
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe evolution and application of RNA-focused small molecule libraries
Amirhossein
Taghavi†
a,
Noah A.
Springer†
ab,
Patrick R. A.
Zanon
 a,
Yanjun
Li
cd,
Chenglong
Li
c,
Jessica L.
Childs-Disney
a and
Matthew D.
Disney
*ab
a,
Yanjun
Li
cd,
Chenglong
Li
c,
Jessica L.
Childs-Disney
a and
Matthew D.
Disney
*ab
aDepartment of Chemistry, The Herbert Wertheim UF Scripps Institute for Biomedical Innovation and Technology, 130 Scripps Way, Jupiter, FL 33458, USA. E-mail: mdisney@ufl.edu
bDepartment of Chemistry, The Scripps Research Institute, 130 Scripps Way, Jupiter, FL 33458, USA
cDepartment of Medicinal Chemistry, Center for Natural Products, Drug Discovery and Development, The University of Florida, Gainesville, FL 32610, USA
dDepartment of Computer & Information Science & Engineering, University of Florida, Gainesville, FL 32611, USA
First published on 13th February 2025
Abstract
RNA structure plays a role in nearly every disease. Therefore, approaches that identify tractable small molecule chemical matter that targets RNA and affects its function would transform drug discovery. Despite this potential, discovery of RNA-targeted small molecule chemical probes and medicines remains in its infancy. Advances in RNA-focused libraries are key to enable more successful primary screens and to define structure–activity relationships amongst hit molecules. In this review, we describe how RNA-focused small molecule libraries have been used and evolved over time and provide underlying principles for their application to develop bioactive small molecules. We also describe areas that need further investigation to advance the field, including generation of larger data sets to inform machine learning approaches.
 Amirhossein Taghavi | Amirhossein Taghavi received his PhD in physics from Luxembourg University under the supervision of Dr. Joshua T. Berryman and Prof. Tanja Schilling investigating the Extensionally Driven Transitions of DNA. He joined the Prof. Disney group in 2021 as a postdoctoral researcher developing computational assisted drug design studying different groups of neurodegenerative diseases associated with RNA expanded repeats. His research is supported by the Muscular Dystrophy Association Development Grant. |
 Noah A. Springer | Noah A. Springer received his BS in Biochemistry from the University of Notre Dame in 2021. The following fall he began doctoral studies in the lab of Prof. Matthew D. Disney, where his research focuses on development of new methods to target disease-causing RNAs with small molecules. His research is supported by the National Science Foundation Graduate Research Fellowship. |
 Patrick R. A. Zanon | Patrick R. A. Zanon received his BSc (2014) and MSc (2017) in Chemistry from the Technical University of Munich, Germany. Under the supervision of Dr Stephan Hacker, he investigated the reactivity of electrophilic probes for proteome-wide profiling and covalent ligand development and was awarded his doctoral degree summa cum laude (2021). Supported by a Walter Benjamin fellowship from the German Research Foundation (DFG), he joined the Disney group in 2022 as a postdoctoral researcher to develop new modalities enhancing the biological acitivity of RNA-targeting small molecules. |
 Yanjun Li | Dr Yanjun Li is an Assistant Professor in the Department of Medicinal Chemistry at the University of Florida (UF). Dr Li earned his PhD in computer science from the UF and then worked as a Senior Research Scientist at the Institute of Deep Learning, Baidu Research USA, and later at Calico Life Sciences. His research expertise and interest span the fields of deep learning, AI-driven drug discovery, AI4Science, and computational biology, with a focus on deep generative modeling for de novo molecule design and geometric deep learning for molecular recognition. |
 Chenglong Li | Dr Chenglong Li obtained his PhD in the field of Molecular Biophysics at Cornell University. He is the Nicholas Bodor Professor for Drug Discovery at the University of Florida; Professor of Medicinal Chemistry, Biochemistry and Biophysics. He is the Director of the NIH/NIGMS Chemistry-Biology Interface (CBI) Predoctoral Training Program at UF; and Associate Director of the Center for Natural Products, Drug Discovery and Development (CNPD3). His main research interest is the theory and practice of molecular recognition from electronic, molecular to organismal levels with combined computational and experimental approaches, with important applications for structure-based AI-enabled drug design and discovery. |
1. Introduction
The dysfunction and aberrant expression of RNA is associated with many diseases,1–8 making this biomolecule a promising therapeutic target.9–11 For example, aberrant splicing events cause diseases including frontotemporal dementia (FTD) and Alzheimer's Disease12 as well as β-thalassemia.13 Aberrant expression of micro (mi)RNAs plays a key role in cancer development by dysregulating the expression of oncogenes or tumor suppressors.14,15 In some cancers, internal ribosome entry sites (IRES) allow certain genes to be translated in a cap-independent manner, bypassing the traditional cap-dependent initiation of translation16,17 and promoting cell survival, angiogenesis, and tumor growth. RNA repeat expansions acquire a gain-of-function and cause microsatellite disorders, a class of neuromuscular diseases.18Different approaches have been adopted to target disease-causing RNAs. Antisense oligonucleotides (ASOs) were among the first modalities19 used for RNA-targeting and have been used in numerous cases.19,20 As ASOs bind to complementary sequences in the target RNA, the most potent oligonucleotides are often those that bind to unstructured regions.21–23 ASOs function via a variety of mechanisms, including RNase H-mediated degradation and steric blockage of RNA binding proteins (RPBs),21 to modulate alternative pre-mRNA splicing, for example. Fomivirsen was the first US Food and Drug Administration (FDA)-approved ASO,24 and since then multiple ASO-based therapeutics have been clinically approved.25–27 Although numerous backbone and base modification have been introduced that improve their stability and chemical properties,28,29 challenges associated with ASO delivery,30,31 stability and trafficking,32 and hepatotoxicity33,34 still hinder their use in the clinic.35
Small molecules provide an alternative for targeting RNA. Although their development towards RNA targets is not as advanced as ASOs, small molecules can have favorable drug-like and physicochemical properties, and these properties can be fine-tuned through conventional medicinal chemistry approaches.36 Folded RNAs provide binding pockets for small molecules including RNA-protein complexes,37–40 and also RNAs with internal loops, bulges, stems, pseudoknots, and junctions.41–45 Designing small molecules to target RNA, however, is fundamentally different and perhaps even more complex than targeting proteins, partly due to the unique features of RNA such as its structural flexibility and dynamics, surface electrostatics, and lower diversity of building blocks (four nucleotides vs. 20 amino acids). Despite these challenges, small molecule–RNA interactions hold the potential to affect biology by various mechanisms, including directing pre-mRNA splicing events, inhibiting precursor miRNA processing, repressing translation, inhibiting RNA–protein complexes, and targeting RNA for degradation (Fig. 1). Additionally, drugging the mRNAs of disease-relevant proteins may provide an alternative method to target “undruggable” proteins,46,47 which comprise a substantial portion of the proteome.
 | ||
| Fig. 1 Therapeutic potential of small molecules targeting RNA. RNA–small molecule interactions have promise to alter biology via a variety of mechanisms, including directing alternative pre-mRNA splicing, inhibiting miRNA processing, disrupting RNA–protein complexes, targeted RNA degradation, and repressing translation. | ||
In this review, the evolution of approaches that have been used to develop RNA-focused libraries will be discussed, including substructure-based libraries, chemical similarity searching, and physicochemical property filtering. Noteworthy developments in the application of machine learning in hit identification and lead optimization are finding their way into the RNA world with promising results. These new approaches combined with more traditional methods such as fragment-based screening can be used to generate more focused libraries.48
2. RNA-focused libraries: past & present
2.1 RNA-targeting drugs: from antibiotics to risdiplam
Although RNA-targeting small molecules have gained attention in recent years, drugs that target RNA have been used for over 70 years (Fig. 2). The oldest and most successfully targeted RNA structure is, perhaps unsurprisingly, ribosomal RNA (rRNA), an ideal target owing to its highly abundant and structured nature. Many classes of antibiotics function by binding and inhibiting the function of the bacterial ribosome, including aminoglycosides (Fig. 2), tetracyclines, amphenicols, macrolides, and oxazolidinones, amongst others.49,50 These ribosome-binding antibiotic classes function by binding various sites within rRNAs and inhibit initiation, elongation, or termination of translation.51 All early ribosome-targeting antibiotics were either natural products or semi-synthetic natural product derivatives. | ||
| Fig. 2 History of RNA-targeting drugs. RNA has been a small molecule drug target for nearly 80 years. In 1946, the first class of ribosome-targeting small molecule antibiotics, aminoglycosides, were introduced clinically. All early small molecules were natural products or derived from natural product scaffolds.50 In 2000, the first fully synthetic RNA-targeting small molecule drug, linezolid,52 was approved, which also targets the ribosome. In 2020, risdiplam became the first non-ribosomal RNA-targeting small molecule drug, with the SMN2/spliceosome complex as the primary target.53 Dates above represent the first clinical use of each drug class, with a representative structure for each. | ||
The discovery and subsequent approval of linezolid in 2000 marked the first fully synthetic (i.e., not derived from a natural product pharmacophore) RNA-targeting antibiotic.54 Linezolid demonstrated that RNA-targeting small molecules could be successfully identified and developed from sources other than natural products. Importantly, the diversity of compound structures, ranging from large, highly charged aminoglycosides to the traditionally drug-like linezolid demonstrated that a wide range of chemical scaffolds can recognize and target RNA with clinical success. For other RNA targets, the development of primary screening hits into drugs has proved more challenging, in some cases due to lower expression levels, the lack of robust or dynamic structures, or inaccessibility due to protein binding.
The approval of risdiplam as an oral medication for the treatment of spinal muscular atrophy (SMA) showed for the first time that small molecule targeting of non-ribosomal RNAs could be therapeutically successful in humans. The SMN-C series of compounds were identified from a phenotypic screen searching for SMN2 pre-mRNA splicing enhancers to compensate for loss-of-function of the highly homologous SMN1.55 This initial hit was optimized via medicinal chemistry to afford the drug risdiplam,53 which gained full FDA approval in 2020. Detailed biophysical and structural studies showed that risdiplam stabilizes an RNA–protein manifold between the SMN2 splice site and a component of the spliceosome, U1 snRNP (small nuclear ribonucleoprotein), to direct splicing.56,57 Risdiplam makes direct contacts to the SMN2 pre-mRNA, inducing stabilization of the RNA by pulling the bulged adenosine within the helical stack and eliminating the clashes with the protein component (U1-C) of the U1 snRNP. A detailed nuclear magnetic resonance (NMR) study revealed that the carbonyl group of risdiplam forms a direct hydrogen bond with the amino group of the unpaired adenine linking the U1 snRNA and the 5′-splice site of SMN2 exon 7, serving as the minimal trans-splicing factor.57 Overall, it was suggested that risdiplam acts through the mechanism of 5′-splice site bulge repair. The extensive work to elucidate risdiplam's mechanism of action provides undeniable evidence that targeting RNA structures with small molecules can transform conventional drug discovery.58–60
2.2 Challenges in targeting RNA with small molecules
Despite these significant advances, small molecule targeting of RNA still faces challenges, including the scarcity of available 3D structures to enable structure-based design (Fig. 3(A)), promiscuous binders, and an incomplete understanding of the privileged chemotypes that drive specific binding to RNA, all of which are key considerations for the creation of RNA-focused small molecule libraries. In 2024, the total number of RNA-only structures in the Protein Data Bank (PDB) was 1830, as compared to 190![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 372 protein-only structures, an approximately 30-year lag (Fig. 3(A)).61 A recent search for small molecule complexes where the small molecule's molecular weight was restrained between 150–500 Da afforded 144482 protein–small molecule complexes and only 862 RNA–small molecule complexes.62 Of these 862 structures, nearly half are of the ribosome or ribosome sub-structures and another ∼25% are aptamers, which were specifically selected to bind the small molecules of interest (Fig. 3(B)). Although these structures could be informative, it is unclear how broadly applicable they are to other types of RNAs.
372 protein-only structures, an approximately 30-year lag (Fig. 3(A)).61 A recent search for small molecule complexes where the small molecule's molecular weight was restrained between 150–500 Da afforded 144482 protein–small molecule complexes and only 862 RNA–small molecule complexes.62 Of these 862 structures, nearly half are of the ribosome or ribosome sub-structures and another ∼25% are aptamers, which were specifically selected to bind the small molecules of interest (Fig. 3(B)). Although these structures could be informative, it is unclear how broadly applicable they are to other types of RNAs.
