
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIdentification of microproteins with transactivation activity by polyalanine motif selection†
Archita
Agrawal
 and
Alan
Saghatelian
*
and
Alan
Saghatelian
*
Clayton Foundation Laboratories for Peptide Biology, Salk Institute for Biological Studies, La Jolla, CA, USA. E-mail: asaghatelian@salk.edu
First published on 6th March 2025
Abstract
Microproteins are an emerging class of proteins that are encoded by small open reading frames (smORFs) less than or equal to 100 amino acids. The functions of several microproteins have been illuminated through phenotypic screening or protein–protein interaction studies, but thousands of microproteins remain uncharacterized. The functional characterization of microproteins is challenging due to a lack of sequence homology. Here, we demonstrate a strategy to enrich microproteins that contain specific motifs as a means to more rapidly characterize microproteins. Specifically, we used the fact that polyalanine motifs are associated with nuclear proteins to select 58 candidate microproteins to screen for transactivation function. We identified three microproteins with transactivation activity when tested as GAL4-fusions in a cell-based luciferase assay. The results support the continued use of the motif selection strategy for the discovery of microprotein function.
Introduction
Microproteins are encoded by small open reading frames (smORFs) in the genome. Microproteins (also known as smORF-encoded peptides SEPs, or micropeptides) were initially overlooked as the conventional gene annotation methods used a length threshold of 100 codons as one of the parameters to define the protein-coding genes.1–4 Other crucial parameters for the annotation of protein-coding genes included the assumption of one protein-coding gene per transcript as well as some degree of comparative sequence-homology, to reduce data analysis complexity and avoid false positive identifications.2,3 The simplistic definition of an open reading frame (ORF) is an in-frame start and stop codon, however without the above-mentioned restrictions, millions of such instances are detected in eukaryotic genomes making the annotation of true protein coding ORFs extremely challenging.3,5 As a consequence, such microproteins that could be encoded by smORFs were disregarded in the initial genome database annotations.The development of ribosome profiling,1,6,7 computational approaches to analyze the genome,2 and proteomics8,9 paved the way for the identification of thousands of previously unknown smORFs as potentially protein-coding.4,10,11 One of the first notable examples of functional characterization of a microprotein came with the discovery of polished rice/tarsal-less (pri/tal) where an 11 amino-acid microprotein was found to be critical for development in flies.12,13 A key example in mammalian biology is myoregulin, an endoplasmic reticulum membrane microprotein that regulates calcium flux to mediate muscle performance.14 As evidence began to mount that smORFs are detected across all living organisms, the functional roles of several microproteins were individually elucidated in a broad range of biological processes.1,15 Consequently, the next big challenge presented itself as how to uncover the possible biological functions of thousands of such uncharacterized microproteins.
The guiding principle of protein-function discovery relies on sequence conservation which signifies biological importance, and provides a starting point for conducting experimental studies. As a class of proteins, microproteins are short sequences and do not align readily with high sequence conservation scores3,11,16 when queried against the canonical proteome databases. Due to these constraints, the prediction and analyses of their probable structural folds at scale by the current computational methods is challenging. Therefore, the studies to experimentally validate the possible biological activities of microproteins have been restricted to select candidates, often that show strict evolutionary conservation of their protein sequences.
It is critical to note that eukaryotic proteomes are rich in protein sequences that lack ordered three-dimensional structural folds or domains, yet possess important molecular functions. Intrinsically disordered regions (IDRs) represent 30–40% of the eukaryotic proteomes.17,18 IDRs exhibit diverse functionality, they can serve as a spacer or flexible sequences residing in between the folded domains of a larger protein, possess regulatory functions such as molecular recognition, scaffolding, and biomolecular condensate formation.17 As opposed to the well-folded protein-domains that mediate functions such as catalysis and molecular binding, IDRs offer functional advantages such as structural adaptability or induced folding, weak but specific interactions, and are often sites for post-translational modifications.17,18 A hallmark feature of IDRs is that they show weaker sequence conservation as compared to the sequences that adopt stable tertiary structures.17
Similar to such IDRs, it is postulated that microproteins may also be comprised of functional sequences despite lacking conservative sequences and definitive structural folds.16 CYREN microprotein is disordered, and immunoprecipitation studies have shown that CYREN can regulate its protein–protein interactions with Ku70/80 and several DNA damage response proteins with distinct short linear motifs (SLiMs).19,20 CYREN localizes to the nucleus and regulates the classical non-homologous end joining pathway.20,21 NBDY is an intrinsically disordered microprotein that can modulate the phase separation properties of P-bodies by its phosphorylation status.22 These case studies represent how intrinsically disordered microproteins can facilitate their molecular function by motifs and/or post-translational modifications, and it is plausible that the biochemical principles that guide IDR-functionality can be applied to microprotein sequences with unknown functions to support their characterization efforts.
We hypothesized that even though thousands of microprotein sequences might not be evolutionarily conserved across their full length, the appearance of specific sequence motifs could predispose them to certain biochemical and cellular activities. If true, this would encourage motif searches across the available microprotein databases to enable the design of focused biofunctional screens, and accelerate the functional characterization of microproteins.
Results
Rationale for the selection of polyalanine-motifs to design an activity-screen for uncharacterized microproteins
Microprotein sequences show differential amino acid composition relative to the reference proteome23 (Fig. 1A and Table S1, ESI†), showing enrichment of alanine, glycine, proline and arginine residues while depletion of lysine, aspartic acid, glutamic acid, glutamine, asparagine, isoleucine, and tyrosine. These would constitute low complexity or low amino acid diversity sequences, that are characteristic sequence features of IDRs.17,24 Protein sequences containing repeated stretches of identical amino acid repeats represent one class of IDRs, and approximately 15% of the human proteome contains such repeats,24–26 as opposed to the 3% estimate by random occurrence.24 Polyalanine repeats are an abundant subclass of amino acid repeats detected in approximately 500 human proteins,25,27,28 and show enrichment for transcription regulatory or nuclear proteins.24,25,27 | ||
Fig. 1 (A) Amino acid composition of microproteins (n = 7655, microprotein database23) relative to the reference human proteome (n = 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 435, UniProtKB), the numerical values are listed in Table S1 (ESI†). (B) GO overrepresentation test for polyalanine protein-subset (n = 415) relative to the reference human proteome. Information relating to the polyalanine subset and GO analysis is presented in Tables S2 and S3 (ESI†). 435, UniProtKB), the numerical values are listed in Table S1 (ESI†). (B) GO overrepresentation test for polyalanine protein-subset (n = 415) relative to the reference human proteome. Information relating to the polyalanine subset and GO analysis is presented in Tables S2 and S3 (ESI†). | ||
Since microproteins contain a higher proportion of alanine residues, we further examined the role and relevance of polyalanine motifs. We reasoned that the random probability of having a minimum of six consecutive alanine residues (6-ala peptide motif) in a protein would be infinitesimally small, and this should help isolate the sequences locally rich in alanine residues. We found 415 proteins that contain the 6-ala peptide motif in the canonical human protein dataset (Table S2, ESI†). Gene ontology analysis showed transcription regulators as the most overrepresented protein class in the polyalanine proteins subset (Fig. 1B and Table S3, ESI†), as well as positive enrichment for the nuclear cellular component (Table S3, ESI†). Additionally, we examined the database of human transcription factors enlisted by Lambert et al., 201829 and 9% of the sequences show a 6-ala motif, a four-fold increase over the expected frequency based on the reference proteome. These observations are consistent with the reports in the literature that polyalanine stretches are often found in transcription regulatory proteins.24,25,27,30 As a consequence, we hypothesized whether selecting microprotein sequences that contain the 6-ala peptide motif could enable the discovery of microproteins with transactivation function.
