
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIdentification and characterization of ternary complexes consisting of FKBP12, MAPRE1 and macrocyclic molecular glues†
Michael
Salcius‡
a,
Antonin
Tutter‡
 a,
Marianne
Fouché‡
b,
Halil
Koc
b,
Dan
King
a,
Anxhela
Dhembi
a,
Andrei
Golosov
a,
Wolfgang
Jahnke
b,
Chrystèle
Henry
b,
Dayana
Argoti
c,
Weiping
Jia
c,
Liliana
Pedro
c,
Lauren
Connor
b,
Philippe
Piechon
b,
Francesca
Fabbiani
b,
Regis
Denay
b,
Emine
Sager
b,
Juergen
Kuehnoel
b,
Marie-Anne
Lozach
b,
Fabio
Lima
b,
Angela
Vitrey
b,
Shu-Yu
Chen
d,
Gregory
Michaud
*a and
Hans-Joerg
Roth
*b
a,
Marianne
Fouché‡
b,
Halil
Koc
b,
Dan
King
a,
Anxhela
Dhembi
a,
Andrei
Golosov
a,
Wolfgang
Jahnke
b,
Chrystèle
Henry
b,
Dayana
Argoti
c,
Weiping
Jia
c,
Liliana
Pedro
c,
Lauren
Connor
b,
Philippe
Piechon
b,
Francesca
Fabbiani
b,
Regis
Denay
b,
Emine
Sager
b,
Juergen
Kuehnoel
b,
Marie-Anne
Lozach
b,
Fabio
Lima
b,
Angela
Vitrey
b,
Shu-Yu
Chen
d,
Gregory
Michaud
*a and
Hans-Joerg
Roth
*b
aNovartis Biomedical Research, 181 Massachusetts Ave, Cambridge, USA
bNovartis Biomedical Research, Fabrikstrasse 2, CH-4056 Basel, Switzerland. E-mail: marianne.fouche@novartis.com; hans-joerg.roth@sunrise.ch
cNovartis Biomedical Research, 5959 Horton Street, Emeryville, USA
dEidgenösisch Technische Hochschule, Inst. Mol. Phys. Wiss, Zürich, Switzerland
First published on 6th March 2025
Abstract
Many disease-relevant and functionally well-validated targets are difficult to drug. Their poorly defined 3D structure without deep hydrophobic pockets makes the development of ligands with low molecular weight and high affinity almost impossible. For these targets, incorporation into a ternary complex may be a viable alternative to modulate and in most cases inhibit their function. Therefore, we are interested in methods to identify and characterize molecular glues. In a protein array screen of 50 different macrocyclic FKBP12 ligands against 2500 different randomly selected proteins, a molecular glue compound was found to recruit a dimeric protein called MAPRE1 to FKBP12 in a compound-dependent manner. The corresponding ternary complex was characterized by TR-FRET proximity assay and native MS spectroscopy. Insights into the 3D structure of the ternary complex were obtained by 2D protein NMR spectroscopy and finally by an X-ray structure, which revealed the ternary complex as a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:2 FKBP12:molecular glue:MAPRE1 complex exhibiting multiple interactions that occur exclusively in the ternary complex and lead to significant cooperativity α. Using the X-ray structure, rationally guided synthesis of a series of analogues led to the cooperativity driven improvement in the stability of the ternary complex. Furthermore, the ternary complex formation of the series was confirmed by cellular NanoBiT assays, whose Amax values correlate with those from the TR-FRET proximity assay. Furthermore, NanoBiT experiments showed the functional impact (inhibition) of these molecular glues on the interaction of MAPRE1 with its intracellular native partners.
:2:2 FKBP12:molecular glue:MAPRE1 complex exhibiting multiple interactions that occur exclusively in the ternary complex and lead to significant cooperativity α. Using the X-ray structure, rationally guided synthesis of a series of analogues led to the cooperativity driven improvement in the stability of the ternary complex. Furthermore, the ternary complex formation of the series was confirmed by cellular NanoBiT assays, whose Amax values correlate with those from the TR-FRET proximity assay. Furthermore, NanoBiT experiments showed the functional impact (inhibition) of these molecular glues on the interaction of MAPRE1 with its intracellular native partners.
Introduction
The modulation of difficult disease-relevant targets by small molecule-induced proximity between a chaperone protein and the target protein has become an important area of drug discovery.1–8 Depending on the type of chaperone recruited to the target, consequences such as ubiquitination, de-ubiquitination, inhibition of the interaction with native protein–protein (PPI) partners, translocation, phosphorylation, de-phosphorylation, glycosylation, chemical modifications by recruited enzymes or other physiological modifications have been reported.9–14 What all these modifications have in common is the formation of a ternary complex consisting of the target protein, the chaperone protein and the compound that induces the corresponding ternary complex.In principle, there are two types of ternary complex-forming compounds. Bifunctional compounds (Bfx) and molecular glues (MG). Ternary complexes formed by Bfx's predominantly derive their free enthalpy from the sum of independent binary affinities of the Bfx to the two proteins. In contrast, ternary complexes formed by molecular glues predominantly derive their free enthalpy from new interactions that occur only in the ternary complex, such as new protein–protein or new MG–protein interactions. The degree of additional stability of the ternary complex resulting from such newly induced interactions is referred to as “cooperativity α”. The cooperativity α can also be described as the degree of additional affinity of a ligand to a protein (the ratio between the binary and the ternary Kd) caused exclusively by the presence of the second protein. Ternary complexes formed by a Bfx may also exhibit cooperativity, but to a lesser extent than ternary complexes of the same thermodynamic stability formed by a MG.15–18
In practice, the distinction between Bfx and MG is often arbitrary, since in practice there is no such thing as a compound without affinity to a protein. It all depends on where the threshold for defining binary affinity to either or both proteins is set. Most ternary complex-forming compounds are therefore hybrids between Bfx and MG. Many of them (e.g. Cyclosporin, Rapamycin, FK506) or the more recent clinical candidate RMC-7977 even have a strong affinity for one of the two proteins. The facts that have led to their classification as typical molecular glues are that they have no or only weakly measurable affinity for the target, and that the ternary complexes that they induce have a very high cooperativity (>10000).19,20
For the same given thermodynamic stability of a ternary complex, MG tend to have a lower molecular weight than Bfx. MG are therefore potentially more drug-like and developable. Additionally, since MG more closely resemble conventional drug-like small molecules, they are more likely to have favorable membrane permeability and cellular uptake. Lastly, the high ternary complex cooperativity enabled by MG and the corresponding absence of a strong target affinity helps to avoid a key challenge inherent to Bfx known as the hook effect,21 whereby ternary complex formation is inhibited at high Bfx concentration. This emphasizes the importance of screening methods that allow hits to be ranked not only by their potency to form ternary complexes, but also by the degree of cooperativity that determines the observed potency. As is well known for binary ligands, the hit with the highest initial potency is not always the most promising. The same is true for ternary complex forming hits. Those that do not have the highest initial potency for ternary complex formation, but a higher cooperativity may have higher potential for drug development and are preferred for optimization.
This explains our interest in drug-like MG starting points for difficult targets. We were interested in exploring the scope of the ubiquitous protein FKBP12 with its – depending on the cell line – high cellular expression levels as a chaperone to recruit targets beyond mTor.22–31 At the time we started our work, mTor and calcineurin were the only targets known to be recruited and inhibited by a binary FKBP12-ligand complex, one with rapamycin, one with FK506 (the target CEP250 was published during our work).32 We wondered whether the FKBP12-Rapamycin-mTor example could be generalized and extended to targets other than mTor. We chose FKBP12 as the chaperone rather than cyclophilin, another proline isomerase with ubiquitous and high expression levels, because of the relatively simple, drug-like binding motif that was known in the literature (the “simplification” of the cyclophilin A binding motif of Sanglifehrin had not been published at that time).33 Also, the “modular synthesis” of macrocycles with the FKBP12 binding motif and a modular and diverse recruitment loop had already been published.23
We decided to explore the two possible dimensions of a chaperone-focused glue screen through two series of experiments. One series was to screen for target recruitment, testing a small number of targets against a higher number of FKBP12 ligands with diverse recruiting loops in the absence and presence of FKBP12. The other series was the reverse, i.e. screening for recruitment a lower number of compounds against a higher number of target proteins in the presence of FKBP12. This publication describes the results of the latter, in which we screened 50 macrocyclic FKBP12 ligands with different recruitment loops on a protein array with a diverse selection of approximately 2500 proteins. From the ∼125000 data points (50 cmpds × ∼2500 proteins), we identified one cmpd that selectively recruits a protein called MAPRE1 to FKBP12 in a compound-dependent manner and with significant cooperativity. We report here the characterization of the corresponding FKBP12:MG:MAPRE1 ternary complex by biophysical and biochemical assays, including the 3D structure obtained by NMR and X-ray crystallography. We also describe our effort to improve the potency by increasing the cooperativity of the molecular glue originally found. Furthermore, we demonstrate the formation of the ternary complexes in cells and show that ternary complex assembly in cells disrupts MAPRE1 association with a known MAPRE1 interacting protein.
Results
Generation of a macrocyclic library scaffold consisting of a FKBP12 binding motif and a modular recruitment loop
At the time we began this work, there was an ongoing internal debate about whether macrocyclic scaffolds have an inherent advantage over linear scaffolds in their ability to recruit target proteins to FKBP12 or other chaperones such as cyclophilin A. The examples of Rapamycin,34 FK506,35 WDB002,32 Cyclosporin A, Sanglifehrin A36 and the Revolution medicine compound RMC-623633 (which recruits RAS to Cyclophilin) suggest that this may be the case. However, even if the closely related linear analogues of these molecular glues no longer show recruitment activity, it could not be excluded that ab initio developed linear FKBP12 ligands may recruit protein targets to FKBP12 (or Cyclosporin) with high cooperativity. Recent results from the Hausch group show that non-macrocyclic compounds can be effective as molecular glues.37 Nevertheless, we decided to use a macrocyclic FKBP12 ligand library consisting of the well-known SLF-FKBP12 binding motif based on the work of Holt et al., who greatly simplified the FKBP12 binding motif of Rapamycin/FK506 (Fig. 1).38 Similar to others23,39 (Fig. 2), we complemented the FKBP12 binding motif with a modular peptide loop consisting of 2–3 amino acid residues (Fig. 3). Using solution phase chemistry, we synthesized approximately 2000 purified compounds, each in amounts of ca. 5 mg, 85% of which had an affinity for FKBP12 of <500 nM as measured in a competitive TR-FRET binding assay. From this library, 50 as diverse as possible compounds with an affinity for FKBP12 of less than 250 nM were selected for MG screening against the ∼2500 proteins on the array. | ||
| Fig. 1 Simplification of the FKBP12 binding motif. Rapamycin and FK506 have the same 9 stereocenter-containing moiety (shown in red, left side), which has been structurally and synthetically simplified to a moiety with only 2 stereocenters, retaining most of the affinity for FKBP12. Changing the stereoconfiguration of the 4-ethyl-1,2-dimethoxybenzene substituent from R to S completely abolishes the binding affinity to FKBP12, giving an impression of the high specificity of even the simplified binding motif (right side). | ||
 | ||
| Fig. 2 Supplementation of the FKBP12 binding motif by a diverse recruiting loop. While the binding of FKBP12 is very sensitive to structural changes in the binding moiety itself, this is not the case for its macrocyclic complementation. Peptidic loops with different numbers of residues, N-methylated or not, are tolerated, maintaining a low nM affinity to FKBP12. | ||
 | ||
| Fig. 3 Macrocyclic scaffolds for FKBP12 ligands with divers and modular recruitment loops. (left) Dipeptidic recruitment loop using Lys side-chain cyclization leads to an “exocyclic” amino group as a diversity vector for functionalization as amides, ureas or amines. (right) The di- or tripeptidic recruitment loop with head-to-tail cyclization leads to an “endocyclic” diversity originating exclusively from the amino acid residues. | ||
Protein array screening and results from hit validation
The protein array screening experiment was performed by incubating a 500 nM solution of biotin-labeled FKBP12 (bio-FKBP12) with the immobilized proteins on the array in the absence and presence of 10 μM compound. Fluorophore-coupled streptavidin was then added to detect the bound bio-FKBP12. Bio-FKBP12 alone showed expected and novel native protein–protein interactions. Of the 50 compounds analyzed, only SLF-1 (a mixture of two epimers denoted later as R,S-SLF-1a and S,S-SLF-1d, see below) showed novel residual fluorescence at several spots on the array after careful washing, which was interpreted as an indication of compound-dependent ternary complex formation (Fig. 4). | ||
| Fig. 4 Schematic representation of the protein array screening. On a protein array glass surface, approximately 2500 randomly collected proteins were incubated with fluorescently labelled FKBP12 (FL-FKBP12). After washing the surface, no novel residual fluorescence was observed on the surface in the absence of any of the 50 selected macrocyclic FKBP12 ligands. In contrast, in the presence of one of the 50 compounds per run, one compound caused novel residual fluorescence on the surface at one position (n = 2), indicating compound-dependent ternary complex formation. | ||
The spots on the array with novel residual fluorescence mapped to replicates of a single protein called MAPRE1, a plus-end tracking protein (+TIPs) that regulates microtubule (MT) behavior and interactions between MTs and other intracellular structures during mitosis. MAPRE1 belongs to the end-binding (EB) family and is also called EB1. MAPRE1 has been shown to bind directly to MTs and to a variety of +TIPs and cytoskeletal proteins, recruiting them to the plus ends. MAPRE1 consists of two domains connected by a flexible linker: the MT-binding domain at the N-terminus (CH), which binds directly to microtubules, and the end-binding homology (EBH) at the C-terminus, the recruitment domain. The EBH domain is structurally a coiled-coil domain. MAPRE1 exists as a homodimer (Fig. 5).40
 | ||
| Fig. 5 MAPRE1 is the only one out of 2500 proteins recruited by 1 out of 50 FKBP12 tested ligands into a ternary complex MAPRE1:R,S-SLF-1a:FKBP12. Model of full length MAPRE1 structure based on 2QJZ (N-term domain) and 1TXQ (C-term domain). MAPRE1 belongs to the family of end-binding proteins (EB) and is a “bifunctional” protein that binds to microtubules (MTs) and brings them into proximity to plus end tracking proteins (+TIPs). | ||
To validate SLF-1 as a ternary complex-forming compound, three different TR-FRET assays were established to demonstrate compound-dependent induced proximity between FKBP12 and (1) full-length MAPRE1, (2) the C-terminal coiled-coil domain, and (3) the N-terminal MT-binding domain. The curves obtained showed very clearly that full-length MAPRE1 and its coiled-coil domain are recruited to FKBP12 in a compound-dependent manner, while the MT-binding domain showed no signs of ternary complex formation (Fig. 6 upper panel). This result validated SLF-1 as a ternary complex forming sample and demonstrated that recruitment of FKBP12 to MAPRE1 occurs via the coiled-coil domain and not via the MT-binding domain.
 | ||
| Fig. 6 TR-FRET proximity assay that validates the ternary complex formation by the screening sample and the resynthesized single isomer involving the coiled coil domain of MAPRE1. The screening sample SLF-1 (diastereomeric mixture) brings full-length MAPRE1 (red) and its coiled-coil-containing N-terminal domain (green) into proximity with FKBP12 in a compound-dependent manner, whereas the C-terminal domain of MAPRE1 (yellow) does not form a ternary complex with SLF-1 and FKBP12 (upper part). Resynthesized R,S-SLF-1a confirms exclusive ternary complex formation with the C-terminus of MAPRE1, even with a higher Amax and assigns ternary complex formation to a single epimer (lower part). | ||
Stereoselective synthesis of R,S-SLF-1a and S,S-SLF-1d and assignment of ternary complex formation to one epimer
The only hit sample SLF-1 found in the protein array screening experiment was a mixture of two epimers. To find out whether the activity is caused by only one of the two and which or by both isomers, we performed a chiral separation of the two enantiomers of the corresponding racemic 4-methylenepiperidine-2-carboxylic acid building block. With both building blocks in hand, but without knowing the absolute stereo configuration, we performed an amide coupling with Boc-L-alanine and subsequent cyclization with each of them, leading to the two corresponding diketopiperazines, one with the two α protons cis and one with them trans to each other. The presence or absence of NOEs between the two α protons allowed the absolute stereochemistry of the isolated enantiomers of the 4-methylenepiperidine-2-carboxylic acid building block to be unambiguously assigned. Peak 1 of the chiral separation corresponded to (S)-4-methylenepiperidine-2-carboxylic acid and peak 2 to (R)-4-methylenepiperidine-2-carboxylic acid (Fig. 7). | ||
| Fig. 7 Assignment of absolute stereochemistry of separated building block enantiomers. The chiral separation of racemic Boc-4-methylenepiperidine-2-carboxylic acid led to two enantiomerically pure amino acids with undetermined absolute stereochemistry. Coupling each of them with L-Ala-OtBu and subsequent cyclization to the two diastereomeric diketopiperazines allowed the assignment of the absolute stereochemistry of the two 4-methylenepiperidine-2-carboxylic acids used. The enantiomer with the earlier retention time (1.42 min) led to the diketopiperazine with two α protons cis to each other, the enantiomer with the later retention time (2.09 min) to the diketopiperazine with the two α protons trans to each other. | ||
With the two enantiomeric building blocks in hand, we were able to synthesize the corresponding epimers from the hit sample SLF-1 with defined stereochemistry, R,S-SLF-1a with the R configuration at the 4-methylenepiperidine-2-carboxylic acid position and the S configuration at the Ala position and S,S-SLF-1d with the S configuration at the methylenepiperidine moiety and the S configuration at the second amino acid (Ala) (Fig. 8).