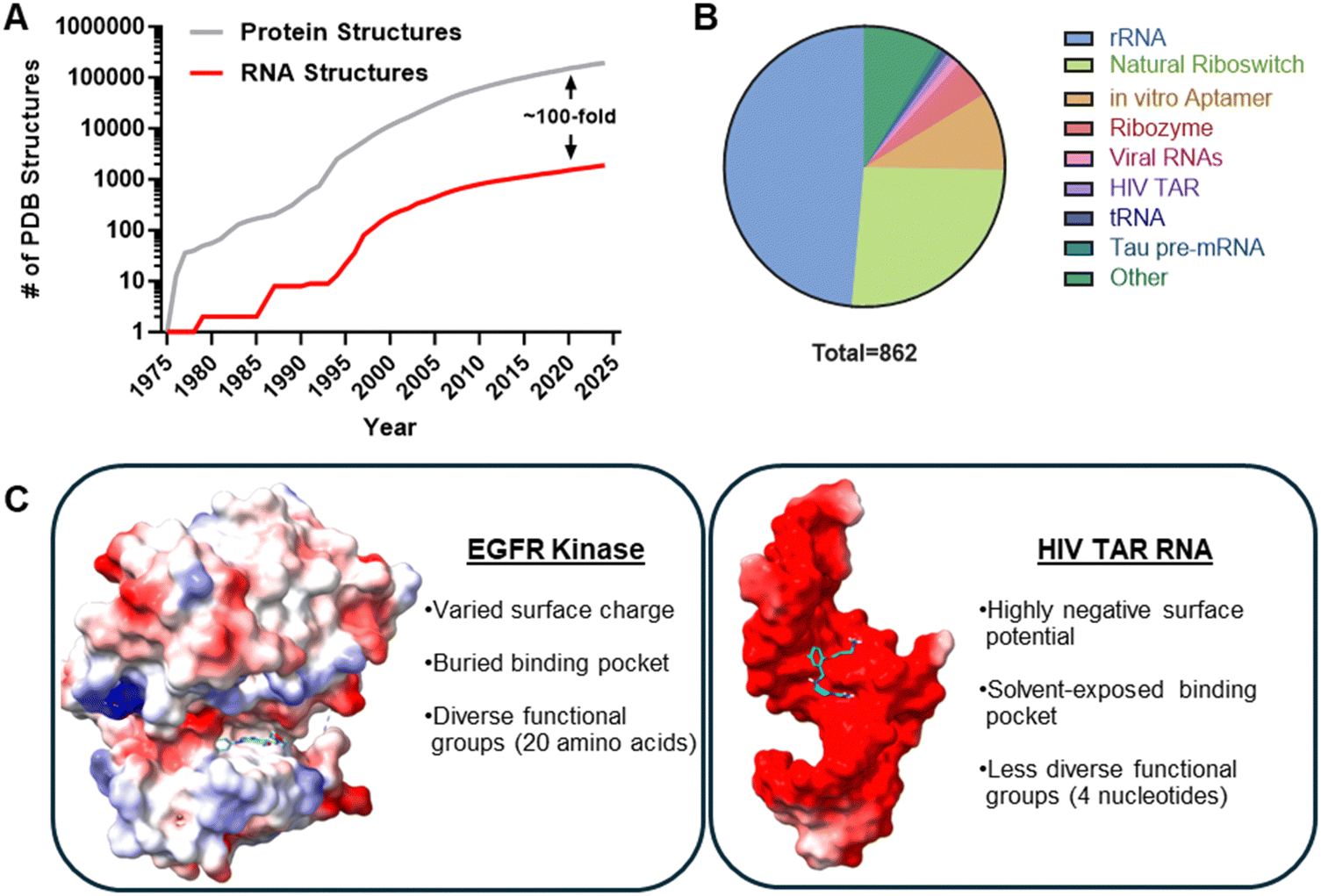 | ||
| Fig. 3 Challenges for RNA-targeted small molecule drugs. (A) A comparison of RNA-only and protein-only structures in the RCSB PDB reveals a large (∼100-fold) discrepancy in the number of three-dimensional structures. (B) Structures containing RNA–small molecule interactions lack diversity, with approximately 80% of all structures belonging to either rRNA, riboswitches, or in vitro selected aptamers. RNA–small molecule structures were collected from the HARIBOSS database.62 (C) A comparison of protein (EGFR Kinase, PDB: 2ITZ) and RNA (HIV TAR, PDB: 1UUI) binding pockets is presented, where red and blue represent negative and positively charged surfaces, respectively, as calculated by UCSF ChimeraX.63 | ||
Small molecules identified from screening efforts are often non-specific or promiscuous binders, related to both RNA's negatively charged backbone and perhaps lack of diversity in its building blocks (Fig. 3(C)). Kelly et al. discuss that electrostatic interactions between the anionic RNA backbone phosphate and cationic functional groups on the small molecule can enhance binding affinity, however they also contribute to non-specific binding.64 Thus, incorporation of positively charged functional groups likely needs to be counter balanced with other target-specific interactions. Perhaps, the most notable example of cationic ligands that target RNA is the aminoglycoside class of antibiotics that bind and inhibit the bacterial ribosome. Its observed side effects, such as ototoxicity, are tied to its promiscuity, hindering clinical application.65,66 However, charge can also be used to improve selectivity. For example, flexible scaffolds with charged centers were specific for an RNA duplex as compared to a DNA duplex where the small molecule could not orient itself in the DNA minor groove.67 Addition of positive charge to diphenylfuran ligands changed an intercalative binding mode to an ionic one, improving specificity.68
RNA contains planar aromatic nucleotide bases that can engage in π–π stacking interactions with aromatic rings in small molecules. While stacking is often specific in defined binding pockets (e.g. in aptamers or riboswitches), it can also occur non-specifically along helical regions or loops of RNA, in DNA, or in proteins. However, various studies have shown that stacking interactions can be used to drive selectivity for RNA.69 One example is an acridine-based ligand discovered to stabilize a G-quadruplex structure found in the long noncoding (lnc)RNA Telomeric Repeat-Containing RNA (TERRA).70 For intercalating small molecules, selectivity can also be improved by designing threading intercalators.71
Another way to increase the binding specificity is to use multivalent compounds to target adjacent binding sites simultaneously, particularly for RNAs with less complex binding pockets.72,73 This multivalency strategy has been used to target expanded repeating RNAs and miRNAs, which are primarily comprised of relatively simple stem-loop structures with internal loops or bulges. Collectively, the design strategy employed for small molecules should be based on the geometrical properties of the target RNA.
Non-specific binding can have broad implications, as exemplified in pentamidine, an FDA-approved antimicrobial drug.74,75 In addition to its antimicrobial function, this compound can bind to the triplet repeat expansion r(CUG)exp that causes myotonic dystrophy type 1 (DM1) and inhibit the binding of the alternative pre-mRNA splicing regulator muscleblind-like 1 (MBNL1), thus improving the splicing defects associated with MBNL1 sequestration.75 However, further investigations showed compound is non-specific for r(CUG)exp and also interacts with DNA and proteins.76
Finally, RNA conformational flexibility is both a challenge and an opportunity for selective binding.77 In solution, both RNAs and proteins exist in multiple different conformations called conformational ensembles.78 While proteins exist in one or a limited number of well-folded structures, RNA exhibits a relatively large number of conformations that have similar stability.79 RNA molecules also have a greater degree of local structural fluctuations compared to proteins,80 posing signficant challenges for structural determination.
Transient RNA conformations observed along the conformational flexibility pathway have important biological functions78 and are also involved in diseases, making them a potential therapeutic target.81,82 These transient conformations provide an opportunity to increase the compound specificity as exemplified in r(G4C2)exp, the hexanucleotide repeat expansion that causes genetically defined amyotrophic lateral sclerosis and frontotemporal dementia, C9orf72 (c9) ALS/FTD.83 The r(G4C2)exp forms two distinct structures: a G-quadruplex or a hairpin structure with a periodic array of 1 × 1 nucleotide GG internal loops.84–87 These two alternative conformations can be targeted with different small molecules that selectively bind either conformation. Interestingly, a small molecule that conformationally selects a hidden, minor conformation, a hairpin that forms 2 × 2 nucleotide GG internal loops, was also discovered.83 This broad conformational flexibility is one of the unique features that distinguishes RNA from protein targets and can be exploited for RNA drug discovery.88
Overall, the adopted design strategy to create RNA-focused libraries, either general or target-specific, with the goal of minimizing non-specific binding, should be initiated with careful examination of the geometrical properties of the binding pocket and then selection of compounds that can form specific interactions with the binding pockets. Thus, compounds are selected based on shape complementarity,89–91 electrostatic complementarity,92 and conformational flexibility.93,94
2.3 Design of early RNA-focused libraries: substructure-based approaches
Design of early RNA-focused libraries relied upon substructure- or pharmacophore-based approaches, where compounds containing previously identified RNA-binding scaffolds were synthesized and screened against RNA targets. Much of this initial work was built upon known nucleic acid-binding compounds such as aminoglycosides (and semi-synthetic derivatives thereof) and DNA intercalators and groove binders, as reviewed previously.95 One of the first RNA-focused libraries was a 55-compound library developed to target rRNA.68 Zhou et al. used 3D structural information of aminoglycosides bound to 16S rRNA to show that 2-deoxystreptamine (2-DOS), a conserved core scaffold in aminoglycosides, binds to rRNA in the same way.96 A synthetic mimetic of 2-DOS (cis-3,5-diamino-piperidine (DAP)) retained RNA-binding functionality and facilitated parallel synthesis and structure–activity relationship (SAR) studies. Since DAP exhibited low binding affinity for rRNA, modeling was used to increase affinity by adding functionality via attachment to the triazine (Fig. 4). Synthesis and biological assays of bacterial growth of more than 55 compounds showed how changes in the headpiece configuration can provide an SAR set for targeting rRNA.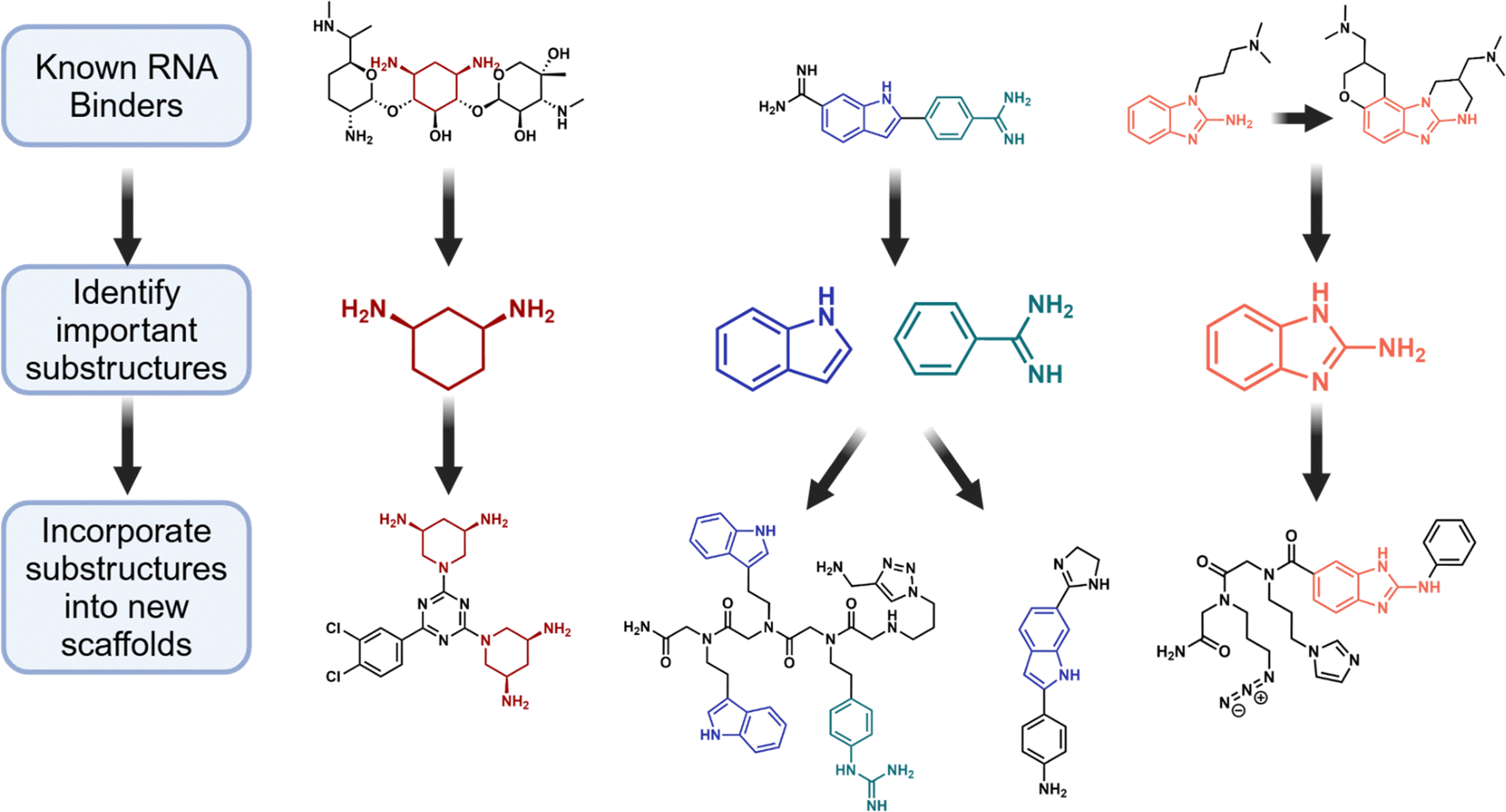 | ||
| Fig. 4 Substructure-based approaches to developing RNA-focused libraries. Early RNA-focused libraries72,96–98 utilized prior knowledge of individual RNA binders such as aminoglycosides (left), 4′,6-diamidino-2-phenylindole (DAPI, middle), and a 2-aminobenzimidazole-based Hepatitis C viral RNA binder. Important substructures from these known RNA binders were extracted and incorporated into new scaffolds, generating early RNA-focused libraries. | ||
This knowledge, the importance of substructures, was applied to design the first RNA-focused library with diverse chemical features, which was synthesized on a peptoid backbone using building blocks that are likely to bind RNA.97 The library used substructures extracted from molecules known to bind RNA (linezolid,52 xanthinol,99 and pentamidine,74 for examples) or building blocks hypothesized to facilitate hydrogen bonding or stacking interactions with RNA, such as the benzene or benzenesulfonaminde (Fig. 4).97 This library comprising 109 compounds was synthesized via a solid-phase approach where each molecule contained an azide handle that was used for site-specific conjugation to alkyne-functionalized agarose microarrays. The microarrays were then screened for binding to the Candida albicans group I intron, a catalytically active RNA molecule (ribozyme) that folds into a tertiary structure with well-defined binding pockets.100 The hit molecules from the binding screen were then tested for their ability to inhibit group I intron self-splicing, resulting in IC50 values ranging from 150 to >5000 μM. The data obtained from this first round of screening were used to identify a set of features that drive binding, incorporating both building block identity and position within the peptoid. These features aided design of a second generation of compounds with overall improved IC50 values for in vitro inhibition of splicing (31–110 μM). This ligand-based approach showed how embedded features in moieties that confer binding to RNA can be harnessed to design an RNA-focused library with improved features and emphasized the importance of the availability of such information. The combinatorial approach adopted in this study showed the importance of feature extraction in disposing the ligands for RNA recognition.