Cell-assay screening with polyalanine microproteins identifies sequences that possess transactivation activity
We identified 58 unique sequences that contain 6-ala from the reported microprotein database23 (Table S4, ESI†). We selected 6-ala as our motif of choice because it is significantly overrepresented in the known transcription regulatory proteins, and represents a pilot library to test our hypothesis. The average length of microprotein in the library was 64 amino acids (aa), with the shortest with 11 aa and the longest with 146 aa. We implemented the robust GAL4/UAS-transactivation system31,32 to test the transactivation potential of microproteins.The microproteins of interest were sequentially named as microprotein transactivator in this study (MPTA-1 to MPTA-58, Table S4, ESI†), and were cloned as C-terminal fusions of the GAL4 DNA-binding domain (GAL4DBD) into the GAL4-vector (Fig. 2). The GAL4-vector contained Renilla luciferase to serve as an internal transfection control. Additionally, estrogen receptor-α (ERα) and p65 transactivation domain 1 (p65_TA1) were included in the experiments as positive controls. The test library was individually co-transfected with the firefly reporter vector containing an upstream 9xUAS element in HEK293T cells (Fig. 2). The DualGlo luciferase assay was used to measure the induction of firefly luciferase by each MPTA relative to the baseline GAL4-activity. The sequences with four-fold or higher induction relative to GAL4-activity in two independent experiments were identified as positive hits (Table 1 and Table S5, ESI†). This screen led to the identification of three microproteins with transactivation activity, MPTA-10 (30 aa), MPTA-17 (37 aa) and MPTA-45 (98 aa).
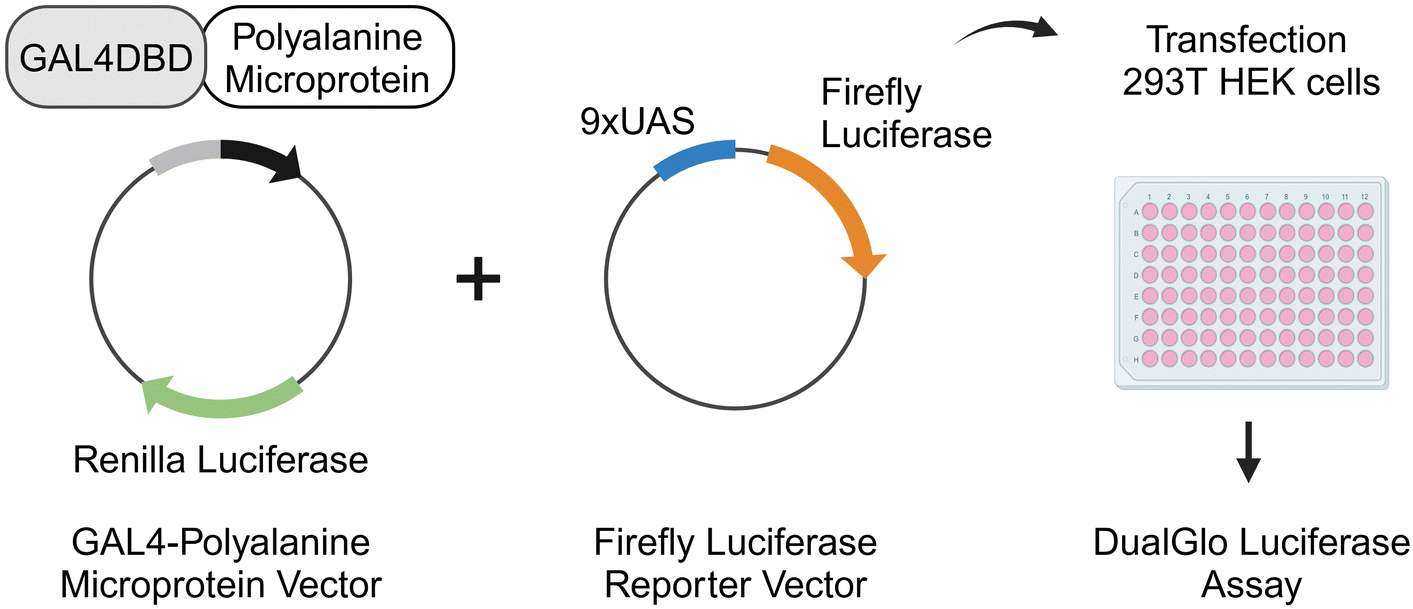 | ||
| Fig. 2 Experimental design to screen polyalanine microproteins for transactivation activity in a cell-based luciferase assay (created with BioRender.com). Microproteins (MPTA-1 to MPTA-58) were synthesized as C-terminal fusions to GAL4DBD and the vector contained renilla luciferase as internal transfection control. The gene expression reporter vector contained a firefly luciferase with an upstream 9xUAS element for the assessment of transactivation activity. HEK293T cells were co-transfected with a test microprotein library plasmid and firefly reporter vector, and 48 hours later the DualGlo Luciferase assay was performed. The sequences are listed in Table S4 (ESI†). | ||
| Replicate 1 | Replicate 2 | Replicate 1 | Replicate 2 | ||
|---|---|---|---|---|---|
| MPTA-1 | 1.29 ± 0.07 | 2.51 ± 0.46 | MPTA-30 | 1.36 ± 0.17 | 0.17 ± 0.07 |
| MPTA-2 | 1.08 ± 0.10 | 1.17 ± 0.10 | MPTA-31 | 1.05 ± 0.11 | 0.66 ± 0.18 |
| MPTA-3 | 0.94 ± 0.14 | 0.73 ± 0.07 | MPTA-32 | 3.29 ± 0.53 | 0.58 ± 0.04 |
| MPTA-4 | 0.51 ± 0.23 | 0.32 ± 0.06 | MPTA-33 | 0.70 ± 0.22 | 1.50 ± 0.20 |
| MPTA-5 | 1.43 ± 0.33 | 1.07 ± 0.06 | MPTA-34 | 0.60 ± 0.12 | 0.31 ± 0.08 |
| MPTA-6 | 0.93 ± 0.38 | 0.60 ± 0.08 | MPTA-35 | 2.92 ± 0.66 | 1.73 ± 0.16 |
| MPTA-7 | 1.04 ± 0.30 | 0.40 ± 0.12 | MPTA-36 | 0.51 ± 0.08 | 0.32 ± 0.04 |
| MPTA-8 | 0.97 ± 0.19 | 0.83 ± 0.19 | MPTA-37 | 0.34 ± 0.05 | 0.47 ± 0.11 |
| MPTA-9 | 2.28 ± 0.31 | 1.16 ± 0.19 | MPTA-38 | 0.65 ± 0.30 | 2.22 ± 0.62 |
| MPTA-10 | 5.41 ± 0.30 | 4.00 ± 0.29 | MPTA-39 | 0.95 ± 0.15 | 0.31 ± 0.04 |
| MPTA-11 | 0.42 ± 0.07 | 0.47 ± 0.06 | MPTA-40 | 1.66 ± 0.36 | 3.27 ± 0.28 |
| MPTA-12 | 0.69 ± 0.15 | 0.10 ± 0.02 | MPTA-41 | 0.92 ± 0.30 | 0.26 ± 0.06 |
| MPTA-13 | 2.60 ± 0.52 | 3.41 ± 0.22 | MPTA-42 | 1.10 ± 0.27 | 0.26 ± 0.04 |
| MPTA-14 | 1.92 ± 0.20 | 1.59 ± 0.13 | MPTA-43 | 2.42 ± 0.54 | 2.41 ± 0.63 |
| MPTA-15 | 1.90 ± 0.26 | 0.53 ± 0.13 | MPTA-44 | 0.55 ± 0.06 | 0.56 ± 0.20 |
| MPTA-16 | 1.49 ± 0.49 | 0.63 ± 0.11 | MPTA-45 | 14.68 ± 2.65 | 9.05 ± 0.91 |
| MPTA-17 | 16.73 ± 4.38 | 12.39 ± 2.01 | MPTA-46 | 0.35 ± 0.10 | 0.19 ± 0.01 |
| MPTA-18 | 1.51 ± 0.37 | 0.28 ± 0.08 | MPTA-47 | 0.20 ± 0.05 | 0.08 ± 0.01 |
| MPTA-19 | 3.17 ± 0.82 | 1.30 ± 0.06 | MPTA-48 | 0.27 ± 0.04 | 1.16 ± 0.31 |