 | ||
| Fig. 8 Relation between enantiomerically pure 4-methylenepiperidine-2-carboxylic acid and corresponding macrocyclic FKBP12 ligands. Enantiomerically pure and assigned (R)- and (S)-Boc-4-methylenepiperidine-2-carboxylic acid enabled the synthesis of the single isomers R,S-SLF-1a (left) and S,S-SLF-1d (right). | ||
Retesting of the two macrocyclic epimers in the TR-FRET proximity assay showed that the observed recruitment activity is exclusively due to R,S-SLF-1a, although S,S-SLF-1d has the higher binary affinity to FKPB12 alone (30 nM) than R,S-SLF-1a (230 nM) (Fig. 6 lower part, Fig. 9).
 | ||
| Fig. 9 Assignment of recruitment activity to the epimer R,S-SLF-1a. Only the single isomer R,S-SLF-1a confirmed the initially observed activity, while S,S-SLF-1d was completely inactive, indicating a high specificity of recruitment of MAPRE1 to the preformed binary R,S-SLF-1a:FKBP12 complex. | ||
R,S-SLF-1a was further investigated by native MS. In native MS experiments of FKBP12 with MAPRE1 only, both monomer and homodimer of MAPRE1 were observed, the homodimer being the primary species. Additionally, FKBP12 was observed as a monomer. There was no evidence of a ternary complex being formed in absence of R,S-SLF-1a.
Performing the same experiments but adding R,S-SLF-1a confirmed compound dependent ternary complex formation. Several bound species were observed in the native MS spectra. The first ligand bound species observed is between HIS-FKBP12 and the ligand, with a composition of 1 FKBP12 + 1 ligand. Additionally, two ternary complexes were confirmed: the first complex containing 1 FKBP12 + 1 ligand + MAPRE1 dimer as major product. The second complex observed was composed of 2 FKBP12 + 2 ligand + MAPRE1 dimer. These results show that the MAPRE1 homodimer can recruit an FKBP12 to each of its monomers, mediated by one molecule of R,S-SLF-1a at a time, leading to a 2:2:2 complex (Fig. 10).
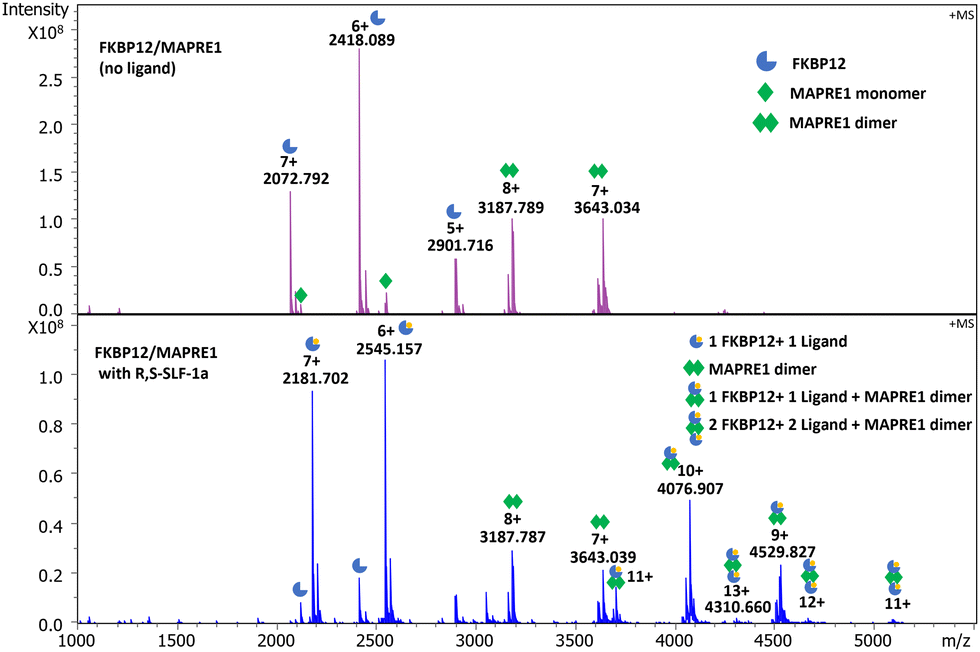 | ||
| Fig. 10 Native MS of the ternary complex FKBP12:R,S-SLF-1a:MAPRE1 (2:2:2). Conditions: 10 μM FKBP and 10 μM MAPRE1 with 25 μM ligand, 1% DMSO. Top panel: FKBP1a with MAPRE1. The primary species observed of MAPRE1 were both monomer (single green diamond) and dimer (double green diamond). FKBP12 monomer was the main species observed for FKBP12 (partial blue circle). Bottom panel: Ternary complex formation mediated by R,S-SLF-1a. The species observed were as follows: free FKBP12 (partial blue circle), FKBP12 + R,S-SLF-1a (partial blue circle with small yellow circle) and MAPRE1 was mainly observed as dimer (double green diamond). Additionally, a ternary complex composed of 1× FKBP12 + 1× R,S-SLF-1a + MAPRE1 dimer (partial blue circle with small yellow circle and double green diamond) and a complex composed of 2× FKBP1a + 2× R,S-SLF-1a + MAPRE1 dimer (2× partial blue circle with small yellow circle and double green diamond). | ||
Protein NMR studies on the ternary complex FKBP12:R,S-SLF-1a:MAPRE1
In order to obtain structural information, we studied the ternary complex by NMR, using 13C–15N-labeled MAPRE1 with unlabeled FKBP12 and R,S-SLF-1a. Since the resonance assignments for MAPRE1 were published by Kanaba et al.,41 we were expecting to delineate the binding site of FKBP12/R,S-SLF-1a on MAPRE1 by observing chemical shift or signal intensity changes in the ternary complex when compared to MAPRE1 in isolation. In NMR, chemical shift or signal intensity changes generally indicate binding of a ligand or another protein, and they can be caused by direct effects (proximity of the ligand) or indirect effects (conformational changes upon binding, thus suggesting allostery).
Fig. 11(A) shows the NMR 15N-HSQC spectrum of MAPRE1 (100 μM, black), and of a 2:2:2 complex of 13C,15N-MAPRE1:R-SLF-1:FKBP12 (100 μM, red). It can be seen that many MAPRE1 signals are affected after addition of FKBP12 and R,S-SLF-1a. While some signals experience chemical shift changes (e.g. E213), most signals experience a strong reduction in signal intensity, essentially getting quenched upon addition of FKBP12 and R,S-SLF-1a. This is probably due to the significantly higher molecular weight and the anisotropic tumbling of the ternary complex. Selected resonance assignments from Kanaba et al. are added to the spectra in Fig. 11(A).