Benzimidazoles are another privileged substructure/scaffold for RNA targets. An early NMR-based approach by Abbott Laboratories identified that simple 2-aminobenzimidazoles bind the bacterial A-site rRNA with Kd values of ∼200 μM.101 Shortly thereafter an “SAR by MS” approach by Ibis Therapeutics identified a 2-aminobenzimidazole with ∼100 μM affinity to the Hepatitis C Virus (HCV) IRES RNA. Medicinal chemistry approaches were able to optimize this compound to afford a restricted, cyclic benzimidazole derivative with <1 μM affinity to the target and cellular activity at single digit micromolar concentrations in an HCV replicon assay.102
Because of the success of 2-aminobenzimidazoles in targeting several RNA structures, a library of 79 compounds containing the substructure (Fig. 4) was synthesized and evaluated in a microarray-based selection named 2-dimensional combinatorial screening (2DCS).103 In 2DCS, a microarray of small molecules is incubated with a library of radiolabeled RNAs – in this case, a library of 4096 RNA hairpin structures containing a randomized region in a 3 × 3 nucleotide internal loop pattern. After washing, the bound RNAs are manually excised from the microarray surface, amplified by RT-PCR, then identified by sequencing. Screening of this library revealed that functionalization of the 2-aminobenzimidazole scaffold both determined whether the compound could bind RNA (only 19 of 79 compounds bound the 4096-member RNA library) and differences in the small molecule's preferred RNA structures.103
In a similar approach, a panel of 43 RNA-focused compounds harboring privileged scaffolds known to bind RNA such as benzimidazoles,104 pentamidine,105 and 4′,6-diamidino-2-phenylindole (DAPI)106 was synthesized and screened for binding to various RNA libraries (Fig. 4).98 Three different RNA motif libraries (containing randomized 6-nucleotide hairpin loops, 3 × 2 nucleotide internal loops, and 4 × 4 nucleotide internal loops) were screened for binding to these RNA-focused small molecules using a fluorescent dye displacement assay. Of the 43 compounds, eight bound one or more of the RNA libraries, affording a hit rate of ∼19%. In contrast, the hit rate for the library of pharmacologically active compounds (LOPAC; designed for protein targets) was only ∼1%.98 Scaffold analysis also showed the importance of indole, 2-phenyl indole, 2-phenyl benzimidazole and pyridinium groups providing invaluable information for building RNA-focused libraries.98 Like the previous study described above, the preferred RNA motifs for hit compounds were determined by 2DCS.
These early attempts to generate RNA-focused libraries demonstrate the value of prior knowledge of RNA-binding chemotypes in their design. However, due to their strict reliance on incorporation of specific substructures, often in a specified arrangement, these early libraries contained minimal diversity and only a handful of novel chemotypes capable of binding RNA were identified. As more information on the types of small molecules that bind RNA is discovered, new and improved RNA-focused libraries will emerge.
2.4 Developing target-specific RNA-focused libraries via similarity searching
Target-specific or target-focused libraries are designed based on either the structural knowledge of a target or target family (structure-based) or the chemical features of small molecules (ligand-based).107–109 Such libraries focus on a subarea of chemical space which confers affinity and specificity toward a target/targets of interest. These libraries have been used extensively in protein-targeting approaches, leveraging the widely available structural information for some targets such as kinases.110–112 Structures of drug targets (especially ligand-bound structures) can also be used in a variety of in silico methods for the virtual screening of large libraries (106–109 compounds). Hits from virtual screening can then be used to prepare a focused library with a significantly smaller size, reducing time and cost of downstream experimental evaluation. This approach has been used successfully in multiple drug discovery projects.113,114In the absence of structural data, as is the case for nearly all RNA targets, ligand-based approaches can be used to create focused libraries. In this approach, the physicochemical properties of known active molecules can be used for a molecular similarity115–117 search campaign against screening libraries (commercial/non-commercial). The ligand-based approaches can leverage either one-dimensional or two-dimensional molecular descriptors,118 which encompass the chemical nature of the small molecules, or three-dimensional descriptors such as pharmacophore properties, shape, or volume.119–121
The accumulated knowledge from other studies122 was carried over to design the first RNA target-specific libraries generated by computational chemical similarity searching. Expanded RNA repeats are causative of microsatellite disorders,123 and expanded r(CUG) repeats [r(CUG)exp] in particular are the toxic agent in myotonic dystrophy type 1 (DM1).124 Small molecule targeting of this disease-relevant RNA has therapeutic potential.125 The 3D shapes of previously identified small molecules targeting this particular RNA105,126–128 were used for screening the National Cancer Institute's (NCI; 250000 compounds) and eMolecules databases (8000000).129 This virtual screening resulted in identifying a bis-benzimidazole scaffold when a Hoechst derivative was used as the query molecule, as it binds the RNA and displaces MBNL1 in vitro.129 The most potent derivative identified, H1, rescued DM1-associated splicing defects and foci formation in a DM1 cell culture model and rescued splicing defects in a DM1 mouse model, albeit with modest potency. This study was the first attempt to apply the ligand-based virtual screening in the creation of a target-specific RNA-focused library, resulting in the identification of lead molecules with improved potency compared to the query molecules.
The identification of bis-benzimidazole as a privileged scaffold for RNA129 initiated the creation of the second target-specific library using the concept of chemical similarity searching.130 A library of structurally diverse yet chemically similar compounds (n = 320) was generated by performing a chemical similarity search of The Scripps Research Institute's (TSRI) drug discovery collection, using H1 (above) as the query molecule. The library was screended for binding to r(CUG) repeats using a time-resolved fluorescence resonance energy transfer (TR-FRET) assay,131 yielding a hit rate of ∼9%. A subsequent substructure analysis showed the abundance of pyridyl, benzimidazole, or imidazole ring systems. Downstream analysis of the bioactive compounds (compounds that improved DM1-associated pre-mRNA alternative splicing defects) and extraction of the physicochemical properties showed the differences between these compounds and the starting library. Bioactive compounds had a larger topological polar surface area (TPSA) compared to the starting library (101 ± 33 Å2vs. 75 ± 25), as well as more hydrogen bond donors (3 ± 1 vs. 2 ± 1,) and acceptors (4 ± 1 vs. 3 ± 1). The majority of these compounds were benzimidazoles with a para-substituted phenyl ring at the 2-position.
2.5 Deriving features of RNA-binding small molecules from HTS of large, diverse libraries against multiple targets
While these early attempts demonstrated the success of pharmacophore-based and target-focused similarity approaches to generate RNA-focused libraries, the libraries oftentimes lacked structural diversity. RNA molecules can adopt a variety of secondary and tertiary structures, which can affect their function and interaction with other molecules. Therefore structural diversity in RNA-focused libraries ensures that a wide range of RNA structures may be targeted by the compounds, increasing the likelihood of identifying RNA-binding molecules. There was a significant need in the field to generate larger datasets to derive general features of RNA-binding small molecules. The primary way this was accomplished was by high throughput screening (HTS). HTS has served as the gold standard for the identification of chemical matter that can modulate a target's activity for the past two decades.132–136 The combination of hit identification through HTS and lead optimization with medicinal chemistry has provided several FDA-approved drugs. Maraviroc, an antiviral drug that inhibits HIV-1 entry into cells,137 resulted from an HTS campaign by Pfizer (a library of ∼500000 compounds) against the CC-chemokine receptor 5. Although the initial hit lacked antiviral activity, it provided the chemical matter for the structure–activity relationship and medicinal chemistry follow-ups, resulting in maraviroc.138 There are multiple examples of FDA-approved drugs confirming the usefulness of HTS.139–141
With the advent of combinatorial chemistry, the size of the screening libraries started to grow, and concepts such as drug-likeness or lead-like properties were later applied to improve the quality of such screening libraries.142 Aided by the development of more accurate and sophisticated cheminformatic tools, higher quality libraries were created by incorporating physicochemical property information such as TPSA and the octanol/water partition coefficient (logP).143 TPSA is a property that affects the ligand's ability to interact with the hydrophilic regions of RNA, for example its backbone or phosphate groups. RNA is often highly hydrated and polar, so ligands with a high enough TPSA may exhibit better water solubility, making them more bioavailable and effective in aqueous environments like the cytoplasm or nuclei. However, too much polar surface area could limit their ability to cross biological membranes, thus it is essential to balance TPSA with logP. While proteins often benefit from high hydrophobicity for membrane binding or intracellular trafficking, RNA-targeting molecules often require a balance of lipophilicity. RNA's negatively charged backbone makes it more hydrophilic, therefore RNA-binding ligands with moderate logP values are more likely to be both water-soluble and able to interact effectively with the RNA target (Table 1). Also, while proteins are highly structured, dynamic molecules with complex three-dimensional shapes, RNA is also structurally diverse but exhibits secondary and tertiary structures that are more flexible and can undergo dynamic conformational changes. This flexibility significantly impacts the chemical motifs used for binding.
| Physicochemical property | Description | Effect on RNA binding |
|---|---|---|
| Topological polar surface area (TPSA) | The surface area of polar atoms | Higher TPSA typically suggests improved aqueous solubility and interaction with the polar RNA backbone. A balance is needed, as a large TPSA may reduce membrane permeability. |
| Octanol–water partition coefficient (logP) | A measure of lipophilicity | A moderate logP is desirable for RNA-targeting ligands. Highly lipophilic compounds may struggle to interact with the negatively charged, hydrophilic RNA backbone, while very hydrophilic compounds may not penetrate cells effectively. |
| Molecular weight | The sum of the atomic weights of all the atoms in the molecule | Larger molecules might have better binding potential due to multiple interaction sites, but they might also face difficulty in membrane penetration. |
| Hydrogen bond donors (HBD) and acceptors (HBA) | Functional groups capable of forming hydrogen bonds | Hydrogen bond donors and acceptors are crucial for achieving selectivity and stability in RNA–ligand complexes, and they are essential for optimizing the efficacy of RNA-targeting small molecules. |
| Conformational flexibility | The ability of a ligand to adopt different conformations | RNA-binding ligands that are flexible may be able to adapt their structure to fit into different RNA target sites. However, flexibility needs to be balanced, as too much flexibility can reduce binding affinity. |
| Planarity | Aromatic rings present in the ligand | Aromatic rings can participate in π–π stacking interactions with RNA bases, a key interaction for small molecules targeting RNA. |
Despite the improvements in the quality and size of screening libraries, the currently available libraries cover a very small part of the drug-like chemical space, comprising an estimated 1030 compounds.144 Because no library can cover this immense druggable space, design of specialized libraries that incorporate both chemical diversity as well as drug-like physicochemical properties can improve the success of screening campaigns without the need for exceedingly large library sizes.
While there have been many HTS campaigns against RNA targets over the past ∼30 years, they primarily focused on single targets and drew few conclusions about general principles of RNA-targeting molecules. In recent years, there has been a more concerted effort to develop principles of RNA-targeting small molecules by screening diverse libraries against multiple RNA targets.
To address this need, Haniff et al. created an RNA-focused library by exploiting the chemical features found in a repository of RNA-binding small molecules found in the literature named Inforna.145 The physicochemical properties of these RNA binders were compared with those of commercially available compounds, affording an RNA-focused library with 3271 compounds. At least 20% of library members were chemically dissimilar from known RNA-binding small molecules. This resulting library was enriched with nitrogen-containing heterocyclic molecules such as phenyl-substituted thiazoles, benzimidazoles, indoles, and quinazolines. Despite the enrichment in these RNA-binding chemotypes, the physicochemical properties of the library more closely resembled those of drugs in DrugBank146 than compounds in the Inforna database. The library was screened against four different RNA targets composed of A–U or G–C base pairs in different arrangements using a fluorescent dye displacement assay. Six structurally distinct classes of small molecules were identified as base pair binders, ranging in affinity from high nanomolar to low micromolar. This study demonstrated that a diverse, drug-like library could successfully identify RNA-binding small molecules.