| MPTA-20 | 1.84 ± 0.38 | 0.73 ± 0.06 | MPTA-49 | 0.16 ± 0.04 | 2.37 ± 0.17 |
| MPTA-21 | 2.24 ± 0.21 | 0.57 ± 0.11 | MPTA-50 | 0.30 ± 0.07 | 0.25 ± 0.01 |
| MPTA-22 | 0.60 ± 0.06 | 0.17 ± 0.03 | MPTA-51 | 0.12 ± 0.06 | 0.38 ± 0.11 |
| MPTA-23 | 1.87 ± 0.13 | 0.87 ± 0.10 | MPTA-52 | 0.15 ± 0.02 | 0.22 ± 0.04 |
| MPTA-24 | 1.52 ± 0.61 | 1.83 ± 0.37 | MPTA-53 | 0.51 ± 0.09 | 0.43 ± 0.09 |
| MPTA-25 | 3.22 ± 0.62 | 1.08 ± 0.17 | MPTA-54 | 0.83 ± 0.12 | 0.36 ± 0.07 |
| MPTA-26 | 0.15 ± 0.05 | 0.21 ± 0.09 | MPTA-55 | 0.28 ± 0.05 | 0.34 ± 0.06 |
| MPTA-27 | 0.32 ± 0.06 | 0.29 ± 0.09 | MPTA-56 | 0.83 ± 0.20 | 0.20 ± 0.07 |
| MPTA-28 | 1.33 ± 0.18 | 0.26 ± 0.03 | MPTA-57 | 1.55 ± 0.25 | 0.71 ± 0.44 |
| MPTA-29 | 1.49 ± 0.88 | 0.17 ± 0.02 | MPTA-58 | 1.01 ± 0.29 | 0.78 ± 0.22 |
MPTA-17 contains a C-terminal 14-amino acid transactivation peptide sequence
We conducted a structure–activity test across the length of MPTA-17 microprotein. As transactivating peptide sequences are typically 15–30 aa,33–35 we wanted to examine if we could identify the specific peptide region of MPTA-17 that is responsible for transactivation function. Four truncated MPTA-17 analogs were prepared, MPTA-17 (1–23), MPTA-17 (1–15), MPTA-17 (16–37), and MPTA-17 (24–37) (Fig. 3A). The analogs were tested in the transactivation assay and the two N-terminal analogs MPTA-17 (1–23) and MPTA-17 (1–15) were found to be inactive (Fig. 3B and Table S6, ESI†). The C-terminal analogs MPTA-17 (16–37) and MPTA-17 (24–37) were fully active. Of note, the shortest analog of 14 aa MPTA-17 (24–37) presented two-fold higher induction of firefly luciferase compared to the native sequence (Fig. 3B and Table S6, ESI†), and did not include the alanine stretch. Additionally, a variant where 7-ala was substituted with 7-ser MPTA-17S was prepared, and it retained the transactivation capability (Table S6, ESI†). | ||
| Fig. 3 (A) Sequence schematic showing different lengths of MPTA-17. (B) HEK293T cells were co-transfected with the respective microprotein variant and the firefly reporter vector, and the transactivation activity was assessed using the DualGlo luciferase assay. The microprotein activities from a representative experiment were plotted as fold change to baseline GAL4-activity (mean ± SD, n = 4), one-way ANOVA with multiple comparison ****p < 0.0001. Each experiment was conducted 3 times and the activity data with respective controls are presented in Table S6 (ESI†). (C) and (D) HEK293T cells were transfected individually with GAL4 or MPTA-17 variants and western blot was performed with GAL4-antibody and β-actin-antibody to detect protein expression, mock transfection without plasmid was used as a control (C) a representative blot is shown, (D) the relative protein band intensities for MPTA-variants in respective lanes were normalized to β-actin and quantified using ImageJ, presented as mean ± SD. Each experiment was conducted 3 times and data are presented in Table S6 (ESI†). | ||
We tested whether the different sequence truncations of the microproteins impacted their overall expression or stability post transfection. To test the protein expression levels of the microprotein variants, western blotting was performed using the GAL4-antibody following transfection in HEK293T cells. The N-terminal analog MPTA-17 (1–23) was present at lower levels compared to the native MPTA-17 (Fig. 3C and D and Table S6, ESI†). This variant was inactive in the transactivation assay (Fig. 3B and Table S6, ESI†), and due to its relatively lower protein level it is unclear whether this peptide region contributes to the transactivation activity of MPTA-17. The C-terminal analogs MPTA-17 (16–37) and MPTA-17 (24–37) were expressed at similar but modestly lower levels than the native protein (Fig. 3D). Importantly, the C-terminal MPTA-17 (24–37) sequence of 14-aa is able to effectively recapitulate the native protein transactivation function and is well-expressed, it is likely the key sequence region that supports the transactivation activity of MPTA-17.