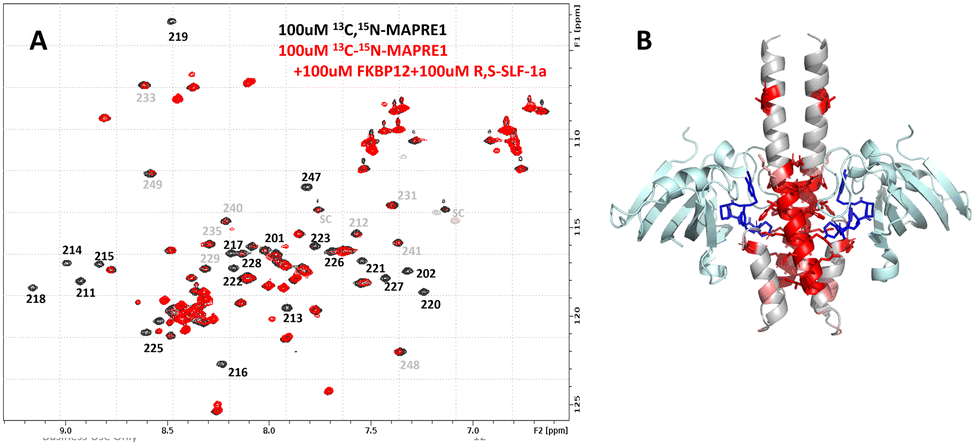 | ||
| Fig. 11 NMR characterization of the ternary complex FKBP12:R,S-SLF-1a:MAPRE1 (2:2:2). (A) 15N-HSQC NMR spectrum of 13C,15N-MAPRE1 (100 μM, black) is superimposed on a 15N-HSQC NMR spectrum of 13C,15N-MAPRE1 in presence of FKBP12:R,S-SLF-1a, generating the 2:2:2 ternary complex. Resonance assignments are given in the figure. (B) MAPRE1 residues that are affected by the addition of FKBP12:R,S-SLF-1a are mapped onto the X-ray structure of the ternary complex. FKBP12 is shown in light cyan, R,S-SLF-1a is shown in blue, and MAPRE1 is shown in gray, except for the residues affected in the ternary complex, which are colored dark red or light red, according to the magnitude of the effect. | ||
After the X-ray structure of the ternary complex was solved (see below), we could map the chemical shift and intensity changes onto the X-ray structure (B). As expected, they mainly cluster around the binding site of FKBP12:R,S-SLF-1a. However, several residues are not in direct contact with FKBP12 or R,S-SLF-1a (>5 A away) and still experience strong effects. These residues are allosterically affected by conformational changes induced by the binding of FKBP12:R,S-SLF-1a.
NMR is known for its unique ability to robustly detect weak affinities. We were thus intrigued to see whether weak intrinsic affinities exist for the respective binary complexes, i.e. between MAPRE1 and R,S-SLF-1a, and between MAPRE1 and FKBP12. The strong binding affinity between FKBP12 and R,S-SLF-1a was already known and not of interest for this investigation. Fig. 12 shows NMR 15N-HSQC spectra of MAPRE1 (black) and MAPRE1 in presence of R,S-SLF-1a (Fig. 12(A), red) and MAPRE1 in presence of FKBP12 (Fig. 12(B), red). While no chemical shift changes were observed in the latter case (indicating no intrinsic affinity for MAPRE1 and FKBP12), two MAPRE1 residues experience small but significant chemical shift changes upon addition of R,S-SLF-1a. Notably, these residues are E213 and Y247 which are in direct contact with R,S-SLF-1a in the crystal structure of the ternary complex, suggesting that the weak binding of R,S-SLF-1a to MAPRE1 in the absence of FKBP12 is specific and at the same site as in the ternary complex. Later, we could confirm this observation by binding studies to MAPRE1 using the spectral shift method.42
 | ||
| Fig. 12 NMR investigation of weak intrinsic interactions in binary complexes. (A) The 15N-HSQC NMR spectrum of 13C,15N-MAPRE1 (50 μM, black) is superimposed on a 15N-HSQC NMR spectrum of 13C,15N-MAPRE1 in presence of 100 μM R,S-SLF-1a, Note that the solubility of R,S-SLF-1a is only about 20 μM. Resonance assignments of the two residues that experience chemical shift changes are given in the figure. (B) The 15N-HSQC NMR spectrum of 13C,15N-MAPRE1 (50 μM, black) is superimposed on a 15N-HSQC NMR spectrum of 13C,15N-MAPRE1 in presence of 350 μM FKBP12, There are no chemical shift changes in the presence of FKBP12, indicating no intrinsic affinity between the two proteins in the absence of R,S-SLF-1a. | ||
X-ray structure of the ternary complex FKBP12:R,S-SLF-1a:MAPRE1
To understand the structural basis for R,S-SLF-1a mediated recruitment of FKBP12 to MAPRE1, we co-crystallized the ternary complex from the separately isolated recombinant flFKBP12 and the C-terminal domain of MAPRE1 composed of the juxtaposed coiled-coil and four helix bundle domains in the presence of R,S-SLF-1a. For structural studies, the synthesized R-enantiomer of SLF-1, R,S-SLF-1a, was used and co-crystals diffracting to 2.7 A were obtained directly from a MCSG space matrix screen. One helix of MAPRE1's coiled-coil was complexed to one molecule of R,S-SLF-1a-bound FKBP12 per asymmetric unit using a symmetry operation to create the biologically relevant dimer. The resultant complex sandwiches two R,S-SLF-1a macrocycles between the centrally placed MAPRE1 coiled-coil and two flanking FKBP12 protamers related by 2-fold rotational symmetry.The macrocycle acts as a molecular glue, recruiting FKBP12 to MAPRE1 via previously-defined interactions between Y82 and I56 of FKBP12 to the diketo functionality and pipecolate moiety of R,S-SLF-1a's FKBP12 binding loop, respectively. The R,S-SLF-1a-FKBP12 complex results in the presentation of the macrocycle's opposite “effector” loop to the coiled-coil with a set of novel interactions to the 4-helix bundle of MAPRE1. The interactions are predominantly hydrophobic, with highly conserved EB1 family residues F218, Y217 and F216 of the coiled-coil working in conjunction with L221, L226, L246 and Y247 of the four-helix bundle, all complexed to the aliphatic loop of R,S-SLF-1a. This set of interactions results in the formation of a “hydrophobic core”, sandwiching the macrocycle between MAPRE1 and FKBP12. Direct interactions are seen between the two proteins. Specifically, H87 and main chain carbonyl of G86 from FKBP12's 80's loop forms a single water-mediated interaction with the side chains of E213 and R214 of MAPRE's coiled-coil (Fig. 13).
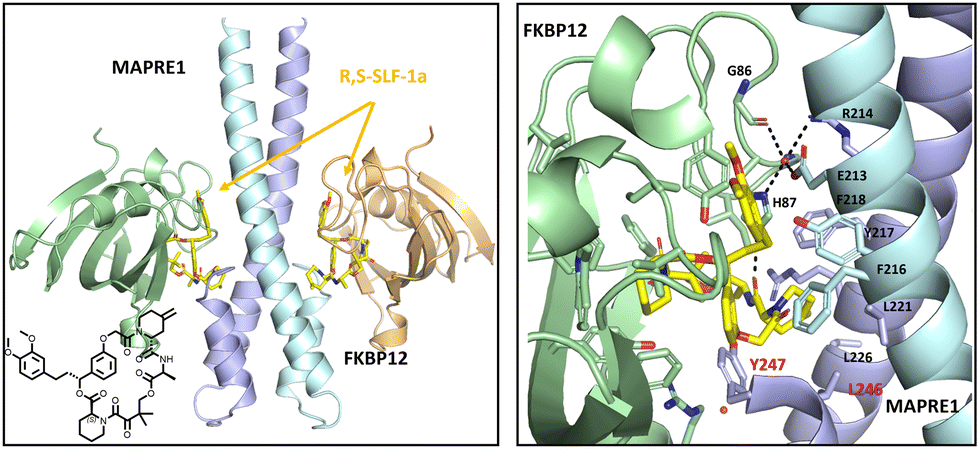 | ||
| Fig. 13 X-ray structure of the ternary complex FKBP12:R,S-SLF-1a:MAPRE1 (2:2:2). R,S-SLF-1a acts as a molecular glue with a predominant hydrophobic interface bridging MAPRE1 and FKBP12. Limited protein–protein interactions flank the R,S-SLF-1a binding pocket. PDB code: 9CO5/DOI: https://doi.org/10.2210/pdb9co5/pdb. | ||
X-ray structure of the binary complex FKBP12:R,S-SLF-1a
In addition, the structure of the binary FKBP12:R,S-SLF-1a complex has also been solved. The FKBP12 binding motif of R,S-SLF-1a without the presence of MAPRE1, shows a similar conformation to that seen in the ternary complex, whereas the MAPRE1 recruitment loop of the R,S-SLF-1a scaffold in the ternary complex shows a very different conformation compared to the conformation in the binary complex (Fig. 14). It is noteworthy that this conformational change is necessary to avoid steric clashes upon MAPRE1 recruitment of the preformed binary FKBP12:R,S-SLF-1a interface. From a thermodynamic point of view, this implies an entropy cost for the formation of the ternary complex, i.e. only part of the conformational ensemble is able to participate in the binding event. | ||
| Fig. 14 X-ray structure of the binary complex FKBP12:R,S-SLF-1a (green and cyan). Purple: R,S-SLF-1a in ternary complex. A conformational change of the solvent exposed loop of R,S-SLF-1a is observed upon MAPRE1 recruitment. | ||
Improvement of ternary complex forming potency of R,S-SLF-1a by close but distinguished analogues
As described above, the absolute stereo-configuration of the initial hit compound at the α position of the 4-methylenepiperidine-2-carboxamide moiety could be assigned to R. The residue at the second position of the recruitment loop was L-Ala (S-configuration), leading to the compound denotation R,S-SLF-1a. Using D-Ala, we synthesized R,R-SLF-1b and – since we had the (S)-4-methylenepiperidine-2-acid at hand – also S,R-SLF-1c (with D-Ala) and finally S,S-SLF-1d (L-Ala). Interestingly, no other stereoisomer showed signs of recruitment activity, despite the reasonable binary affinities of the other isomers to FKBP12 (Table 1 entry 1–4). To determine the role of the exocyclic methylene group of the 4-piperidine carboxamide moiety for the recruitment of MAPRE1, we synthesized the corresponding R- and S-methyl compounds with the “active” R,S stereochemistry on the scaffold (R,S-SLF-2a and R,S-SLF-2b, respectively, entries 5 and 6). Both derivatives completely lost their potency for ternary complex formation. From a structural view point, the failure of recruiting MAPRE1 by R,S-SLF-2a/b suggests that modifying the methylene group of 4-piperidine which is in close contact with the backbone of the MAPRE1 helical domain might disturbs the complementarity of the interface of the pre-formed binary complex (Fig. 13). In a next step, we attempted to modify the methyl group of the alanine in order to capture new interactions on the MAPRE1 side of the ternary complex or at least to explore the tolerated scope for modification. As long as the original stereochemistry R,S was retained, the recruitment activity could also be maintained or even improved. R,S-SLF-3 (S-ethyl instead of S-methyl) showed an improved Amax of 93% compared to 51% in the recruitment assay (entry 7). A slightly lower Amax of 67% was observed for L-valine (R,S-SLF-4a, entry 8), but again all activity was lost when the stereochemistry at this position was changed from S to R (S,S-SLF-4b, entry 9). Exchanging the isopropyl group for a cyclopropyl group gave only weak activity (Amax = 24%, entry 10). The linear extension of the alkyl moiety to S-propyl led to an almost complete recovery of activity (Amax = 46%, R,S-SLF-5a, entry 11), while the same derivative with the stereochemistry S at the position α of the piperidine moiety again led to a complete loss of activity (S,S-SLF-5b, entry 12). A significant improvement to an Amax of 255% was achieved by replacing L-Ala with L-Ser (R,S-SLF-6, entry 13). The analogous derivative with the S-CH2NH2 residue had a potency in the same range as the other active compounds (Amax = 45%, R,S-SLF-7, entry 14). Replacing the ethyl residue with a CH2CF3 group reduces the activity back to an Amax of 27% (R,S-SLF-8, entry 15), while the introduction of the CH2CH2OMe residue restores the original potency level (R,S-SLF-9, entry 16). The introduction of the α,α methyl residue significantly improved the potency (Amax = 135%, R,--SLF-10, entry 17) and bridging the two methyl groups to a cyclopropyl residue, directly attached to the scaffold backbone gave the strongest recruitment of this series (Amax = 350%, R,--SLF-11, entry 18) (Fig. 15).| Entry | Compound denotation | Stereochemistry at α-position of piperidine-2-carboxy amide | 4-Piperidine substitution and stereochemistry | Stereochemistry and substituent at α-position of amino acid | TR-FRET recruitment assay (%Amax) | TR-FRET binary FKBP12 binding (nM) | NanoBiT (%Amax) |
|---|---|---|---|---|---|---|---|
| 1 | R,S-SLF-1a | R | ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 CH2 |
(S)-CH3 | 51 | 230 | 774 |
| 2 | R,R-SLF-1b | R | CH2 |
(R)-CH3 | 0 | 720 | 627 |
| 3 | S,R-SLF-1c | S | CH2 |
(R)-CH3 | 0 | 680 | 11 |
| 4 | S,S-SLF-1d | S | CH2 |
(S)-CH3 | 0 | 30 | 11 |
| 5 | R,S-SLF-2a | R | (R)-CH3 | (S)-CH3 | 0 | 140 | 177 |
| 6 | R,S-SLF-2b | R | (S)-CH3 | (S)-CH3 | 0 | 210 | Not done |
| 7 | R,S-SLF-3 | R | CH2 |
(S)-CH2CH3 | 93 | 370 | 987 |
| 8 | R,S-SLF-4a | R | CH2 |
(S)-CH(CH3)2 | 67 | 360 | 830 |
| 9 | S,S-SLF-4b | S | CH2 |
(S)-CH(CH3)2 | 0 | 300 | 20 |
| 10 | R,S-SLF-4c | R | CH2 |
(S)-CH2-cPropyl | 24 | 560 | 731 |
| 11 | R,S-SLF-5a | R | CH2 |
(S)-CH2CH2CH3 | 46 | 140 | 795 |
| 12 | S,S-SLF-5b | S | CH2 |
(S)-CH2CH2CH3 | 0 | 110 | 25 |
| 13 | R,S-SLF-6 | R | CH2 |
(S)-CH2OH | 255 | 540 | 1315 |
| 14 | R,S-SLF-7 | R | CH2 |
(S)-CH2NH2 | 45 | 1240 | 748 |
| 15 | R,S-SLF-8 | R | CH2 |
(S)-CH2CF3 | 27 | 610 | 777 |
| 16 | R,S-SLF-9 | R | CH2 |
(S)-CH2CH2OMe | 51 | 440 | 933 |
| 17 | R,--SLF-10 | R | CH2 |
–CH3, CH3 | 135 | 350 | 1024 |
| 18 | R,--SLF-11 | R | CH2 |
cPropyl | 350 | 610 | 1244 |
 | ||
| Fig. 15 Structural modifications around the original hit compound R,S-SLF-1a. | ||
In order to assign the observed differences in Amax values to the corresponding changes in cooperativity α and/or to changes in the binary affinity of the corresponding glues to FKBP12 and/or MAPRE1, we performed an in-depth biophysical binding study, the results of which we publish simultaneously with this work.42 Furthermore, to rationalize the observed differences, and particularly the gain in potency for specific interactions in the ternary complex, which leads to an increase in measurable cooperativity, we have started an in-depth molecular dynamics study of the corresponding complexes. We will publish the results of this study in due course.
Cellular validation of ternary complex formation with a NanoBiT assay and correlation of Amax with TR-FRET Amax values
In order to validate our biochemical TR-FRET MAPRE1 recruitment results in a cellular context, we used a cellular NanoBiT assay that works on the principle of split-nanoluciferase complementation (Fig. 16). We fused the large fragment of the split nanoluciferase (LgBiT) to FKBP12, and the small fragment (SmBiT) to the coiled-coil domain of MAPRE1 (N183-Y268) (Fig. 16(A)). We transiently co-transfected the two BiT-fusion constructs into HEK293T cells and measured the luminescence signal following a dose–response treatment of R,S-SLF-1a and related analogs (Fig. 16(B)), to ask whether the biochemically determined structure–activity-relationship (SAR) transferred to the orthogonal cellular assay. As can be seen in Fig. 16(C), the cellular recruitment Amax values correlate well with the biochemical recruitment Amax values. This indicates not only that these compounds are cell permeable and form ternary complexes in cells, but also that cellular uptake of these compounds within the incubation time of the proximity assay is not rate-limiting. | ||
| Fig. 16 Cellular recruitment of MAPRE1 to FKBP12 by R,S-SLF-1a analogs correlates with biochemical results. (A) Schematic of the cellular NanoBiT assay. (B) NanoBiT dose response curves with R,S-SLF-1a and analogues. (C) Scatter plot of TR-FRET recruitment Amaxversus NanoBiT recruitment Amax. (D) Dependency on FKBP12 ligand binding site for MAPRE1 recruitment. Dose response of the 12 SLF analogs with the highest %Amax values from panel (A) but in the presence of 80 μM of the indicated competitive non-recruiting FKBP12 ligand or DMSO. | ||
We choose the Amax values in both assays as a benchmark for comparison because the Amax values were expected to correlate with the concentration of ternary complex formed and thus – in absence of a strong binary target affinity – with cooperativity α, since cooperativity α determines the amount of ternary complex formed but not the concentration of ligand at which this occurs. Thus, a higher binary target and/or chaperone affinity shifts the formation of the ternary complex to lower required ligand concentrations, but may not necessarily reflect a change in cooperativity α.
To confirm that the NanoBiT assay is an accurate readout of the specific recruitment between MAPRE1 and FKBP12 by the respective glue molecules via the FKBP12 ligand pocket and not an artifact of a complex cellular environment, we designed a ligand competition experiment. By incubating the cells with a high concentration of a ligand with high affinity for FKBP12 but with no measurable glue recruitment activity toward MAPRE1, we should be able to out-compete the glue molecules for occupancy of the FKBP12 ligand pocket. We chose two FKBP12 ligands with which to do this experiment, neither of which measurably recruits MAPRE1 to FKBP12: S,S-SLF-1d (30 nM FKBP12 EC50, Table 1, entry 4) and Rapamycin (5 nM FKBP12 EC50, data not shown). By contrast, R,S-SLF-1a has a 230 nM FKBP12 EC50 (Table 1, entry 1). A dose response of the 12 MAPRE1 recruiting compounds with significant %Amax values (Table 1 and Fig. 16(B)) was repeated, but in presence of 80 μM of the competitive FKBP12 ligands S,S-SLF-1d or Rapamycin (or DMSO control), added to all doses of recruiting compounds (Fig. 16(D)). Both competitive but non-recruiting FKBP12 ligands abrogated the dose response signals for all 12 MAPRE1 recruiting compounds, whereas DMSO had no significant effect on recruitment. Interestingly, whereas Rapamycin was able to outcompete even the highest dose of all glue recruiters tested (20 μM), S,S-SLF-1d was not able to completely outcompete the highest dose of the more potent recruiting molecules, consistent with its ∼6-fold weaker affinity for FKBP12 relative to Rapamycin.
Functional validation of MARPRE1 recruitment with a NanoBiT assay
Next, we asked whether recruitment of MAPRE1 to FKBP12 results in a biological consequence. MAPRE1 is reported to have many binding partners according to BioGrid (https://thebiogrid.org). One of these, CEP215 (Uniprot Q96SN8, also known as CDK5RAP2), is a high-confidence binding partner as evidenced by multiple orthogonal experimental methods indicated in BioGrid. Importantly, interaction between MAPRE1 and CEP215 maps to the same coiled-coil region of MAPRE1 that recruited to R,S-SLF-1a-FKBP12.43 Therefore we tested whether we could competitively disrupt MAPRE1 recruitment to FKBP12 by overexpression of CEP215. To do this, we modified the design of the NanoBiT assay (Fig. 17). We included a third construct in the transfection mix, either WT CEP215 or a double point-mutant (L938A/P939A) which does not interact with MAPRE1.43 In this setup, FKBP12 is tagged with LgBiT and expressed weakly via the HSVTK promoter, while either WT CEP215 or mutant CEP215 is expressed strongly via the CMV promoter and without a tag. The rationale behind this experiment is that MAPRE1 recruitment to FKBP12 in the presence of R,S-SLF-1a should be competitively blocked by overexpression of untagged WT CEP215 but not by overexpression of untagged mutant CEP215. When WT CEP215 was included, we observed an 84% decrease of MAPRE1 recruitment to FKBP12 relative to when mutant CEP215 was included. As a negative signal control, S,S-SLF-1d was also included. This compound was shown not to recruit MAPRE1 by either TR-FRET or NanoBiT despite having high binding affinity to FKBP12 (Table 1). This demonstrates that recruitment of MAPRE1 to FKBP12 by R,S-SLF-1a can be competed by MAPRE1 binding to CEP215 in cells.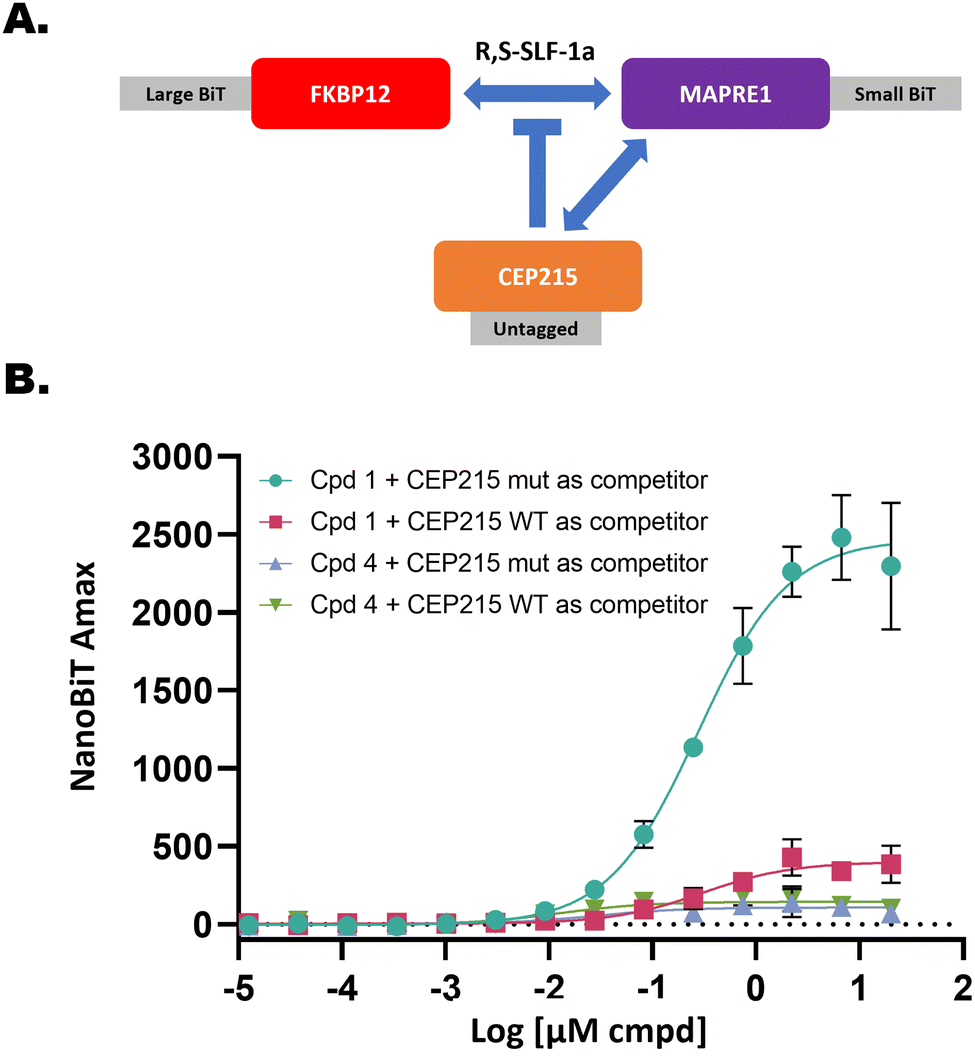 | ||
| Fig. 17 Recruitment of MAPRE1 to FKBP12 by R,S-SLF-1a is disrupted by forced MAPRE1-CEP215 interaction in cells. (A) Schematic of NanoBiT competition experiment. LgBiT-FKBP12 and SmBiT-MAPRE1 create a signal only in the presence of R,S-SLF-1a, which can be competed by co-expressing untagged CEP215 binding to MAPRE1. (B) Overexpressed WT CEP215, but not mutant CEP215, strongly diminishes recruitment of MAPRE1 to FKBP12 (R,S-SLF-1a = strong recruiter; Cpd 4 = R,S-SLF-1d = strong ligand of FKBP12 but non-recruiting epimer of R,S-SLF-1a). | ||
Discussion
Many disease-relevant and functionally well-validated targets are difficult to drug. Their poorly defined 3D structure without deep hydrophobic pockets makes the development of ligands with low molecular weight and high affinity very challenging. For these targets, incorporation into a ternary complex can be a viable alternative to modulate and in most cases inhibit their function. The interacting species in such cases is not just a low molecular weight ligand, but a “ligand” that is a molecular glue that only binds to the target if it is already bound to the chaperone protein. The resulting ternary complex offers the possibility of engaging a much larger surface area with better opportunities for interactions within the target protein than is possible with a low molecular weight compound alone. Since the target is complexed by a binary species consisting of the molecular glue and the chaperone protein, the requirement for a strong independent binary affinity to the target is eliminated. An additional advantage of this modality is that – due to the larger surface area covered, remote differences in the target, e.g. those in target isoforms, can be significantly involved in the ternary complex, providing therefore opportunities for selectivity's that cannot be achieved with low molecular weight ligands. This and the generally lower molecular weight of molecular glues compared to bifunctional ternary complexing compounds for the same inhibitory effect explains our interest and that of many others in developing methods to regularly find molecular glues that form ternary complexes with a wide range of targets and a chaperone protein.As far as the chaperone is concerned, FKBP12 was chose due to its attractive properties. Since it is found in all tissues of the body, it can in principle be used as a chaperone for any target, regardless of where it is located (except in cell nuclei). This property would allow generalization of this modality so that FKBP12 could be the workhorse for addressing many difficult intracellular targets. The high cellular concentration of FKBP12 (up to 1 μM, data not shown) thermodynamically drives the formation of binary chaperone-glue complexes, which are the species that recruit the target, resulting in a high concentration of the ternary complex formed for a given concentration of molecular glue. Depending on the target and the corresponding therapy, the high occurrence of FKBP12 throughout the body can also be a disadvantage, as this leads to large amounts of the molecular glue being buffered and retained throughout the body. To address this limitation, a chaperone would ideally be located only at the target site to which it is recruited by the molecular glue. This would lead to a selective accumulation of the glue in the target tissue and prevent its distribution throughout the body. The emerging development of RIPTACs is aimed precisely in this direction.44
Furthermore, a scaffold protein such as FKBP12, which by virtue of its function must have the ability to interact with many different native partners, may be inherently better suited to a role as a chaperone for multiple targets than a protein that has essentially only one native interaction partner. The use of cyclophilin or the hub protein 14-3-3, both of which also have many interaction partners, as chaperones could at least support this argument. The fact that FKBP12 selectively recruits mTor, calcineurin or CEP250 – depending on whether it is bound to rapamycin, FK506 or WDB002 – also suggests the potential of FKBP12 as a chaperone for other targets if a suitable molecular glue can be found.