In a similar but expanded approach, another ∼2000 compound RNA-focused library was created by comparing features of the Inforna library to AstraZeneca's corporate collection (Fig. 5(A)).147 This library was screened against three RNA libraries comprising a total of 21504 unique RNA structures using 2DCS. In all, 27 compounds (1.4% hit rate) bound RNA and contained five key scaffolds: phenyl-bis-benzimidazoles, phenyl-benzimidazoles, 2-aminoquinazolines, 4,6-diaminopyrimidines, and 2-guanidino-3-methylthiazoles. By comparing the physicochemical properties of binders to non-binders, computational analysis revealed that the RNA binders were more lipophilic, had fewer rotatable bonds, more hydrogen bond donors, greater polar surface area, and fewer sp3 carbons. Though structurally distinct from them, the hit molecules had similar physicochemical properties as compounds in two other repositories of known small molecules that bind RNA.148,149
 | ||
| Fig. 5 Examples of the applicability of HTS to identify RNA-binding chemotypes against diverse RNA targets. (A) Haniff et al.145 utilized molecular descriptors of Inforna RNA binders to generate a ∼2000 compound library which was screened against >20000 RNA targets (1.4% hit rate). The hits were then used to generate a second-generation, 980 compound RNA-focused library via chemical similarity techniques. (B) Rizvi et al.150 first screened two non-RNA focused libraries (hit rates of 0.01% and 0.04%). Three models were utilized to generate a new RNA-focused library, which showed a markedly increased hit rate. | ||
In an alternative approach, an RNA-focused library was created by first screening two diverse, protein-targeting libraries (∼55000 compounds) against 42 different disease-relevant RNA targets with the affinity mass spectrometry method dubbed Automated Ligand Identification System (ALIS).151,152 ALIS employs sequential size exclusion and reverse-phase chromatography followed by mass spectrometry to identify RNA binders. Perhaps unsurprisingly, this initial screen had low hit rates for the RNA targets, 0.04% and 0.01% for the two libraries, much lower than the 1.5% and 0.05% hit rates observed for proteins screened against the same libraries (Fig. 5(B)).
By generating a machine learning (ML) model based upon calculated molecular fingerprints, chemical features that discriminated between RNA binders and non-binders were identified. The model was then used to select molecules from Merck's compound collection to create an RNA-focused library with 3700 compounds.150 This new RNA-focused library was re-screened against 32 of the 42 RNA targets and showed a markedly increased hit rate of 0.32%. Additionally, this increased hit rate was seen across most RNA targets (24 out of the 32 tested RNAs had a higher hit rate in the RNA-focused screen than the initial screen), suggesting that the new library contains features generally associated with RNA binding. Importantly, the screening hits identified were largely specific, with 66% of identified hits only binding one of the 42 RNA targets. Further, of the compounds that specifically recognized one RNA structure, nearly 58% did not bind proteins from Merck's prior in-house ALIS screens, suggesting these compounds preferentially bind specific RNA targets. PCA (principal component analysis) of physicochemical properties of RNA binders vs. protein binders revealed that although the chemical space of RNA-binding and protein-binding small molecules overlap largely within the drug-like space, the chemical motifs that contribute to RNA and protein binding are distinct. Examples of these discriminatory characteristics include aromatic amine-containing heterocycles and amidine-like motifs.
In another campaign against diverse RNA targets, a small molecule microarray library containing nearly 25000 compounds was screened for binding 27 RNAs (including G-quadruplexes, hairpins, pseudoknots, three-way junctions, and triple helices) and nine DNA targets, with several individual screens published previously.41,153 The RNA targets had hit rates between 0.48% (hairpins) and 0.85% (pseudoknots); when only considering selective small molecules, the hit rate ranged from 0.16% to 0.33%.154 Perhaps it is not surprising that pseudoknots, the most structurally complex of the targets, had the highest hit rate. The 2188 small molecules that bound at least one of these nucleic acid targets, of which 2003 bound one of the RNAs, were compiled in the Repository Of BInders to Nucleic acids (ROBIN) library.154 As will be discussed in more detail later, several machine learning models were developed and assessed to identify discriminatory features between RNA binders and non-binders, revealing that nitrogen content and aromaticity were important features.
Overall, primary screening of general-purpose screening libraries against RNA targets or target classes can be adopted as a strategy to identify the properties that discriminate RNA binders from non-binders or protein binders. These features can then be applied to create RNA-focused libraries that are enriched in properties that facilitate binding to RNA. In the following sections, alternative methods to identify RNA-binding small molecules, such as fragment screening, DNA-encoded libraries (DELs), and virtual screening, are discussed.
2.6 Design of RNA-focused fragment libraries
Fragment screening prioritizes up-front screening of small (<300 Da) fragment molecules to identify substructures that recognize the target of interest. Fragments capable of binding are then optimized by “growing” into larger molecules or “merging” with other identified fragments to generate higher affinity ligands.155 This approach has been successfully applied in the protein-targeting space, exemplified by the FDA-approved drugs vemurafenib156 and venetoclax.157While prior studies had utilized fragment screening methods against RNA, the first attempt to generate a library of RNA-focused fragments was in 2009. Bodoor et al. chose fragments by first collecting known RNA binders from literature, prioritizing compounds with Kd values less than 50 μM.158 In total, 120 molecules were identified, and analysis of the physicochemical properties showed the similarities between these molecules and kinase and protease binders. The overlap of the chemical space between known RNA binders and proteins such as kinases provides guidelines to discriminate RNA from protein binders.159 These 120 molecules were fragmented in silico to generate 250 fragments. These fragments were then clustered using their molecular fingerprints and chemical descriptors, and 102 commercially available fragments were chosen to represent each cluster. This library was then screened against the ribosomal decoding site (“A-Site”) RNA using different NMR experiments, which resulted in five hits, including two hit fragments chemically dissimilar from known A-site RNA binders. This study also showed that a detailed analysis of already known RNA binders is a logical starting point to generate a library of RNA-focused fragments.
The exploitation of the chemical features of known drugs to find similar molecules, named analogues, has been a successful approach.160–162 The chemical features of known RNA binders collected in Inforna were used to identify enriched scaffolds that afford RNA binding.163 Scaffold extraction showed high enrichment of triazole, thiazole, furan, and quinoline, along with other scaffolds. These scaffolds were then virtually screened against a library of 11788 small molecules, building an RNA-focused fragment library with 2500 compounds. The Rule of Three was generally followed in building the library, wherein fragments had a molecular weight (MW) below 300 Da and three or fewer hydrogen bond donors and acceptors.164 The library was also drug-like, with an average quantitative estimate of drug-likeness (QED)165 score of 0.75 ± 0.10. The RNA binding propensity of this library was assessed using 2DCS, where one fragment was identified to prefer an A-bulge found in the Dicer processing side of pre-miR-372 (KD = 300 ± 140 nM). Although this approach did not identify new scaffolds for RNA binding, it represents a successful workflow in design of RNA-focused fragment libraries.
Fragment-based hit discovery or lead optimization has several advantages regarding the ease of synthesis, characterization, and high success rate.162–167 This study showed that despite the differences between targeting proteins and RNA, many established approaches can be adapted for RNA targets.
2.7 DNA-encoded libraries (DELs)
DELs are a powerful tool in drug discovery, allowing screening of vast chemical libraries with high efficiency.168 DEL screening involves synthesizing large libraries of small molecules, where each building block is encoded by a unique DNA barcode, added orthogonally during library synthesis. This DNA barcoding allows for the rapid identification of hits using next-generation sequencing (NGS) techniques. Although DELs were not initially applied towards RNA targets for fear of its interaction with the DNA tags, advances in the field, including ensuring that the loading of the DNA tag comprises <1% of bead loading169 and “patches”,170 have enabled DEL screening for RNA.Two different approaches have been developed recently to address the concerns of the RNA target of interest interacting with DNA tags in DEL screens. In the first example, Benhamou et al.169 utilized a one-bead one-compound (OBOC) solid-phase DEL library, rather than the more traditional solution-phase DEL. This enabled sub-stoichiometric loading of DNA onto the bead, where the compound was in ∼250-fold excess relative to the DNA barcode, reducing the potential effect of RNA–DNA hybridization. The authors integrated 2DCS with this solid-phase DEL in a massively parallel screening pipeline to probe affinity landscapes between RNA folds and small molecules. Fluorescence-activated cell sorting (FACS) was used to identify the DEL beads which preferentially bind to a library of RNAs containing 3 × 3 nucleotide internal loops relative to a fully base-paired control RNA. Hit molecules from the DEL screen were then resynthesized and screened using 2DCS to identify which of the 4096 3 × 3 internal loops preferentially bind the small molecules. Though the library was synthesized using a racemic mixture of proline derivatives, resynthesis of hits as pure diastereomers revealed that some, but not all, hits preferred different RNA structures dependent upon stereochemical identity. These results suggested that future design of RNA-targeting DELs may benefit from the incorporation of compounds with defined stereochemistry to improve or alter specificity.
The Inforna platform was then utilized to identify disease-relevant pre-miRNAs that overlapped with the DEL selection, affording a nanomolar binder for the primary transcript of oncogenic miR-27a (pri-miR-27a). This study demonstrated the power of DEL screening in combination with selection-based methods to target RNA.
Shortly thereafter, Chen et al. utilized an alternative, solution-phase DEL approach that incorporated patches to reduce interactions between the RNA targets and the DNA tags.170 The authors noted significant extents of false positives when screening their ∼10 billion member DEL library against HIV TAR RNA using standard techniques. In their optimized method, non-specific RNA–DNA hybridization was essentially ablated upon utilization of a combination of pre-incubation with RNA “patches” containing the same sequences as the RNA target and competitive elution using known ligands that bind HIV TAR. The authors then used their approach to identify two new binders of the FMN riboswitch with affinity of <20 nM towards the target.170
While these initial RNA-targeting DELs demonstrated the utility of the approach towards RNAs, the libraries were not enriched in known RNA-binding substructures. Such a library was developed by enriching the DEL building blocks with RNA-focused motifs, including benzimidazoles, azaindoles, pyrazoles, and others.171 The 12672-member DEL library was then screened for binding specifically to r(CUG) repeats by co-incubation with a fully base-paired RNA. As expected, hit compounds shared commonalities with previously known RNA binders, for example the number of hydrogen bond acceptors and the number of tertiary amines, but also showed discriminatory properties such as a lack of positive charge. Subsequent studies for target engagement and bioactivity showed that one DEL compound improved aberrant alternative splicing associated with DM1. This study demonstrated that utilization of prior knowledge in the design of an RNA-focused DEL can identify bioactive RNA-targeting ligands from relatively small, focused libraries.
Overall, the examples discussed above delineate a general strategy in building an RNA-focused library: (i) screening of general-purpose libraries against a variety of RNA structures (or a specific class of RNAs) and then leveraging the obtained knowledge of RNA binders to create an RNA-focused library; or (ii) exploiting the chemical features of known RNA binders to create RNA-biased libraries to narrow down the very specific features that can discriminate an RNA binder from protein binders.
3. The future of RNA-focused libraries: methods to enable the identification and prediction of RNA-binding small molecules
3.1 Docking-based virtual screening to identify RNA-targeting small molecules
Molecular docking is one of the most widely used tools for structure-based drug design, and its success rate depends on the availability of high-resolution 3D structures.172,173 However, the higher conformational dynamics of RNAs compared to proteins has been a major hurdle in elucidating the 3D structure of RNA targets alone and in complex with small molecules.174 Although single-particle cryogenic electron microscopy (cryo-EM) has emerged as a powerful technique to provide 3D structures of RNAs,175 the field is significantly lagging behind its implementation for proteins.To overcome the lack of available 3D structures for RNA targets, methods for molecular docking for hit identification through virtual screening have been developed.176 For example, Shi et al. identified a potent inhibitor of miR-21 through virtual screening of 1990 compounds from the National Cancer Institute's (NCI) diversity dataset against a computationally predicted structure of miR-21's Dicer processing site (using MC-Fold/MC-Sym177).178 In another study, selective molecules targeting RNA tetraloops (arginine–RNA aptamer complex, a biotin–RNA pseudoknot complex, and a theophylline–RNA complex) were identified.179 After establishing that docking can recapitulate the experimentally determined pose of an aminoacridine derivative (AD1) bound to a hairpin, virtual screening identified AD2 with a binding specificity for tetraloops (Kd = ∼1 μM), as compared to double stranded RNA (Kd = ∼25 μM).
The same group applied this approach to identify small molecules targeting the GGAG tetraloop, a highly conserved stem-loop (SL-3) in the HIV-1 genome.180 Molecular docking of 1367 compounds identified two compounds (compounds 5 and 9) as selective binders of SL-3 RNA. The compounds showed noticeable specificity for tetraloops over double-stranded RNAs (∼3.5- and ∼6-fold, respectively) and single-stranded RNA (∼50- and ∼25-fold, respectively).180 It should be noted that docking was combined with a short (5 ns) molecular dynamics simulation to account for the flexibility of RNA, and the final hits were identified among the compounds that formed a stable complex with SL-3. Such an approach has also been successfully implemented for other viral targets including HIV-1 TAR,181 a pseudoknot present in the SARS-CoV genome,43 the HCV IRES subdomain IIa,182 and a cis-acting regulatory stem-loop RNA of hepatitis B virus (HBV).183 Nine of the molecules identified in this study bound RNA with micromolar affinity, most of which had not been shown previously to bind RNA, demonstrating that the docking methods can identify new RNA-binding ligands.
In a particularly interesting example, ∼100000 compounds were screened for binding HIV-1 TAR RNA using a Tat peptide displacement assay, affording seven hit molecules and, importantly, over 100000 non-hit molecules or “decoys”, both of which were employed in a subsequent virtual screening campaign.184 The training set for the virtual screen comprised these seven hits plus an additional 78 experimentally validated small molecules and ∼100000 decoys. Prior to virtual screening, this library of 100085 compounds was filtered to provide two distinct libraries: (i) in one library, the molecules with outlier physicochemical properties were removed; and (ii) in the other, the DUD-E protocol was used to select a subset of both the hits and property-matched non-hits. Virtual screening of these libraries against an ensemble of 20 molecular dynamics-generated HIV-1 TAR RNA structures demonstrated enrichment of the true, experimentally validated hits among the virtual screening hits. Importantly, screening against the full ensemble of 20 RNA structures was key, as screening against fewer structures resulted in the identification of fewer hits. This study showed that including experimental data to refine virtual screening efforts can significantly improve the identification of RNA-binding chemical matter.