MPTA-45 sequence truncation reveals its 24-amino acid transactivation peptide sequence
To identify the minimal transactivating peptide sequence of MPTA-45, and see whether it requires alanine stretch for its activity, we prepared a series of analogs with the presence or absence of the alanine stretch (Fig. 4A). MPTA-45 (1–75) and MPTA-45 (1–69) had diminished activity as compared to the native sequence in the GAL4-transactivation assay (Fig. 4B and Table S7, ESI†). MPTA-45 (68–98) showed reduced activity compared to the native sequence, and MPTA-45 (76–98) was inactive (Fig. 4B and Table S7, ESI†). We created an additional analog MPTA-45 (68–91) that was closer at recapitulating the activity of the native protein (Fig. 4B and Table S7, ESI†). As a control, MPTA-45 (1–91) was also tested and found to be fully active (Table S7, ESI†). These data suggest that 68–91 sequence of 24-aa within the MPTA-45 is the minimal peptide sequence that possesses transactivation activity. To examine the relevance of alanine stretch within MPTA-45 (68–91), the alanine stretch was removed, yielding inactive MPTA-45 (76–91) (Table S7, ESI†).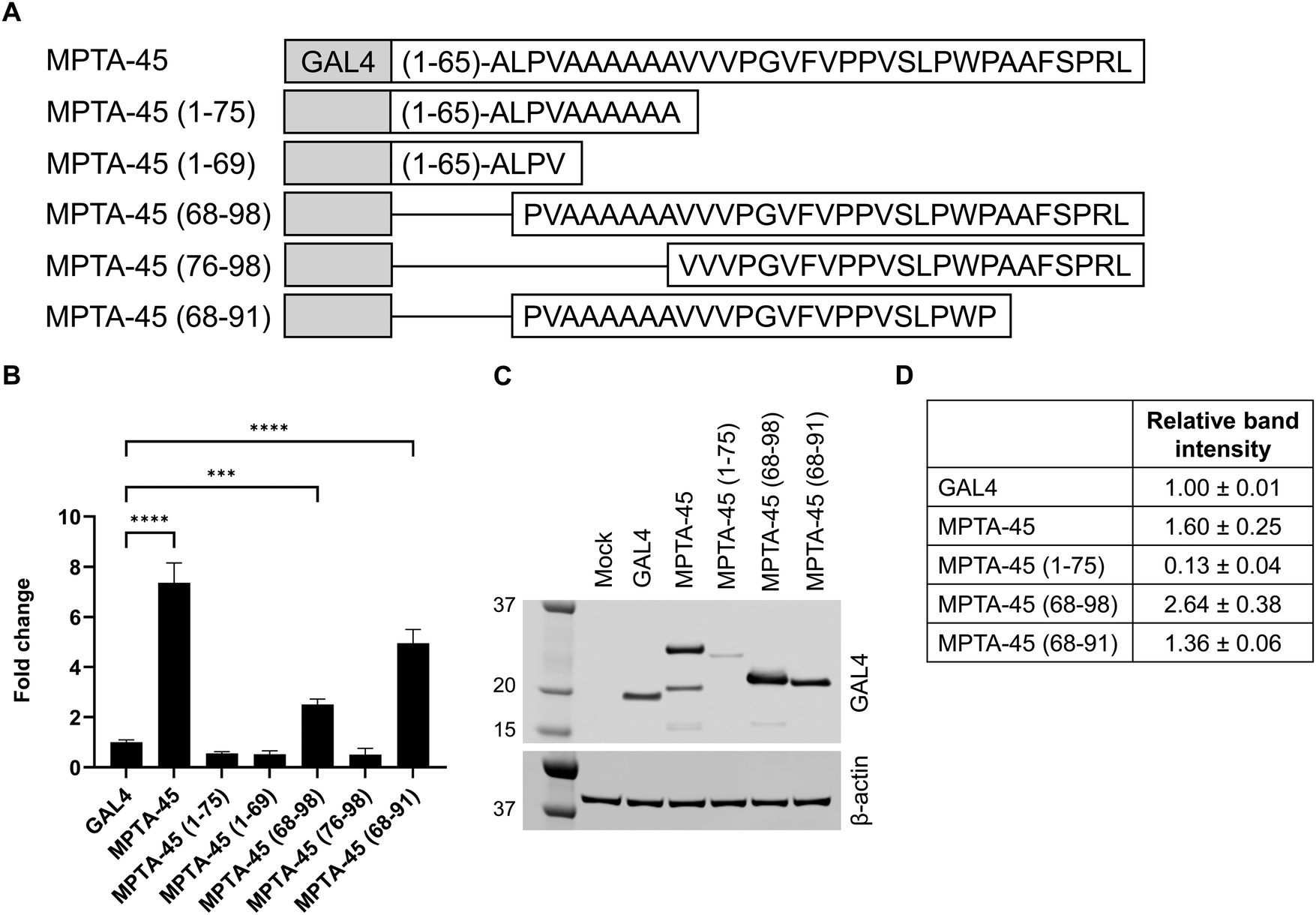 | ||
| Fig. 4 (A) Sequence schematic showing different lengths of MPTA-45. (B) HEK293T cells were co-transfected with the respective microprotein variant and the firefly reporter vector, and the transactivation activity was assessed using the DualGlo luciferase assay. The microprotein activities from a representative experiment were plotted as fold change to baseline GAL4-activity (mean ± SD, n = 4), one-way ANOVA with multiple comparisons ****p < 0.0001, ***p = 0.0001. Each experiment was conducted 2 times and the activity data with respective controls are presented in Table S7 (ESI†). (C) and (D) HEK293T cells were transfected with GAL4 or MPTA-45 variants and western blot was performed with GAL4-antibody and β-actin-antibody to detect protein expression, mock transfection without plasmid was done as a control (C) a representative blot is shown, (D) the relative protein band intensities for MPTA-variants in respective lanes were normalized to β-actin and quantified using ImageJ, presented as mean ± SD. Each experiment was conducted 3 times and data are presented in Table S7 (ESI†). | ||
The protein expression test for selected MPTA-45 analogs was performed by western blotting. MPTA-45 (1–75) analog showed lower expression compared to the native protein (Fig. 4C and D and Table S7, ESI†). MPTA-45 (68–98) was expressed at a higher level, while MPTA-45 (68–91) and MPTA-45 (76–98) showed relatively comparable levels to the native protein (Fig. 4C and D, Fig. S1A and B and Table S7, ESI†). A protein of lower molecular weight than expected was detected for MPTA-45, however, the quantification and comparison of the protein expression among the different analogs was done based on the observed band of the correct molecular weight. In this set of variants, MPTA-45 (68–91) was the shortest peptide with transactivation activity that was well-expressed relative to the native MPTA-45, and contained the alanine stretch.
Discussion
Microproteins represent a new class of proteins and the application of genetic screening and chemical biological approaches to characterize their possible functions is gaining momentum.11 Defining the possible functions of microproteins is challenging due to their short length, lack of sequence homology, as well as limited structural definitions. To advance this objective, chemical biology tools such as affinity-immunoprecipitation proteomics and proximity-labeling technologies have proven resourceful, enabling microprotein characterization. Microproteins can localize into specific subcellular regions to direct their biological activities, and this information can be leveraged towards functional discovery efforts.36,37 MicroID platform38 based on the proximity biotinylation technology was recently developed to identify the subcellular location of microproteins in cell lines and mouse tissue. The work led to the identification of 154 previously unannotated microproteins (or alt proteins) associated with the nucleus compartment, and of note, 16 candidates (10%) contained a 5 or 6-ala stretch in their sequences. Therefore, it is plausible that several microproteins that are alanine-rich could indeed be nuclear. A SEHBP microprotein is a recently identified microprotein that can localize to the nucleus, interact with chromatin-associated proteins, and possesses transcription regulatory function upon overexpression in cells.39 These case studies demonstrate that several microproteins could indeed be nuclear proteins, and some microproteins may possess transcription regulatory functions.In this study, we designed a transactivation screen to find functional microprotein sequences by a motif-selection approach. Based on the significant enrichment of alanines observed in human transcription factors and nuclear proteins, we formulated our hypothesis to select candidate microproteins with a 6-ala motif and search for bioactive sequences. We successfully identified three microproteins possessing transactivating activity, MPTA-10, MPTA-17 and MPTA-45 (5% hits) with the GAL4-transactivation assay from a pilot library of 58 microproteins. These sequences show short hydrophobic clusters, one or more tryptophan residues and a few polar residues (Fig. S2, ESI†), which align with the expected sequence features of transactivating peptides.33–35 Prior transactivation screens noted that no specific consensus motif is deterministic of the transactivation function, though some features such as the presence of hydrophobic residues in short clusters interspersed with polar or charged residues are more common.33–35 They are in agreement with the idea that transactivation peptides within the transcription factors typically reside in IDRs and are short, <30 amino acids.33–35
A random DNA library encoding for 67000+ peptides of ≤20 aa length was tested as a fusion to the yeast heat shock factor-1, to identify transactivating sequences and yielded 1% positive hits.34 Additional high-throughput studies conducted to identify and characterize transactivating peptide sequence properties have noted positive hit rates ranging from 0.1% to 4%.33,40 The direct comparison of the positive hit rates observed in large-scale studies with randomized peptide sequences and a proof-of-concept screen presented here is not ideal, however, it is safe to state that a polyalanine motif-selection strategy to discover transactivation peptides from microprotein sequences is no worse than such endeavors.
Polyalanine domains in transcription factors have been defined in the context of at least nine human congenital diseases,24,25,41 where the expansion of the polyalanine tract leads to loss-of-function or abnormal function of the encoded protein. HOXD13 is one example where the expansion of native polyalanine tract by additional 7–14 alanines results in synpolydactyly syndrome.42,43 The elongated alanine stretch in the disease variant affects the biomolecular co-condensate formation of HOXD13 with the transcriptional coactivator mediator.44,45 Polyalanine sequences can influence the biophysical properties of the proteins including their secondary structure, protein aggregation, phase separation and cellular localization.24,46,47 Alanine-rich sequences were initially recognized in the transcriptional repressors of insect proteins such as Kruppel48 and Hox,49 and FEV50 is one example of a human repressor protein. The molecular rationale to justify the high prevalence of polyalanine stretches in transcription regulatory proteins across eukaryotic proteomes remains to be fully defined, and no generalized molecular mechanism of their specific function has been proposed.24,25,27,28 However, there is sufficient evidence of enrichment of alanine repeats in nuclear as well as transcription regulatory proteins to warrant continued studies on this subject.
Structure–activity testing for MPTA-17 showed that MPTA-17 (24–37), did not require an alanine stretch for transactivation (Fig. 3B), while the MPTA-45 (68–91) contained the alanine stretch to assist its activity (Fig. 4B). Further work will be required to understand how alanine rich motifs may contribute to the transactivation function directly or indirectly, by affecting the biophysical characteristics or subcellular localization of microproteins in the cellular context. We note that some microprotein sequences or variants may be more prone to proteolytic degradation, or otherwise have variable expression profiles when transfected in cellular assays which could lead to false negatives during preliminary screening. Protein sequences terminating with C-terminal alanine repeats have been demonstrated to target proteins for proteolytic degradation by E3 ligases,51,52 and it is possible that MPTA-17 (1–23) and MPTA-45 (1–75) with C-terminal alanine residues are more prone to degradation in comparison to the respective native sequences (Fig. 3C, D and 4C, D). Additional studies will be required to ascertain whether the microproteins identified in the synthetic GAL4-transactivation assay could be endogenous regulators of transcription.
IDRs are known to possess SLiMs of 3–10 residues to facilitate their functionality.17,53 The annotation of functional SLiMs in the eukaryotic linear motif (ELM) database has been rising steadily.53 It is proposed that such sequences can rapidly evolve by the use of motifs or specific sequence features instead of absolute sequence homology at the level of amino acids.17 Microproteins are thought to represent products from de novo gene creation, are less conserved than canonical genes54,55 and also possess features similar to IDRs. A high-throughput protein interaction screen on a peptide matrix was conducted for peptide sequences derived from microproteins to identify their protein interaction partners, and led to the identification of dozens of microprotein interactions that are likely to be governed by SLiMs.55 It is predictable that with the growing microprotein database availability and illustrations of how SLiMs in microproteins can drive molecular functions, the motif-guided screening approach presented here can be expanded to design broader test libraries to explore the functional space of microproteins.
Conclusions
smORF-encoded microproteins offer a rich resource to search for new biologically active peptides. The work presented here extends the application of biochemical strategies to design activity screens for microproteins, as thousands of microproteins await functional characterization. Recent publications11,38,55 demonstrate the topical importance of how chemical biology strategies are advancing microprotein research. We hope that this study will serve as a proof-of-concept to encourage further investigations with motif-selection strategies that are independent of strict evolutionary conservation to explore the diverse biological functionality of microproteins.Experimental
Cloning
BioXP platform (TelesisBio) was used to synthesize gene fragments corresponding to the polyalanine microprotein sequences and p65_TA1 positive control. Each gene fragment contained a minimum of 40 bp overlap on 5′ and 3′ ends to facilitate Gibson assembly. GAL4-ERα (pBIND ERα Vector, Promega #E1390) was used as the backbone vector. Restriction digestion for GAL4-ERα was performed using PvuI (NEB #R0150S) and DraI (NEB #R0129S) to remove the ERα-encoding fragment, and the desired vector product was gel purified for use as a backbone vector for Gibson assembly. The Gibson assembly products obtained were transformed in either of the chemical competent E. coli (TOP10 Invitrogen #C409601, DH5α T1R Invitrogen #C44812-01, max efficiency DH5α Invitrogen #18258012, RapidTrans Tam1, ActiveMotif #11096) or electrocompetent E. coli (Ecloni 10G Elite Lucigen #60051-1). GAL4 encoded protein sequence corresponding to the DNA-binding domain (MKLLSSIEQACDICRLKKLKCSKEKPKCAKCLKNNWECRYSPKTKRSPLTRAHLTEVESRLERLEQLFLLIFPREDLDMILKMDSLQDIKALLTGLFVQDNVNKDAVTDRLASVETDMPLTLRQHRISATSSSEESSNKGQRQLTV) followed by linker sequence (AIPSTPPTPSPAIA) – this vector (referred to as GAL4-vector) was designated as baseline control for all experiments. All microproteins were cloned as C-terminal fusion to this GAL4-vector. Methionine was added at the end of the linker sequence for specific microproteins and the truncated variants that did not contain start methionine. ERα encoded protein sequence corresponding to the ligand binding domain (KRSKKNSLALSLTADQMVSALLDAEPPILYSEYDPTRPFSEASMMGLLTNLADRELVHMINWAKRVPGFVDLTLHDQVHLLECAWLEILMIGLVWRSMEHPGKLLFAPNLLLDRNQGKCVECMVEIFDMLLATSSRFRMMNLQGEEFVCLKSIILLNSGVYTFLSSTLKSLEEKDHIHRVLDKITDTLIHLMAKAGLTLQQQHQRLAQLLLILSHIRHMSNKGMEHLYSMKCKNVVPLYDLLLEMLDAHRLHAPTSRGGASVEETDQSHLATAGSTSSHSLQKYYITGEAEGFPATV). p65_TA1 encoded protein sequence (PGLPNGLLSGDEDFSSIADMDFSALLSQISS). Microprotein mutation or deletion analogs were prepared by the standard mutagenesis PCR method using Phusion Hot Start II High Fidelity PCR Master mix (Thermo Scientific #F565) with respective oligos (Eton Biosciences) followed by Dpn1 digestion (NEB #R0176S). All the plasmid or PCR products were individually transformed in E. coli, and single colonies were grown for plasmid isolation (Qiagen Miniprep) and verified by Sanger sequencing. pGL4.35 (luc2P/9XUASGAL4/Hygro) (Promega #E1370) was used as the firefly reporter vector containing 9xUAS element upstream of the minimal adenoviral promoter.DualGlo luciferase assay
HEK293T cells were cultured in DMEM (Corning #10-013-CV) with 10% FBS (Gibco) in a 5% CO2 humidified incubator at 37 °C. HEK293T cells were seeded in poly-L-lysine coated 96-well plates at 20000 cells per well. The next day, the cells co-transfected with a test GAL4-MPTA plasmid, or respective control plasmid GAL4, GAL4-ERα, p65-TA1 and firefly luciferase reporter plasmid using lipofectamine LTX reagent with PLUS reagent (Invitrogen #15338100) in the Opti-MEM I reduced serum medium (Gibco #31985062). Each treatment was done in quadruplicates. 24 hours post-treatment, the selected wells treated with GAL4-ERα as the positive control were supplemented with 30 nM Estradiol (Sigma-Aldrich #E1024, CAS No. 50-28-2). 48-Hours post-transfection, Dual-Glo® luciferase assay system (Promega #E2940) was performed as described. The medium was aspirated, 75 μL per well PBS and 75 μL DualGlo reagent were added, and the plate was incubated for 15 minutes at room temperature on a shaker. The cell lysate solution (120 μL per well) was transferred to a 96-well black plate, and firefly luminescence was recorded with a BioTek synergy plate reader. The StopGlo reagent (60 μL per well) was added to the plate and incubated for 15 minutes at room temperature on the shaker, and Renilla luminescence was recorded.
Western blotting
HEK293T cells were seeded at 0.4 × 106 cells per well in a 6-well plate coated with poly-L-lysine. The next day, each well was transfected with 2 μg GAL4 or GAL4-MPTA plasmid with the lipofectamine LTX reagent with PLUS reagent (Invitrogen #15338100). 48 hours post-transfection, cells were lysed or frozen at −80 °C until further processing. Cells were collected with a scraper and lysed using cell lysis buffer (NP-40 Thermo Scientific #28324; buffer composition – 0.5% NP-40, 50 mM Tris, 150 mM NaCl, 5 mM EDTA, 1 mM DTT, pH 7.6) with the 1× halt protease inhibitor cocktail (Thermo Scientific #78430). The lysate was centrifuged at 21000× g for 25 minutes at 4 °C, and the supernatant was collected. The total protein concentration of each lysate sample was determined using Pierce Microplate BCA Protein Assay Kit – reducing agent compatible (Thermo Scientific #23252). Samples were prepared for gel-loading at equal protein concentration (22 μg per lane) with bolt LDS sample buffer (Invitrogen #B0008) and heated for 10 minutes at 70 °C. A Bolt 4-12% Bis-Tris Plus WedgeWell (Invitrogen #NW04120BOX) gel was run with 1× Bolt MES SDS buffer (Invitrogen #B0002) with the samples and a protein ladder (Biorad #1610377). The protein was transferred to a PVDF membrane (Invitrogen #IB24002) with the iBlot2 system. The membrane was blocked using intercept blocking buffer TBS (Licor #927-60001) and incubated with GAL4(DBD) antibody (SantaCruz #sc-510 RK5C1) overnight at 4 °C. The membrane was washed three times with TBST (with 0.1% Tween-20), followed by Goat anti-Mouse IgG (IRDye 800CW, Licor #926-32210) incubation for 2 hours at room temperature, and washed 5 times with TBST. The membrane was imaged using an Odyssey CLx scanner. The same membrane was incubated with β-actin rabbit monoclonal antibody (Licor #926-42210) followed by Goat anti-Rabbit IgG (Alexa Fluor 680, Invitrogen #A21109) and imaged. Each plasmid transfection was done three times, and respectively three western blot experiments were performed unless noted otherwise. MPTA-45 (76–98) plasmid transfection and respective western blot was performed once.
Data analysis
Amino acid composition of the reference human proteome (sequences retrieved from UniProtKB Homo sapiens) and microproteins database23 were calculated as the sum of individual amino acids divided by the total number of amino acids in the entire dataset respectively.Gene Ontology analysis for the polyalanine proteins subset was done by statistical overexpression test with Panther Protein Class and GO cellular component complete (https://www.pantherdb.org/).
For the DualGlo luciferase assay performed in 96-well plates, the firefly luminescence signal was divided by renilla luminescence for each well to obtain the transactivation activity ratio, which was then normalized with baseline GAL4 Activity. Data obtained was analyzed in this manner consistently across all experiments and the raw data from one experiment are shown in Table S8 (ESI†). The data are presented as mean ± SD (n = 4) for each plasmid treatment, and the experiment was performed two or three times as noted. One-way ANOVA with multiple comparisons (Dunnett's) statistical test was performed using GraphPad Prism 10. All the graphs were plotted using GraphPad Prism 10.
