated with conjugate 1-RGD or complex 2 (5 μM, 4 h), followed by incubation of BCN-OH (250 μM, 4 h) and MitoTracker Deep Red (100 nM, 20 min). LSCM images revealed that the luminescent products were specifically localised in the mitochondria with Pearson's correlation coefficients (PCC's) of 0.96 and 0.97, respectively (Fig. S12, ESI†). The cellular localisation of conjugate 1-RGD and complex 2 after photoirradiation was also studied. Incubation of U87-MG cells with conjugate 1-RGD or complex 2 (5 μM, 4 h), followed by photoirradiation at 450 nm (10 mW cm−2) for 20 min and then co-staining with MitoTracker Deep Red (100 nM, 20 min), gave rise to substantial image overlap, with PCC values of 0.92 and 0.96, respectively (Fig. 5). The intracellular localisation of their photoproducts was examined by co-staining experiments involving complexes 3 and 4 (5 μM, 4 h), which showed significant mitochondrial accumulation (PCCs = 0.96 and 0.97, respectively) (Fig. 5). The high mitochondria specificity of the conjugate and complexes can be attributed to their high lipophilicity and monocationic charge.39,40
 |
| | Fig. 3 LSCM images of live U87-MG, MCF-7 and HEK293 cells pretreated without (upper) or with (lower) RGD peptide (50 μM, 30 min), incubated with conjugate 1-RGD (5 μM, 4 h; λex = 405 nm, λem = 600–700 nm) and followed by continuous photoirradiation at 450 nm (10 mW cm−2) for 20 min. Scale bar = 20 μm. | |
 |
| | Fig. 4 LSCM images of U87-MG, MCF-7 and HEK293 cells incubated with conjugate 1-RGD (5 μM, 4 h; λex = 405 nm, λem = 600–700 nm) without (upper) or with (lower) continuous photoirradiation at 450 nm (10 mW cm−2) for 20 min. Scale bar = 20 μm. | |
 |
| | Fig. 5 LSCM images of live U87-MG cells incubated with conjugate 1-RGD or complex 2 (5 μM, 4 h; λex = 405 nm, λem = 600–700 nm) and photoirradiated at 450 nm (10 mW cm−2) for 20 min or incubated with complexes 3 or 4 (5 μM, 4 h; λex = 405 nm, λem = 600–700 nm) in the dark and then treated with MitoTracker Deep Red (100 nM, 20 min; λex = 635 nm, λem = 650–680 nm). Scale bar = 20 μm. PCC = 0.92 (1-RGD); 0.96 (2); 0.96 (3) and 0.97 (4). | |
Mitochondria play vital roles in cellular energy production, maintaining calcium levels and regulating programmed cell death within the cells.41,42 Given the high mitochondria-targeting ability of conjugate 1-RGD and complexes 2–4, their (photo)cytotoxicity towards cancerous (U87-MG and MCF-7) and normal (HEK293) cells was evaluated using the MTT assay. Notably, conjugate 1-RGD exhibited higher dark cytotoxicity towards U87-MG cells (IC50,dark = 26 μM) but no dark cytotoxicity towards MCF-7 and HEK293 cells (IC50,dark > 50 μM) (Table 3), which is consistent with its cellular uptake efficiencies (Table S3, ESI†) and integrin-targeting properties. Additionally, the conjugate showed the highest photocytotoxicity towards U87-MG cells (IC50,light = 0.08 μM) with a photocytotoxicity index (PI) value of 325 (Table 3), indicating the excellent targeting capability towards cancerous cells with integrin αvβ3 overexpression. Conjugate 1-RGD also displayed high photocytotoxic activity towards cancerous MCF-7 cells (IC50,light = 0.22 μM; PI = 236) with much lower photocytotoxicity towards normal HEK293 cells (IC50,light = 1.3 μM; PI > 38). Complexes 2–4, which did not exhibit any integrin-targeting behaviour, showed high dark cytotoxicity (IC50,dark = 2.5–15 μM) and increased photocytotoxicity upon light irradiation (IC50,light = 0.02–0.10 μM) towards all the cell lines. Therefore, it can be inferred that the excellent photoinduced cytotoxicity of conjugate 1-RGD is attributed to its targeting properties towards integrin-overexpressing cancer cells, the release of highly cytotoxic photoproducts (complexes 3 and 4) and subsequent enhanced 1O2 photosensitisation effect upon photoirradiation. Calcein-AM and propidium iodide double staining assay was used to further investigate the therapeutic efficacy of conjugate 1-RGD towards U87-MG cells (Fig. S13, ESI†). Cells treated with conjugate 1-RGD exhibited intense fluorescence from Calcein-AM with no emission from propidium iodide. After light irradiation, the green emission from Calcein-AM diminished, while strong red emission from the propidium dye was detected for the 1-RGD-treated cells. This observation confirms the biocompatibility of the conjugate under dark conditions and highlights its potent cytotoxic activity upon light activation.
Table 3 (Photo)cytotoxicity of conjugate 1-RGD and complexes 2–4 towards U87-MG, MCF-7 and HEK293 cells. The cells were first incubated with the conjugate or complexes in the dark for 4 h, then incubated in the dark or irradiated at 450 nm (10 mW cm−2) for 20 min and kept in the dark for 16 h. PI is the ratio of IC50,dark/IC50,light
| Complex/conjugate |
U87-MG |
MCF-7 |
HEK293 |
| IC50,dark/μM |
IC50,light/μM |
PI |
IC50,dark/μM |
IC50,light/μM |
PI |
IC50,dark/μM |
IC50,light/μM |
PI |
|
1-RGD
|
26 ± 1 |
0.08 ± 0.01 |
325 |
52 ± 1 |
0.22 ± 0.01 |
236 |
>50 |
1.3 ± 0.1 |
>38 |
|
2
|
11 ± 1 |
0.08 ± 0.01 |
138 |
15 ± 1 |
0.10 ± 0.01 |
150 |
12 ± 1 |
0.09 ± 0.01 |
133 |
|
3
|
2.7 ± 0.1 |
0.02 ± 0.01 |
135 |
2.5 ± 0.1 |
0.02 ± 0.01 |
125 |
3.5 ± 0.2 |
0.04 ± 0.01 |
88 |
|
4
|
4.1 ± 0.2 |
0.04 ± 0.01 |
103 |
4.2 ± 0.1 |
0.04 ± 0.01 |
105 |
5.0 ± 0.1 |
0.05 ± 0.01 |
100 |
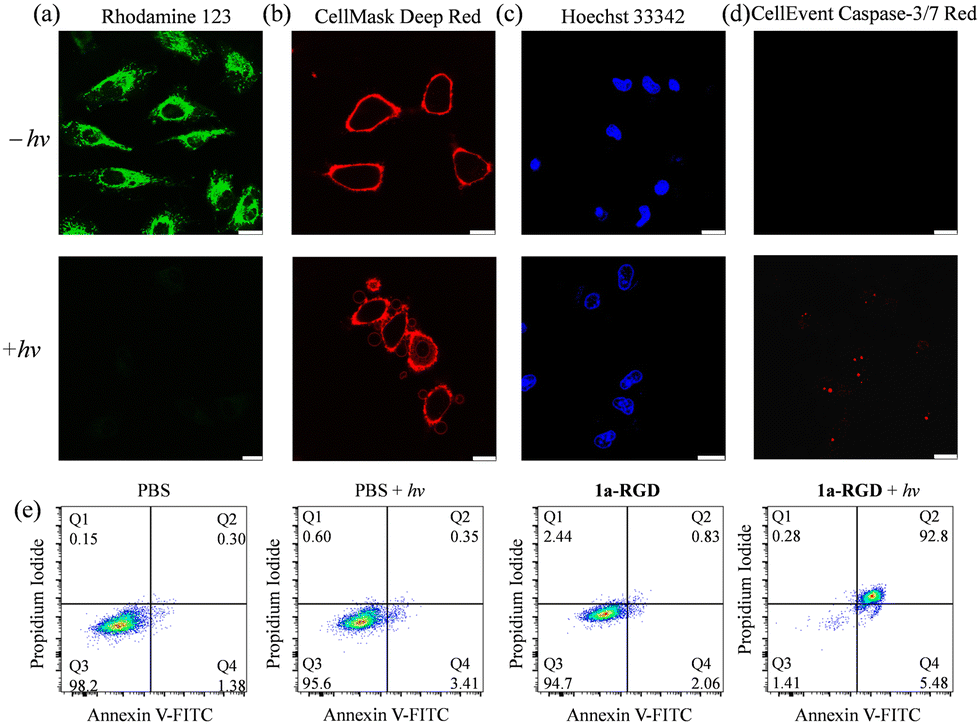
Based on the impressive cancer cell selectivity and photoactivatable property of conjugate 1-RGD, the photoinduced cell death pathway was further examined. LSCM images revealed that U87-MG cells treated with conjugate 1-RGD and reactive oxygen species (ROS) indicator CM-H2DCFDA displayed strong emission under light conditions compared to cells under dark treatment, indicative of the effective intracellular ROS generation by the conjugate upon photoactivation (Fig. S14, ESI†). The mitochondrial membrane potential (MMP) was characterised by staining the cells with rhodamine 123 after incubation of conjugate 1-RGD without or with light irradiation. Intense emission was detected for cells under the dark condition, which significantly diminished in intensity after light irradiation, suggesting the opening of the mitochondrial permeability transition pores (Fig. 6a). Additionally, morphological features for apoptotic cells, such as plasma membrane blebbing and nuclear condensation were also observed when the cells were stained with CellMask Deep Red and Hoechst 33342 (Fig. 6b and c), respectively. The activation of caspase-3/7 was also observed, implying an apoptotic pathway for cells treated with the conjugate upon photoactivation (Fig. 6d). The cell death mechanism of the conjugate was further examined using Annexin V/propidium iodide staining assays. As shown in Fig. 6e, there were very low populations of apoptotic cells (2.89%) in cells treated with the conjugate under the dark condition, which is comparable to untreated cells (1.68%) or cells subjected to light irradiation alone (3.76%). However, the population of apoptotic cells sharply increased to 98.28% after photoirradiation of the 1-RGD-incubated cells, confirming that apoptosis is the primary cell death pathway induced by the conjugate. All these results highlight the use of S,S-tetrazine-bearing iridium(III)–RGD conjugates as photoactivatable reagents for cancer-targeted imaging and therapy.
 |
| | Fig. 6 LSCM images of (a) MMP, (b) plasma membrane, (c) nuclear morphology, or (d) caspase-3/7 activity of U87-MG cells upon pretreatment with conjugate 1-RGD (5 μM, 4 h) without (upper) or with (lower) light irradiation at 450 nm (10 mW cm−2) for 20 min. All the samples were stained with rhodamine 123 (5 μM, 15 min; λex = 488 nm, λem = 500–550 nm), CellMask Deep Red (5 μM, 15 min; λex = 635 nm, λem = 650–700 nm), Hoechst 33342 (5 μM, 15 min; λex = 405 nm, λem = 415–495 nm), or CellEvent Caspase-3/7 Red (20 μL, 1:100, 1 h; λex = 590 nm, λem = 610–630 nm) prior to microscope imaging. Scale bar = 20 μm. (e) Flow cytometric analysis of U87-MG cells pretreated with conjugate 1-RGD (1 μM, 4 h) without or with continuous photoirradiation at 450 nm (10 mW cm−2) for 20 min, followed by staining with Alexa Fluor 647–Annexin V conjugate (5 μL, 15 min; λex = 638 nm) and propidium iodide (2 μL, 100 μg mL−1, 15 min; λex = 561 nm). | |
Conclusions
We developed a new platform for the preparation of a cancer-selective photoactivatable anticancer prodrug with controllable emission and photosensitisation of 1O2. The prodrug was prepared by conjugation of an iridium(III) Cl,S-tetrazine complex with a cancer-targeting cyclic RGD peptide. The resulting conjugate 1-RGD initially displayed low emission intensity and 1O2 generation efficiency due to the quenching tetrazine unit. However, upon photoactivation, significant emission enhancement and lifetime extension were observed for the conjugate due to the photodissociation of the S,S-tetrazine unit, accompanied by improved 1O2 generation. The IEDDA cycloaddition reaction of the conjugate with BCN-OH also led to substantial emission enhancement. Importantly, conjugate 1-RGD demonstrated the highest cellular uptake in U87-MG cells overexpressing integrin αvβ3 and exhibited remarkable photoinduced anticancer properties resulting from the synergistic effects of the release of the highly cytotoxic iridium(III) thiocyanate and amide complexes and enhanced 1O2 photogeneration. We believe that the introduction of a Cl,S-tetrazine moiety to transition metal complexes or other molecular scaffolds will be a versatile paradigm for the development of innovative anticancer agents or drug delivery systems.
Author contributions
L. H.: conceptualisation, data curation, formal analysis, investigation, writing – original draft, writing – review & editing; J. S.: data curation, formal analysis, writing – original draft, writing – review & editing; L. C.-C. L.: data curation, formal analysis, writing – original draft, writing – review & editing; G.-X. Xu.: data curation, formal analysis, writing – original draft, writing – review & editing; P. K.-K. L.: data curation, formal analysis, writing – original draft, writing – review & editing; K. K.-W. L.: conceptualisation, funding acquisition, project administration, resources, supervision, writing – original draft, writing – review & editing.
Data availability
The data supporting this article have been included as part of the ESI.†
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank the Hong Kong Research Grants Council (Project No. CityU 11301121, CityU 11317022, CityU 11309423 and C7075-21GF) and the Hong Kong Research Grants Council and National Natural Science Foundation of China (Project No. N_CityU104/21) for financial support. We also thank the funding support from “Laboratory for Synthetic Chemistry and Chemical Biology” under the Health@InnoHK Programme launched by the Innovation and Technology Commission, The Government of Hong Kong SAR, P. R. China. L. H. acknowledges the receipt of a Postgraduate Studentship and Research Tuition Scholarship, both administered by City University of Hong Kong.
References
- J. L. Lau and M. K. Dunn, Bioorg. Med. Chem., 2018, 26, 2700 CrossRef CAS PubMed.
- R. A. Petros and J. M. DeSimone, Nat. Rev. Drug Discovery, 2010, 9, 615 CrossRef CAS PubMed.
- R.-M. Lu, Y.-C. Hwang, I. J. Liu, C.-C. Lee, H.-Z. Tsai, H.-J. Li and H.-C. Wu, J. Biomed. Sci., 2020, 27, 1 CrossRef CAS PubMed.
- Y. T. Lee, Y. J. Tan and C. E. Oon, Eur. J. Pharmacol., 2018, 834, 188 CrossRef CAS PubMed.
- E. Pérez-Herrero and A. Fernández-Medarde, Eur. J. Pharm. Biopharm., 2015, 93, 52 CrossRef PubMed.
- Y. Li, K. Xiao, W. Zhu, W. Deng and K. S. Lam, Adv. Drug Delivery Rev., 2014, 66, 58 CrossRef CAS PubMed.
- M. Karimi, A. Ghasemi, P. Sahandi Zangabad, R. Rahighi, S. M. Moosavi Basri, H. Mirshekari, M. Amiri, Z. Shafaei Pishabad, A. Aslani, M. Bozorgomid, D. Ghosh, A. Beyzavi, A. Vaseghi, A. R. Aref, L. Haghani, S. Bahrami and M. R. Hamblin, Chem. Soc. Rev., 2016, 45, 1457 RSC.
- J. M. Silva, E. Silva and R. L. Reis, J. Controlled Release, 2019, 298, 154 CrossRef CAS PubMed.
- R. Weinstain, T. Slanina, D. Kand and P. Klán, Chem. Rev., 2020, 120, 13135 CrossRef CAS PubMed.
- X. Ai, J. Mu and B. Xing, Theranostics, 2016, 6, 2439 CrossRef CAS PubMed.
- N. A. Smith and P. J. Sadler, Philos. Trans. R. Soc., A, 2013, 371, 20120519 CrossRef PubMed.
- K. K.-W. Lo, Acc. Chem. Res., 2015, 48, 2985 CrossRef CAS PubMed.
- C. Imberti, P. Zhang, H. Huang and P. J. Sadler, Angew. Chem., Int. Ed., 2020, 59, 61 CrossRef CAS PubMed.
- L. C.-C. Lee and K. K.-W. Lo, Chem. Rev., 2024, 124, 8825 CrossRef CAS PubMed.
- N. J. Farrer, L. Salassa and P. J. Sadler, Dalton Trans., 2009, 10690 RSC.
- U. Schatzschneider, Eur. J. Inorg. Chem., 2010, 2010, 1451 CrossRef.
- B. S. Howerton, D. K. Heidary and E. C. Glazer, J. Am. Chem. Soc., 2012, 134, 8324 CrossRef CAS PubMed.
- L. N. Lameijer, D. Ernst, S. L. Hopkins, M. S. Meijer, S. H. C. Askes, S. E. Le Dévédec and S. Bonnet, Angew. Chem., Int. Ed., 2017, 56, 11549 CrossRef CAS PubMed.
- K. K.-S. Tso, K.-K. Leung, H.-W. Liu and K. K.-W. Lo, Chem. Commun., 2016, 52, 4557 RSC.
- N. K. Devaraj, S. Hilderbrand, R. Upadhyay, R. Mazitschek and R. Weissleder, Angew. Chem., Int. Ed., 2010, 49, 2869 CrossRef CAS PubMed.
- K. Lang, L. Davis, J. Torres-Kolbus, C. Chou, A. Deiters and J. W. Chin, Nat. Chem., 2012, 4, 298 CrossRef CAS PubMed.
- S. P.-Y. Li, A. M.-H. Yip, H.-W. Liu and K. K.-W. Lo, Biomaterials, 2016, 103, 305 CrossRef CAS PubMed.
- J. Shum, L. C.-C. Lee, M. W.-L. Chiang, Y.-W. Lam and K. K.-W. Lo, Angew. Chem., Int. Ed., 2023, 62, e202303931 CrossRef CAS PubMed.
- M. J. Tucker, M. Abdo, J. R. Courter, J. Chen, A. B. Smith III and R. M. Hochstrasser, J. Photochem. Photobiol., A, 2012, 234, 156 CrossRef CAS PubMed.
- M. J. Tucker, J. R. Courter, J. Chen, O. Atasoylu, A. B. Smith III and R. M. Hochstrasser, Angew. Chem., Int. Ed., 2010, 49, 3612 CrossRef CAS PubMed.
- J. Waluk, J. Spanget-Larsen and E. W. Thulstrup, Chem. Phys., 1995, 200, 201 CrossRef CAS.
- C. Canovas, M. Moreau, C. Bernhard, A. Oudot, M. Guillemin, F. Denat and V. Goncalves, Angew. Chem., Int. Ed., 2018, 57, 10646 CrossRef CAS PubMed.
- S. P.-Y. Li, V. M.-W. Yim, J. Shum and K. K.-W. Lo, Dalton Trans., 2019, 48, 9692 RSC.
- J.-H. Zhu, G.-X. Xu, J. Shum, L. C.-C. Lee and K. K.-W. Lo, Chem. Commun., 2021, 57, 12008 RSC.
- K. Suzuki, A. Kobayashi, S. Kaneko, K. Takehira, T. Yoshihara, H. Ishida, Y. Shiina, S. Oishi and S. Tobita, Phys. Chem. Chem. Phys., 2009, 11, 9850 RSC.
- S. Hehlgans, M. Haase and N. Cordes, Biochim. Biophys. Acta, 2007, 1775, 163 CAS.
- E. Ruoslahti and M. D. Pierschbacher, Science, 1987, 238, 491 CrossRef CAS PubMed.
- A. Meyer, J. Auernheimer, A. Modlinger and H. Kessler, Curr. Pharm. Des., 2006, 12, 2723 CrossRef CAS PubMed.
- H. Hamidi and J. Ivaska, Nat. Rev. Cancer, 2018, 18, 533 CrossRef CAS PubMed.
- S. L. Goodman, H. J. Grote and C. Wilm, Biol. Open, 2012, 1, 329 CrossRef CAS PubMed.
- H. A. Kim, K. Nam and S. W. Kim, Biomaterials, 2014, 35, 7543 CrossRef CAS PubMed.
- D. Ding, J. Liang, H. Shi, R. T. K. Kwok, M. Gao, G. Feng, Y. Yuan, B. Z. Tang and B. Liu, J. Mater. Chem. B, 2014, 2, 231 RSC.
- Z. Zhao, K. Qiu, J. Liu, X. Hao and J. Wang, Chem. Commun., 2020, 56, 12542 RSC.
- S. P.-Y. Li, C. T.-S. Lau, M.-W. Louie, Y.-W. Lam, S. H. Cheng and K. K.-W. Lo, Biomaterials, 2013, 34, 7519 CrossRef CAS PubMed.
- C. Caporale and M. Massi, Coord. Chem. Rev., 2018, 363, 71 CrossRef CAS.
- N. Joza, S. A. Susin, E. Daugas, W. L. Stanford, S. K. Cho, C. Y. J. Li, T. Sasaki, A. J. Elia, H.-Y. M. Cheng, L. Ravagnan, K. F. Ferri, N. Zamzami, A. Wakeham, R. Hakem, H. Yoshida, Y.-Y. Kong, T. W. Mak, J. C. Zúñiga-Pflücker, G. Kroemer and J. M. Penninger, Nature, 2001, 410, 549 CrossRef CAS PubMed.
- E. Verdin, M. D. Hirschey, L. W. S. Finley and M. C. Haigis, Trends Biochem. Sci., 2010, 35, 669 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Guang-Xi
Xu
a,
Peter Kam-Keung
Leung
ac and
Kenneth Kam-Wing
Lo
a,
Guang-Xi
Xu
a,
Peter Kam-Keung
Leung
ac and
Kenneth Kam-Wing
Lo