Overall, these attempts showcased the applicability and usefulness of virtual screening in hit identification targeting different RNAs. Of particular promise are the ensemble-based approaches, which may better account for the inherent flexibility of RNA targets which is otherwise difficult to account for in high-throughput virtual screening campaigns. Although no follow-up analyses were performed to extract the chemical features of virtual screening hits compared to non-hits, the lead small molecules could be used to design target-specific libraries in future studies. Molecular docking, therefore, may provide a means to generate libraries enriched in RNA-binding compounds, although subsequent experimental validation is always required.
3.2 Machine learning (ML) to optimize RNA-binding small molecules
ML methods depend on large amounts of data for an efficient learning process. The accumulated data for either RNA structures alone or RNA–small molecule complexes during the past decades have provided the training resources for ML-based methods development. These methods have been applied for RNA structure prediction (secondary or tertiary), prediction of RNA–small molecule interactions, or identification of the binding pocket. One of the first applications of ML methods in the RNA field was the prediction of secondary structures.185 Later on, deep learning methods were introduced and their use in secondary structure predictions showed better performance.186,187 These methods have also been used in tertiary structure predictions, a significant challenge due to conformational flexibility of RNA.188,189 In particular, geometric deep learning methods have been successful in blind prediction of tertiary structures.190,191One of the challenges in using molecular docking is the inaccuracy of docking poses.192,193 ML-based methods can be trained even on the limited available data that describes RNA–small molecule complexes and can help to separate accurate vs. non-accurate poses. In one of the first examples, it was shown that using the random forest classifier in RNAPosers could separate the accurate RNA–small molecules poses from decoys.194 AnnapuRNA is another example that uses supervised ML models, K Nearest Neighbors (a simple, yet powerful, supervised machine learning algorithm used for both classification and regression tasks), and multi-layer feedforward artificial neural network, achieving high accuracy in prediction of bound poses of small molecules.195 These augmentation approaches can be combined with conventional molecular docking to increase the efficiency of hit identification.
As one of the only attempts to use primitive machine learning models in the evaluation of RNA-focused libraries, Yazdani et al. determined features that differentiate RNA binders from non-binders incorporated in the ROBIN library by using a class-weighted logistic least absolute shrinkage and selection operator (LASSO) regression model.154 To generate a model that can separate RNA binders from non-binders, 1664 molecular descriptors were calculated using Mordred196 (an open-source software tool designed to calculate molecular descriptors), and these features were used to train the LASSO regression model. The analysis found that features related to nitrogen content and aromaticity favor RNA binding. The performance of the LASSO model was not ideal (AUPRC score of 0.37, where AUPRC stands for Area Under the Precision–Recall Curve, a performance metric often used in evaluating binary classification models, especially for imbalanced datasets where the focus is on the minority class), as expected due to its several disadvantages, particularly when dealing with large datasets, high-dimensional data, or outliers. The computational cost, sensitivity to feature scaling, and difficulty with imbalanced data also make it less suitable for complex or large-scale problems without careful tuning and pre-processing. However, the authors showed that application of more advanced techniques like feedforward neural networks can significantly improve the model performance (AUPRC score of 0.78).154
The main power of ML methods is their predictive capabilities, and they can be trained on almost any class of data, not solely structural data, particularly important due to the scarcity of available RNA–small molecule complexes. RNA 3D structure information, however, has also been employed in building predictive models to identify new small molecules.197,198 ML-based methods have also been used for the de novo generation of small molecules by exploiting the chemical features that drive specificity toward RNA. Such approaches can significantly decrease the search of the chemical space to identify target-specific hit molecules.199 Several ML-based models have been developed around miRNAs to identify novel small molecules.200–202
Overall, ML approaches can make important contributions in the RNA therapeutics field, from predicting 3D structures of RNA molecules to small molecule design and lead optimization. Although none of these methods has been used to create an RNA-focused library with experimental validation, we anticipate such a library will be forthcoming. As each method depends on large amounts of data for training purposes, the quality of predictions will improve as more data becomes available.
4. Other considerations & conclusions
The accumulated knowledge gathered for targeting RNA with small molecules has helped to understand some of the driving factors in binding specificity and target selection. RNA-focused libraries are now available from commercial sources for HTS efforts. Notably, these libraries remain limited in the chemical space they cover and hence are of insufficient diversity at present.Despite the tremendous progress made in the RNA-targeted small molecule field, several factors have hampered its advancement. Assay development for small molecule screening has been mainly focused on proteins and extensively optimized over the years. For example, SPR (surface plasmon resonance), which is now a standard technique for screening small molecules and extracting kinetic data for proteins, still faces several challenges when applied to RNA. The different kinetic behavior of RNA and in some cases significant conformational changes after binding to the small molecule and lower affinity poses several challenges ranging from immobilization and mass transport203 to data analysis.203,204
Thermodynamic data has been essential for lead optimization during the drug discovery process.205 It has been shown that like proteins, enthalpically driven binding is more favorable for RNA-targeting small molecules than entropically driven ones.206–210 Moreover, idiosyncrasies have been observed within an RNA-binding chemotype. For example, electrostatic interactions play an important role in the binding of aminoglycosides to RNA, contributing >50% of the total free energy of binding.211 The same analysis for deoxystreptamine dimers binding to RNA hairpin loops revealed only ∼20% of the total free energy of binding is due to electrostatic interactions, and in contrast to aminoglycosides (enthalpic), binding of dimers is an entropically driven interaction.212 There is a dearth of these types of thermodynamic measurements, which are especially challenging when characterizing RNA–small molecule interactions. These difficulties arise due to weak interactions of initial hits which necessitates using of high concentrations of small molecules to detect binding, where aggregation may also occur; likewise achieving such concentrations may require higher amounts of DMSO that affects RNA stability/structure.213 Nonetheless, elucidation of these forces can help in the design of RNA-focused libraries and to discriminate RNA-binding small molecules from protein binders.
Despite these challenges, significant progress has been made to identify small molecules that target different classes of RNA, as manifested in publicly available databases such SM2miR214 (a database of the experimentally validated small molecules that affect microRNA expression), R-SIM215 (a database for binding affinities for RNA–small molecule interactions), R-BIND216 (Database of Bioactive RNA-Targeting Small Molecules and Associated RNA Secondary Structures), Inforna217 (a database of experimentally determined RNA–small molecule interactions that enables sequence-based design), NoncoRNA218 (a database of experimentally supported non-coding RNAs and drug targets in cancer), ROBIN,154 and RNAmigos219 (a combination of machine learning and molecular docking to identify RNA targeting small molecules).
The future of RNA-targeted small molecule libraries is rapidly evolving, fueled by advances in drug discovery, high-throughput screening, AI-driven design, and structural biology. The current developments in RNA 3D structure determination at high resolutions has become increasingly promising which may help overcome hurdles in structure-based drug design toward RNA targets.220
As researchers continue to recognize the therapeutic potential of RNA molecules in treating diseases, including cancer, viral infections, and neurodegenerative disorders, RNA-targeted small molecule libraries are becoming crucial resources for identifying effective modulators. Developing libraries with RNA-biased scaffolds and functional groups that specifically favor interactions with RNA structures could improve hit rates in RNA-targeted screening, and AI can optimize RNA-targeted libraries by predicting binding affinity, selectivity, and even RNA-binding motifs, enabling the design of novel small molecules with high RNA specificity. For example, by using generative models like Variational Autoencoders (VAEs)221 and Generative Adversarial Networks (GANs),222 new small molecules tailored to bind specific RNA motifs or structures can be designed. This approach allows the exploration of novel chemistries outside conventional chemical spaces.
Considering RNA structure and dynamics for therapeutic applications, conformation adopted by an RNA in vivo may be very different than the folds adopted in vitro. RNA molecules in the cell are typically involved in dynamic processes such as transcription, splicing, translation, and interaction with RNA-binding proteins (RBPs). These interactions cause the RNA to adopt a variety of conformational states throughout its life cycle. In vitro experiments, however, often employ isolated RNAs or fragments of a transcript that are possibly in a more static, simplified state. The conformations and dynamics in vivo may be far more complex, with RNA undergoing multistate folding, binding events, or undergoing conformational transitions triggered by specific cellular factors. Therefore, for therapeutic applications, it is crucial that small molecules designed to interact with RNA can bind and modulate RNA conformations that are relevant to its function in the natural cellular environment, not just in its static in vitro state.223,224
The future of RNA-targeted small molecule libraries will be defined by greater specificity, structural diversity, and an enhanced ability to interact with complex RNA structures and RNA–protein complexes. Leveraging AI-driven molecular design, 3D structural data, and high-throughput RNA-specific screening technologies will be key to accelerating discovery. These advancements could open doors to novel RNA-targeted therapeutics for a wide array of diseases, including cancers, viral infections, and rare genetic disorders.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
M. D. D. is a founder of Expansion Therapeutics. M. D. D. and J. L. C. are founders of Ribonaut Therapeutics.Acknowledgements
This work was funded by the National Institutes of Health (R35 NS116846 and R01 CA249180 to M. D.D.), the Department of Defense (HT94252310336 to M. D. D.), the Florida Department of Health (Florida Cancer Innovation Fund grant #MOABO to J. L. C.), the Muscular Dystrophy Association (grant ID #1069959 to M. D. D.; Development grant ID #963835 to A. T.), and the German Research Foundation (DFG) through a Walter Benjamin Programme postdoctoral fellowship (grant ID #515396515 to P.R.A.Z). This material is based upon work supported by the National Science Foundation Graduate Research Fellowship under grant no. 2235200 (to N. A. S.). Any opinion, findings, and conclusions or recommendations expressed in this material are those of the authors(s) and do not necessarily reflect the views of the National Science Foundation. Figures created in Biorender https://BioRender.com.References
- T. A. Cooper, L. Wan and G. Dreyfuss, Cell, 2009, 136, 777–793 CrossRef CAS PubMed.
- L. P. Ranum and J. W. Day, Trends Genet., 2004, 20, 506–512 CrossRef CAS PubMed.
- Y. Yuan, S. A. Compton, K. Sobczak, M. G. Stenberg, C. A. Thornton, J. D. Griffith and M. S. Swanson, Nucleic Acids Res., 2007, 35, 5474–5486 CrossRef CAS PubMed.
- M. Wojciechowska and W. J. Krzyzosiak, Hum. Mol. Genet., 2011, 20, 3811–3821 CrossRef CAS PubMed.
- S. Lefebvre, L. Bürglen, S. Reboullet, O. Clermont, P. Burlet, L. Viollet, B. Benichou, C. Cruaud, P. Millasseau and M. Zeviani, Cell, 1995, 80, 155–165 CrossRef CAS PubMed.
- S. Toden, T. J. Zumwalt and A. Goel, Biochim. Biophys. Acta, Rev. Cancer, 2021, 1875, 188491 CrossRef CAS PubMed.
- E. J. Wild and S. J. Tabrizi, Lancet Neurol., 2017, 16, 837–847 CrossRef CAS PubMed.
- R. J. Osborne and C. A. Thornton, Hum. Mol. Genet., 2006, 15, R162–R169 CrossRef CAS PubMed.
- T. R. Damase, R. Sukhovershin, C. Boada, F. Taraballi, R. I. Pettigrew and J. P. Cooke, Front. Bioeng. Biotechnol., 2021, 9, 628137 CrossRef PubMed.
- F. Wang, T. Zuroske and J. K. Watts, Nat. Rev. Drug Discovery, 2020, 19, 441–442 CrossRef CAS PubMed.
- R. Kole, A. R. Krainer and S. Altman, Nat. Rev. Drug Discovery, 2012, 11, 125–140 CrossRef CAS PubMed.
- R. Rademakers, M. Cruts and C. Van Broeckhoven, Hum. Mutat., 2004, 24, 277–295 CrossRef CAS PubMed.
- M. M. Scotti and M. S. Swanson, Nat. Rev. Genet., 2016, 17, 19–32 CrossRef CAS PubMed.
- D. D. Vo, T. P. Tran, C. Staedel, R. Benhida, F. Darfeuille, A. Di Giorgio and M. Duca, Chemistry, 2016, 22, 5350–5362 CrossRef CAS PubMed.
- A. Esquela-Kerscher and F. J. Slack, Nat. Rev. Cancer, 2006, 6, 259–269 CrossRef CAS PubMed.
- A. A. Komar and M. Hatzoglou, Front. Oncol., 2015, 5, 233 Search PubMed.
- R. Marques, R. Lacerda and L. Romão, Biomedicines, 2022, 10, 1865 CrossRef CAS PubMed.
- L. P. Ranum and T. A. Cooper, Annu. Rev. Neurosci., 2006, 29, 259–277 CrossRef CAS PubMed.
- M. L. Stephenson and P. C. Zamecnik, Proc. Natl. Acad. Sci. U. S. A., 1978, 75, 285–288 CrossRef CAS PubMed.
- A. A. Levin, N. Engl. J. Med., 2019, 380, 57–70 CrossRef PubMed.