ImageJ was used to analyze the relative band intensities for western blots. Rectangular boxes around the desired bands were made, and intensity peak plots were obtained with the Analyze Gels option. The ratio of GAL4 or GAL4-MPTA band intensity to the respective β-actin in individual lanes was calculated. The obtained ratio was normalized to GAL4 to represent the relative band intensity of each tested GAL4-MPTA construct and data are presented as Mean ± SD from three experiments unless noted otherwise.
Table of content – Created in BioRender. Agrawal, A. (2025) https://BioRender.com/m76s319, Fig. 2 – Created in BioRender. Agrawal, A. (2025) https://BioRender.com/i24z440.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by Frederik Paulsen Chair (A. S.), and the NIH (P30 CA014195, R01GM102491, A. S.). The authors thank Dr Eduardo Vieira de Souza for assistance with examining the proteomic datasets for microprotein sequences.References
- A. Saghatelian and J. P. Couso, Nat. Chem. Biol., 2015, 11, 909–916 CrossRef PubMed.
- S. D. Mackowiak, H. Zauber, C. Bielow, D. Thiel, K. Kutz, L. Calviello, G. Mastrobuoni, N. Rajewsky, S. Kempa, M. Selbach and B. Obermayer, Genome Biol., 2015, 16, 179 CrossRef PubMed.
- J. P. Couso, Genome Biol., 2015, 16, 189 CrossRef PubMed.
- M. A. Brunet, S. A. Levesque, D. J. Hunting, A. A. Cohen and X. Roucou, Genome Res., 2018, 28, 609–624 CrossRef PubMed.
- M. A. Basrai, P. Hieter and J. D. Boeke, Genome Res., 1997, 7, 768–771 CrossRef PubMed.
- N. T. Ingolia, S. Ghaemmaghami, J. R. Newman and J. S. Weissman, Science, 2009, 324, 218–223 CrossRef PubMed.
- N. T. Ingolia, G. A. Brar, S. Rouskin, A. M. McGeachy and J. S. Weissman, Nat. Protoc., 2012, 7, 1534–1550 CrossRef PubMed.
- S. A. Slavoff, A. J. Mitchell, A. G. Schwaid, M. N. Cabili, J. Ma, J. Z. Levin, A. D. Karger, B. A. Budnik, J. L. Rinn and A. Saghatelian, Nat. Chem. Biol., 2013, 9, 59–64 CrossRef PubMed.
- J. Ma, C. C. Ward, I. Jungreis, S. A. Slavoff, A. G. Schwaid, J. Neveu, B. A. Budnik, M. Kellis and A. Saghatelian, J. Proteome Res., 2014, 13, 1757–1765 CrossRef PubMed.
- J. M. Mudge, J. Ruiz-Orera, J. R. Prensner, M. A. Brunet, F. Calvet, I. Jungreis, J. M. Gonzalez, M. Magrane, T. F. Martinez, J. F. Schulz, Y. T. Yang, M. M. Alba, J. L. Aspden, P. V. Baranov, A. A. Bazzini, E. Bruford, M. J. Martin, L. Calviello, A. R. Carvunis, J. Chen, J. P. Couso, E. W. Deutsch, P. Flicek, A. Frankish, M. Gerstein, N. Hubner, N. T. Ingolia, M. Kellis, G. Menschaert, R. L. Moritz, U. Ohler, X. Roucou, A. Saghatelian, J. S. Weissman and S. van Heesch, Nat. Biotechnol., 2022, 40, 994–999 CrossRef PubMed.
- D. Schlesinger and S. J. Elsasser, FEBS J., 2022, 289, 53–74 CrossRef PubMed.
- M. I. Galindo, J. I. Pueyo, S. Fouix, S. A. Bishop and J. P. Couso, PLoS Biol., 2007, 5, e106 CrossRef PubMed.
- T. Kondo, Y. Hashimoto, K. Kato, S. Inagaki, S. Hayashi and Y. Kageyama, Nat. Cell Biol., 2007, 9, 660–665 CrossRef CAS PubMed.
- D. M. Anderson, K. M. Anderson, C. L. Chang, C. A. Makarewich, B. R. Nelson, J. R. McAnally, P. Kasaragod, J. M. Shelton, J. Liou, R. Bassel-Duby and E. N. Olson, Cell, 2015, 160, 595–606 CrossRef CAS PubMed.
- B. W. Wright, Z. Yi, J. S. Weissman and J. Chen, Trends Cell Biol., 2022, 32, 243–258 CrossRef.
- J. J. Mohsen, A. A. Martel and S. A. Slavoff, Proteomics, 2023, 23, e2100211 CrossRef.
- A. S. Holehouse and B. B. Kragelund, Nat. Rev. Mol. Cell Biol., 2024, 25, 187–211 CrossRef.
- P. Tompa, Trends Biochem. Sci., 2012, 37, 509–516 CrossRef PubMed.
- L. Xie, M. E. Bowman, G. V. Louie, C. Zhang, M. S. Ardejani, X. Huang, Q. Chu, C. J. Donaldson, J. M. Vaughan, H. Shan, E. T. Powers, J. W. Kelly, D. Lyumkis, J. P. Noel and A. Saghatelian, Biochemistry, 2023, 62, 3050–3060 CrossRef PubMed.
- P. J. Hung, B. Johnson, B. R. Chen, A. K. Byrum, A. L. Bredemeyer, W. T. Yewdell, T. E. Johnson, B. J. Lee, S. Deivasigamani, I. Hindi, P. Amatya, M. L. Gross, T. T. Paull, D. J. Pisapia, J. Chaudhuri, J. J. H. Petrini, N. Mosammaparast, G. K. Amarasinghe, S. Zha, J. K. Tyler and B. P. Sleckman, Mol. Cell, 2018, 71, 332–342 CrossRef e338.
- N. Arnoult, A. Correia, J. Ma, A. Merlo, S. Garcia-Gomez, M. Maric, M. Tognetti, C. W. Benner, S. J. Boulton, A. Saghatelian and J. Karlseder, Nature, 2017, 549, 548–552 CrossRef PubMed.
- Z. Na, Y. Luo, D. S. Cui, A. Khitun, S. Smelyansky, J. P. Loria and S. A. Slavoff, J. Am. Chem. Soc., 2021, 143, 12675–12687 CrossRef PubMed.
- T. F. Martinez, Q. Chu, C. Donaldson, D. Tan, M. N. Shokhirev and A. Saghatelian, Nat. Chem. Biol., 2020, 16, 458–468 CrossRef PubMed.
- S. Chavali, A. K. Singh, B. Santhanam and M. M. Babu, Nat. Rev. Chem., 2020, 4, 420–434 CrossRef.
- J. Amiel, D. Trochet, M. Clement-Ziza, A. Munnich and S. Lyonnet, Hum Mol Genet, 2004, 13(Spec No 2), R235–R243 CrossRef PubMed.
- A. S. Kumar, D. T. Sowpati and R. K. Mishra, PLoS One, 2016, 11, e0166854 CrossRef PubMed.
- H. Lavoie, F. Debeane, Q. D. Trinh, J. F. Turcotte, L. P. Corbeil-Girard, M. J. Dicaire, A. Saint-Denis, M. Page, G. A. Rouleau and B. Brais, Hum. Mol. Genet., 2003, 12, 2967–2979 CrossRef.
- P. Mier, C. A. Elena-Real, J. Cortes, P. Bernado and M. A. Andrade-Navarro, Bioinformatics, 2022, 38, 4851–4858 CrossRef.
- S. A. Lambert, A. Jolma, L. F. Campitelli, P. K. Das, Y. Yin, M. Albu, X. Chen, J. Taipale, T. R. Hughes and M. T. Weirauch, Cell, 2018, 172, 650–665 CrossRef.
- N. Rado-Trilla, K. Arato, C. Pegueroles, A. Raya, S. de la Luna and M. M. Alba, Mol. Biol. Evol., 2015, 32, 2263–2272 CrossRef.
- A. H. Brand and N. Perrimon, Development, 1993, 118, 401–415 CrossRef.
- A. Paguio, P. Stecha, K. V. Wood and F. Fan, Curr. Chem. Genomics, 2010, 4, 43–49 CrossRef.
- A. Erijman, L. Kozlowski, S. Sohrabi-Jahromi, J. Fishburn, L. Warfield, J. Schreiber, W. S. Noble, J. Soding and S. Hahn, Mol. Cell, 2020, 78, 890–902 CrossRef e896.
- C. N. Ravarani, T. Y. Erkina, G. De Baets, D. C. Dudman, A. M. Erkine and M. M. Babu, Mol. Syst. Biol., 2018, 14, e8190 CrossRef.
- M. V. Staller, A. S. Holehouse, D. Swain-Lenz, R. K. Das, R. V. Pappu and B. A. Cohen, Cell Syst., 2018, 6, 444–455 CrossRef PubMed e446.
- K. R. Hassel, O. Brito-Estrada and C. A. Makarewich, iScience, 2023, 26, 106781 CrossRef.
- A. M. Whited, I. Jungreis, J. Allen, C. L. Cleveland, J. M. Mudge, M. Kellis, J. L. Rinn and L. E. Hough, Biophys. Rep., 2024, 4, 100167 Search PubMed.
- Z. Na, X. Dai, S. J. Zheng, C. J. Bryant, K. H. Loh, H. Su, Y. Luo, A. F. Buhagiar, X. Cao, S. J. Baserga, S. Chen and S. A. Slavoff, Mol. Cell, 2022, 82, 2900–2911 CrossRef e2907.
- M. Koh, I. Ahmad, Y. Ko, Y. Zhang, T. F. Martinez, J. K. Diedrich, Q. Chu, J. J. Moresco, M. A. Erb, A. Saghatelian, P. G. Schultz and M. J. Bollong, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2021943118 CrossRef PubMed.
- M. Abedi, G. Caponigro, J. Shen, S. Hansen, T. Sandrock and A. Kamb, BMC Mol. Biol., 2001, 2, 10 CrossRef.
- L. Y. Brown and S. A. Brown, Trends Genet., 2004, 20, 51–58 CrossRef.
- Y. Muragaki, S. Mundlos, J. Upton and B. R. Olsen, Science, 1996, 272, 548–551 CrossRef PubMed.
- F. R. Goodman, S. Mundlos, Y. Muragaki, D. Donnai, M. L. Giovannucci-Uzielli, E. Lapi, F. Majewski, J. McGaughran, C. McKeown, W. Reardon, J. Upton, R. M. Winter, B. R. Olsen and P. J. Scambler, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 7458–7463 CrossRef.
- S. Basu, S. D. Mackowiak, H. Niskanen, D. Knezevic, V. Asimi, S. Grosswendt, H. Geertsema, S. Ali, I. Jerkovic, H. Ewers, S. Mundlos, A. Meissner, D. M. Ibrahim and D. Hnisz, Cell, 2020, 181, 1062–1079 CrossRef PubMed e1030.
- A. N. Albrecht, U. Kornak, A. Boddrich, K. Suring, P. N. Robinson, A. C. Stiege, R. Lurz, S. Stricker, E. E. Wanker and S. Mundlos, Hum. Mol. Genet., 2004, 13, 2351–2359 CrossRef PubMed.
- S. Caburet, A. Demarez, L. Moumne, M. Fellous, E. De Baere and R. A. Veitia, J. Med. Genet., 2004, 41, 932–936 CrossRef PubMed.
- C. Messaed and G. A. Rouleau, Neurobiol. Dis., 2009, 34, 397–405 CrossRef PubMed.
- J. D. Licht, M. J. Grossel, J. Figge and U. M. Hansen, Nature, 1990, 346, 76–79 CrossRef PubMed.
- R. Galant and S. B. Carroll, Nature, 2002, 415, 910–913 CrossRef.
- P. Maurer, F. T'Sas, L. Coutte, N. Callens, C. Brenner, C. Van Lint, Y. de Launoit and J. L. Baert, Oncogene, 2003, 22, 3319–3329 CrossRef.
- P. R. Patil, A. M. Burroughs, M. Misra, F. Cerullo, C. Costas-Insua, H. C. Hung, I. Dikic, L. Aravind and C. A. P. Joazeiro, Cell Rep., 2023, 42, 113100 CrossRef PubMed.
- X. Wang, Y. Li, X. Yan, Q. Yang, B. Zhang, Y. Zhang, X. Yuan, C. Jiang, D. Chen, Q. Liu, T. Liu, W. Mi, Y. Yu and C. Dong, Nat. Commun., 2023, 14, 2474 CrossRef PubMed.
- M. Kumar, S. Michael, J. Alvarado-Valverde, A. Zeke, T. Lazar, J. Glavina, E. Nagy-Kanta, J. M. Donagh, Z. E. Kalman, S. Pascarelli, N. Palopoli, L. Dobson, C. F. Suarez, K. Van Roey, I. Krystkowiak, J. E. Griffin, A. Nagpal, R. Bhardwaj, F. Diella, B. Meszaros, K. Dean, N. E. Davey, R. Pancsa, L. B. Chemes and T. J. Gibson, Nucleic Acids Res., 2024, 52, D442–D455 CrossRef PubMed.
- N. Vakirlis, Z. Vance, K. M. Duggan and A. McLysaght, Cell Rep., 2022, 41, 111808 Search PubMed.
- C. L. Sandmann, J. F. Schulz, J. Ruiz-Orera, M. Kirchner, M. Ziehm, E. Adami, M. Marczenke, A. Christ, N. Liebe, J. Greiner, A. Schoenenberger, M. B. Muecke, N. Liang, R. L. Moritz, Z. Sun, E. W. Deutsch, M. Gotthardt, J. M. Mudge, J. R. Prensner, T. E. Willnow, P. Mertins, S. van Heesch and N. Hubner, Mol. Cell, 2023, 83, 994–1011 CrossRef CAS PubMed e1018.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cb00277f |
| This journal is © The Royal Society of Chemistry 2025 |