For this reason, we exposed FKBP12 to 2500 different proteins and 50 macrocyclic FKBP12 ligands with differences in the recruitment loop, resulting in 125000 combinations in one experiment. Molecular glues with a high cooperativity typically have very high recruitment selectivity, which means that structures that look similar behave very differently in the environment of a ternary complex. Selectivity, and therefore diversity, depends on the criteria applied, meaning that they are very different under different criteria applied.45 The fact that only one in 125000 combinations was productive indicates the highly specific requirements for a productive interaction.
The original hit shows medium affinity for FKBP12 (230 nM), very low affinity for MAPRE1 (mM), while FKBP12 and MAPRE1 show no detectable affinity for each other. According to our classification, R,S-SLF-1a is therefore a type I molecular glue.15 However, the question of how subtle modifications at the 4-piperidine and the α position of 4-methylenepiperidine-2-carboxamide lead to different cooperativities remains unanswered. Furthermore, the question of what role the observed conformational change of the MAPRE1 recruitment loop in R,S-SLF-1a and related compounds plays in ternary complex formation requires a more detailed investigation of the structural dynamics. A corresponding study is in progress and the results will be reported in due course.
Conclusions
In a protein array screen of 50 different macrocyclic FKBP12 ligands against 2500 different randomly selected proteins, a hit was found that recruits a dimeric protein called MAPRE1 to FKBP12 (2:2:2 complex) in a compound-dependent manner. Identifying the required stereochemistry from the hit mixture of diastereomers yielded the active epimer (R,S-SLF-1a). The corresponding ternary complex was characterized by TR-FRET proximity assay and native MS spectroscopy. Insights into the 3D structure of the ternary complex were obtained by 2D protein NMR spectroscopy and finally an X-ray structure of the corresponding FKBP12:R,S-SLF-1a:MAPRE1 ternary complex. R,S-SLF-1a showed only a very weak affinity to the recruited target MAPRE1, which in turn also showed no signs of intrinsic affinity to the chaperone protein FKBP12, i.e. the free energy of ternary complex formation originates from considerable cooperativity α. The results of an extensive study allowed the quantification of the cooperativity α of several of the reported compounds and is discussed in a separate publication.42
A comparison of R,S-SLF-1a bound only to FKBP12 with that bound in the ternary complex shows that the macrocyclic scaffold undergoes a significant conformational change when the ternary is formed from the binary complex.
The synthesis of a small series of analogs of R,S-SLF-1a showed a very high specificity for the ternary complex forming molecular glues. In particular, any change in the stereochemistry of the scaffold immediately led to a complete loss of activity. The X-ray structure of the ternary complex could be used to improve the initial Amax value in the TR-FRET proximity assay by modifying the L-Ala position from 51% (R,S-SLF-1a) to 350% (R,--SLF-11). Despite many other synthesized compounds (not shown in this work), further optimization was not possible, although the X-structure provided a strong rationale for capturing further interactions. This leads us to the conclusion that further optimization requires the inclusion of the molecular dynamics of the entire ternary complex. Corresponding work is the subject of a manuscript in preparation.
Author contributions
Design and synthesis of macrocyclic compounds were performed by H. K., M. F., A. G. Synthesis, chiral separation, X-ray and NMR work of specific building blocks were performed by F. L., A. V., L. C., P. P., M. A. L., R. S., E. S. LC-HRMS was performed by J. K. Protein array screening and TR-FRET assays were performed by M. S. Native MS analysis was performed by D. A., W. J., L. P. Protein NMR was performed by C. H., W. J. X-ray work on ternary complexes were performed by M. S., A. D., D. K. Cell assays were performed by A. T. Project lead and conceptual input by G. M., H. J. R. The manuscript was written by H. J. R.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- Z. Kozicka, D. J. Suchyta, V. Focht, G. Kempf, G. Petzold, M. Jentzsch, C. Zou, C. Di Genua, K. A. Donovan, S. Coomar, M. Cigler, C. Mayor-Ruiz, J. L. Schmid-Burgk, D. Haussinger, G. E. Winter, E. S. Fischer, M. Slabicki, D. Gillingham, B. L. Ebert and N. H. Thoma, Nat. Chem. Biol., 2024, 20, 93–102 Search PubMed.
- F. Li, I. A. M. Aljahdali and X. Ling, Int. J. Mol. Sci., 2022, 23, 6206 CrossRef CAS.
- H. Wu, H. Yao, C. He, Y. Jia, Z. Zhu, S. Xu, D. Li and J. Xu, Acta Pharm. Sin. B, 2022, 12, 3548–3566 CrossRef CAS PubMed.
- E. G. Weagel, J. M. Foulks, A. Siddiqui and S. L. Warner, Med. Chem. Res., 2022, 22, 1068 CrossRef.
- T. M. Geiger, S. C. Schäfer, J. K. Dreizler, M. Walz and F. Hausch, Curr. Res. Chem. Biol., 2022, 2, 100018 CrossRef CAS.
- A. Poso, J. Med. Chem., 2021, 64, 10680 Search PubMed.
- J. Liu, H. Chen, H. U. Kaniskan, L. Xie, X. Chen, J. Jin and W. Wei, J. Am. Chem. Soc., 2021, 143, 8902 CrossRef CAS.
- J. A. Dewey, C. Delalande, S. A. Azizi, V. Lu, D. Antonopoulos and G. Babnigg, J. Med. Chem., 2023, 66, 9278–9296 CrossRef CAS PubMed.
- S. Yamazoe, J. Tom, Y. Fu, W. Wu, L. Zeng, C. Sun, Q. Liu, J. Lin, K. Lin, W. J. Fairbrother and S. T. Staben, J. Med. Chem., 2020, 63, 2807–2813 Search PubMed.
- L. Hua, Q. Zhang, X. Zhu, R. Wang, Q. You and L. Wang, J. Med. Chem., 2022, 65, 8091–8112 CrossRef CAS PubMed.
- S. Lai, A. E. Modell and A. Choudhary, Proximity Inducing Bifunctional Molecules Beyond PROTACs, 2022 DOI:10.1002/9781119774198.ch12.
- S. U. Siriwardena, D. N. P. Munkanatta Godage, V. M. Shoba, S. Lai, M. Shi, P. Wu, S. K. Chaudhary, S. L. Schreiber and A. Choudhary, J. Am. Chem. Soc., 2020, 142, 14052–14057 CrossRef CAS PubMed.
- W. J. Gibson, A. Sadagopan, V. M. Shoba, A. Choudhary, M. Meyerson and S. L. Schreiber, J. Am. Chem. Soc., 2023, 146, 26028 CrossRef PubMed.
- G. S. Tender and C. R. Bertozzi, RSC Chem. Biol., 2023, 4, 986–1002 RSC.
- R. R. Stein, M. Fouche, J. D. Kearns and H. J. Roth, RSC Chem. Biol., 2023, 4, 512–523 RSC.
- J. A. Ward, C. Perez-Lopez and C. Mayor-Ruiz, ChemBioChem, 2023, 24, e202300163 CrossRef CAS.
- D. Park, J. Izaguirre, R. Coffey and H. Xu, ACS Bio & Med Chem Au, 2023, 3, 74 Search PubMed.
- C. J. Gerry and S. L. Schreiber, Nat. Chem. Biol., 2020, 16, 369–378 CrossRef CAS PubMed.
- S. Gaali, R. Gopalakrishnan, Y. Wang, C. Kozany and F. Hausch, Curr. Med. Chem., 2011, 18, 5355–5379 Search PubMed.
- M. Holderfield, B. J. Lee, J. Jiang, A. Tomlinson, K. J. Seamon, A. Mira, E. Patrucco, G. Goodhart, J. Dilly, Y. Gindin, N. Dinglasan, Y. Wang, L. P. Lai, S. Cai, L. Jiang, N. Nasholm, N. Shifrin, C. Blaj, H. Shah, J. W. Evans, N. Montazer, O. Lai, J. Shi, E. Ahler, E. Quintana, S. Chang, A. Salvador, A. Marquez, J. Cregg, Y. Liu, A. Milin, A. Chen, T. B. Ziv, D. Parsons, J. E. Knox, J. E. Klomp, J. Roth, M. Rees, M. Ronan, A. Cuevas-Navarro, F. Hu, P. Lito, D. Santamaria, A. J. Aguirre, A. M. Waters, C. J. Der, C. Ambrogio, Z. Wang, A. L. Gill, E. S. Koltun, J. A. M. Smith, D. Wildes and M. Singh, Nature, 2024, 629, 919–926 CrossRef CAS PubMed.
- R. Casement, A. Bond, C. Craigon and A. Ciulli, Methods Mol. Biol., 2021, 2365, 79–113 Search PubMed.
- P. S. Marinec, L. Chen, K. J. Barr, M. W. Mutz, G. R. Crabtree and J. E. Gestwicki, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 1336–1341 Search PubMed.
- Z. Guo, S. Y. Hong, J. Wang, S. Rehan, W. Liu, H. Peng, M. Das, W. Li, S. Bhat, B. Peiffer, B. R. Ullman, C. M. Tse, Z. Tarmakova, C. Schiene-Fischer, G. Fischer, I. Coe, V. O. Paavilainen, Z. Sun and J. O. Liu, Nat. Chem., 2019, 11, 254–263 CrossRef CAS.
- S. L. Schreiber, Science, 1991, 251, 283–287 CrossRef CAS PubMed.
- G. Baughman, G. J. Wiederrecht, F. Chang, M. M. Martin and S. Bourgeois, Biochem. Biophys. Res. Commun., 1997, 232, 437–443 CrossRef CAS PubMed.
- T. Liu, J. Xiong, S. Yi, H. Zhang, S. Zhou, L. Gu and M. Zhou, Oncogene, 2017, 36, 1678–1686 CrossRef CAS PubMed.
- K. H. Schreiber, D. Ortiz, E. C. Academia, A. C. Anies, C. Y. Liao and B. K. Kennedy, Aging Cell, 2015, 14, 265–273 CrossRef CAS PubMed.
- M. Sasaki, S. Nishimura, Y. Yashiroda, A. Matsuyama, H. Kakeya and M. Yoshida, iScience, 2022, 25, 105659 CrossRef CAS.
- J. M. Bonner and G. L. Boulianne, Cell. Signalling, 2017, 38, 97–105 CrossRef CAS.
- J. M. Kolos, A. M. Voll, M. Bauder and F. Hausch, Front. Pharmacol., 2018, 9, 1425 CrossRef CAS PubMed.
- M. Xing, J. Wang, Q. Yang, Y. Wang, J. Li, J. Xiong and S. Zhou, Cancer Chemother. Pharmacol., 2019, 84, 861–872 Search PubMed.
- U. K. Shigdel, S. J. Lee, M. E. Sowa, B. R. Bowman, K. Robison, M. Zhou, K. H. Pua, D. T. Stiles, J. A. V. Blodgett, D. W. Udwary, A. T. Rajczewski, A. S. Mann, S. Mostafavi, T. Hardy, S. Arya, Z. Weng, M. Stewart, K. Kenyon, J. P. Morgenstern, E. Pan, D. C. Gray, R. M. Pollock, A. M. Fry, R. D. Klausner, S. A. Townson and G. L. Verdine, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 17195 CrossRef CAS.
- C. J. Schulze, K. J. Seamon, Y. Zhao, Y. C. Yang, J. Cregg, D. Kim, A. Tomlinson, T. J. Choy, Z. Wang, B. Sang, Y. Pourfarjam, J. Lucas, A. Cuevas-Navarro, C. Ayala-Santos, A. Vides, C. Li, A. Marquez, M. Zhong, V. Vemulapalli, C. Weller, A. Gould, D. M. Whalen, A. Salvador, A. Milin, M. Saldajeno-Concar, N. Dinglasan, A. Chen, J. Evans, J. E. Knox, E. S. Koltun, M. Singh, R. Nichols, D. Wildes, A. L. Gill, J. A. M. Smith and P. Lito, Science, 2023, 381, 794–799 CrossRef CAS PubMed.
- J. Choi, J. Chen, S. L. Schreiber and J. Clardy, Science, 1996, 273, 239–242 CrossRef CAS.
- J. P. Griffith, J. L. Kim, E. E. Kim, M. D. Sintchak, J. A. Thomson, M. J. Fitzgibbon, M. A. Fleming, P. R. Caron, K. Hsiao and M. A. Navia, Cell, 1995, 82, 507–522 CrossRef CAS PubMed.
- Y. Che, A. M. Gilbert, V. Shanmugasundaram and M. C. Noe, Bioorg. Med. Chem. Lett., 2018, 28, 2585–2592 CrossRef CAS.
- F. H. Knaup, C. Meyners, W. O. Sugiarto, S. Wedel, M. Springer, C. Walz, T. M. Geiger, M. Schmidt, M. Sisignano and F. Hausch, J. Med. Chem., 2023, 66, 5965 CrossRef CAS.
- D. A. Holt, J. I. Luengo, D. S. Yamashita, H. J. Oh, A. L. Konialian, H. K. Yen, L. W. Rozamus, M. Brandt and M. J. Bossard, J. Am. Chem. Soc., 1993, 115, 9925–9938 CrossRef CAS.
- X. Wu, L. Wang, Y. Han, N. Regan, P. K. Li, M. A. Villalona, X. Hu, R. Briesewitz and D. Pei, ACS Comb. Sci., 2011, 13, 486–495 CrossRef PubMed.
- T. B. Almeida, A. J. Carnell, I. L. Barsukov and N. G. Berry, Sci. Rep., 2017, 7, 15533 CrossRef CAS PubMed.
- T. Kanaba, R. Maesaki, T. Mori, Y. Ito, T. Hakoshima and M. Mishima, Biochim. Biophys. Acta, 2013, 1834, 499–507 CrossRef CAS.
- J. Schnatwinkel, R. R. Stein, M. Salcius, J. Wong, S.-Y. Chen, M. Fouché and H.-J. Roth, bioRxiv, 2025, preprint DOI:10.1101/2025.01.15.631501v1.
- K. W. Fong, S. Y. Hau, Y. S. Kho, Y. Jia, L. He and R. Z. Qi, Mol. Biol. Cell, 2009, 20, 3660–3670 CrossRef CAS PubMed.
- K. Raina, C. D. Forbes, R. Stronk, J. P. Rappi, K. J. Eastman, S. W. Gerritz, X. Yu, H. Li, A. Bhardwaj, M. Forgione, A. Hundt, M. P. King, Z. M. Posner, A. Denny, A. McGovern, D. E. Puleo, E. Garvin, R. Chenard, N. Zaware, J. J. Mousseau, J. Macaluso, M. Martin, K. Bassoli, K. Jones, M. Garcia, K. Howard, L. M. Smith, J. M. Chen, C. A. De Leon, J. Hines, K. J. Kayser-Bricker and C. M. Crews, bioRxiv, 2023, preprint DOI:10.1101/2023.01.01.522436.
- H. J. Roth, Curr. Opin. Chem. Biol., 2005, 9, 293–295 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2372678 and 2372681. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cb00279b |
| ‡ Equal contributors. |
| This journal is © The Royal Society of Chemistry 2025 |