- M. A. Havens, D. M. Duelli and M. L. Hastings, Wiley Interdiscip. Rev.: RNA, 2013, 4, 247–266 CrossRef CAS PubMed.
- D. H. Mathews, M. E. Burkard, S. M. Freier, J. R. Wyatt and D. H. Turner, RNA, 1999, 5, 1458–1469 CrossRef CAS PubMed.
- M. Menzi, B. Wild, U. Pradère, A. L. Malinowska, A. Brunschweiger, H. L. Lightfoot and J. Hall, Chem. Eur. J., 2017, 23, 14221–14230 CrossRef CAS PubMed.
- B. Roehr, J. Int. Assoc. Physicians AIDS Care, 1998, 4, 14–16 CAS.
- J. R. Mendell, L. R. Rodino-Klapac, Z. Sahenk, K. Roush, L. Bird, L. P. Lowes, L. Alfano, A. M. Gomez, S. Lewis and J. Kota, Ann. Neurol., 2013, 74, 637–647 CrossRef CAS PubMed.
- E. W. Ottesen, Transl. Neurosci., 2017, 8, 1–6 CrossRef CAS PubMed.
- M. D. Benson, M. Waddington-Cruz, J. L. Berk, M. Polydefkis, P. J. Dyck, A. K. Wang, V. Planté-Bordeneuve, F. A. Barroso, G. Merlini and L. Obici, N. Engl. J. Med., 2018, 379, 22–31 CrossRef CAS PubMed.
- P. Y. Ho and A. M. Yu, Wiley Interdiscip. Rev.: RNA, 2016, 7, 186–197 CrossRef CAS PubMed.
- A. Khvorova and J. K. Watts, Nat. Biotechnol., 2017, 35, 238–248 CrossRef CAS PubMed.
- R. L. Juliano, Nucleic Acids Res., 2016, 44, 6518–6548 CrossRef PubMed.
- E. Linnane, P. Davey, P. Zhang, S. Puri, M. Edbrooke, E. Chiarparin, A. S. Revenko, A. R. Macleod, J. C. Norman and S. J. Ross, Nucleic Acids Res., 2019, 47, 4375–4392 CrossRef CAS PubMed.
- O. S. Thomas and W. Weber, Front. Bioeng. Biotechnol., 2019, 7, 415 CrossRef PubMed.
- S. A. Burel, C. E. Hart, P. Cauntay, J. Hsiao, T. Machemer, M. Katz, A. Watt, H.-H. Bui, H. Younis and M. Sabripour, Nucleic Acids Res., 2016, 44, 2093–2109 CrossRef CAS PubMed.
- E. E. Swayze, A. M. Siwkowski, E. V. Wancewicz, M. T. Migawa, T. K. Wyrzykiewicz, G. Hung, B. P. Monia and C. F. Bennett, Nucleic Acids Res., 2007, 35, 687–700 CrossRef CAS PubMed.
- M. Gagliardi and A. T. Ashizawa, Biomedicines, 2021, 9, 433 CrossRef CAS PubMed.
- W. Yin and M. Rogge, Clin. Transl. Sci., 2019, 12, 98–112 CrossRef CAS PubMed.
- S. E. Butcher and A. M. Pyle, Acc. Chem. Res., 2011, 44, 1302–1311 CrossRef CAS PubMed.
- C. P. Jones and A. R. Ferré-D’Amaré, Trends Biochem. Sci., 2015, 40, 211–220 CrossRef CAS PubMed.
- I. Tinoco Jr and C. Bustamante, J. Mol. Biol., 1999, 293, 271–281 CrossRef CAS PubMed.
- P. Brion and E. Westhof, Annu. Rev. Biophys. Biomol. Struct., 1997, 26, 113–137 CrossRef CAS PubMed.
- F. A. Abulwerdi, W. Xu, A. A. Ageeli, M. J. Yonkunas, G. Arun, H. Nam, J. S. Schneekloth Jr, T. K. Dayie, D. Spector and N. Baird, ACS Chem. Biol., 2019, 14, 223–235 CrossRef CAS PubMed.
- J. D. Dinman, M. J. Ruiz-Echevarria and S. W. Peltz, Trends Biotechnol., 1998, 16, 190–196 CrossRef CAS PubMed.
- S.-J. Park, Y.-G. Kim and H.-J. Park, J. Am. Chem. Soc., 2011, 133, 10094–10100 CrossRef CAS PubMed.
- M. Garavís, B. López-Méndez, A. Somoza, J. Oyarzabal, C. Dalvit, A. Villasante, R. Campos-Olivas and C. González, ACS Chem. Biol., 2014, 9, 1559–1566 CrossRef PubMed.
- K. Furukawa, H. Gu, N. Sudarsan, Y. Hayakawa, M. Hyodo and R. R. Breaker, ACS Chem. Biol., 2012, 7, 1436–1443 CrossRef CAS PubMed.
- K. T. Samarasinghe and C. M. Crews, Cell Chem. Biol., 2021, 28, 934–951 CrossRef CAS PubMed.
- X. Xie, T. Yu, X. Li, N. Zhang, L. J. Foster, C. Peng, W. Huang and G. He, Signal Transduct. Target. Ther., 2023, 8, 335 CrossRef PubMed.
- J. Lim, S.-Y. Hwang, S. Moon, S. Kim and W. Y. Kim, Chem. Sci., 2020, 11, 1153–1164 RSC.
- B. Weisblum and J. Davies, Bacteriol. Rev., 1968, 32, 493–528 CrossRef CAS PubMed.
- M. I. Hutchings, A. W. Truman and B. Wilkinson, Curr. Opin. Microbiol., 2019, 51, 72–80 CrossRef CAS PubMed.
- J. Poehlsgaard and S. Douthwaite, Nat. Rev. Microbiol., 2005, 3, 870–881 CrossRef CAS PubMed.
- D. Clemett and A. Markham, Drugs, 2000, 59, 815–827 CrossRef CAS PubMed.
- H. Ratni, M. Ebeling, J. Baird, S. Bendels, J. Bylund, K. S. Chen, N. Denk, Z. Feng, L. Green and M. Guerard, J Med. Chem., 2018, 61(15), 6501–6517 CrossRef CAS PubMed.
- S. M. R. Hashemian, T. Farhadi and M. Ganjparvar, Drug Des., Dev. Ther., 2018, 1759–1767 CrossRef CAS PubMed.
- N. A. Naryshkin, M. Weetall, A. Dakka, J. Narasimhan, X. Zhao, Z. Feng, K. K. Ling, G. M. Karp, H. Qi and M. G. Woll, Science, 2014, 345, 688–693 CrossRef CAS PubMed.
- S. Campagne, S. Boigner, S. Rüdisser, A. Moursy, L. Gillioz, A. Knörlein, J. Hall, H. Ratni, A. Cléry and F. H.-T. Allain, Nat. Chem. Biol., 2019, 15, 1191–1198 CrossRef CAS PubMed.
- F. Malard, A. C. Wolter, J. Marquevielle, E. Morvan, A. Ecoutin, S. H. Rüdisser, F. H. Allain and S. Campagne, Nucleic Acids Res., 2024, 52, 4124–4136 CrossRef CAS PubMed.
- Z. Tang, S. Akhter, A. Ramprasad, X. Wang, M. Reibarkh, J. Wang, S. Aryal, S. S. Thota, J. Zhao and J. T. Douglas, Nucleic Acids Res., 2021, 49, 7870–7883 CrossRef CAS PubMed.
- E. W. Ottesen, N. N. Singh, D. Luo, B. Kaas, B. J. Gillette, J. Seo, H. J. Jorgensen and R. N. Singh, Nucleic Acids Res., 2023, 51, 5948–5980 CrossRef CAS PubMed.
- Y. Ishigami, M. S. Wong, C. Martí-Gómez, A. Ayaz, M. Kooshkbaghi, S. M. Hanson, D. M. McCandlish, A. R. Krainer and J. B. Kinney, Nat. Commun., 2024, 15, 1880 CrossRef CAS PubMed.
- H. M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, I. N. Shindyalov and P. E. Bourne, Nucleic Acids Res., 2000, 28, 235–242 CrossRef CAS PubMed.
- F. P. Panei, R. Torchet, H. Menager, P. Gkeka and M. Bonomi, Bioinformatics, 2022, 38, 4185–4193 CrossRef CAS PubMed.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS PubMed.
- M. L. Kelly, C.-C. Chu, H. Shi, L. R. Ganser, H. P. Bogerd, K. Huynh, Y. Hou, B. R. Cullen and H. M. Al-Hashimi, RNA, 2021, 27, 12–26 CrossRef CAS PubMed.
- G. G. Jayaraj, S. Ramasamy, D. Bose, H. Suryawanshi, M. Lalwani, S. Sivasubbu and S. Maiti, bioRxiv, 2017, preprint, 137935 DOI:10.1101/137935.
- S. Hong, K. A. Harris, K. D. Fanning, K. L. Sarachan, K. M. Frohlich and P. F. Agris, J. Biol. Chem., 2015, 290, 19273–19286 CrossRef CAS PubMed.
- C. Zimmer and U. Wähnert, Prog. Biophys. Mol. Biol., 1986, 47, 31–112 CrossRef CAS PubMed.
- M. Zhao, L. Ratmeyer, R. G. Peloquin, S. Yao, A. Kumar, J. Spychala, D. W. Boykin and W. D. Wilson, Biorg. Med. Chem., 1995, 3, 785–794 CrossRef CAS PubMed.
- G. Padroni, N. Patwardhan, M. Schapira and A. Hargrove, RSC Med. Chem., 2020, 11, 802–813 RSC.
- G. W. Collie, S. Sparapani, G. N. Parkinson and S. Neidle, J. Am. Chem. Soc., 2011, 133, 2721–2728 CrossRef CAS PubMed.
- M. Krishnamurthy, K. Simon, A. M. Orendt and P. A. Beal, Angew. Chem., Int. Ed., 2007, 46, 7044–7047 CrossRef CAS PubMed.
- S. P. Velagapudi, S. M. Gallo and M. D. Disney, Nat. Chem. Biol., 2014, 10, 291–297 CrossRef CAS PubMed.
- S. G. Rzuczek, L. A. Colgan, Y. Nakai, M. D. Cameron, D. Furling, R. Yasuda and M. D. Disney, Nat. Chem. Biol., 2017, 13, 188–193 CrossRef CAS PubMed.
- M. Sands, M. A. Kron and R. B. Brown, Rev. Infect. Dis., 1985, 7, 625–6344 CrossRef CAS PubMed.
- F. Doua, T. Miezan, S. Singaro Jr and T. Baltz, Am. J. Trop. Med. Hyg., 1996, 55, 586–588 CAS.
- L. A. Coonrod, M. Nakamori, W. Wang, S. Carrell, C. L. Hilton, M. J. Bodner, R. B. Siboni, A. G. Docter, M. M. Haley and C. A. Thornton, ACS Chem. Biol., 2013, 8, 2528–2537 CrossRef CAS PubMed.
- T. Hermann, Biochimie, 2002, 84, 869–875 CrossRef CAS PubMed.
- L. R. Ganser, M. L. Kelly, D. Herschlag and H. M. Al-Hashimi, Nat. Rev. Mol. Cell Biol., 2019, 20, 474–489 CrossRef CAS PubMed.
- D. Herschlag, S. Bonilla and N. Bisaria, Cold Spring Harbor Perspect. Biol., 2018, 10, a032433 CrossRef CAS PubMed.
- A. Ke and J. A. Doudna, Methods, 2004, 34, 408–414 CrossRef CAS PubMed.
- P. Dallaire, H. Tan, K. Szulwach, C. Ma, P. Jin and F. Major, Nucleic Acids Res., 2016, 44, 9956–9964 Search PubMed.
- L. R. Ganser, M. L. Kelly, N. N. Patwardhan, A. E. Hargrove and H. M. Al-Hashimi, J. Mol. Biol., 2020, 432, 1297–1304 CrossRef CAS PubMed.
- A. Ursu, J. T. Baisden, J. A. Bush, A. Taghavi, S. Choudhary, Y.-J. Zhang, T. F. Gendron, L. Petrucelli, I. Yildirim and M. D. Disney, ACS Chem. Neurosci., 2021, 12, 4076–4089 CrossRef CAS PubMed.
- A. Maity, F. R. Winnerdy, G. Chen and A. T. Phan, Biochemistry, 2021, 60, 1097–1107 CrossRef CAS PubMed.
- K. Mulholland, H.-J. Sullivan, J. Garner, J. Cai, B. Chen and C. Wu, ACS Chem. Neurosci., 2019, 11, 57–75 CrossRef PubMed.
- X. Wang, K. J. Goodrich, E. G. Conlon, J. Gao, A. H. Erbse, J. L. Manley and T. R. Cech, RNA, 2019, 25, 935–947 CrossRef CAS PubMed.
- A. Taghavi, J. T. Baisden, J. L. Childs-Disney, I. Yildirim and M. D. Disney, Nucleic Acids Res., 2023, 51, 5325–5340 CrossRef CAS PubMed.
- N. I. Orlovsky, H. M. Al-Hashimi and T. G. Oas, J. Am. Chem. Soc., 2019, 142, 907–921 CrossRef PubMed.
- A. Nicholls, G. B. McGaughey, R. P. Sheridan, A. C. Good, G. Warren, M. Mathieu, S. W. Muchmore, S. P. Brown, J. A. Grant and J. A. Haigh, J. Med. Chem., 2010, 53, 3862–3886 CrossRef CAS PubMed.
- J. A. Grant, M. A. Gallardo and B. T. Pickup, J. Comput. Chem., 1996, 17, 1653–1666 CrossRef CAS.
- G. M. Sastry, S. L. Dixon and W. Sherman, J. Chem. Inf. Model., 2011, 51, 2455–2466 CrossRef CAS PubMed.
- E. Kangas and B. Tidor, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 1999, 59, 5958 CrossRef CAS PubMed.
- T. A. Fritz, D. Tondi, J. S. Finer-Moore, M. P. Costi and R. M. Stroud, Chem. Biol., 2001, 8, 981–995 CrossRef CAS PubMed.
- P. Cozzini, G. E. Kellogg, F. Spyrakis, D. J. Abraham, G. Costantino, A. Emerson, F. Fanelli, H. Gohlke, L. A. Kuhn and G. M. Morris, J. Med. Chem., 2008, 51, 6237–6255 CrossRef CAS PubMed.
- J. R. Thomas and P. J. Hergenrother, Chem. Rev., 2008, 108, 1171–1224 CrossRef CAS PubMed.
- Y. Zhou, V. E. Gregor, B. K. Ayida, G. C. Winters, Z. Sun, D. Murphy, G. Haley, D. Bailey, J. M. Froelich and S. Fish, Bioorg. Med. Chem. Lett., 2007, 17, 1206–1210 CrossRef CAS PubMed.
- L. P. Labuda, A. Pushechnikov and M. D. Disney, ACS Chem. Biol., 2009, 4, 299–307 CrossRef CAS PubMed.
- T. Tran and M. D. Disney, Nat. Commun., 2012, 3, 1125 CrossRef PubMed.
- E. Davis and H. Rozov, Practitioner, 1975, 215, 793–798 CAS.
- Y. Zhang and M. J. Leibowitz, Nucleic Acids Res., 2001, 29, 2644–2653 CrossRef CAS PubMed.
- L. Yu, T. K. Oost, J. M. Schkeryantz, J. Yang, D. Janowick and S. W. Fesik, J. Am. Chem. Soc., 2003, 125, 4444–4450 CrossRef CAS PubMed.
- P. P. Seth, A. Miyaji, E. A. Jefferson, K. A. Sannes-Lowery, S. A. Osgood, S. S. Propp, R. Ranken, C. Massire, R. Sampath, D. J. Ecker, E. E. Swayze and R. H. Griffey, J. Med. Chem., 2005, 48, 7099–7102 CrossRef CAS PubMed.
- S. P. Velagapudi, A. Pushechnikov, L. P. Labuda, J. M. French and M. D. Disney, ACS Chem. Biol., 2012, 7, 1902–1909 CrossRef PubMed.
- Y. He, J. Yang, B. Wu, D. Robinson, K. Sprankle, P.-P. Kung, K. Lowery, V. Mohan, S. Hofstadler and E. E. Swayze, Bioorg. Med. Chem. Lett., 2004, 14, 695–699 CrossRef CAS PubMed.
- M. B. Warf, M. Nakamori, C. M. Matthys, C. A. Thornton and J. A. Berglund, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 18551–18556 CrossRef CAS PubMed.
- M. D. Disney, J. L. Childs and D. H. Turner, ChemBioChem, 2004, 5, 1647–1652 CrossRef CAS PubMed.
- M. Feher, Y. Gao, J. C. Baber, W. A. Shirley and J. Saunders, Biorg. Med. Chem., 2008, 16, 422–427 CrossRef CAS PubMed.
- J. L. Jenkins, M. Glick and J. W. Davies, J. Med. Chem., 2004, 47, 6144–6159 CrossRef CAS PubMed.
- C. John Harris, R. D. Hill, D. W. Sheppard, M. J. Slater and P. F. W. Stouten, Comb. Chem. High Throughput Screen., 2011, 14, 521–531 CrossRef PubMed.
- Y. Liu and N. S. Gray, Nat. Chem. Biol., 2006, 2, 358–364 CrossRef CAS PubMed.
- R. DeSimone, K. Currie, S. Mitchell, J. Darrow and D. Pippin, Comb. Chem. High Throughput Screen., 2004, 7, 473–493 CrossRef CAS PubMed.
- V. Pogacic, A. N. Bullock, O. Fedorov, P. Filippakopoulos, C. Gasser, A. Biondi, S. Meyer-Monard, S. Knapp and J. Schwaller, Cancer Res., 2007, 67, 6916–6924 CrossRef CAS PubMed.
- M. H. Howard, T. Cenizal, S. Gutteridge, W. S. Hanna, Y. Tao, M. Totrov, V. A. Wittenbach and Y.-J. Zheng, J. Med. Chem., 2004, 47, 6669–6672 CrossRef CAS PubMed.
- C. N. Cavasotto, M. A. Ortiz, R. A. Abagyan and F. J. Piedrafita, Bioorg. Med. Chem. Lett., 2006, 16, 1969–1974 CrossRef CAS PubMed.
- J.-L. Reymond, Chimia, 2022, 76, 1045–1051 CrossRef CAS PubMed.
- G. Maggiora, M. Vogt, D. Stumpfe and J. Bajorath, J. Med. Chem., 2014, 57, 3186–3204 CrossRef CAS PubMed.
- A. Bender and R. C. Glen, Org. Biomol. Chem., 2004, 2, 3204–3218 RSC.
- A. M. Helguera, R. D. Combes, M. P. Gonzalez and M. Cordeiro, Curr. Top. Med. Chem., 2008, 8, 1628–1655 CrossRef CAS PubMed.
- D. C. Kombo, K. Tallapragada, R. Jain, J. Chewning, A. A. Mazurov, J. D. Speake, T. A. Hauser and S. Toler, J. Chem. Inf. Model., 2013, 53, 327–342 CrossRef CAS PubMed.
- S. Mapari and K. V. Camarda, Curr. Opin. Chem. Eng., 2020, 27, 60–64 CrossRef.
- J. Meyers, M. Carter, N. Y. Mok and N. Brown, Future Med. Chem., 2016, 8, 1753–1767 CrossRef PubMed.
- M. D. Disney, L. P. Labuda, D. J. Paul, S. G. Poplawski, A. Pushechnikov, T. Tran, S. P. Velagapudi, M. Wu and J. L. Childs-Disney, J. Am. Chem. Soc., 2008, 130, 11185–11194 CrossRef CAS PubMed.
- I. Malik, C. P. Kelley, E. T. Wang and P. K. Todd, Nat. Rev. Mol. Cell Biol., 2021, 22, 589–607 CrossRef CAS PubMed.
- A. J. Angelbello, S. G. Rzuczek, K. K. Mckee, J. L. Chen, H. Olafson, M. D. Cameron, W. N. Moss, E. T. Wang and M. D. Disney, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 7799–7804 CrossRef CAS PubMed.
- T. M. Wheeler, K. Sobczak, J. D. Lueck, R. J. Osborne, X. Lin, R. T. Dirksen and C. A. Thornton, Science, 2009, 325, 336–339 CrossRef CAS PubMed.
- A. Pushechnikov, M. M. Lee, J. L. Childs-Disney, K. Sobczak, J. M. French, C. A. Thornton and M. D. Disney, J. Am. Chem. Soc., 2009, 131, 9767–9779 CrossRef CAS PubMed.
- M. M. Lee, J. L. Childs-Disney, A. Pushechnikov, J. M. French, K. Sobczak, C. A. Thornton and M. D. Disney, J. Am. Chem. Soc., 2009, 131, 17464–17472 CrossRef CAS PubMed.
- J. F. Arambula, S. R. Ramisetty, A. M. Baranger and S. C. Zimmerman, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 16068–16073 CrossRef CAS PubMed.
- R. Parkesh, J. L. Childs-Disney, M. Nakamori, A. Kumar, E. Wang, T. Wang, J. Hoskins, T. Tran, D. Housman and C. A. Thornton, J. Am. Chem. Soc., 2012, 134, 4731–4742 CrossRef CAS PubMed.
- S. G. Rzuczek, M. R. Southern and M. D. Disney, ACS Chem. Biol., 2015, 10, 2706–2715 CrossRef CAS PubMed.
- C. Z. Chen, K. Sobczak, J. Hoskins, N. Southall, J. J. Marugan, W. Zheng, C. A. Thornton and C. P. Austin, Anal. Bioanal. Chem., 2012, 402, 1889–1898 CrossRef CAS PubMed.
- P. Szymański, M. Markowicz and E. Mikiciuk-Olasik, Int. J. Mol. Sci., 2011, 13, 427–452 CrossRef PubMed.
- E. Martis, R. Radhakrishnan and R. Badve, J. Appl. Pharm. Sci., 2011, 02–10 Search PubMed.
- D. C. Fara, T. I. Oprea, E. R. Prossnitz, C. G. Bologa, B. S. Edwards and L. A. Sklar, Drug Discov. Today Technol., 2006, 3, 377–385 CrossRef PubMed.
- L. M. Mayr and P. Fuerst, SLAS Discov., 2008, 13, 443–448 CrossRef CAS PubMed.
- R. Macarron, M. N. Banks, D. Bojanic, D. J. Burns, D. A. Cirovic, T. Garyantes, D. V. Green, R. P. Hertzberg, W. P. Janzen and J. W. Paslay, Nat. Rev. Drug Discovery, 2011, 10, 188–195 CrossRef CAS PubMed.
- S. M. Woollard and G. D. Kanmogne, Drug Des., Dev. Ther., 2015, 5447–5468 CAS.
- P. Dorr, M. Westby, S. Dobbs, P. Griffin, B. Irvine, M. Macartney, J. Mori, G. Rickett, C. Smith-Burchnell and C. Napier, Antimicrob. Agents Chemother., 2005, 49, 4721–4732 CrossRef CAS PubMed.
- K. J. Duffy, A. N. Shaw, E. Delorme, S. B. Dillon, C. Erickson-Miller, L. Giampa, Y. Huang, R. M. Keenan, P. Lamb and N. Liu, J. Med. Chem., 2002, 45, 3573–3575 CrossRef CAS PubMed.
- K. J. Duffy, A. T. Price, E. Delorme, S. B. Dillon, C. Duquenne, C. Erickson-Miller, L. Giampa, Y. Huang, R. M. Keenan and P. Lamb, J. Med. Chem., 2002, 45, 3576–3578 CrossRef CAS PubMed.
- J. A. Lemm, D. O'Boyle, M. Liu, P. T. Nower, R. Colonno, M. S. Deshpande, L. B. Snyder, S. W. Martin, D. R. St. Laurent and M. H. Serrano-Wu, J. Virol., 2010, 84, 482–491 CrossRef CAS PubMed.
- S. J. Lane, D. S. Eggleston, K. A. Brinded, J. C. Hollerton, N. L. Taylor and S. A. Readshaw, Drug Discovery Today, 2006, 11, 267–272 CrossRef CAS PubMed.
- D. M. Volochnyuk, S. V. Ryabukhin, Y. S. Moroz, O. Savych, A. Chuprina, D. Horvath, Y. Zabolotna, A. Varnek and D. B. Judd, Drug Discov. Today, 2019, 24, 390–402 CrossRef CAS PubMed.
- J.-L. Reymond, Acc. Chem. Res., 2015, 48, 722–730 CrossRef CAS PubMed.
- H. S. Haniff, A. Graves and M. D. Disney, ACS Comb. Sci., 2018, 20, 482–491 CrossRef CAS PubMed.
- D. S. Wishart, C. Knox, A. C. Guo, S. Shrivastava, M. Hassanali, P. Stothard, Z. Chang and J. Woolsey, Nucleic Acids Res., 2006, 34, D668–672 CrossRef CAS PubMed.
- H. S. Haniff, L. Knerr, X. Liu, G. Crynen, J. Boström, D. Abegg, A. Adibekian, E. Lekah, K. W. Wang, M. D. Cameron, I. Yildirim, M. Lemurell and M. D. Disney, Nat. Chem., 2020, 12, 952–961 CrossRef CAS PubMed.
- B. S. Morgan, B. G. Sanaba, A. Donlic, D. B. Karloff, J. E. Forte, Y. Zhang and A. E. Hargrove, ACS Chem. Biol., 2019, 14, 2691–2700 CrossRef CAS PubMed.
- A. Mehta, S. Sonam, I. Gouri, S. Loharch, D. K. Sharma and R. Parkesh, Nucleic Acids Res., 2014, 42, D132–141 CrossRef CAS PubMed.
- N. F. Rizvi, J. P. Santa Maria Jr, A. Nahvi, J. Klappenbach, D. J. Klein, P. J. Curran, M. P. Richards, C. Chamberlin, P. Saradjian and J. Burchard, SLAS Discov., 2020, 25, 384–396 CrossRef CAS PubMed.
- N. F. Rizvi, J. A. Howe, A. Nahvi, D. J. Klein, T. O. Fischmann, H. Y. Kim, M. A. McCoy, S. S. Walker, A. Hruza, M. P. Richards, C. Chamberlin, P. Saradjian, M. T. Butko, G. Mercado, J. Burchard, C. Strickland, P. J. Dandliker, G. F. Smith and E. B. Nickbarg, ACS Chem. Biol., 2018, 13, 820–831 CrossRef CAS PubMed.
- N. F. Rizvi and E. B. Nickbarg, Methods, 2019, 167, 28–38 CrossRef CAS PubMed.
- D. R. Calabrese, K. Zlotkowski, S. Alden, W. M. Hewitt, C. M. Connelly, R. M. Wilson, S. Gaikwad, L. Chen, R. Guha and C. J. Thomas, Nucleic Acids Res., 2018, 46, 2722–2732 CrossRef CAS PubMed.
- K. Yazdani, D. Jordan, M. Yang, C. R. Fullenkamp, D. R. Calabrese, R. Boer, T. Hilimire, T. E. Allen, R. T. Khan and J. S. Schneekloth Jr, Angew. Chem., Int. Ed., 2023, 135, e202211358 CrossRef.
- A. Bancet, C. Raingeval, T. Lomberget, M. Le Borgne, J.-F. Guichou and I. Krimm, J. Med. Chem., 2020, 63, 11420–11435 CrossRef CAS PubMed.
- K. T. Flaherty, U. Yasothan and P. Kirkpatrick, Nat. Rev. Drug Discovery, 2011, 10, 811–813 CrossRef CAS PubMed.
- L. M. Juárez-Salcedo, V. Desai and S. Dalia, Drugs context, 2019, 8, 212574 Search PubMed.
- K. Bodoor, V. Boyapati, V. Gopu, M. Boisdore, K. Allam, J. Miller, W. D. Treleaven, T. Weldeghiorghis and F. Aboul-ela, J. Med. Chem., 2009, 52, 3753–3761 CrossRef CAS PubMed.
- S. M. Meyer, T. Tanaka, A. Taghavi, J. T. Baisden, M. Grefe and M. D. Disney, ACS Chem. Biol., 2023, 18, 2336–2342 CrossRef CAS PubMed.
- C. G. Wermuth, Drug Discov. Today, 2006, 11, 348–354 CrossRef CAS PubMed.
- Q. Gao, L. Yang and Y. Zhu, Curr. Comput.-Aided Drug Des., 2010, 6, 37–49 CrossRef CAS PubMed.
- D. C. Rees, M. Congreve, C. W. Murray and R. Carr, Nat. Rev. Drug Discov., 2004, 3, 660–672 CrossRef CAS PubMed.
- B. M. Suresh, Y. Akahori, A. Taghavi, G. Crynen, Q. M. Gibaut, Y. Li and M. D. Disney, J. Am. Chem. Soc., 2022, 144, 20815–20824 CrossRef CAS PubMed.
- H. Jhoti, G. Williams, D. C. Rees and C. W. Murray, Nat. Rev. Drug Discovery, 2013, 12, 644 CrossRef CAS PubMed.
- G. R. Bickerton, G. V. Paolini, J. Besnard, S. Muresan and A. L. Hopkins, Nat. Chem., 2012, 4, 90–98 CrossRef CAS PubMed.
- K. P. Lundquist, V. Panchal, C. H. Gotfredsen, R. Brenk and M. H. Clausen, ChemMedChem, 2021, 16, 2588–2603 CrossRef CAS PubMed.
- B. M. Suresh, A. Taghavi, J. L. Childs-Disney and M. D. Disney, J. Med. Chem., 2023, 66, 6523–6541 CrossRef CAS PubMed.
- L. Mannocci, M. Leimbacher, M. Wichert, J. Scheuermann and D. Neri, Chem. Commun., 2011, 47, 12747–12753 RSC.
- R. I. Benhamou, B. M. Suresh, Y. Tong, W. G. Cochrane, V. Cavett, S. Vezina-Dawod, D. Abegg, J. L. Childs-Disney, A. Adibekian and B. M. Paegel, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2114971119 CrossRef CAS PubMed.
- Q. Chen, Y. Li, C. Lin, L. Chen, H. Luo, S. Xia, C. Liu, X. Cheng, C. Liu, J. Li and D. Dou, Nucleic Acids Res., 2022, 50, e67 CrossRef CAS PubMed.
- Q. M. R. Gibaut, Y. Akahori, J. A. Bush, A. Taghavi, T. Tanaka, H. Aikawa, L. S. Ryan, B. M. Paegel and M. D. Disney, J. Am. Chem. Soc., 2022, 144, 21972–21979 CrossRef CAS PubMed.
- P. H. Torres, A. C. Sodero, P. Jofily and F. P. Silva Jr, Int. J. Mol. Sci., 2019, 20, 4574 CrossRef CAS PubMed.
- J. De Ruyck, G. Brysbaert, R. Blossey and M. F. Lensink, Adv. Appl. Bioinform. Chem., 2016, 1–11 Search PubMed.
- S. L. Bonilla and J. S. Kieft, J. Mol. Biol., 2022, 434, 167802 CrossRef CAS PubMed.
- R. M. Glaeser, Annu. Rev. Biophys., 2019, 48, 45–61 CrossRef CAS PubMed.
- N. Moitessier, E. Westhof and S. Hanessian, J. Med. Chem., 2006, 49, 1023–1033 CrossRef CAS PubMed.
- M. Parisien and F. Major, Nature, 2008, 452, 51–55 CrossRef CAS PubMed.
- Z. Shi, J. Zhang, X. Qian, L. Han, K. Zhang, L. Chen, J. Liu, Y. Ren, M. Yang and A. Zhang, Cancer Res., 2013, 73, 5519–5531 CrossRef CAS PubMed.
- Z. Yan, S. Sikri, D. L. Beveridge and A. M. Baranger, J. Med. Chem., 2007, 50, 4096–4104 CrossRef CAS PubMed.
- D. M. Warui and A. M. Baranger, J. Med. Chem., 2009, 52, 5462–5473 CrossRef CAS PubMed.
- A. C. Stelzer, A. T. Frank, J. D. Kratz, M. D. Swanson, M. J. Gonzalez-Hernandez, J. Lee, I. Andricioaei, D. M. Markovitz and H. M. Al-Hashimi, Nat. Chem. Biol., 2011, 7, 553–559 CrossRef CAS PubMed.
- E. Kallert, L. A. Rodriguez, J.-Å. Husmann, K. Blatt and C. Kersten, RSC Med. Chem., 2024, 15, 1527–1538 RSC.
- L. T. Olenginski, W. K. Kasprzak, S. K. Attionu, B. A. Shapiro and T. K. Dayie, Molecules, 2023, 28, 1803 CrossRef CAS PubMed.
- L. R. Ganser, J. Lee, A. Rangadurai, D. K. Merriman, M. L. Kelly, A. D. Kansal, B. Sathyamoorthy and H. M. Al-Hashimi, Nat. Struct. Mol. Biol., 2018, 25, 425–434 CrossRef CAS PubMed.
- Q. Zhao, Z. Zhao, X. Fan, Z. Yuan, Q. Mao and Y. Yao, J. Phys. Chem. B, 2021, 17, e1009291 CAS.
- W. Lu, Y. Tang, H. Wu, H. Huang, Q. Fu, J. Qiu and H. Li, BMC Bioinf., 2019, 20, 1–10 CrossRef CAS PubMed.
- L. Wang, Y. Liu, X. Zhong, H. Liu, C. Lu, C. Li and H. Zhang, Front. Genet., 2019, 10, 440502 Search PubMed.
- J. A. Cruz and E. Westhof, Nat. Methods, 2011, 8, 513–519 CrossRef CAS PubMed.
- C. Theis, C. Höner Zu Siederdissen, I. L. Hofacker and J. Gorodkin, Nucleic Acids Res., 2013, 41, 9999–10009 CrossRef CAS PubMed.
- R. J. Townshend, S. Eismann, A. M. Watkins, R. Rangan, M. Karelina, R. Das and R. O. Dror, Science, 2021, 373, 1047–1051 CrossRef CAS PubMed.
- W. Wang, C. Feng, R. Han, Z. Wang, L. Ye, Z. Du, H. Wei, F. Zhang, Z. Peng and J. Yang, Nat. Commun., 2023, 14, 7266 CrossRef CAS PubMed.
- Y.-C. Chen, Trends Pharmacol. Sci., 2015, 36, 78–95 CrossRef PubMed.
- S. Saikia and M. Bordoloi, Curr. Drug Targets, 2019, 20, 501–521 CrossRef CAS PubMed.
- S. Chhabra, J. Xie and A. T. Frank, J. Phys. Chem. B, 2020, 124, 4436–4445 CrossRef CAS PubMed.
- F. Stefaniak and J. M. Bujnicki, J. Phys. Chem. B, 2021, 17, e1008309 CAS.
- H. Moriwaki, Y.-S. Tian, N. Kawashita and T. Takagi, J. Cheminform., 2018, 10, 1–14 CrossRef PubMed.
- C. Oliver, V. Mallet, R. S. Gendron, V. Reinharz, W. L. Hamilton, N. Moitessier and J. Waldispühl, Nucleic Acids Res., 2020, 48, 7690–7699 CrossRef CAS PubMed.
- K. Wang, R. Zhou, Y. Wu and M. Li, Brief. Bioinform., 2023, 24, bbac486 CrossRef PubMed.
- H. Grimberg, V. S. Tiwari, B. Tam, L. Gur-Arie, D. Gingold, L. Polachek and B. Akabayov, J. Cheminform., 2022, 14, 4 CrossRef CAS PubMed.
- C. Shen, J. Luo, Z. Lai and P. Ding, J. Chem. Inf. Model., 2020, 60, 4085–4097 CrossRef CAS PubMed.
- Z. Zhuo, Y. Wan, D. Guan, S. Ni, L. Wang, Z. Zhang, J. Liu, C. Liang, Y. Yu and A. Lu, Adv. Sci., 2020, 7, 1903451 CrossRef CAS PubMed.
- C.-C. Wang, X. Chen, J. Qu, Y.-Z. Sun and J.-Q. Li, J. Chem. Inf. Model., 2019, 59, 1668–1679 CrossRef CAS PubMed.
- P. Schuck and H. Zhao, Surface plasmon resonance: methods and protocols, 2010, pp. 15–54 Search PubMed.
- P. B. Palde, L. O. Ofori, P. C. Gareiss, J. Lerea and B. L. Miller, J. Med. Chem., 2010, 53, 6018–6027 CrossRef CAS PubMed.
- G. G. Ferenczy and G. M. Keserű, Drug Discovery Today, 2010, 15, 919–932 CrossRef CAS PubMed.
- J. E. Ladbury, G. Klebe and E. Freire, Nat. Rev. Drug Discovery, 2010, 9, 23–27 CrossRef CAS PubMed.
- P. Giri and G. S. Kumar, Mol. BioSyst., 2008, 4, 341–348 RSC.
- G. S. Kumar and A. Basu, Biochim. Biophys. Acta, Gen. Subj., 2016, 1860, 930–944 CrossRef CAS PubMed.
- B. Saha and G. S. Kumar, J. Photochem. Photobiol., B, 2017, 167, 99–110 CrossRef CAS PubMed.
- L. Pascale, S. Azoulay, A. Di Giorgio, L. Zenacker, M. Gaysinski, P. Clayette and N. Patino, Nucleic Acids Res., 2013, 41, 5851–5863 CrossRef CAS PubMed.
- M. Kaul and D. S. Pilch, Biochemistry, 2002, 41, 7695–7706 CrossRef CAS PubMed.
- J. R. Thomas, X. Liu and P. J. Hergenrother, Biochemistry, 2006, 45, 10928–10938 CrossRef CAS PubMed.
- T. W. Mayhood and W. T. Windsor, Anal. Biochem., 2005, 345, 187–197 CrossRef CAS PubMed.
- X. Liu, S. Wang, F. Meng, J. Wang, Y. Zhang, E. Dai, X. Yu, X. Li and W. Jiang, Bioinformatics, 2013, 29, 409–411 CrossRef CAS PubMed.
- S. R. Krishnan, A. Roy and M. M. Gromiha, J. Mol. Biol., 2023, 435, 167914 CrossRef PubMed.
- A. Donlic, E. G. Swanson, L.-Y. Chiu, S. L. Wicks, A. U. Juru, Z. Cai, K. Kassam, C. Laudeman, B. G. Sanaba and A. Sugarman, ACS Chem. Biol., 2022, 17, 1556–1566 CrossRef CAS PubMed.
- M. D. Disney, A. M. Winkelsas, S. P. Velagapudi, M. Southern, M. Fallahi and J. L. Childs-Disney, ACS Chem. Biol., 2016, 11, 1720–1728 CrossRef CAS PubMed.
- L. Li, P. Wu, Z. Wang, X. Meng, C. Zha, Z. Li, T. Qi, Y. Zhang, B. Han and S. Li, J. Hematol. Oncol., 2020, 13, 1–4 CrossRef PubMed.
- J. G. Carvajal-Patino, V. Mallet, D. Becerra, L. F. Nino, C. Oliver and J. Waldispuhl, bioRxiv, 2023, preprint, 2023.2011.2023.568394 DOI:10.1101/2023.11.23.568394.
- C. J. Langeberg and J. S. Kieft, Nucleic Acids Res., 2023, 51, e100–e100 CrossRef CAS PubMed.
- Q. Liu, M. Allamanis, M. Brockschmidt and A. Gaunt, Adv. Neural Inf. Process. Syst., 2018, 31, 7795–7804 Search PubMed.
- S. Tripathi, A. I. Augustin, A. Dunlop, R. Sukumaran, S. Dheer, A. Zavalny, O. Haslam, T. Austin, J. Donchez and P. K. Tripathi, Artif. Intell. Life Sci., 2022, 2, 100045 Search PubMed.
- G. Zhu and X. Chen, Adv. Drug Delivery Rev., 2018, 134, 65–78 CrossRef CAS PubMed.
- J. Gallego and G. Varani, Acc. Chem. Res., 2001, 34, 836–843 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |