
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRaman signatures of type A and B influenza viruses: molecular origin of the “catch and kill” inactivation mechanism mediated by micrometric silicon nitride powder†
Giuseppe
Pezzotti
 *abcdef,
Yoshiki
Yasukochi
g,
Eriko
Ohgitani
b,
Maiko
Nakashio
bh,
Masaharu
Shin-Ya
b,
Tetsuya
Adachi
bdi,
Toshiro
Yamamoto
d,
Saki
Ikegami
ij,
Wenliang
Zhu
j,
Koichiro
Higasa
g,
Kazu
Okuma
i and
Osam
Mazda
*b
*abcdef,
Yoshiki
Yasukochi
g,
Eriko
Ohgitani
b,
Maiko
Nakashio
bh,
Masaharu
Shin-Ya
b,
Tetsuya
Adachi
bdi,
Toshiro
Yamamoto
d,
Saki
Ikegami
ij,
Wenliang
Zhu
j,
Koichiro
Higasa
g,
Kazu
Okuma
i and
Osam
Mazda
*b
aBiomedical Engineering Center, Kansai Medical University, 1-9-11 Shin-machi, Hirakata, Osaka 573-1191, Japan. E-mail: pezzotti@hirakata.kmu.ac.jp
bDepartment of Immunology, Graduate School of Medical Science, Kyoto Prefectural University of Medicine, Kamigyo-ku, 465 Kajii-cho, Kyoto 602-8566, Japan. E-mail: mazda@koto.kpu-m.ac.jp
cDepartment of Orthopedic Surgery, Tokyo Medical University, 6-7-1 Nishi-Shinjuku, Shinjuku-ku, 160-0023 Tokyo, Japan
dDepartment of Dental Medicine, Graduate School of Medical Science, Kyoto Prefectural University of Medicine, Kamigyo-ku, Kyoto 602-8566, Japan
eDepartment of Molecular Science and Nanosystems, Ca’ Foscari University of Venice, Via Torino 155, 30172 Venice, Italy
fDepartment of Applied Science and Technology, Politecnico di Torino, Corso Duca degli Abruzzi 24, 10129 Torino, Italy
gDepartment of Genome Analysis, Institute of Biomedical Science, Kansai Medical University, 2-3-1 Shin-machi, Hirakata, Osaka 573-1191, Japan
hDepartment of Emergency and Critical Care Medicine, Faculty of Medicine, Fukuoka University, Fukuoka 814-0180, Japan
iDepartment of Microbiology, Kansai Medical University, School of Medicine, 2-5-1 Shinmachi, Hirakata, 573-1010 Osaka Prefecture, Japan
jCeramic Physics Laboratory, Kyoto Institute of Technology, Sakyo-ku, Matsugasaki, 606-8585 Kyoto, Japan
First published on 22nd January 2025
Abstract
A multiomic study of the structural characteristics of type A and B influenza viruses by means of highly spectrally resolved Raman spectroscopy is presented. Three virus strains, A H1N1, A H3N2, and B98, were selected because of their known structural variety and because they have co-circulated with variable relative prevalence within the human population since the re-emergence of the H1N1 subtype in 1977. Raman signatures of protein side chains tyrosine, tryptophan, and histidine revealed unequivocal and consistent differences for pH characteristics at the virion surface, while different conformations of two C–S bond configurations in gauche and trans methionine rotamers provided distinct low-wavenumber fingerprints for different virus lineages/subtypes. Short-term exposure to a few percent fraction of silicon nitride (Si3N4) micrometric powder in an aqueous environment completely inactivated the influenza virions, independent of lineage/subtype dependent characteristics. The molecular-scale details of the inactivation process were studied by Raman spectroscopy and interpreted in terms of a “catch and kill” mechanism, in which the hydrolyzing ceramic surface first attracts virions with high efficiency through electrochemical interactions (mimicking cellular sialic acid) and then “poisons” the viruses by local hydrolytic elution of ammonia and nitrogen radicals. The latter event causes severe damage to the virions’ structures, including structural degradation of RNA purines, rotameric scrambling of methionine residues, formation of sulfhydryl and ionized carboxyl groups, and deprotonation/torsional deformation of tyrosine, tryptophan, and histidine residues. This study confirmed the antiviral effectiveness of Si3N4 powder, which is safe to the human body and simply activated by water molecules. Raman spectroscopy was confirmed as a powerful tool in molecular virology, complementary to genomics and unique in providing direct information on virus structures at the molecular scale.
1. Introduction
Seasonally recurrent influenza epidemics, which systematically originate from the emergence of antigenic viral variants, have so far comprised the concurrent circulation of two influenza A subtypes and of the antigenically distinct lineages of type B.1 These lineages have so far given rise to genetic reassortment with antigenic drifts leading to recombinants of both A- and B-type viruses.1,2 Influenza viruses have so far been identified and thoroughly investigated by assessing their genome sequences3–5 and imaging their morphological characteristics by means of high-resolution cryo-electron microscopy and tomography.6–8 However, multiomic structural assessments of influenza virions have so far been less commonly reported in the published literature.9,10 In the stream of pioneering studies conducted by Thomas and coworkers11–13 and Takeuchi and coworkers,14–16 extensive multiomic Raman analyses have recently been developed and have found applications in virus identification,17–19 structural analysis20,21 and detection,22,23 monitoring viral replication,24,25 and supporting vaccine development and control.26The uniqueness of Raman spectroscopy in virology consists of providing a nearly instantaneous multiomic snapshot of the virion molecular structure, which could be used as a fingerprint unique to virus types and subtypes. Raman analysis of viruses not only enables the identification/classification of viral strains, but also helps to interpret their virulence, replication, and inactivation characteristics.27 For example, Raman spectroscopy can provide insights into structural properties that reveal conformational changes in viral proteins upon interaction with host cells or therapeutic agents. This information could help understanding virus–host interactions and developing antiviral drugs. Moreover, Raman spectroscopy has the potential for enabling on-site detection of viruses in clinical samples, although these developments are yet in embryo. For example, upon analyzing Raman spectra of virus-infected saliva, blood cells, or tissues, it has been possible to distinguish between infected and uninfected samples.28 This approach could be particularly useful in promptly diagnosing viral infections in resource-limited settings or in circumstances where traditional diagnostic methods are unavailable. In more basic research approaches,24,25 Raman spectroscopy has been employed to monitor the replication dynamics of viruses in real-time. Upon tracking changes in the Raman spectra of infected cells in time lapse, it has been possible to gain insights into the kinetics of viral replication and the effectiveness of antiviral treatments. Raman spectroscopy could also be used to analyse the structural integrity of viral antigens and to monitor the stability of vaccine formulations.29,30 Accordingly, once appropriately developed and validated, Raman analysis could be key in ensuring the efficacy and safety of vaccines before they are administered to the public.
In the present work, we document a Raman multiomic approach to characterize three influenza viral strains: A H1N1, A H3N2, and B98. It is stated at the outset that a complete understanding of all Raman band components in different viral strains is a quite challenging task because of the molecular complexity of the viral structures, in turn leading to strong overlaps of multiple Raman signals. Therefore, in a number of contexts related to RNA and amino acid signals, we could only offer here the most probable interpretation of the observed Raman behaviors. First, we studied and compared structural differences among the above three strains by interpreting the main Raman fingerprints of RNA bases and aromatic and sulphur-containing amino acids. As an additional goal, we studied the process of inactivation of the three examined viral strains after short-term exposure to micrometric Si3N4 ceramic powder in aqueous solution. In the latter effort, emphasis was placed on the effects of hydrolytic reactions taking place at the immediate surface of the ceramic powder, leading to concurrent environmental alkalinization and ammonia elution. In particular, the following aspects were elucidated: (i) ring damage to RNA purine bases; (ii) scrambling of S-containing methionine rotamers upon contact with surface silanols and their successive oxidation by nitrogen radicals; (iii) hydration levels of “buried” tyrosine as a pH sensor and the formation of tyrosinate anions; and (iv) structural variations of aromatic amino acids tryptophan (in terms of the molecular torsional angle) and histidine (in terms of tautomers) upon pH alkalinization. Despite the complexity of signal assignments, we shall show in the remainder of this paper that Raman spectroscopy is capable of providing molecular-scale features specific to each viral strain at a level of depth and thoroughness that are unachievable by other analytical methods. In this study, multiomic analyses of Raman spectra have enabled us to accurately speciate different viral strains and to deepen our understanding of their molecular structures, while providing direct evidence at the molecular level for the mechanism exerted on virions by Si3N4 ceramic powder independent of viral strain. We define this mechanism as a “catch and kill” two-step machinery that first attracts the virions towards the Si3N4 surface through the exploitation of electrochemical interactions mimicking those of cellular sialic acid and then “poisons” the viruses through hydrolytically eluted ammonia molecules entering the viral membrane and subsequently forming nitrogen radicals.
2. Experimental procedures
2.1 Description of influenza strains and inoculated cells
Three influenza strains were all obtained from the Virus Research Center, National Institute of Infectious Diseases: A/Puerto Rico/8/34 (H1N1), A/Panama/2007/1999 (H3N2), and B/Yamanashi/166/1998 (B98). All strains were enveloped viruses with the negative-sense, single-stranded, and segmented RNA genome (belonging to the Orthomyxoviridae family). They were selected since they represent an exemplification of both structural and genetic diversities among influenza viruses. Different molecular compositions, both in capsid proteins and RNA structure, are known to confer on influenza virions different specificity and infectivity characteristics.31 The studied virions have also been reported to possess different surface charges32 and differences in surface chemistry and electrochemistry characteristics, playing important roles in their level of infectivity and immune escape. The two studied subtypes of influenza A viruses differed from each other in the properties of their surface antigens, hemagglutinin (HA) and neuraminidase (NA), which are also the main targets in developing antiviral drugs.33 Such differences make H1N1 and H3N2 viruses differ in their epidemiological characteristics and contribute to variations in the levels of infectivity, virulence, and ability to cause seasonal outbreaks. It should be noted that influenza viruses of types A and B are morphologically indistinguishable under the transmission electron microscope, since they share a similar spherical and filamentous morphology, with ∼100 nm diameter and ∼300 nm length for spherical structures and filamentous shapes, respectively.34 The virions are covered with viral surface glycoproteins, HA and NA (about 300–400 and 40–50 HA and NA sites, respectively), sticking out from the lipid bilayer, although the numbers of each protein vary between different subtypes.7,35,36 The structures of HA and NA glycoproteins represent the basis for further subdivisions of influenza A virus. Together with the ion-channel M2 protein, they are the essential envelope proteins regulating viral entry, subsequent budding within, and release from host cells.37 The creation of novel strains, which occurs upon small structural changes in HA and/or NA, takes place under the effects of both immunological and vaccine responses across the human population.38 Antigenic drifts also include structural modifications of the M1 matrix protein that encircles the virion core and whose structure drifts despite no specific hydrophobic domain being apparently required for membrane binding. On the other hand, the structure of the M2 ion channel, responsible for ion transport towards the inside of the virions, was found to drift in response to vaccines and the neuraminidase inhibitor oseltamivir in avian influenza A viruses, although it is yet unclear whether such mutations originated in patients or were brought in from an avian source.39,40 On the other hand, the influenza B viral strain has been known to evolve and mutate slower than A viruses.41,42Fig. 1a shows the phylogenetic tree of the three studied strains with respect to maximum likelihood (ML) according to the Whelan and Goldman (WAG) model,43 based on amino acid sequences in HA. The multiple alignment of their amino acid sequences was implemented in TM-align Server,44 while the construction of phylogenetic trees and the estimation of the best-fit substitution model were conducted in MEGA v.11.45 The same figure also summarizes the antigenic characteristics of the studied viral strains with emphasis on the HA receptor-binding domain (RBD, binding to cell sialic acid; Fig. 1b–d, cf. labels in the inset), M1 matrix proteins (Fig. 1e–g), and M2 channels (Fig. 1h–j). Key amino acid features, as discussed in the following sections, are emphasized in the inset to each figure. The M1 structures of A H3N2 and B98 strains and the M2 structure of the A H1N1 strain were predicted by means of AlphaFold2 via ColabFold v1.5.546 and AlphaFold2 v2.3.2 via Multimer mode in AlphaFold Colab,47 respectively. The AlphaFold and AlphaFold2 utilize an artificial intelligence system that can predict three-dimensional structures of proteins from amino acid sequences with atomic-level accuracy.48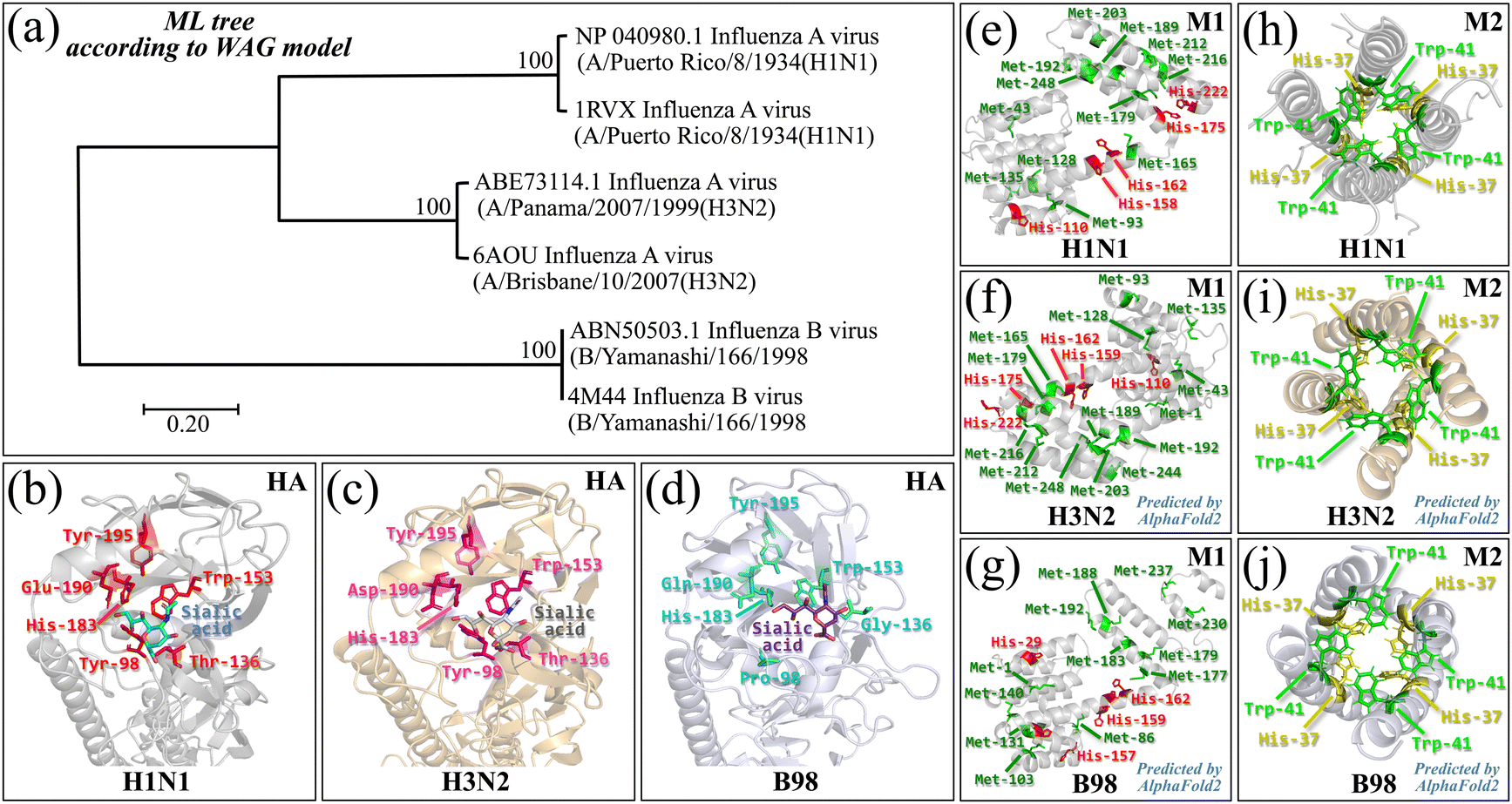 | ||
| Fig. 1 (a) Phylogenetic tree of the three studied influenza strains according to maximum likelihood (ML) with the WAG model, based on HA amino acid sequences; (b)–(d) their HA RBDs binding to cell sialic acid (cf. labels in the inset); (e)–(g) M1 matrix proteins with emphasis on the locations of histidine and methionine residues; and (h)–(j) M2 channels with emphasis on the locations of histidine and tryptophan residues. Amino acid sequences with the protein Data Bank (PDB) ID (1RVX, 6AOU, and 4M44) in (a) were obtained from the PDB database (https://www.rcsb.org/), whereas the sequences with GenBank accession numbers NP_040980.1, ABE73114.1, and ABN50503.1 were retrieved from the National Center for Biotechnology Information (NCBI) database (https://www.ncbi.nlm.nih.gov/). The protein 3D structure data in (b)–(j) were obtained from the PDB database (6Z5l for M1 in A H1N1, 2MUV for M2 in A H3N2, and 2KIX for M2 in B/Taiwan/70061/2006) or predicted by AlphaFold or AlphaFold2 (cf. insets). | ||
Madin-Darby canine kidney (MDCK) cells were purchased from DS Pharma Biomedical Co., Ltd (Osaka Prefecture, Suita, Japan). The cells were cultured in DMEM (Nacalai Tesque, Kyoto, Japan) supplemented with 4% FBS, 100 U per ml penicillin, and 100 μg ml−1 streptomycin (Complete Medium) and plated in a 6-well plate at 6 × 105 cells/well for plaque assay, or loaded on glass based dishes (TECHNO GLASS Co., Shizuoka, Japan) at 1 × 106 cells per dish for immunochemistry assays. Cells were cultured in complete medium at 37 °C in an atmosphere containing 5% CO2. MDCK cells have been widely used to examine the infectivity of various viral strains of influenza. Their suitability arises from both being highly susceptible to influenza virus and having replication kinetics similar to that of human cells.
2.2 Immunochemistry characterization
An aqueous solution containing a concentration of ∼2.5 vol% Si3N4 micrometric powder was added to an aqueous solution containing influenza virions (virus/solution 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), leading to a final concentration of Si3N4 particles of 2.5 vol%. Influenza virus isolated from infected cells had a titer of 3 × 105 pfu/150 μl. Mixing was applied for times variable between 1 and 30 min (referred to as virus inactivation time, henceforth) at room temperature using slowly rotating equipment. The ceramic particles were then separated by filtration and centrifugation (at 12000 rpm for 2 minutes at 4 °C). The viral infectivity of the supernatant was compared with that of sham samples (negative control; simply referred to as the control sample, henceforth) by plaque assay. The number of plaque forming units (PFU/100 μL) was then counted as a function of virus inactivation time. Specifications of the Si3N4 particle size and surface chemistry characteristics before/after exposure to the aqueous environment have been given in ref. 49, together with a schematic draft of inoculation and testing/characterization procedures. In the same context, additional evidence of viral inactivation was given by means of fluorescence microscopy.
:1), leading to a final concentration of Si3N4 particles of 2.5 vol%. Influenza virus isolated from infected cells had a titer of 3 × 105 pfu/150 μl. Mixing was applied for times variable between 1 and 30 min (referred to as virus inactivation time, henceforth) at room temperature using slowly rotating equipment. The ceramic particles were then separated by filtration and centrifugation (at 12000 rpm for 2 minutes at 4 °C). The viral infectivity of the supernatant was compared with that of sham samples (negative control; simply referred to as the control sample, henceforth) by plaque assay. The number of plaque forming units (PFU/100 μL) was then counted as a function of virus inactivation time. Specifications of the Si3N4 particle size and surface chemistry characteristics before/after exposure to the aqueous environment have been given in ref. 49, together with a schematic draft of inoculation and testing/characterization procedures. In the same context, additional evidence of viral inactivation was given by means of fluorescence microscopy.
In plaque assay, a confluent monolayer of MDCK cells was set in a 6-well plate and washed twice with serum-free DMEM (SF DMEM). Infection followed with 100 μl of virus suspension in a tenfold serial dilution. After incubation at 37 °C for 1 h with tilting every 10 min, unabsorbed inoculum was removed, and infected cells were overlaid with 4 ml of DMEM containing 2.5 μg ml−1 trypsin (Sigma-Aldrich Co. LLC, Saint Louis, USA) and 0.2% albumin (Wako Pure Chemical Industries, Ltd, Osaka, Japan). The plate was incubated at 37 °C in an atmosphere of 5% CO2 for 2 days. For plaque counting, the cells were fixed with 5% glutaraldehyde solution for 2 h, the agarose medium was then removed, and the cells were stained with 1% crystal violet before being subjected to plaque counting.
2.3 Statistical analysis
The statistical relevance of the experimental data was analyzed by computing mean values and standard deviations. Statistical validity was evaluated by applying the unpaired Student t-test. Values p < 10−2 and p < 10−3 were considered as statistically significant and labeled with two and three asterisks, respectively.2.4 Raman spectroscopic analysis
Raman spectra were collected using a highly spectrally resolved and sensitive instrument (LabRAM HR800, Horiba/Jobin-Yvon, Kyoto, Japan) with a 20× optical lens. The spectroscope was operated in confocal imaging configuration, using sub-micrometric shifts along a direction perpendicular to the sample plane in order to maximize the signal intensity at each spotted location. Full details about virus sample configuration and virus/probe interaction were given in a previous paper.19 A holographic notch filter within the optical circuit was used to concurrently achieve highly sensitive and highly resolved spectral acquisitions. The spectral resolution of 1.5 cm−1 was obtained using a 532 nm excitation source operating at 10 mW. The Raman scattered light was monitored using a single monochromator connected to an air-cooled charge-coupled device (CCD) detector (Andor DV420-OE322; 1024 × 256 pixels). The acquisition time was fixed at 10 s. Thirty spectra were collected and averaged at each location/condition. Raman spectra were deconvoluted into Gaussian–Lorentzian sub-bands using commercially available software (LabSpec 4.02, Horiba/Jobin-Yvon, Kyoto, Japan).3. Experimental results
3.1 Immunochemistry assessments
Results of plaque counting and the virus reduction rate as a function of exposure time to 2.5 vol% Si3N4 ceramic particles in aqueous solution are shown in Fig. 2a by conducting inoculation experiments with the A H1N1 strain at 4 °C. As seen, the PFU counts decreased and the reduction rate increased as compared to the control sample with increasing exposure time of the virus to the Si3N4-containing solution. The reduction rate was ∼82% (p < 10−2; n = 3), ∼98% (p < 10−2; n = 3), and 100% (p < 10−3; n = 3) after 1 min, 5 min, and ≥10 min exposure, respectively. In other words, no infection occurred in cells inoculated with the A H1N1 virus samples preliminary exposed to Si3N4 for times equal or longer than 10 min. Experiments conducted using the same protocol, but limited to 10 min Si3N4/virus exposure, were then applied to quantitatively assess the inactivation effect of the Si3N4 powder in the cases of A H3N2 and B98 strains. As shown in Fig. 2b, this additional set of experiments proved that, independent of influenza virus being of A or B type, the Si3N4 powder fully inactivated the virus within an exposure time as short as 10 min. Given the quite small volumetric fraction (i.e., 2.5 vol%) of Si3N4 particles dispersed in aqueous solution, the above data support the hypothesis of a strong attraction and successive destructive chemical interaction between influenza viruses and the Si3N4 surface. This hypothesis will be substantiated by Raman analysis comparing viral strains before and after Si3N4 exposure, as described in the following sections. | ||
| Fig. 2 (a) Results of plaque counting (left side) and the virus reduction rate (right side) as a function of exposure time to 2.5 vol% Si3N4 micrometric particles (n = 3; inoculation experiments conducted at 4 °C; cf. labels in the inset) and (b) comparison among the three investigated influenza strains after 10 min-exposure experiments conducted with the same protocol in (a). The latter data show that, independent of influenza viruses being of A- or B-type, exposure to Si3N4 powder led to their full inactivation. | ||
3.2 Raman spectroscopic results
Fig. 3a and b show average Raman spectra in the wavenumber interval 600–1800 cm−1, which were collected on the three investigated viral strains before and after exposure for 10 min to 2.5 vol% Si3N4 in aqueous solution, respectively (cf. strain labels in the inset). As evident at a first glance, the average Raman spectra of non-treated strains were clearly different from each other, thus revealing major differences in molecular conformation. Moreover, virus spectra collected after Si3N4 exposure strongly differed from those of the respective non-treated strains. Such differences represent a clear proof for the impact that exposure to Si3N4 in aqueous solution had on the molecular structure of different virions. The investigated spectral interval was divided into six zones (referred to as Zones I–VI henceforth). In each zone, high-resolution spectra were newly collected, which revealed with better sharpness the specific vibrational features of interest. Fig. 4–9 show the high-resolution (average) Raman spectra recorded in the wavenumber intervals 600–800 cm−1 (Zone I), 800–1000 cm−1 (Zone II), 1000–1200 cm−1 (Zone III), 1200–1400 cm−1 (Zone IV), 1400–1600 cm−1 (Zone V), and 1550–1750 cm−1 (Zone VI), respectively, for A H1N1, A H3N2, and B98 viral strains before/after 10 min-exposure to Si3N4 powder in aqueous solution (cf. also labels in the inset). High-resolution spectra in each spectral zone were deconvoluted into a series of Gaussian–Lorentzian sub-band components, which were numbered (cf. labels in the inset) and assigned to vibrational origins. In the case of non-treated viral strains, we located a total number of 67 Raman bands in the wavenumber interval 600–1800 cm−1. However, several additional bands appeared in Si3N4-exposed virions as a result of oxidation and other post-translational and degradation phenomena due to the interaction between Si3N4 hydrolytic moieties and viral molecules. Raman bands in RNA purines/pyrimidines and in key amino acid residues (relevant to the discussions in this paper) are reviewed in Fig. S1 and S2 (ESI†), respectively; wavenumbers at maximum and tentative assignments for the numbered Raman sub-band components are given in Tables S-I–S-VI (cf. ESI†). In a preliminary effort to understand whether or not the spectra of different influenza variants could be distinguished by examining their mere morphology, we performed PCA analysis (cf. Fig. S3 in the ESI†). However, the PCA analysis was not capable of giving clear-cut variant speciation, suggesting that advanced machine learning procedures should be developed in future work for achieving this specific purpose. Nevertheless, with the main focus of this paper being multiomic analyses of virions’ structures, we attempt hereafter to provide a thorough spectroscopic view of Raman fingerprints of key-molecules and to explain how and why they differ among different influenza variants.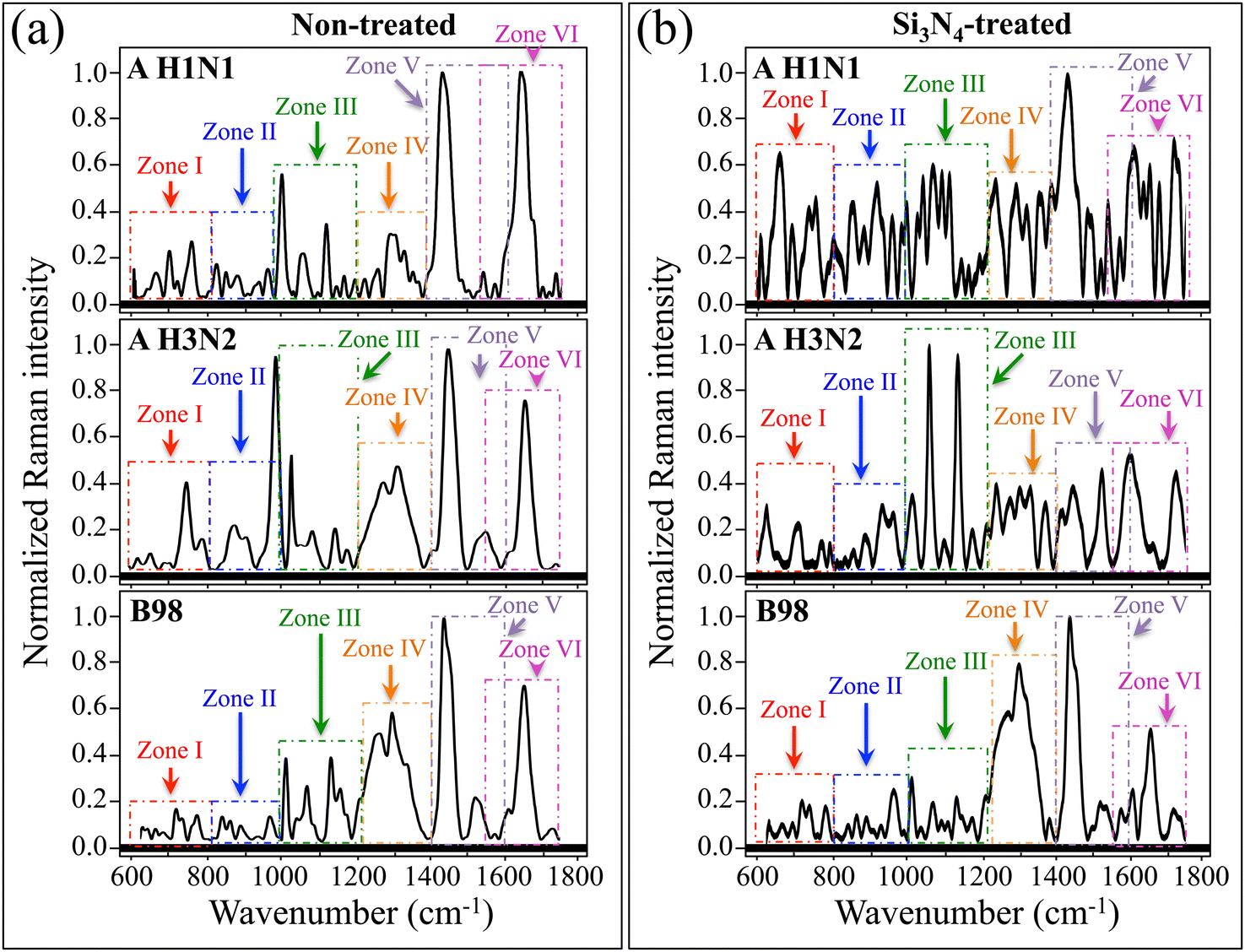 | ||
| Fig. 3 Average Raman spectra recorded in the wavenumber interval 600–1800 cm−1 for the three investigated viral strains (a) before (non-treated) and (b) after (Si3N4-treated) exposure for 10 min to 2.5 vol% Si3N4 in aqueous solution (strains located by labels in the inset). The investigated spectral interval is divided into six zones (labeled as Zones I–VI) in which high-resolution spectra were collected to reveal specific vibrational features of interest. | ||
 | ||
| Fig. 4 High-resolution Raman spectra recorded in the interval 600–800 cm−1 (Zone I), for A H1N1, A H3N2, and B98 viral strains before (labeled as non-treated in (a), (c) and (e), respectively) and after (labeled as Si3N4-treated in (b), (d) and (f), respectively) 10 min-exposure to Si3N4 powder in aqueous solution. Spectra are deconvoluted into a series of numbered Gaussian–Lorentzian sub-band components (precise wavenumber positions and vibrational origins are given in Table S-I, ESI†). Different colors are used to emphasize specific components and to locate common molecular origins. Asterisks locate new bands that only appear after Si3N4 exposure. | ||
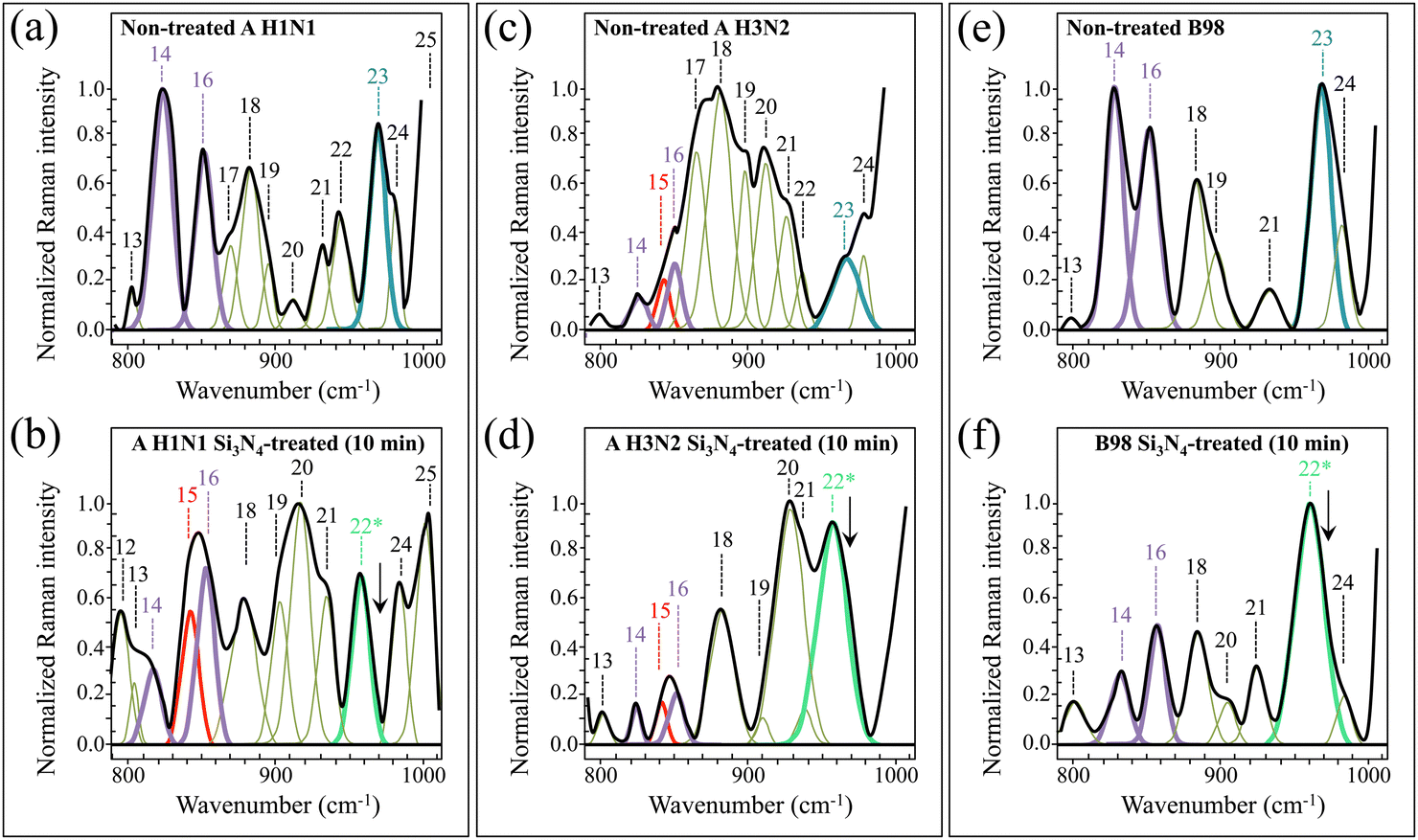 | ||
| Fig. 5 High-resolution Raman spectra recorded in the interval 800–1000 cm−1 (Zone II), for A H1N1, A H3N2, and B98 viral strains before (labelled as non-treated in (a), (c) and (e), respectively) and after (labelled as Si3N4-treated in (b), (d) and (f), respectively) 10 min-exposure to Si3N4 powder in aqueous solution. Spectra are deconvoluted into a series of numbered Gaussian–Lorentzian sub-band components (precise wavenumber positions and vibrational origins are given in Table S-II, ESI†). Different colours are used to emphasize specific components and to locate common molecular origins. Asterisks locate new bands that only appear after Si3N4 exposure. | ||
 | ||
| Fig. 6 High-resolution Raman spectra recorded in the interval 1000–1200 cm−1 (Zone III), for A H1N1, A H3N2, and B98 viral strains before (labelled as non-treated in (a), (c) and (e), respectively) and after (labelled as Si3N4-treated in (b), (d) and (f), respectively) 10 min-exposure to Si3N4 powder in aqueous solution. Spectra are deconvoluted into a series of numbered Gaussian–Lorentzian sub-band components (precise wavenumber positions and vibrational origins are given in Table S-III, ESI†). Different colours are used to emphasize specific components and to locate common molecular origins. Asterisks locate new bands that only appear after Si3N4 exposure. | ||
 | ||
| Fig. 7 High-resolution Raman spectra recorded in the interval 1200–1400 cm−1 (Zone IV), for A H1N1, A H3N2, and B98 viral strains before (labelled as non-treated in (a), (c) and (e), respectively) and after (labelled as Si3N4-treated in (b), (d) and (f), respectively) 10 min-exposure to Si3N4 powder in aqueous solution. Spectra are deconvoluted into a series of numbered Gaussian–Lorentzian sub-band components (precise wavenumber positions and vibrational origins are given in Table S-IV, ESI†). Different colours are used to emphasize specific components and to locate common molecular origins. Asterisks locate new bands that only appear after Si3N4 exposure. | ||
 | ||
| Fig. 8 High-resolution Raman spectra recorded in the interval 1400–1600 cm−1 (Zone V), for A H1N1, A H3N2, and B98 viral strains before (labelled as non-treated in (a), (c) and (e), respectively) and after (labelled as Si3N4-treated in (b), (d) and (f), respectively) 10 min-exposure to Si3N4 powder in aqueous solution. Spectra are deconvoluted into a series of numbered Gaussian–Lorentzian sub-band components (precise wavenumber positions and vibrational origins are given in Table S-V, ESI†). Different colours are used to emphasize specific components and to locate common molecular origins. Asterisks locate new bands that only appear after Si3N4 exposure. | ||
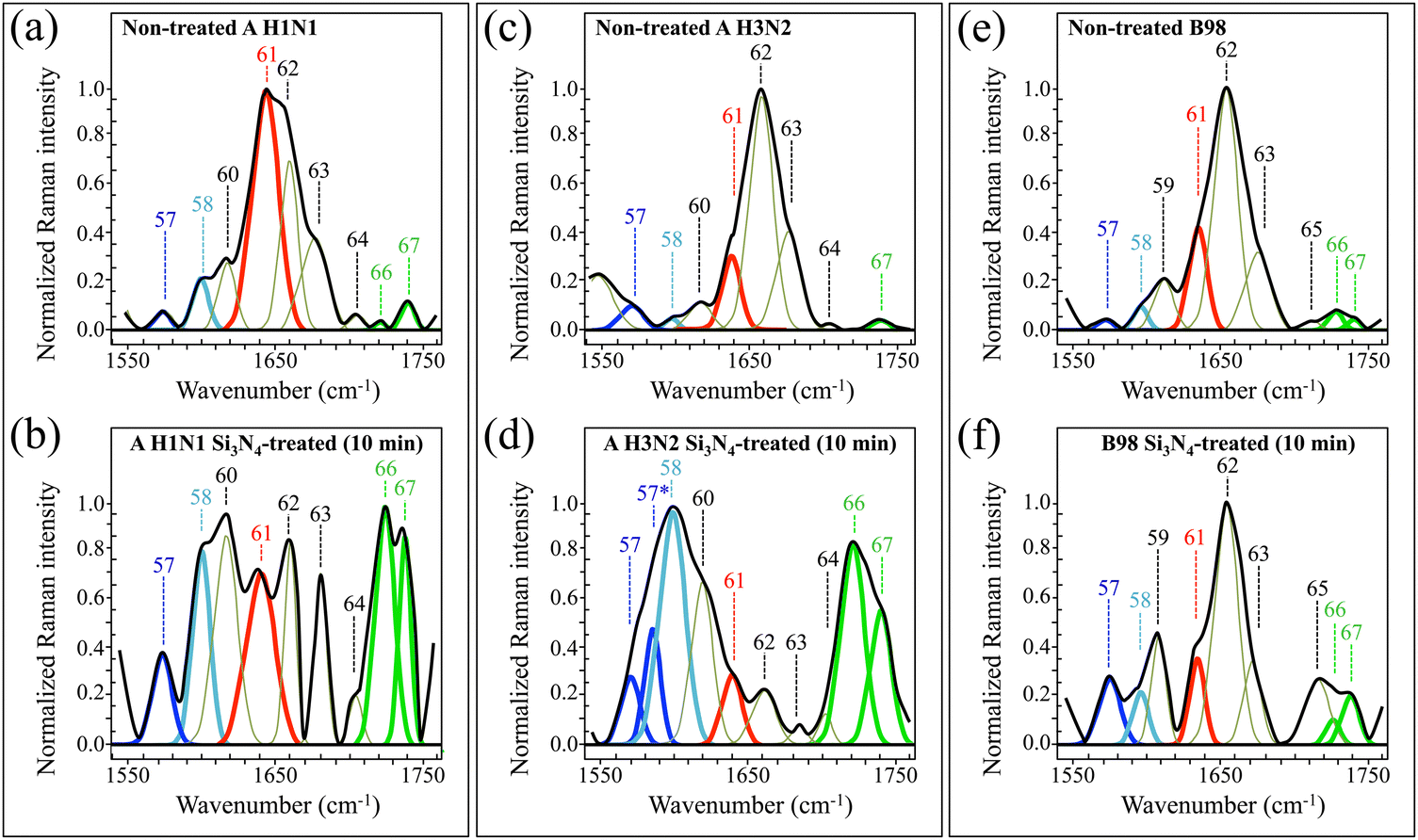 | ||
| Fig. 9 High-resolution Raman spectra recorded in the interval 1550–1750 cm−1 (Zone VI), for A H1N1, A H3N2, and B98 viral strains before (labelled as non-treated in (a), (c) and (e), respectively) and after (labelled as Si3N4-treated in (b), (d) and (f), respectively) 10 min-exposure to Si3N4 powder in aqueous solution. Spectra are deconvoluted into a series of numbered Gaussian–Lorentzian sub-band components (precise wavenumber positions and vibrational origins are given in Table S-VI, ESI†). Different colours are used to emphasize specific components and to locate common molecular origins. | ||
As an anticipation of the discussion in Section 4, we shall first introduce the peculiar signals of interest included in the above 6 Raman spectral regions, as follows: C–S stretching from S-containing amino acids and ring vibrations from RNA purine bases (Zone I), ring vibrations from both tyrosine (and tyrosinate) residues and RNA purine bases (Zone II), S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching in oxidized S-containing amino acids (Zone III), C–N stretching and C–H vibrations from the tryptophan ring (Zone IV), CC stretching in deprotonated/partly protonated histidine and tryptophan, and stretching of –COO− deprotonated terminal bonds (Zone V); and CC stretching vibrations from fully protonated histidine and carbonyl stretching vibrations (Zone VI).
O stretching in oxidized S-containing amino acids (Zone III), C–N stretching and C–H vibrations from the tryptophan ring (Zone IV), CC stretching in deprotonated/partly protonated histidine and tryptophan, and stretching of –COO− deprotonated terminal bonds (Zone V); and CC stretching vibrations from fully protonated histidine and carbonyl stretching vibrations (Zone VI).
In Zone I (Fig. 4), the major spectral differences among different strains could be seen in the relative intensity of Bands 3–5, 7, and 8. These signals represent C–S stretching vibrations from different methionine rotamers.50 Significant intensity variations in C–S bands were also noticed for each strain after exposure to Si3N4 powder, an observation that can be interpreted by invoking the occurrence of significant scrambling in methionine rotameric structures upon interaction with the ceramic particles. In addition, there is also strong spectroscopic evidence in support of the occurrence of oxidation in S-containing amino acids. One striking evidence resides in the appearance of two new bands for the Si3N4-exposed A H1N1 virus (cf. Bands 4* and 8* at 662 and 725 cm−1, respectively; cf.Fig. 4b). These relatively strong signals could be assigned to stretching of –C(H2)–S– bonds (with the H atom at the trans position with respect to the sulfur atom) and stretching of –S–CH3 bonds, respectively, in methionine (cf. also Table SI, ESI†).51 However, they are also strongly contributed by C–S stretching in methionine sulfoxide on the CH2 side (Band 4*) and the CH3 side (Band 8*).52 None of these two signals could be seen in the exposed A H3N2 strain (cf.Fig. 4d) and only the 8* signal in the exposed B98 one (cf.Fig. 4f). On the other hand, the exposed B98 strain was the only strain presenting a new strong signal at 764 cm−1 (Band 10* in Fig. 4f), which arises from stretching of –C(H2)–S– bonds with the C atom at the trans position with respect to the sulfur atom. The latter spectral change can be related to modifications in the number and vibrational boundary condition of CH2 groups that occur as a consequence of both desulfurization and decarboxylation processes due to radical attack. The first leads to the formation of CH3SH moieties and the second to the corresponding formation of amine and aldehyde groups.52 The change in CH2 configuration should also lead to an enhancement in its rocking signal, which also contributes to the band at 764 cm−1, as an index of relevant modifications in the methylene structure and content upon Si3N4 exposure.52 Additional discussion on the spectral configuration of Zone I in different strains is given in the forthcoming section (Section 4.2). A final important feature is regarding the signal of benzene ring breathing in RNA adenine, which appears at 738 cm−1 (Band 9 in Fig. 4).53 This band conspicuously disappeared upon Si3N4 exposure for both A H3N2 and B98 strains, while it remained almost unchanged for the A H1N1 strain. This feature will further be discussed later in Section 4.1.
Interesting spectral differences among non-treated viral strains and their modifications upon exposure to Si3N4 powder could also be seen in Zone II (800–100 cm−1; cf.Fig. 5 and Table S-II, ESI†). The most striking difference in this zone resided in the morphology of the tyrosine doublet (cf. Bands 14 and 16 located at 824 and 852 cm−1, respectively). The doublet arises from out-of-plane C–H bending in tyrosine (Band 14) and in-plane ring breathing (Band 16).54 The relative intensity of these band components greatly varied among different variants and, for the same virus variant, after exposure to Si3N4. Variations in the relative intensity of the tyrosine doublet are related to environmental pH in the immediate neighborhood of the virion surface and give us the possibility to obtain accurate information on the state of ionization of tyrosine hydroxyl groups in proteins,54 as discussed in detail in the forthcoming section (Section 4.3). An additional band appearing in between the two components of the doublet has been assigned to ring deformation in tyrosinate, namely, the anionic form of tyrosine.55 Moreover, a new band at 959 cm−1 (labeled as Band 22*) was seen in all strains after exposure to Si3N4 powder (cf.Fig. 5b, d and f). The appearance of this new strong signal can be assigned to S–H in-plane bending vibrations in homocysteine.56 The latter observation, together with variations observed in Zone I, supports the hypothesis of post-translational oxidative processes in viral proteins upon exposure to Si3N4 powder. In other words, the S-containing (unburied) residues of HA in all influenza strains are the ones that most promptly adsorb on the Si3N4 surface and undergo fundamental modifications in their thioether groups and terminal structures. A probable scenario for such an efficient reaction might consist of an electrostatic attraction between the Si3N4 surface and the envelope of influenza A virions, as further discussed later in Sections 4.2 and 4.4. Finally, Band 23 at 965 cm−1, assigned to 5-ring deformation in RNA guanine,53 completely disappeared for all strains after exposure to Si3N4 powder (cf. Band 23 in Fig. 6a, c and evs. arrows indicating this missing band in Fig. 6b, d and f). A discussion about the latter spectroscopic feature is given later in Section 4.1.
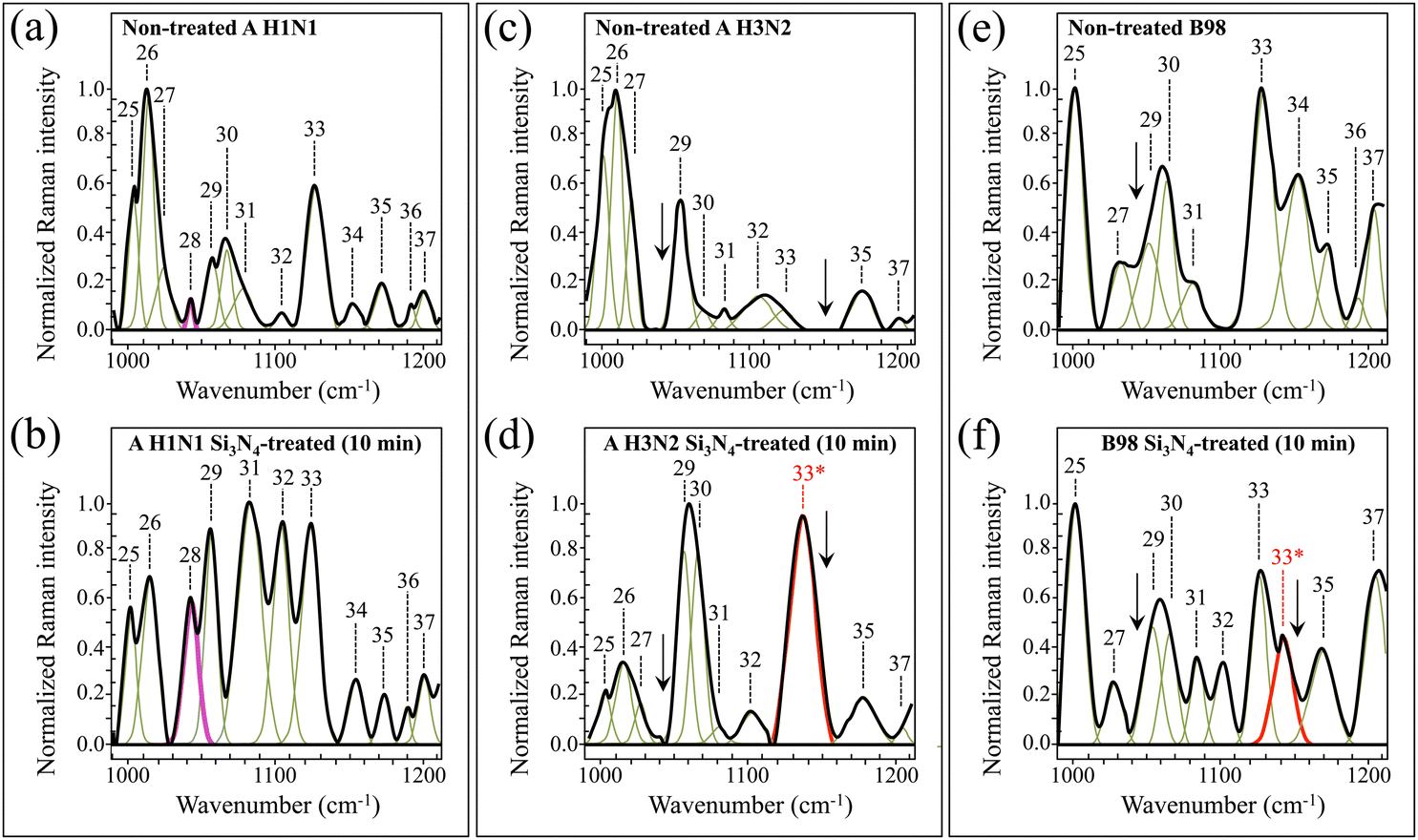
The main characteristics of Zone III (1000–1200 cm−1) resided in the presence of a strong signal at 1043 cm−1 from SO stretching in oxidized methionine (methionine sulfoxide),52 a peculiar characteristic of the Si3N4-exposed A H1N1 strain (cf. Band 28 in Fig. 6a and b and arrows for the same (missing) band in Fig. 6c–f). This band was very weak in the non-treated A H1N1 virions, and its significant increase upon exposure to Si3N4 powder supports the hypothesis of methionine oxidation. On the other hand, exposure of the A H3N2 and B98 strains to Si3N4 promoted the appearance of a new band at 1141 cm−1 (labeled as Band 33* in Fig. 6d and f), which was not seen in the spectrum of the exposed A H1N1 strain (cf.Fig. 6b). The newly appeared Band 33* could be assigned to C–S–C stretching in the thioether configuration of methionine (cf. Table S-III, ESI†).52 Thioether cleavage can be triggered by protonated amino groups, which act as hydrogen bond donors and form strong hydrogen bonds with hydrogen bond acceptor silylamine sites on the Si3N4 surface.57 Note that the potential ability of secondary silylamines to bond to carbon likely involves the possibility of direct links with the methionine methyl group CH3 to form a quaternary amine with a positive charge. The rationale for the above peculiar features regarding methionine and related compounds will be given in Sections 4.2 and 4.4.
Besides the presence of Amide III bands between 1220 and 1270 cm−1 (cf. Bands 30–42 in Fig. 7a–c and Table S-IV, ESI†), the spectral morphology of Zone IV (1200–1400 cm−1) included a number of interesting features. Such additional signals provided insights into differences among non-treated strains and the degradation phenomenon upon contact with Si3N4 particles. The most important spectral characteristic in Zone IV was the tryptophan doublet at 1345 and 1366 cm−1 (Bands 47 and 48 in Fig. 7; arising from C–H bending vibrations and indole ring C–N stretching, respectively).50 This doublet senses surface pH and is related to torsional angles of the tryptophan molecule (as shown later in Zone V), thus providing unique insight into both virions’ surface pH (to be compared to the output of tyrosine doublet analysis from Zone II) and functionality of viral proton channels before/after exposure to Si3N4 powder. Similar to the case of the tyrosine doublet, the intensity ratio of the tryptophan doublet has been studied in detail by several authors.14,16 An interpretation of the Raman behavior of the tryptophan doublet for different viral strains will be given in the forthcoming sections (Sections 4.3 and 4.4). As a further issue seen in Fig. 7b and d, both A-type viral strains presented two new bands (labeled as 39* and 42*, at 1236 and 1278 cm−1, respectively) after exposure to Si3N4. These additional bands were not seen in the Si3N4-exposed B98 strain (cf.Fig. 7f). On the other hand, the exposed B98 strain showed a new signal at 1380 cm−1 (labeled as Band 48* in Fig. 7f), which could not be resolved in neither of the A-type strains. The 39*, 42*, and 48* signals were related to the degradation phenomena of amino acid residues (cf. vibrational origins in Table S-IV, ESI†), as discussed in the forthcoming sections (Sections 4.2 and 4.3). Zone V (1400–1600 cm−1), depicted in Fig. 8 (cf. also Table S-V, ESI†), contained two types of signals intrinsically capable of clarifying the degree of protonation in the aromatic amino acid residues histidine and tryptophan contained in viral proteins. As discussed later, the protonation of these two residues is key to the functionality of the viruses. Bands labeled as 55, 57, and 57* (at 1528, 1575, and 1584 cm−1, respectively) indicate stretching vibrations of the C4C5 bond in the imidazole ring of deprotonated histidine and partially protonated τ and π histidine tautomers, respectively.14 Different tautomeric structures, whose Raman signatures are seen in Zone V through their C4C5 stretching signal, develop in strongly alkaline (i.e., the deprotonated structure) and slightly acidic-to-slightly alkaline (τ and π histidine tautomers) environments. As can be seen in Fig. 8, the intensities of Raman signals from different histidine tautomers, which are directly related to their volume fractions, differed in different influenza strains. Moreover, tautomeric fractions were strongly altered upon exposure to Si3N4 powder. This information adds upon pH assessments using tyrosine and tryptophan doublets, as reported above and further discussed in the forthcoming section (Section 4.3). Another important signal in Zone V is represented by Band 56 (at 1546 cm−1), which arises from C2C3 bond stretching in the pyrrole ring of tryptophan.14 The wavenumber location of this band has been related to the torsional angle, χ2,1, of the indolyl side chain, which is affected by the degree of deprotonation of the tryptophan molecule, and thus depends on environmental pH. The χ2,1 angle in unexposed and Si3N4-exposed strains varied in the intervals 1544–1548 and 1547–1550 cm−1, respectively, thus revealing important differences and fluctuations in both molecular conformation and environmental pH. It was also noticed that the C2C3 stretching signal disappeared in Si3N4-exposed A H3N2 virions (cf.Fig. 8d), this being an experimental evidence hinting to tryptophan ring breaking in this strain under Si3N4 hydrolytic reactions. More details of this point will be given in Section 4.3. Finally, two signals at 1414 and 1598 cm−1 (Bands 49* and 58, respectively) were assigned to symmetric and antisymmetric stretching in the deprotonated –COO− group of homocysteine, thus confirming the presence of this molecule after Si3N4 exposure, as already reported for Zone II. Note that Band 58 is also contributed by NH3 bending in methionine sulfoxide as an additional product of oxidation.
Zone VI (1550–1750 cm−1), shown in Fig. 9 and Table S-VI (ESI†) with respect to its band components, is characteristic of the Amide I vibrational mode as a composite band assembly from different secondary structures of viral proteins (cf. Bands 61–65).58 All three non-treated strains showed similar Raman morphological profiles in their Amide I interval at 1630–1715 cm−1. The A H3N2 and B98 non-treated strains both experienced the most prominent Amide I signal at 1657 cm−1 (i.e., α-helix configuration; cf. Band 62 in Fig. 9c and e, and Table S-VI, ESI†). On the other hand, the non-treated strain A H1N1 presented its strongest signal at 1638 cm−1, namely, in the wavenumber interval of the Amide I β-sheet configuration (cf. Band 61 in Fig. 9a and Table S-VI, ESI†). All non-treated strains showed relatively weak (and partly missing) signals from β-turn configurations (cf. Bands 64 and 65 in Fig. 9a, c and e). The anomaly of an apparently stronger Amide I signal from the β-sheet configuration only in the A H1N1 non-treated strain could be interpreted by taking into consideration the overlap of this signal with the C4C5 stretching mode of biprotonated histidine (imidazolium).14 As explained in more detail later in Section 4.3, the A H1N1 strain presented the most acidic pH at its environmental interface among other strains, which justifies a stronger signal from the imidazolium molecule.
Another striking feature of Zone VI resided in the strong amplification, detected for all strains, of the carbonyl-stretching doublet located at 1727 and 1742 cm−1 upon exposure to Si3N4 powder (cf. Bands 66 and 67 in Fig. 9). Although also contributed by CO stretching in lipids,59 those two signals are likely to be strongly enhanced by carbonyl stretching at the oxidized C8 atom of guanine and adenine RNA purines.60 Such oxidation, only possible due to radical interactions, results in the opening of the imidazole ring to form 2,6-diamino-4-oxy-5-formamidopyrimidine and 4,6-diamino-5-formamidopyrimidine molecules.61,62 Such oxidized compounds are usually referred to as Fapy-guanine and Fapy-adenine (simply FapyGua and FapyAde, henceforth).63,64 Note that the observation of enhanced carbonyl stretching bands as a consequence of imidazole ring breakage also matches the finding of a complete disappearance of Band 23 at 965 cm−1 (cf. above description in Zone II) mainly contributed by 5-ring deformation in RNA guanine.53 Further discussion about the latter spectroscopic feature will be given in the next section (Section 4.1).
4. Discussion
4.1 Raman fingerprints of RNA and its degradation patterns
RNA purine and pyrimidine present distinct markers in the Raman spectra of viruses.11–13,19,21,41,49,53 As shown in Fig. S1 (ESI†), specific Raman signals could be selected to univocally (or, at least, predominantly) mark RNA in the Raman spectrum of viruses. Regarding purines, the 5-ring deformation signal in guanine (at 965 cm−1; Band 23 in Fig. 5 and 10a) and the ring-breathing signal in adenine (at 734 cm−1; Band 9 in Fig. 4 and 10b) were suitable choices for RNA Raman analysis of influenza viruses. Possible pyrimidine markers were located at 760 cm−1 (ring breathing) in cytosine and 802 cm−1 (ring breathing) in uracil (cf. Bands 10 and 13 in Fig. 4 and 5, respectively, and Fig. S1, ESI†). However, only the latter signal, although quite weak in the non-treated strains, is mainly contributed by uracil, while the former suffers severe overlap by a significantly stronger tryptophan band, which gives the preponderant contribution to this signal (cf. Fig. S2, ESI†). In previous work,19 we defined the concept of “Raman genome” in order to emphasize differences among RNA of different viral strains assessed by means of Raman spectroscopy, in comparison with other well-established methods of genomic analysis in virology. It was found that the relative intensity of RNA Raman signals not only depended on the fractions of RNA bases, but also, and in a complex way, on the specific RNA sequences, since the vibrational intensity of each molecule also depends on neighboring conditions. Here, we focus on the impact that hydrolytic reactions at the interface between Si3N4 and virions had on the structure of RNA purines, as summarized in Fig. 10c–h. The guanine signal at 965 cm−1 completely disappeared from the spectrum of all Si3N4-exposed influenza strains (cf. Band 23 in Fig. 10f–h). The selected spectroscopic marker for the adenine base, seen at 738 cm−1 (labeled as Band 9), was found unchanged in the A H1N1 strain, but it almost disappeared in both A H3N2 and B98 strains upon exposure to Si3N4. A different pattern in the above prospect of RNA damage was recorded for the uracil signal located at 802 cm−1 (cf. Band 13 in Fig. 5). This signal was generally weak in the non-treated strains, but tended to increase in intensity and to slightly shift towards higher wavenumbers upon Si3N4 exposure. | ||
| Fig. 10 Structure of RNA purines and their oxidized structures: (a) guanine and FapyGua and (b) adenine and FapyAde; the 5-ring deformation signal from guanine at 965 cm−1 and the ring breathing signal from adenine at around 734 cm−1 were taken as markers of the two RNA bases, respectively (cf. labels in the inset). Both oxidized molecules present carbonyl stretching bands at around 1727 and 1742 cm−1. Band components depicted in (a) and (b) were extracted from deconvoluted spectra in Fig. 4, 5 and 9, and are shown in (c), (d) and (e) for non-treated and in (f), (g) and (h) for Si3N4-exposed A H1N1, A H3N2, and B98, respectively. Note that the 965 cm−1 5-ring deformation band of guanine systematically disappeared in all strains and the carbonyl stretching bands significantly enhanced after exposure to Si3N4 (cf. (f)–(h)), which is spectroscopic proof of 5-ring breakage and degradation into Fapy molecules, respectively. | ||
RNA viruses are the most prone to ammonia attack, because their genome type is more susceptible to degradation, while other more stable genome types present a slower inactivation, mainly driven by degradation of viral proteins.65 The complete or almost complete disappearance of ring vibrations in both guanine and adenine purines, as observed here, obviously arises from breakage of the imidazole ring with formation of the respective formamidopyrimidines (Fapy structures).66 However, ring breakage cannot be simply assigned to a direct interaction with ammonia molecules, but instead requires the occurrence of off-stoichiometric reactions with oxygen and/or nitrogen radicals. Peroxynitrite anions (ONOO−) exhibit pronounced oxidizing properties toward purine moieties, which in turn lead to the production of 8-oxo-7,8-dihydro-2′-deoxyguanosine (8-oxo-dGuo) in a very low yield. Then, the 8-oxo-dGuo molecules further react to form 2,6-diamino-4-hydroxy-5-formamidopyrimidine (FapyGua). Similarly, the 4,6-diamino-5-formamidopyrimidine molecule (FapyAde) is formed as an oxidation product of the adenine base. The structures of FapyGua and FapyAde are schematically shown in Fig. 10a and b, respectively. A strong hint in support of the hypothesized purine oxidation mechanism is given by the significant enhancement of CO stretching signals in the spectral interval 1720–1740 cm−1 (cf.Fig. 10f–h and Bands 66 and 67 in Fig. 9), which are strong Raman signals characteristic of Fapy and other oxidized molecules.
Despite being in contrast to the evidence of ring disruption in purines, the recorded behavior of uracil RNA pyrimidine upon exposure to Si3N4 powder could also be considered as evidence in support of the hypothesis of a radical species effecting RNA degradation. Ring degradation of pyrimidines can only arise from photohydration reactions,67 through a mechanism that requires high-energy radiative and oxygen-independent processes, which are obviously not involved in the hydrolytic reactions studied here. Moreover, Nonoyama et al.68 have reported that both cytosine and uracil pyrimidines are conspicuously unaffected by exposure to peroxynitrite anions, thus supporting the present finding of uracil inertness in a hydrolytic Si3N4 environment. Note, however, that the uracil structure, which primarily exists in the lactam (amide) form in neutral aqueous solution,69,70 is ionized in an alkaline environment to form hydroxyl groups to replace the double-bonded oxygens forming the lactim (imidic acid) structure.71 The hydrolytic reactions taking place at the solid surface of Si3N4 in an aqueous environment induce a robust pH buffering at the biological interface with virions;72–74 according to such reactions, the local pH values become comparable with that of uracil ionization (pK ∼ 9.2). Since uracil lacks aromatic properties, it is prone to amide-imide tautomeric shift.75 Under the high-pH environmental conditions developed at the interface with Si3N4 particles, the uracil structure could thus undergo amide-imidic acid tautomeric shift, a structural change that could in turn explain the observed variations of Band 13 in both intensity and wavenumber shift, as reported above.
4.2 The spectroscopic role of S-containing amino acid residues
In the stream of our previous Raman studies of viral strains,19,21,49,76 this study focused on and confirmed how the molecular symmetry properties of S-containing amino acid residues could be exploited in order to unequivocally classify different viral strains. In particular, we focused here on the low-wavenumber C–S stretching signals of trans and gauche methionine rotamers (cf. Zone I in Fig. 3 and 4). As shown in Fig. 11a, the different molecular symmetry of methionine rotamers leads to clear spectral differences in their vibrational signals. C–S stretching Raman signals extracted from the respective spectra in the spectral zone between 600 and 750 cm−1 are shown for the three investigated influenza strains in Fig. 11b–d (cf. labels in the inset). An examination of C–S band fractions in the non-treated strains revealed remarkable differences, which could be classified according to the fraction of S-containing amino acid rotamers. trans and gauche methionine rotamers share a common C–S stretching band on the CH2 side of the molecule at 653 ± 2 cm−1 (Band 4 in Fig. 4 and Table SI, ESI†). An additional stretching component for C–S + S–C bonds is seen at 669 ± 1 and 640 ± 3 cm−1 (Bands 5 and 3 in Fig. 4 and Table SI, ESI†) in trans and gauche rotameric configurations, respectively. On the other hand, C–S and C–S–C stretching components on the CH3 carboxyl side in trans and gauche configurations can be found at 713 ± 2 and 696 ± 2 cm−1, respectively (cf. Bands 7 and 8 in Fig. 4 and Table SI, ESI†).50,51,77–79 The large variations in both the spectral position and the relative intensity of C–S bond-related Raman bands among different (non-treated) strains reflect compositional differences in the fractions of methionine rotamers, which in turn are the consequence of the different amino acid sequences to which methionine residues link. Such differences in molecular symmetry characteristics are of fundamental importance in virus classification and response to external stress, since they play key roles in a number of functions during the virus lifetime.78,79 The methionine trans-to-gauche ratio, Rt/g, namely, the ratio of the cumulative areal intensity C–S bands from trans and gauche rotamers, can be selected as a parameter that swiftly represents the structural difference between influenza strains. The Rt/g values computed for the three non-treated strains, given in the inset in Fig. 11b–d, showed relatively close (>1) values for the two strains of type A, while the ratio for the type B strain was <1 and 40–50% lower than those of both type A strains. Such a big difference in the Rt/g ratio between type A and type B strains reflects their different chirality characteristics and could be used as an effective fingerprint in type speciation of influenza virus. Methionine residues are strongly hydrophobic and are located at buried sites in the core of globular proteins. Fig. 1e–g emphasizes the location of buried methionine residues in the M1 matrix protein of H1N1, H3N2, and B98 strains, respectively. Nevertheless, they showed large structural variations upon exposure to the Si3N4-surface environment, thus hinting at a strong interaction with Si3N4 hydrolytic products and their radical sub-products, as discussed in Section 4.4. From the present data, it is immediately clear that environmental effects heavily impacted the methionine structure by inducing a scrambling drift in the population ratio of trans and gauche rotamers. The Rt/g value for all three investigated strains was 0.7, lowered by 40–50%, while the B98 strain remained the same as that of the non-treated strain. Additional fingerprints of environmental interaction could also be found in the methionine spectrum of Si3N4-exposed virions. In A H1N1 and B98 strains, a more than two fold increase was recorded in the relative intensity of both C–S and C–S + S–C stretching signals on the CH2 side (cf. Bands 7 and 8 in Fig. 11b). Concurrently, three new or strongly intensified bands appeared at 662, 725, and 1043 cm−1 (cf. also bands 4*, 8*, and 28; the first two in Fig. 4b and the latter in Fig. 6b). According to Torreggiani et al.,52 the above three signals could be assigned to C–S stretching on the CH2 side, C–S stretching on the CH3 side, and SO stretching in methionine sulfoxide, respectively; the first two bands were completely missing in the spectrum of all three unexposed virions, while the latter could be seen with very low intensity only in the A H1N1 strain; however, it increased in relative intensity by more than one order of magnitude after Si3N4 exposure (cf.Fig. 11b). The methionine sulfoxide molecule, which is depicted in Fig. 11e, is a post-translational product of methionine, which forms upon oxidation and occurs as a consequence of environmental stress exposure operated by both non-radical and free radical species. Note that the marked shift and signal intensification of C–S and of C–S + S–C stretching in the sulfoxide molecule as compared to non-oxidized methionine are the consequences of variations in both stiffness and flexibility of chemical bonds upon oxidation. A further proof of the formation of methionine sulfoxide is the appearance or the significant enhancement of two bands at 1414 and 1598 cm−1 (cf. bands 49* and 58 in Fig. 8b), which could be seen in all exposed strains. These bands are assigned to symmetric and antisymmetric stretching of –COO− terminal bonds, respectively (cf.Fig. 11e). The signal at 1598 cm−1 might be overlapped and, thus, be contributed by in-plane bending vibrations of the NH3+ terminal group of methionine sulfoxide.52 Note that vibrational signals from –COO− terminal bonds are also present in the spectrum of homocysteine (Fig. 11f), whose presence in the spectrum of the Si3N4-exposed A H1N1 strain seems to be also substantiated by a relatively strong signal at 959 cm−1 from S–H in-plane bending (band labeled 22* in Fig. 5b, not detectable in the non-treated virus sample; cf.Fig. 11b).52 Note that the A H1N1 strain was the only one showing the sulfoxide SO stretching signal at 1043 cm−1 after exposure to Si3N4, while all strains showed the homocysteine S–H bending signal at 959 cm−1. In other words, only the A H1N1 strain underwent oxidation of the buried methionine residues, while all strains experienced oxidation of environmentally exposed cysteine residues upon interaction with Si3N4 hydrolytic products.
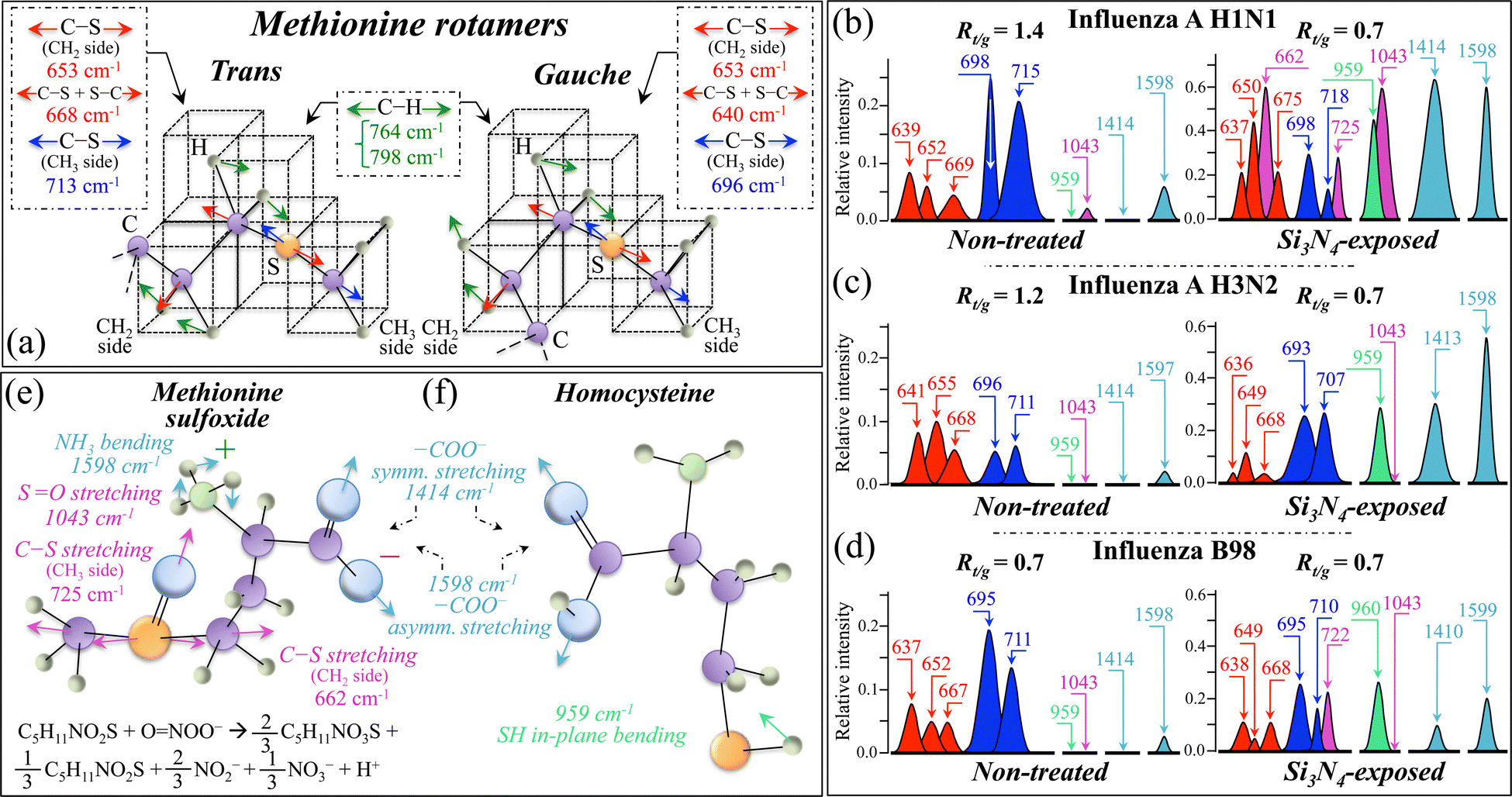 | ||
| Fig. 11 (a) Different molecular symmetries of trans and gauche methionine rotamers leading to different wavenumbers for C–S stretching signals with the same vibrational origin (cf. labels in the inset); on the left side of (b), (c) and (d), band components extracted from the respective spectra in Zone I are plotted for non-treated A H1N1, A H3N2, and B98 strains, respectively. In (e) and (f), the structures and main spectroscopic markers of methionine sulfoxide and homocysteine are shown as oxidation products (cf. labels and chemical reaction with peroxynitrite in the inset). On the right side of (b), (c) and (d), C–S band components extracted from the respective spectra in Zone I are plotted for Si3N4-exposed A H1N1, A H3N2, and B98 strains, respectively, together with spectroscopic markers of the oxidized molecules shown in (e) and (f). Note how C–S band fractions both in non-treated and Si3N4-exposed strains revealed marked structural differences (classified according to the trans–to-gauche ratio, Rt/g; cf. values in the inset). | ||
Oxidation of methionine residues within proteins takes place in the presence of a variety of oxygen and nitrogen radicals, including peroxynitrite, ONOO−.80 The proposed radical reaction causing methionine oxidation at the surface of Si3N4 bioceramics is given as follows:80–82
| C5H11NO2S + ONOO− → 2/3C5H11NO3S + 1/3C5H11NO2S + 2/3NO2− + 1/3NO3− + H+ | (1) |
4.3 Raman signals from aromatic amino acids as environmental sensors
The phenyl ring of tyrosine residues is known to generate a Raman doublet of relatively high intensity at around 850 and 820 cm−1, which has been ascribed to Fermi interaction.83,84 The doublet bands, governed by Fermi coupling, arise from fundamental ring-stretching vibrations (at 850 cm−1) and the first overtone of the phenolic ring deformation mode (at 820 cm−1) (cf. Bands 16 and 14, respectively, in Fig. 5 and 12a). The intensity ratio of such a doublet, RTyr = I850/I820, is related to the hydrogen-bonding state of the phenolic OH group, according to a relationship that was initially quantified by Siamwiza et al.84 and later extended to the non-hydrogen-bonded phenoxyl state by Arp et al.85 A more recent study conducted by Hernandez et al.55 revealed all characteristic Raman markers of tyrosine and tyrosinate phenol rings as a function of environmental pH. In an acidic environment, the tyrosine ratio of fully protonated tyrosine is RTyr <1, then, at increasing pH, RTyr reaches values increasingly larger than 1 for zwitterionic, partly hydrated, and fully hydrated configurations (Fig. 12a–d). Raman analysis using the tyrosine doublet has been quite popular in studying the protein structure of viral strains.21,86–91 Since viral proteins contains tyrosine residues at multiple sites, the Raman spectrum obviously reads an RTyr value that reflects the average hydrogen-bonding state of all tyrosine phenoxyls (i.e., “buried” vs. “unburied” configurations). For tyrosine residues in “buried” configuration, the non-hydrogen-bonded phenoxyl state is often encountered in the tightly packed α-helical subunits of viral capsids.85,86,90 The above observation implies that values RTyr < 1 prove that the majority of tyrosine residues are in a “buried” state.92Fig. 12e shows the band components, the exact wavenumbers and the values of the tyrosine ratio as recorded for the three influenza strains studied in this paper, before and after exposure to Si3N4 aqueous solutions (cf. labels in the inset). As seen, the viral strains differ from each other in their non-treated state, with types A H1N1 and B98 mainly showing hydrophobic and “buried” tyrosine residues (RTyr = 0.8 and 0.9, respectively), while type A H3N2 virions mainly include zwitterionic tyrosine residues (RTyr = 1.3). After exposure to Si3N4 powder dispersed in aqueous solution, all virion types similarly shifted toward a hydrophilic “unburied” configuration, characteristic of high pH values. Upon plotting these results in terms of pH values (Fig. 12f),93 A H1N1 and B98 virions are both found to display surface pH values at around 4, while the A H3N2 stems at neutral pH (∼7). After Si3N4 exposure all strains reached a fully hydrated “unburied” state with environmental pH between 8 and 10. | ||
| Fig. 12 Tyrosine structure and its characteristic Raman doublet (doublet ratio, RTyr = I850/I820) as a function of environmental pH: (a) fully protonated structure in an acidic environment, (b) zwitterionic structure in a neutral environment, and (c) partly and (d) fully hydrated structures in an increasingly alkaline environment. In (e), tyrosine doublets of A H1N1, A H3N2, and B98 strains as extracted from the respective Zone II spectra (Fig. 5) before and after exposure to Si3N4 particles (cf. tyrosine ratios in the inset). Broken lines represent an intermediate ring deformation band of tyrosinate anions located at 842 cm−1 (cf. explanations in Section 4.3). The plot in (f) gives the relationship between the tyrosine ratio, RTyr, and the pH value at the virion surface (from ref. 92). Note how all strains after exposure to Si3N4 revealed high surface pH values as a consequence of irreversible modifications to the tyrosine structure. | ||
An additional characteristic in the spectral zone of the tyrosine doublet was the presence of a relatively strong band in between the doublet (at 842 cm−1), which only appeared in the two type A viral strains at RTyr > 1 (cf. broken-line peak components in Fig. 5 and 12e). In order to explain this additional feature, we hypothesized that high pH leads to deprotonated tyrosine (tyrosinate), which in turn alters the morphology of the Raman doublet with the appearance of an intermediate band. The presence of the 842 cm−1 band thus suggests the coexistence of tyrosine and tyrosinate moieties in the protein structure of influenza type A virions. This interpretation is supported by the fact that the RTyr value observed for strain type A, when the intermediate band appears, is ∼1.25, as reported in the literature for the phenolic OH group acting as both the donor and the acceptor of moderate hydrogen bonds.86,94 In summary, one could state that the presence of the 842 cm−1 band indicates a tendency to deprotonate and is symptomatic of an increased environmental pH. Tyrosine residues (specifically those at position 76), which are conserved in >99% of influenza type A virus strains sequenced to date, are critical for the formation of infectious virus particles.95 Therefore, their deprotonation should be a critical circumstance in their inactivation. Note that the presence of tyrosinate molecules at high pH could be a consequence of higher fractions of “buried” locations for tyrosine residues, which could be deprotonated upon alkalinization while yet remaining dehydrated. In other words, tyrosine residues “buried” in the three-dimensional structure of proteins of influenza A type are damaged, but yet remain “buried”.91 On the other hand, the absence of any intermediate peak between the doublet components in type B virions, despite their high surface pH after exposure to Si3N4 powder, could be a consequence of a higher fraction of environmentally exposed tyrosine residues, which thus become promptly hydrated after being deprotonated by alkalinization.
Raman signals from tryptophan have also been extensively investigated since they, similar to the case of tyrosine, can provide useful information about side chains in proteins.16,96–102 One of the main tryptophan markers lies in the wavenumber interval 1540–1560 cm−1 and arises from stretching of the C2C3 double bond in the indole ring (Fig. 13a). This Raman signal is indicative of the amount of torsional rotation of the indolyl side chain, which in turn links to the degree of deprotonation and thus to environmental pH. Torsional displacements are represented by the torsional angle, χ2,1, of the C2–C3–Cβ–Cα linkage (cf.Fig. 13a). The wavenumber of the C2C3 stretching, νCC, Raman signal correlates with the absolute value of the torsional angle, χ2,1, in the range 60°–120°, according to the following equation:98
| νCC = 1541.8 + [6.9cos(3|χ2,1|) + 1]1.2 | (2) |
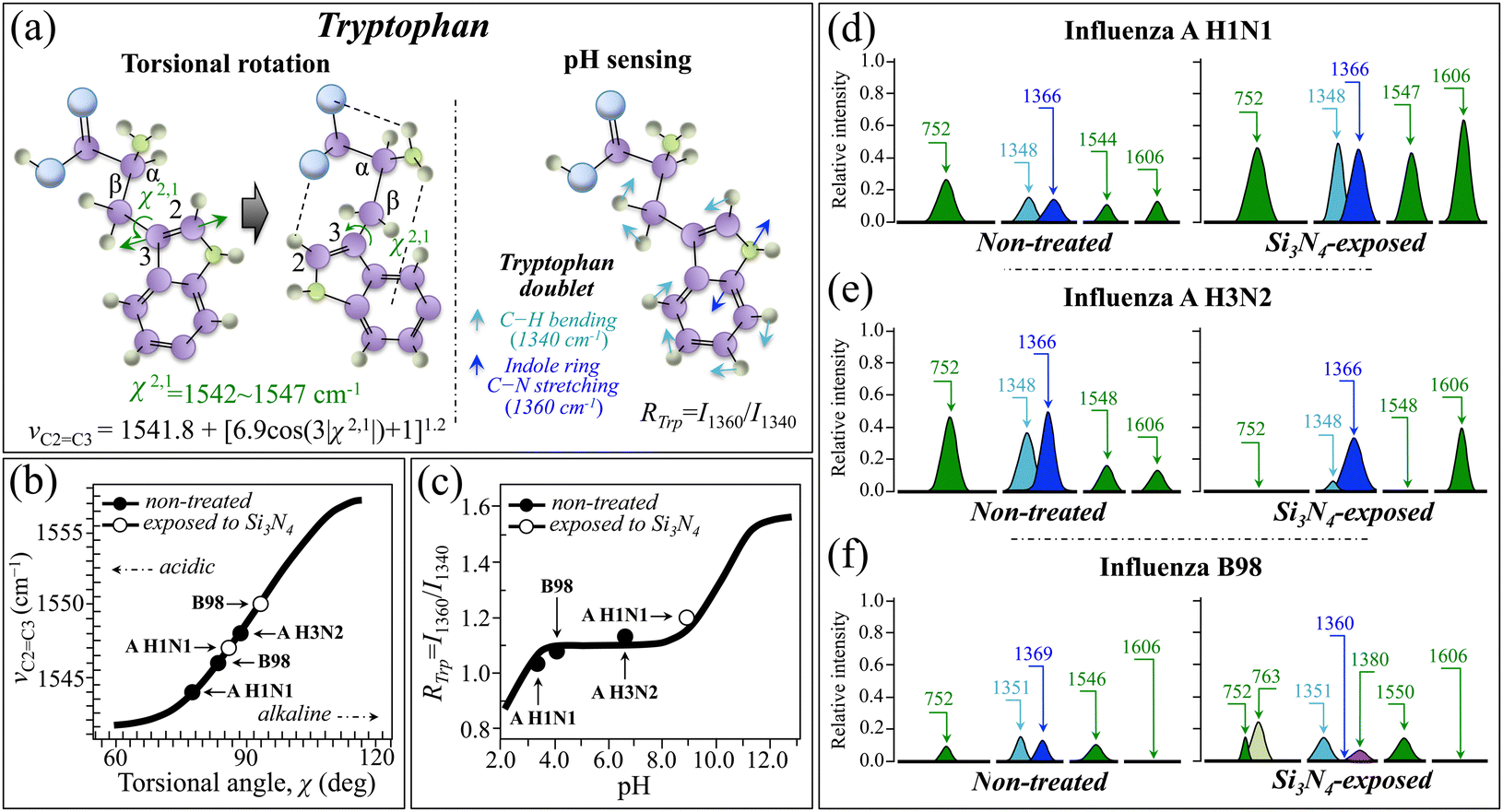 | ||
| Fig. 13 (a) Raman spectroscopic markers of tryptophan: (i) stretching of the C2C3 double bond in the indole ring (wavenumber interval 1540–1560 cm−1), and (ii) Fermi doublet at around 1340 cm−1 (C–H vibrations) and 1360 cm−1 (C–N stretching). Section (b) gives the relationships between molecular torsional displacements, χ2,1, of the C2–C3–Cβ–Cα linkage and the wavenumber of the C2C3 stretching signal (from ref. 97), while section (c) shows the relationship between the tryptophan intensity ratio, RTrp = I1360/I1340, and virions’ surface pH values (from ref. 16). In (d), (e) and (f), C2C3 stretching bands and tryptophan doublets of A H1N1, A H3N2, and B98 strains are shown, respectively, as extracted from spectral Zones IV and V before and after exposure to Si3N4 particles (cf.Fig. 7 and 8, respectively). The computed values are then plotted in (b) and (c). Additional signals of interest are also shown as discussed in the text. | ||
In the Raman spectrum of tryptophan, a Fermi doublet is also present with components at 1340 cm−1 (C–H vibrations) and 1360 cm−1 (C–N stretching) (Fig. 13a).102 Such a doublet represents an additional sensor for the local environment around the indolyl ring: the tryptophan intensity ratio, RTrp = I1360/I1340, increases with decreasing pH (i.e., with increasing hydrophobicity) and vice versa. The relationship between RTrp and pH was precisely established in a basic study conducted by Harada et al.16 and is shown in Fig. 13c. Fig. 13d–f show the band components and the exact wavenumbers for the C2C3 stretching mode (expected in the wavenumber interval 1540–1560 cm−1) and the Fermi doublet (expected at around 1340/1360 cm−1) as recorded for the three influenza strains studied in this paper, before and after exposure to Si3N4 aqueous solutions (cf. labels in the inset). The computed values for the torsional angle, χ2,1, (from experimental νCC values) and pH (from experimental RTrp values) were retrieved for different viral strains, as shown in Fig. 13b and c, respectively. Notably both plots are basically consistent with the tyrosine plot in Fig. 12f, showing an increasing alkaline order and consistent pH values at the surface of non-treated A H1N1, B98, and A H3N3 virions (Fig. 13b and c, respectively). Additional proof for Raman pH sensing could be found upon monitoring the intensity of the Raman band at 752 cm−1 (cf. Band 10 in Fig. 4; from breathing vibration of the indolyl ring),102 whose intensity is inversely related to the hydrophobicity of the local ring environment.99 The trend for the latter parameter was consistent with the above pH estimations made by using both tyrosine and tryptophan doublets: the intensity for the 752 cm−1 band was lower in the spectra of A H1N1 and B98, which possessed lower surface pH (i.e., higher hydrophobicity) as compared to A H3N2. Upon exposure to Si3N4, all three tryptophan pH-sensitive sensors consistently indicated a shift to an increasingly alkaline environment. However, after exposure to Si3N4, only the A H1N1 strain maintained a spectral morphology fully compatible with the above-discussed algorithms, while several anomalies were noticed for both A H3N2 and B98 strains, as follows:
(i) The C2C3 stretching band was missing in the A H3N2 strain (cf. missing Band 56 as emphasized by an arrow in Fig. 8d).
(ii) In the Fermi doublet of the A H2N3 strain, the lower wavenumber component (seen at 1348 cm−1) was so low in intensity to lead to unrealistically high values of the RTrp ratio (cf. Band 47 in Fig. 7d).
(iii) In the Fermi doublet of the B98 strain, the low-wavenumber component (seen at 1351 cm−1) was maintained at wavenumber/intensity similar to the non-treated state, but the high-wavenumber component completely disappeared, while a new band component appeared at around 1380 cm−1 (cf. Band 48* in Fig. 7f).
(iv) The indole ring-breathing band at 752 cm−1 completely disappeared in the spectrum of the exposed A H3N2 strain (cf. absence of Band 10 as emphasized by an arrow in Fig. 4d).
A consistent explanation for all these characteristics could be given by considering the impact that the presence of radical species could have on the tryptophan structure.103 The tryptophan spectral markers after exposure to Si3N4 unequivocally hints at tryptophan residues having been heavily damaged in their original structure for both A H2N3 and B98 strains. This conclusion can be drawn because a complete or almost complete vanishing is observed for the indole ring vibration at 752 cm−1 (cf.Fig. 13e and f). In addition, the conspicuous disappearance of the stretching signals of the C2C3 bond (in the range of 1540–1560 cm−1), concurrent with a remarkable increase in intensity of the band at 1606 cm−1 (contributed by C–C stretching in the benzene rings of different aromatic amino acids; cf. also Table SVI, ESI†),102 points to breakage of the indole ring and could be interpreted as proof of the formation of degradation products (e.g., N-formylkynurenine).104 For exposed A H1N1 and A H3N2, additional proof in support of the formation of N-formylkynurenine could be found in the appearance or significant intensification of two new bands at 1239 and 1685 cm−1 (labeled as 39* and 66, respectively), both of which can be assigned to vibrations of the N-formyl group and can superimpose the amide regions.104 The appearance of a new band at 1380 cm−1 (Band 48* in Fig. 7f), only seen for the Si3N4-exposed B98 strain, could indicate the presence of strongly deprotonated tryptophan or of an intermediate compound in the tryptophan degradation pathway towards the formation of N-formylkynurenine.24,29 In this context, the significant broadening of the high-wavenumber components of the tryptophan doublet (seen at 1366 cm−1) in the exposed A H3N2 strain also appears to be a consequence of this band incorporating additional spectral components from kynurenine.
The pKa of the side-chain amino acid histidine (∼6) is the closest to physiological pH among all amino acids.105,106 This characteristic makes histidine extremely sensitive to even small changes in environmental pH, whose structural variations produce charged states reflecting a series of tautomeric structures (Fig. 14a). At low pH, both imidazole nitrogens are in the protonated state to give a structure referred to as cationic imidazolium (Fig. 14a, top). Near neutral pH, two uncharged rotameric structures can be found, namely, the Nε2-protonated τ and the Nδ1-protonated π tautomers (cf.Fig. 14a, middle). With further increasing pH (mildly alkaline), the backbone nitrogen becomes first deprotonated to give anionic histidine, while conserving a neutral side chain in either the τ or the π tautomeric state. In addition, at higher pH, the imidazole ring loses additional protons to become an imidazolate ion (Fig. 14a, bottom).106,107 Despite the high sensitivity to pH, the Raman bands of histidine tautomers are generally relatively weak as compared with those of other aromatic amino acids. According to Takeuchi,14 a powerful Raman structural marker available to unveil tautomeric conformation, hydrogen bonding state, and hydrophobic interaction of histidine residues can be found in the wavenumber interval 1520–1640 cm−1 and arises from C4C5 stretching vibrations. This vibrational marker senses the presence of hydrogen at the nitrogen sites of the imidazole ring. Fig. 14b compares the C4C5 stretching wavenumbers in various histidine tautomers. As seen, the wavenumber of the C4C5 signal increases with increasing number of protonated nitrogen atoms, namely, with decreasing pH. In Fig. 14c, d and e, intensities are compared for the C4C5 Raman components of different tautomers in the spectra of A H1N1, A H3N2, and B98 strains, respectively. Information that can be deduced from this analysis is in line with the outputs of the Raman behavior of tyrosine and tryptophan bands, as described above. Regarding non-treated strains, A H1N1 was the strain with the strongest Raman signal at 1638 cm−1 (93%; Fig. 14c), which symbolizes its highest level of surface acidity among the studied strains (cf. also Fig. 12f and 13c). On the other hand, the lowest intensity of the 1638 cm−1 imidazolium signal, and thus the strongest shift toward surface basicity, was recorded in the non-treated A H3N2 strain (61%; Fig. 14d), again in agreement with analysis based on tyrosine and tryptophan Raman signals (cf. also Fig. 12f and 13c). The non-treated B98 strain maintained an intermediate level of surface pH with its 71% fraction of biprotonated histidine side-chain residues. After exposure to Si3N4 aqueous solution, all strains shifted towards alkaline surface-pH values, with their Raman spectra showing a decrease in the relative intensity of the imidazolium C4C5 high-wavenumber band with respect to the same signal from both neutral and imidazolate tautomers. The alkaline shift appeared somewhat related to the initial degree of surface acidity with the A H1N1 and A H3N2 strains, i.e., the most and least acidic strains in the non-treated state, respectively, yet maintaining the lowest and the highest pH levels, respectively, after Si3N4 exposure (cf. tautomer fractions in Fig. 14c–e). An additional interesting feature was that the A H3N2 strain was the only strain to display a relatively strong signal from the π tautomeric state after exposure to Si3N4 aqueous solution (i.e., the signal at 1584 cm−1; cf.Fig. 14d). Although the present Raman analysis cannot explain the reason(s) for such peculiarity, it is believed that it is a consequence of the peculiar arrangement of histidine in the M2 channel,108 as further discussed in the next section. Note that cationic histidine can serve as a general acid and hydrogen bond donor, with its deprotonation chemistry combining tautomerization with ring flips, as a consequence of interconversion of protonated and unprotonated nitrogens and according to protein function.108
 | ||
| Fig. 14 (a) Histidine tautomeric structures: biprotonated (cationic) imidazolium (top) at low pH, uncharged Nε2-protonated τ and Nδ1-protonated π tautomers (middle) at near neutral pH, and deprotonated (anionic) imidazolate (bottom) at high pH. In (b), plot of the C4C5 stretching marker of tautomeric conformation in the wavenumber interval 1520–1640 cm−1 (according to ref. 14). In (c), (d) and (e), C4C5 Raman signals of different tautomeric structures are compared as extracted from the spectra of A H1N1, A H3N2, and B98 strains, respectively (cf.Fig. 8 and 9). Fractions of tautomeric structures are computed as areal fractions and shown in the inset. The information deduced from tautomeric analysis of histidine is compared with the outputs of similar analysis for tyrosine and tryptophan, as described in the text. | ||
In summary, Raman markers from three distinct aromatic amino acid residues, namely, tyrosine, tryptophan, and histidine, consistently provided clear hints about molecular-scale differences at the biological interface of different influenza viral strains, as well as their modifications upon short-term exposure to Si3N4 powder in aqueous solution. Such differences have a profound impact on virus/cell interaction and on the successive conduction mechanisms of biological proton channels that are fundamentally important for the virus life cycle. These points are further discussed in the next section.
4.4 Molecular scale mechanisms of the Si3N4 “catch and kill” effect
The present paper, in line with our previously published studies on silicon nitride bioceramics,49,91,94,123 demonstrates that this compound in the solid state can operate, in the presence of water, a competitive binding mechanism against the HA trimer. In particular, the originality of the present study lies in the fact that the proposed competitive binding mechanism unambiguously applies to influenza strains with quite different HA trimeric structures. Note also that, as discussed in more detail later in this section, silicon nitride also dramatically alters the environmental pH at the virion/solid interface, thus irreversibly altering the successive step of conformational change needed for membrane fusion and requiring endosome acidification (cf. above point (ii)). Fig. 15a and b show the chemical structure of sialic acid and a schematic draft of the interaction between influenza virions and the sialic acid terminals of glycoproteins in the cellular plasma membrane, respectively. In a similar way, Fig. 15c and d schematically describe the structure of surface silanols (deprotonated by pH alkalinization at the Si3N4 surface) and their interactions with the influenza virions, which mimics the role of sialic acid in cells according to the proposed competitive binding mechanism. An additional attraction spot is also proposed in Fig. 15e, which configures nitrogen vacancy sites (charging 3 +) in their interaction with deprotonated histidine tautomers. The latter binding mechanism is determined by the pronounced structural modifications of the histidine structure revealed by Raman analysis after contact with the highly alkaline interface of Si3N4 powder (cf.Fig. 14). Note, finally, that, besides the outputs of the Raman analysis (further discussed in the following), the presence of a strong electrostatic attraction between influenza virions and Si3N4 micrometric grains is also supported by the fact that a quite small fraction of ceramic particles in solution is capable of interacting within a short interval of time with almost the totality of virions, as demonstrated by the immunochemistry data in Fig. 2.
 | ||
| Fig. 15 (a) Chemical structure of sialic acid and (b) schematic draft of the interaction between influenza virions and the sialic acid terminals of glycoproteins in the cellular plasma membrane; (c) structure of surface silanols (deprotonated by pH alkalinization at the Si3N4 surface) and (d) their interactions with the influenza virions, which mimics the role of sialic acid in cells according to the proposed competitive binding mechanism. In (e), a nitrogen vacancy site (charging 3+) interacts with a fully deprotonated histidine tautomeric molecule as developed at the highly alkaline interface of Si3N4 powder. | ||
 | ||
| Fig. 16 (a) Schematic draft of HA/silanol locking, namely, the proposed “catch” mechanism, and hydrolytic elution of NH3 gas and NH4+ ions with strong environmental alkalinization and formation of nitrogen radicals at the virion/solid interface, namely, the proposed “kill” mechanism. In (b), a schematic draft is shown of the M1 protein linking the viral lipid envelope (containing HA) to the nucleocapsid RNP. The M1 methionine-aromatic motif, which plays a fundamental role in protein–protein binding interactions, could be disrupted and M1 destabilized upon interaction with peroxynitrite (cf. oxidation reaction to form methionine sulfoxide in (c)). The schematic drafts in (d) and (e) show the molecular conformations of tryptophan 41 and histidine 37 residues in the AM2 channel in alkaline and acidic environments, respectively. In the former case (d), histidine is in the imidazolate state and the completely deprotonated and strongly negatively charged rings lock the M2 channel. On the other hand, at low pH (e), the protonated histidine 37 residues flip away from each other and allow proton conductivity in the AM2 ion channel. The strong environmental alkalinization induced at the virion surface by Si3N4 hydrolysis is thus expected to lock the M2 proton channels, thus impeding viral reproduction and propagation. | ||
A further important aspect of the homolytic cleavage of Si–N bonds at the solid surface of Si3N4 is the concurrent formation of free electrons (cf.Fig. 16a). Free electrons can split water molecules and react with adsorbed oxygen to yield oxygen radical anions and other highly oxidative protonated radicals. The availability of free electrons enables oxidation of NH3 into hydroxylamine molecules (NH2OH), which represents the starting point for the formation of nitric oxide (NO), the concurrent production of three hydrogen protons, and the liberation of three additional free electrons. NO radicals then promptly react with oxygen leading to additional radical species such as nitrogen dioxide (NO2), nitrite (NO2−) and nitrate anions (NO3−), higher oxides (N2O3), and peroxides (peroxynitrite, ONOO−, and its conjugate peroxynitrous acid, ONOOH). Among the above-mentioned reactive nitrogen radicals, ONOO− is by far the chemical species most lethal to virions.124
In summary, the above-discussed three chemical events, namely, initial HA/silanol locking (i.e., the above-discussed “catch” mechanism), hydrolytic elution of NH3 gas and NH4+ ions, strong environmental alkalinization at the virion/solid interface, and successive formation of nitrogen radicals poisoning the virions represent the main circumstances behind the observed antiviral behavior of Si3N4. Such a set of events are schematically shown in Fig. 16a. Highly spectrally resolved Raman analysis has clearly shown the consequences at the molecular level of the effect of ammonia and nitrogen radicals on influenza virions, with slightly different details but equal effectiveness against both A- and B-type strains. As shown in Fig. 10–14 and discussed in the previous sections, the main observed structural damage is related to structural degradation of RNA purines, methionine rotameric scrambling, formation of sulfhydryl and ionized carboxyl groups, and deprotonation/torsional deformation of tyrosine, tryptophan, and histidine molecules. Structural damage to key amino acids is discussed in some further detail below.
Key methionine residues have long been known to play a fundamental role in viral propagation of influenza virus by functioning in its biosynthesis. It was also reported that their oxidation drastically interferes with viral propagation.125 The strong propensity of methionine to interact with aromatic-containing residues, including tryptophan, tyrosine, and phenylalanine, is key in viral assembly. The so-called methionine-aromatic motif is prevalent in many viral protein structures, is common to all types of influenza viruses, and acts as a stabilizer in protein–protein binding interactions.126 Accordingly, methionine oxidation, as observed in this study upon surface contact with Si3N4 powder, is expected to disrupt key methionine-aromatic motifs and destabilize protein binding interactions.
M1 is the most abundant protein in the influenza virus particles and plays a critical role in their assembly and budding processes.127,128 As schematically shown in Fig. 16b, M1 juxtaposes between the viral lipid envelope and the nucleocapsid, thus interacting with the cytoplasmic tail of transmembrane viral proteins (i.e., HA, NA, and the M2 channel) on the outer side and the viral nucleocapsid, namely, the viral ribonucleoprotein (RNP) on the inner side. These interactions are fundamental in triggering the budding process responsible for both formation and release of virus particles. Ali et al.129 demonstrated that M1 specifically interacts with the transmembrane domain of influenza HA at the initial stage of the budding process. Gomez-Puertas et al.130 confirmed this hypothesis by showing that the M1 protein, even in the absence of viral polypeptides other than HA, can assemble into virus-like budding particles, which are then released into the culture medium. These results suggest that co-expression of the HA glycoprotein is a key-factor in modulating self-association and membrane-binding properties of the M1 polypeptide. Other authors have later reported results consistent with the above interpretation of the M1 protein functionality, including the interaction of methionine-rich hydrophobic clusters at the interfaces between C-terminal domains along the M1 oligomer,131,132 and cryo-microscopy shows that the association between most of the M1 structure and the virus membrane occurs through electrostatic interactions.133 Among a number of key-locations in the M1 structure of influenza A virus, the methionine 165 residue was reported to fundamentally contribute to the nuclear import and to be essential for viral replication.134 As mentioned above, the influenza M1 protein constitutes a layer under the viral lipid membrane and consists of the N-domain and the C-domain connected by a loop region including the sequence arginine 163, glutamine 164, methionine 165, and valine 166 (referred to as the RQMV motif).133 M165 stabilizes the structure of M1 protein and affects its interaction with other cellular proteins. Among several other functions, M1 binds to the cytoplasmic tails of HA and serves as a docking site in the interaction between M1 and nucleoproteins, which mainly relies on the positive and negative charges of the two counterparts (cf.Fig. 16b).135,136 Moreover, inside the viral envelope, M1 provides structural stability to the virion and bridges the interactions between the viral lipid membrane and the ribonucleoprotein core. M165, which is not transported into but stays near the nucleus, once oxidized, hinders the transport of clustered mitochondria homolog proteins and impedes the successive translocation of chromosome region maintenance 1 proteins, thus blocking viral replication.134
Another key position for methionine residues in the M1 matrix protein is methionine 43. The amino acid position 43 is located in the N-terminal domain and folds into the third helix of the M1 protein.137,138 Based on data from the genome sequence database, the methionine amino acid at position 43 of the M1 protein is highly conserved among influenza A viruses.139 This fact strongly hints at the importance of methionine 43 in the viral life cycle. In other words, besides the above-discussed functions of the M1 protein, which include its roles in viral assembly associated with RNP and RNA, and the control of RNP nuclear import and export, the M1 protein also contributes through the methionine 43-residue to the pathogenicity of influenza virus. Although some rare mutations showing a different amino acid at the 43-position yet retain high pathogenicity and replication capacity, substitutions at this key position affect the fundamental functions of the M1 protein.
According to the above reasoning, the formation of methionine sulfoxide, which we have observed in the Raman spectrum of Influenza A H1N1, if extended to the methionine 165 residue in the RQMV motif and the to methionine 43 residue in the N-terminal domain of the M1 matrix protein, is expected to lead to destabilization and dysfunction of the viral matrix protein M1. The highly volatile peroxynitrite molecules, which form in the cascade reactions originating from Si3N4 hydrolysis, could easily reach all buried methionine positions in the virion structure and compromise their functions. Fig. 16c schematically shows the chemical reaction between methionine and peroxynitrite (or peroxynitrous acid) molecules to form methionine sulfoxide, additional nitrite and nitrate radicals, and hydrogen ions.140
Another important spectroscopic aspect, which directly relates to structural changes in aromatic amino acid residues, is the presence of deprotonated configurations of tryptophan and the tautomeric modifications of histidine residues in the structure of the M2 channel in influenza A virions (AM2). The AM2 protein forms a homotetramer ion channel in the virion lipid membrane, which is specific for proton conductance and is activated by low pH with a transition midpoint at pH 5.7.15 The pH regulation in the golgi vesicular compartments is necessary for the protection of the acid-sensitive HA trimer from denaturing during transport of glycoproteins to the host cell surface prior to virus assembly.141–143
In influenza A type the polypeptide chain of the AM2 protein consists of 97 amino acid residues, which include a putative 19-residue single transmembrane domain (residues 25–43) positioned between the N- and C-terminal hydrophilic regions.144 The transmembrane domain is rich in hydrophobic residues including tryptophan 41 residues with rings external to the channel; it also contains histidine 37 residues with rings oriented internally to the channel. Fig. 16d schematically shows tryptophan 41 and histidine 37 locations with respect to the AM2 channel, drawn on the basis of the rigorous plot of the M2 channel shown in Fig. 1h and i for H1N1 and H3N2, respectively. As previously discussed, the imidazole group of histidine is positively charged, polar, and hydrophobic at pH = 5, while it turns into neutral, apolar, and hydrophobic at pH = 7.4.145 At higher pH, the imidazolate state occurs with the aromatic ring completely deprotonated and strongly negatively charged (cf.Fig. 14a). As schematically shown in Fig. 16d and e, the four histidine 37 residues are sensitive to environmental pH and obey a ring-flip-assisted imidazole deprotonation mechanism, which regulates proton conductivity in the AM2 ion channel.15 The channel is in its closed state at high pH, with the four histidine residues forming a flat and stacked structure preventing the formation of the hydrogen-bonded water chain that conducts protons (Fig. 16d). On the other hand, the AM2 channel turns into a conducting state at low pH, with the imidazolium residues acquiring hydrogen protons, promptly moving away from each other under the effect of electrostatic repulsion among their positively charged imidazole rings, and thus opening the channel to a flow of water molecules that in turn transports protons (Fig. 16e).146–150 The M2 protein of influenza B (BM2) has also been reported as consisting of a tetrameric proton channel embedded in the lipid membrane of B-type virions (cf.Fig. 1j).151 BM2 is yet less known with respect to its complete structure and functions as compared to AM2, and no drug is so far available for its inhibition. However, it is known that BM2 conserves the proton-selective residue histidine and the gating residue tryptophan.152,153 Mutation of histidine 19 in BM2 abolishes proton conduction154 exactly in the same way as mutation of histidine 37 in AM2 disrupts proton selectivity and acid activation.155–157 Nevertheless, despite the structural and functional similarities, AM2 and BM2 yet exhibit important differences in their proton conduction profiles. BM2 was found to conduct protons at twice as fast as AM2.158 Moreover, BM2 is capable of conducting protons outward, whereas AM2 is not.159 As a consequence of these functional differences, histidine 19 in BM2 protonates with a significantly lower proton-dissociation equilibrium constant as compared to histidine 37 in AM2.160
Raman data on aromatic residues discussed in Fig. 12–14 consistently showed that exposure to Si3N4 altered the surface pH of influenza virions causing a significant shift towards alkaline values, independent of strain being A or B type. In such a high-pH environmental situation, the four internal histidine residues form a strong edge-face π-stacked structure, preventing the formation of a hydrogen-bonded water chain to conduct protons. In other words, environmental alkalinization by Si3N4 hydrolytic reactions tightly locks either AM2 or BM2 channels in a configuration similar to that depicted in Fig. 16d. Such ionic barrier hampers the proton transport function of both types of M2 channels and, with it, precludes acidification inside the virions and viral uncoating. While the scientific community is yet waiting for complete elucidation of the three-dimensional BM2 structure to facilitate drug design and inhibit influenza B virus, the present Raman spectroscopic data confirmed that histidine 37 (in AM2) and histidine 19 (in BM2) residues could be completely deprotonated and the M2 channel completely deactivated upon short-term exposure to micrometric Si3N4 powder, a powerful and widely effective solid-state inactivator of influenza viruses.
Note finally that, in previous studies on viral inactivation, we have compared Si3N4 with different antipathogenic or inert compounds such as aluminium nitride, alumina/zirconia, and metallic copper.161,162 Such comparisons demonstrated that the “catch and kill” mechanism is peculiar to Si3N4. More importantly, the high antiviral effect of Si3N4 does not require pushing the particle size to a nanometric level. Accordingly, despite its high antiviral effectiveness, Si3N4 is the only so far known inorganic compound that, effective on a micrometric size, does not damage eukaryotic cells.74
5. Conclusions
In the present study, we have investigated the structural differences of influenza viruses of types A and B by means of highly spectrally resolved Raman spectroscopy. Within the main structural motifs, which were conserved in all influenza strains, we located fundamental differences in methionine rotameric fractions, tyrosine hydration levels, tryptophan torsional configurations, and histidine tautomeric structures, which are peculiar to each viral strain and crucially define its function in the virus life cycle. This study emphasizes the importance of Raman multiomic analysis of viruses in clarifying the details of their molecular structures that could not be accessible by other analytical techniques. The Raman spectrum could also capture fine structural variations that enable the unfolding of mechanisms of viral inactivation, thus providing a unique path to develop drugs and vaccines with high efficiency.The present study also confirmed Si3N4 as a potent solid-state virus inactivator, which merges the effects of competitive HA binding, oxidation and thioether cleavage of methionine residues, M2 channel deprotonation, and RNA purine damage. The pronounced structural modifications of HA, RNA, M1 and M2 channels, unequivocally revealed by large variations in the Raman spectra, provided the rationale for the mechanisms by which Si3N4 particles almost instantaneously deactivate influenza virions, independent of their type/subtype. Control experiments conducted by following exactly the same procedure, except for the lack of a minor fraction of Si3N4 powder in solution, proved that the molecular changes observed in the presence of powder in solution could only be ascribed to Si3N4-particle/virion chemical interactions. The initial action of competitive binding displayed by deprotonated silanols, which mimic cellular sialic acid sites, greatly enhanced the probability of particle/virion encounter, while the following elution of NH3 and nitrogen radical molecules poisons the virions. Since the inactivation kinetics was the same among different influenza viruses, the interaction between Si3N4 particles and virions was figured out as a “catch and kill” sequence. From a technological point of view, electrical attraction of virions towards the inactivating Si3N4 particles appeared to be a key factor since only a small volumetric fraction of particles sufficed to fully inactivate a high concentration of viral strains. Finally, it should be noted that a large part of the structural information obtained here is largely invisible to conventional techniques of virus analysis. This demonstrates the unique ability of Raman spectroscopy to elucidate functionally important aspects of viral structure and inactivation.
Author contributions
Conceptualization, G. P., T. A., T. Y., and O. M.; data curation, Y. Y., E. O., M. N., M. S.-Y., T. A., S. I., W. Z., K. H., K. O., and O. M.; formal analysis, G. P., Y. Y., and W. Z.; funding acquisition, T. A. and T. Y.; methodology, G. P., E. O., M. N., and M. S.-Y.; project administration, G. P.; resources, K. H., K. O., and O. M.; software, Y. Y.; visualization, G. P.; writing – original draft, G. P.; and writing – reviewing and editing, W. Z.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
Giuseppe Pezzotti is a consultant of Denka Co. Ltd. Research Center and YKK Corporation. Other authors declare that they have no conflicts of interest.Acknowledgements
The JSPS KAKENHI Grants-in-Aid for Scientific Research (C) (23K09359), the Japanese Association for Dental Science, Sponsored research 2022, and the Shimadzu Diagnostics Corporation Open Innovation NeyeS 2022 supported this research. The authors thank Mr Y. Yamamoto and Mr N. Tomari from the Kyoto Integrated Science & Technology Bio-Analysis (Shimogyo-ku, Kyoto, Japan) for their support in the use of the LabRAM ARAMIS Raman device.References
- A. J. Hay, V. Gregory, A. R. Douglas and Y. P. Lin, The evolution of human influenza viruses, Philos. Trans. R. Soc. London, Ser. B, 2001, 356, 1861–1870 Search PubMed.
- V. N. Petrova and C. A. Russell, The evolution of seasonal influenza viruses, Nat. Rev. Microbiol., 2018, 16, 47–60 Search PubMed.
- J. Piasecka, A. Jarmolowicz and E. Kierzek, Organization of the influenza A virus genomic RNA in the viral replication cycle – structure, interactions, and implications for the emergence of new strains, Pathogens, 2020, 9, 951 CrossRef CAS PubMed.
- R. P. Chauhan and M. L. Gordon, A systematic review analyzing the prevalence and circulation of influenza viruses in swine population worldwide, Pathogens, 2020, 9, 355 CrossRef CAS PubMed.
- K. Imai, K. Tamura, T. Tanigaki, M. Takizawa, E. Nakayama, T. Taniguchi, M. Okamoto, Y. Nishiyama, N. Tarumoto, K. Mitsutake, T. Murakami, S. Maesaki and T. Maeda, Whole genome sequencing of influenza A and B viruses with the MinION Sequencer in the clinical setting: a pilot study, Front. Microbiol., 2018, 9, 2748 CrossRef.
- J. Fontana, G. Cardone, J. B. Heymann, D. C. Winkler and A. C. Steven, Structural changes in influenza virus at low pH characterized by cryo-electron tomography, J. Virol., 2012, 86, 2919–2929 CrossRef CAS.
- A. K. Harris, G. Cardone, D. C. Winkler, J. B. Heymann, M. Brecher, J. White and A. Steven, Influenza virus pleiomorphy characterized by cryoelectron tomography, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 19123–19127 Search PubMed.
- Q. J. Huang, K. Song, C. Xu, D. N. A. Bolon, J. P. Wang, R. W. Finberg, C. A. Schiffer and M. Somasundaran, Quantitative structural analysis of influenza virus by cryo-electron tomography and convolutional neural networks, Structure, 2022, 30, 777–786 Search PubMed.
- Z. Zhu, R. You, H. Li, S. Feng, H. Ma, C. Tuo, X. Meng, S. Feng and Y. Peng, Multi-omics data integration reveals the complexity and diversity of host factors associated with influenza virus infection, PeerJ, 2023, 11, e16194 CrossRef PubMed.
- L. Lecoq, M.-L. Fogeron, B. H. Meier, M. Nassal and A. Boeckmann, Solid-state NMR for studying the structure and dynamics of viral assemblies, Viruses, 2020, 12, 1069 CrossRef CAS.
- G. J. Thomas, Jr., Raman spectroscopy and virus research, Appl. Spectrosc., 1976, 30, 483–494 CrossRef.
- G. J. Thomas, Jr., Laser-excited spectra of ribosomal RNA, Biochim. Biophys. Acta, 1970, 213, 417–423 CrossRef PubMed.
- G. J. Thomas, Jr., G. C. Medeiros and K. A. Hartman, The dependence of Raman scattering on the conformation of ribosomal RNA, Biochem. Biophys. Res. Commun., 1971, 44, 587–592 CrossRef.
- H. Takeuchi, Raman structural markers of tryptophan and histidine side chain proteins, Biopolymers, 2003, 72, 305–317 CrossRef CAS PubMed.
- A. Okada, T. Miura and H. Takeuchi, Protonation of histidine and histidine-tryptophan interaction in the activation of the M2 ion channel from influenza A virus, Biochemistry, 2001, 40, 6053–6060 CrossRef CAS PubMed.
- I. Harada, T. Miura and H. Takeuchi, Origin of the doublet at 1360 and 1340 cm−1 in the Raman spectra of tryptophan and related compounds, Spectrochim. Acta, 1986, 42A, 307–312 CrossRef CAS.
- J. Lukose, A. K. Barik, N. Mithun, M. Sanoop Pavithran, S. D. George, V. M. Murukeshan and S. Chidangil, Raman spectroscopy for viral diagnostics, Biophys. Rev., 2023, 15, 199–221 CrossRef.
- J. Ye, Y.-T. Yeh, Y. Xue, Z. Wang, N. Zhang, H. Liu, K. Zhang, R. Ricker, Z. Yu, A. Roder, N. Perea Lopez, L. Organtini, W. Greene, S. Hafenstein, H. Lu, E. Ghedin, M. Terrones, S. Huang and S. X. Huang, Accurate virus identification with interpretable Raman signatures by machine learning, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2118836119 CrossRef CAS.
- G. Pezzotti, F. Boschetto, E. Ohgitani, Y. Fujita, M. Shin-Ya, T. Adachi, T. Yamamoto, N. Kanamura, E. Marin, W. Zhu, I. Nishimura and O. Mazda, Raman molecular fingerprints of SARS-CoV-2 British variant and the concept of Raman barcode, Adv. Sci., 2022, 9, 2103287 CrossRef CAS PubMed.
- S. R. Fish, K. A. Hartman, G. J. Stubbs and G. J. Thomas, Jr., Structural studies of tobacco mosaic virus and its components by laser Raman spectroscopy, Biochemistry, 1981, 20, 7449–7457 CrossRef CAS PubMed.
- G. Pezzotti, E. Ohgitani, H. Imamura, S. Ikegami, M. Shin-Ya, T. Adachi, K. Adachi, T. Yamamoto, N. Kanamura, E. Marin, W. Zhu, K. Higasa, Y. Yasukochi, K. Okuma and O. Mazda, Raman multi-omic snapshot and statistical validation of structural differences between Herpes Simplex Type I and Epstein–Barr viruses, Int. J. Mol. Sci., 2023, 24, 15567 CrossRef PubMed.
- C. Carlomagno, D. Bertazioli, A. Gualerzi, S. Picciolini, P. I. Banfi, A. Lax, E. Messina, J. Navarro, L. Bianchi, A. Caronni, F. Marenco, S. Monteleone, C. Arienti and M. Bedoni, COVID-19 salivary Raman fingerprint: innovative approach for the detection of current and past SARS-CoV-2 infections, Sci. Rep., 2021, 11, 4943 Search PubMed.
- S. Desai, S. V. Mishra and A. Joshi, Raman spectroscopy-based detection of RNA viruses in saliva: a preliminary report, J. Biophotonics, 2020, 13, e202000189 Search PubMed.
- G. Pezzotti, W. Zhu, T. Adachi, S. Horiguchi, E. Marin, F. Boschetto, E. Ohgitani and O. Mazda, Metabolic machinery encrypted in the Raman spectrum of influenza A virus-inoculated mammalian cells, J. Cell. Physiol., 2020, 235, 5146–5170 Search PubMed.
- D. Tiwari, S. Jakhmola, D. K. Pathak, R. Kumar and H. Chandra Jha, Temporal in vitro Raman spectroscopy for monitoring replication kinetics of Epstein-Barr virus infection in glial cells, ACS Omega, 2020, 5(45), 29547–29560 CrossRef PubMed.
- A. Silge, T. Bocklitz, B. Becker, W. Matheis, J. Popp and I. Bekeredjian-Ding, Raman spectroscopy-based identification of toxoid vaccine products, NPJ Vaccines, 2018, 3, 50 CrossRef PubMed.
- D. Nemecek and G. J. Thomas, Jr., Raman spectroscopy of viruses and viral proteins, in Frontiers of Molecular Spectroscopy, ed. J. Laane, Elsevier, Amsterdam, The Netherlands, 2009, pp. 553–595 Search PubMed.
- S. D. Dryden, S. Anastasova, G. Satta, A. J. Thompson, D. R. Leff and A. Darzi, Rapid uropathogen identification using surface enhanced Raman spectroscopy active filters, Sci. Rep., 2021, 11, 8802 CrossRef.
- B. S. McAvan, L. A. Bowsher, T. Powell, J. F. O’Hara, M. Spitali, R. Goodacre and A. J. Doig, Raman spectroscopy to monitor post-translational modification and degradation in monoclonal antibody therapeutics, Anal. Chem., 2020, 92, 10381–10389 CrossRef PubMed.
- L. M. Almehmadi, S. V. Reverdatto, V. V. Ermolenkov, A. Shekhtman and I. K. Lednev, In situ stability test for mRNA vaccines based on deep-UV resonance Raman spectroscopy, Anal. Chem., 2023, 96, 1003–1008 CrossRef PubMed.
- L. Byrd-Leotis, S. E. Galloway, E. Agbogu and D. A. Steinhauer, Influenza hemagglutinin (HA) stem region mutations that stabilize or destabilize the structure of multiple HA subtypes, Virology, 2015, 89, 4504–4516 CrossRef PubMed.
- Influenza Research Database (file EF541421), https://www.fludb.org/brc/fluSegmentDetails.spg?ncbiGenomicAccession=EF541421&decorator=influenza.
- S. Skalickova, Z. Heger, L. Krejcova, V. Pekarik, K. Bastl, J. Janda, F. Konstolansky, E. Vareckova, O. Zitka, V. Adam and R. Kizek, Perspective of use of antiviral peptides against influenza virus, Viruses, 2015, 7, 5428–5442 CrossRef PubMed.
- J. Seladi-Schulman, J. Steel and A. C. Lowen, Spherical influenza viruses have a fitness advantage in embryonated eggs, while filament-producing strains are selected in vivo, J. Virol., 2013, 87, 13343–13353 CrossRef.
- A. Gaymard, N. Le Briand, E. Frobert, B. Lina and V. Escuret, Functional balance between neuraminidase and hemagglutinin in influenza viruses, Clin. Microbiol. Infect., 2016, 22, 975–983 CrossRef PubMed.
- V. Moulès, O. Terrier, M. Yver, B. Riteau, C. Moriscot, O. Ferraris, T. Julien, E. Giudice, J.-P. Rolland, A. Erny, M. Bouscambert-Duchamp, E. Frobert, M. Rosa-Calatrava, Y. P. Lin, A. Hay, D. Thomas, G. Schoehn and B. Lina, Importance of viral genomic composition in modulating glycoprotein content on the surface of influenza virus particles, Virology, 2011, 414, 51–62 CrossRef.
- G. P. Leser and R. A. Lamb, Influenza virus assembly and budding in raft-derived microdomains: a quantitative analysis of the surface distribution of HA, NA and M2 proteins, Virology, 2005, 342, 215–227 CrossRef PubMed.
- R. Fajri Nuwarda, A. A. Alharbi and V. Kayser, An overview of influenza viruses and vaccines, Vaccines, 2021, 9, 1032 CrossRef.
- L. V. Gubareva, L. Kaiser and F. G. Hayden, Influenza virus neuraminidase inhibitors, Lancet, 2000, 355, 827–835 CrossRef.
- G. G. Hayden, Amantadine and rimantadine: clinical aspects, in Antiviral drug resistance, ed. D. D. Richman, Wiley, New York, 1996, pp. 59–77 Search PubMed.
- M. Yamashita, M. Krystal, W. M. Fitch and P. Palese, Influenza B virus evolution: co-circulating lineages and comparison of evolutionary pattern with those of influenza A and C viruses, Virology, 1988, 163, 112–122 CrossRef.
- E. Nobusawa and K. Sato, Comparison of mutation rates of human influenza A and B viruses, J. Virol., 2006, 80, 3675–3678 CrossRef PubMed.
- S. Whelan and N. Goldman, A general empirical model of protein evolution derived from multiple protein families using a maximum-likelihood approach, Mol. Biol. Evol., 2001, 18, 691–699 CrossRef PubMed.
- Y. Zhang and J. Skolnick, TM-align: A protein structure alignment algorithm based on TM-score, Nucleic Acids Res., 2005, 33, 2302–2309 CrossRef.
- K. Tamura, G. Stecher and S. Kumar, MEGA 11: Molecular Evolutionary Genetics Analysis Version 11, Mol. Biol. Evol., 2021, 38, 3022–3027 CrossRef.
- M. Mirdita, K. Schütze, Y. Moriwaki, H. Heo, S. Ovchinnikov and M. Steinegger, ColabFold: making protein folding accessible to all, Nat. Methods, 2022, 19, 679–682 CrossRef.
- R. Evans, M. O’Neill, A. Pritzel, N. Antropova, A. Senior, T. Green, A. Zidek, R. Bates, S. Blackwell, J. Yim, O. Ronneberger, S. Bodenstein, M. Zielinski, A. Bridgland, A. Potapenko, A. Cowie, K. Tunyasuvunakool, R. Jain, E. Clancy, P. Kohli, J. Jumper and D. Hassabis, Protein complex prediction with AlphaFold-Multimer, bioRxiv, 2022, preprint DOI:10.1101/2021.10.04.463034.
- Z. Yang, X. Zheng, Y. Zhao and R. Chen, AlphaFold2 and its applications in the fields of biology and medicine, Signal Transduction Targeted Ther., 2023, 8, 115 CrossRef PubMed.
- G. Pezzotti, F. Boschetto, E. Ohgitani, Y. Fujita, W. Zhu, E. Marin, B. J. McEntire, B. S. Bal and O. Mazda, Silicon nitride: a potent solid-state bioceramic inactivator of ssRNA viruses, Sci. Rep., 2021, 11, 2977 CrossRef PubMed.
- G. Zhu, X. Zhu, Q. Fan and X. Wan, Raman spectra of amino acids and their aqueous solutions, Spectrochim. Acta, Part A, 2011, 78, 1187–1195 CrossRef.
- E. Podstawka, Y. Ozaki and L. M. Proniewicz, Part II: Surface-enhanced Raman spectroscopy investigation of methionine containing heterodipeptides adsorbed on colloidal silver, Appl. Spectrosc., 2004, 58, 581–590 CrossRef PubMed.
- A. Torreggiani, S. Barata-Vallejo and C. Chatgilialoglu, Combined Raman and IR spectroscopic study on the radical-based modifications of methionine, Anal. Bioanal. Chem., 2011, 401, 1231–1239 CrossRef.
- F. Madzharova, Z. Heiner, M. Gühlke and J. Kneipp, Surface-enhanced hyper-Raman spectra of adenine, guanine, cytosine, thymine, and uracil, J. Phys. Chem. C, 2016, 120, 15415–15423 CrossRef.
- J. L. McHale, Fermi resonance of tyrosine and related compounds. Analysis of the Raman doublet, J. Raman Spectrosc., 1982, 13, 21–24 CrossRef.
- B. Hernandez, Y.-M. Coic, F. Pflueger, S. G. Kruglik and M. Ghomi, All characteristic Raman markers of tyrosine and tyrosinate originate from phenol ring fundamental vibrations, J. Raman Spectrosc., 2016, 47, 210–220 CrossRef.
- S. Gunasekaran, A. Bright, T. S. Renuga Devi, R. Arunbalaji, G. Anand, J. Dhanalakshmi and S. Kumaresan, Experimental and semi-empirical computations of the vibrational spectra of methionine, homocysteine and cysteine, Arch. Phys. Res., 2010, 1, 12–26 Search PubMed.
- N. K. Penta, B. C. Peethala, H. P. Amanapu, A. Melman and S. V. Babu, Role of hydrogen bonding on the adsorption of several amino acids on SiO2 and Si3N4 and selective polishing of these materials using ceria dispersions, Colloids Surf., A, 2013, 429, 67–73 CrossRef.
- M. Voicescu, S. Ionescu and C. I. Nistor, Spectroscopic study of 3-hydroxyflavone – protein interaction in lipidic bi-layers immobilized on silver particles, Spectrochim. Acta, Part A, 2017, 170, 1–8 CrossRef PubMed.
- K. Czamara, K. Majzner, M. Z. Pacia, K. Kochan, A. Kaczor and M. Baranska, Raman spectroscopy of lipids: a review, J. Raman Spectrosc., 2015, 46, 4–20 CrossRef.
- P. Cysewski, Theoretical studies on the tautomeric properties of diamino-5-formamidopyrimidines, Z. Naturforsch., C: J. Biosci., 1998, 53, 1027–1036 CrossRef.
- T. R. O’Connor, S. Boiteux and J. Laval, Ring-opened 7-methylguanine residues in DNA are a block to in vitro DNA synthesis, Nucleic Acids Res., 1988, 16, 5879–5894 CrossRef.
- J. B. Neto, A. Gentil, R. E. Cabral and A. Sarasin, Mutation spectrum of heat-induced abasic sites on a single-stranded shuttle vector replicated in mammalian cells, J. Biol. Chem., 1992, 267, 19718–19723 CrossRef CAS.
- J. T. Lutgerink, E. van den Akker, I. Smeets, D. Pachen, P. van Dijk, J. M. Aubry, H. Joenje, M. V. Lafleur and J. Retel, Interaction of singlet oxygen with DNA and biological consequences, Mutat. Res., 1992, 275, 377–386 CAS.
- K. Muller, P. Leukel, K. K. Mayer and W. Wiegrebe, Modification of DNA bases by anthralin and related compounds, Biochem. Pharmacol., 1995, 49, 1607–1613 CrossRef CAS.
- L. Decrey, S. Kazama and T. Kohn, Ammonia as an in situ sanitizer: influence of virus genome type on inactivation, Appl. Environ. Microbiol., 2016, 82, 4909–4920 CrossRef CAS.
- T. Douki and J. Cadet, Peroxynitrite mediated oxidation of purine bases in nucleosides and DNA, Free Radical Res. Commun., 1996, 24, 369–380 CrossRef CAS PubMed.
- R. J. Boorstein, T. P. Hilbert, J. Cadet, R. P. Cunningham and G. W. Teebor, UV-induced pyrimidine hydrates in DNA are repaired by bacterial and mammalian DNA glycosylase activities, Biochemistry, 1989, 28, 6164–6170 CrossRef CAS.
- N. Nonoyama, H. Oshima, C. Shoda and H. Suzuki, The reaction of peroxynitrite with organic molecules bearing a biologically important functionality. The multiplicity of reaction modes as exemplified by hydroxylation, nitration, nitrosation, dealkylation, oxygenation, and oxidative dimerization and cleavage, Bull. Chem. Soc. Jpn., 2001, 74, 2385–2395 CrossRef CAS.
- D. Shugar and J. J. Fox, Spectrophotometric studies of nucleic acid derivatives and related compounds as a function of pH. I. Pyrimidines, Biochim. Biophys. Acta, 1952, 9, 199–218 CrossRef CAS PubMed.
- J. Lang, J. Strum and R. Zana, Ultrasonic absorption in aqueous solutions of nucleotides and nucleosides. I. Effect of pH and concentration, J. Phys. Chem., 1973, 77, 2329–2334 CrossRef CAS.
- D. J. Brown, R. F. Evans, W. B. Cowden and M. D. Fenn, The Pyrimidine, John Wiley & Sons, New York, USA, 1994, pp. 96–106 Search PubMed.
- G. Pezzotti, A spontaneous solid-state NO donor to fight antibiotic resistant bacteria, Mater. Today Chem., 2018, 9, 80–90 CrossRef CAS.
- G. Pezzotti, Surface chemistry of bioceramics: The missing key, in Bioceramics from macro to nanoscale, ed. A. Osaka and R. Narayan, Elsevier, Amsterdam, The Netherlands, 2021, pp. 297–324 Search PubMed.
- G. Pezzotti, Silicon nitride: A bioceramic with a gift, ACS Appl. Mater. Interfaces, 2019, 11, 26619–26636 CrossRef CAS.
- Y. D. Hartono, M. Ito, A. Villa and L. Nilsson, Computational study of uracil tautomeric forms in the ribosome: the case of uracil and 5-oxyacetic acid uracil in the first anticodon position of tRNA, J. Phys. Chem. B, 2018, 122, 1152–1160 CrossRef CAS PubMed.
- G. Pezzotti, E. Ohgitani, Y. Fujita, H. Imamura, F. Pappone, A. Grillo, M. Nakashio, M. Shin-Ya, T. Adachi, T. Yamamoto, N. Kanamura, E. Marin, W. Zhu, T. Inaba, Y. Tanino, Y. Nukui, K. Higasa, Y. Yasukochi, K. Okuma and O. Mazda, Raman fingerprints of SARS-CoV-2 Omicron subvariants: Molecular roots of virological characteristics and evolutionary directions, ACS Infect. Dis., 2023, 9, 2226–2251 CrossRef CAS PubMed.
- X. Cao and G. Fischer, Conformational and infrared spectral studies of L-methionine and its N-deuterated isotopomer as isolated zwitterions, J. Phys. Chem. A, 2002, 106, 41–50 CrossRef CAS.
- P. Švancǎrová and T. Betáková, Conserved methionine 165 of matrix protein contributes to the nuclear import and is essential for influenza A virus replication, Virol. J., 2018, 15, 187 CrossRef PubMed.
- Z. Ye, T. Liu, D. P. Offringa, J. McInnis and R. A. Levandowski, Association of influenza virus matrix protein with ribonucleoproteins, J. Virol., 1999, 73, 7467–7473 CrossRef CAS PubMed.
- D. Perrin and W. H. Koppenol, The quantitative oxidation of methionine to methionine sulfoxide by peroxynitrite, Arch. Biochem. Biophys., 2000, 377, 266–272 CrossRef CAS PubMed.
- J. T. Brosnan and M. E. Brosnan, The sulfur-containing amino acids: an overview, J. Nutr., 2006, 136, 16365–16405 Search PubMed.
- B. C. Lee and V. N. Gladyshev, The biological significance of methionine sulfoxide stereochemistry, Free Radical Biol. Med., 2011, 50, 221–227 CrossRef CAS.
- R. C. Lord and N. T. Yu, Laser-excited Raman spectroscopy of biomolecules. I. Native lysozyme and its constituent amino acids, J. Mol. Biol., 1970, 50, 509–524 CrossRef CAS PubMed.
- M. N. Siamwiza, R. C. Lord, M. C. Chen, T. Takamatsu, I. Harada, H. Matsuura and T. Shimanouchi, Interpretation of the doublet at 850 and 830 cm−1 in the Raman spectra of tyrosyl residues in proteins and certain model compounds, Biochemistry, 1975, 14, 4870–4876 CrossRef CAS PubMed.
- Z. Arp, D. Autrey, J. Laane, S. A. Overman and G. J. Thomas, Jr., Tyrosine Raman signatures of the filamentous virus Ff are diagnostic of non-hydrogen-bonded phenoxyls: demonstration by Raman and infrared spectroscopy of p-cresol vapor, Biochemistry, 2001, 40, 2522–2529 CrossRef CAS PubMed.
- S. A. Overman, K. L. Aubrey, N. S. Vispo, G. Cesareni and G. J. Thomas, Jr., Novel tyrosine markers in Raman spectra of wild-type and mutant (Y21M and Y24M) Ff virions Indicate unusual environments for coat protein phenoxyls, Biochemistry, 1994, 33, 1037–1042 CrossRef CAS.
- Z. Q. Wen, S. A. Overman and G. J. Thomas, Jr., Structure and interactions of the single-stranded DNA genome of filamentous virus fd: investigation by ultraviolet resonance Raman spectroscopy, Biochemistry, 1997, 36, 7810–7820 CrossRef CAS.
- M. Tsuboi, Y. Kubo, T. Ikeda, S. A. Overman, O. Osman and G. J. Thomas, Jr., Protein and DNA residue orientations in the filamentous virus Pf1 determined by polarized Raman and polarized FTIR spectroscopy, Biochemistry, 2003, 42, 940–950 CrossRef CAS.
- S. A. Overman and G. J. Thomas, Jr., Raman spectroscopy of the filamentous virus Ff (fd, f1, M13): structural interpretation for coat protein aromatics, Biochemistry, 1995, 34, 5440–5451 CrossRef CAS.
- M. Tsuboi, K. Ushizawa, K. Nakamura, J. M. Benevides, S. A. Overman and G. J. Thomas, Jr., Orientations of Tyr 21 and Tyr 24 in the capsid of filamentous virus Ff deter- mined by polarized Raman spectroscopy, Biochemistry, 2001, 40, 1238–1247 CrossRef CAS.
- G. Pezzotti, E. Ohgitani, S. Ikegami, M. Shin-Ya, T. Adachi, T. Yamamoto, N. Kanamura, E. Marin, W. Zhu, K. Okuma and O. Mazda, Instantaneous inactivation of herpes simplex virus by silicon nitride bioceramics, Int. J. Mol. Sci., 2023, 24, 12657 CrossRef CAS.
- Y. Xu and C. Lu, Raman spectroscopic study on structure of human immunodeficiency virus (HIV) and hypericin-induced photosensitive damage of HIV, Sci. China, Ser. C: Life Sci., 2005, 48, 117–132 CAS.
- H. Van Dael, J. P. Lafant and F. Van Cauwelaert, Tyrosine group behaviour in bovine α-lactalbumin as revealed by its Raman effect, Eur. Biophys. J., 1987, 14, 409–414 CrossRef CAS.
- G. Pezzotti, E. Ohgitani, Y. Fujita, H. Imamura, M. Shin-Ya, T. Adachi, T. Yamamoto, N. Kanamura, E. Marin, W. Zhu, I. Nishimura and O. Mazda, Raman fingerprints of SARS-CoV-2 Delta variant and mechanisms of its instantaneous inactivation by silicon nitride bioceramics, ACS Infect. Dis., 2022, 8, 1563–1581 CrossRef CAS.
- M. L. Grantham, S. M. Stewart, E. N. Lalime and A. Pekosz, Tyrosines in the influenza A virus M2 protein cytoplasmic tail are critical for production of infectious virus particles, J. Virol., 2010, 84, 8765–8776 CrossRef CAS.
- H. Takeuchi and I. Harada, Normal coordinate analysis of the indole ring, Spectrochim. Acta, 1986, 42A, 1069–1078 CrossRef CAS.
- T. Miura, H. Takeuchi and I. Harada, Characterization of individual tryptophan side chains in proteins using Raman spectroscopy and hydrogen-deuterium exchange kinetics, Biochemistry, 1988, 27, 88–94 CrossRef CAS PubMed.
- T. Miura, H. Takeuchi and I. Harada, Tryptophan Raman bands sensitive to hydrogen bonding and side-chain conformation, J. Raman Spectrosc., 1989, 20, 667–671 CrossRef CAS.
- H. Takeuchi, Raman structural markers of tryptophan and histidine side chains in proteins, Biopolymers, 2003, 72, 305–317 CrossRef CAS PubMed.
- A. Combs, K. McCann, D. Autrey, J. Laane, S. A. Overman and G. J. Thomas, Jr., Raman signature of the non-hydrogen-bonded tryptophan side chain in proteins: experimental and ab initio spectra of 3-methylindole in the gas phase, J. Mol. Struct., 2005, 735–736, 271–278 CrossRef CAS.
- S. Bhattacharya, N. Vyas, A. K. Ojha, S. Dasgupta and A. Roy, Surface-enhanced Raman measurements and DFT calculations for L-tryptophan of varying pH in silver soil, J. Raman Spectrosc., 2012, 43, 718–723 CrossRef CAS.
- B. Hernandez, F. Pflueger, A. Adenier, S. G. Kruglik and M. Ghomi, Vibrational analysis of amino acids and short peptides in hydrated media. VIII. Amino acids with aromatic side chains: L-phenylalanine, L-tyrosine, and L-tryptophan, J. Phys. Chem. B, 2010, 114, 15319–15330 CrossRef CAS PubMed.
- T. Suzuki, H. F. Mower, M. D. Friesen, I. Gilibert, T. Sawa and H. Ohshima, Nitration and nitrosation of N-acetyl-L-tryptophan and tryptophan residues in proteins by various reactive nitrogen species, Free Radical Biol. Med., 2004, 37, 671–681 CrossRef CAS.
- L. Bieker and H. Schmidt, Raman spectra of N-formylkynurenine derivatives of lysozyme produced by ozone oxidation, FEBS Lett., 1979, 106, 268–270 CrossRef CAS PubMed.
- S. Li and M. Hong, Protonation, tautomerization, and rotameric structure of histidine: a comprehensive study by magic-angle-spinning solid-state NMR, J. Am. Chem. Soc., 2011, 133, 1534–1544 CrossRef CAS.
- K. G. Strothkamp and S. J. Lippard, Chemistry of the imidazole-bridge bimetallic center in the Cu-Zn. SOD and its model compounds, Acc. Chem. Res., 1982, 15, 318–326 CrossRef CAS.
- J. E. Morgan, M. I. Verkhovsky and M. J. Wikstroem, The histidine cycle: A new model for proton translocation in the respiratory heme-copper oxidases, J. Bioenerg. Biomembr., 1994, 26, 599–608 CrossRef CAS PubMed.
- F. Hu, W. Luo and M. Hong, Mechanisms of proton conduction and gating in influenza M2 proton channels from solid-state NMR, Science, 2010, 330, 505–508 CrossRef CAS.
- K. J. Cross, L. M. Burleigh and D. A. Steinhauer, Mechanisms of cell entry by influenza virus, Expert Rev. Mol. Med., 2001, 3, 1–18 CrossRef CAS.
- L. Byrd Leotis, S. E. Galloway, E. Agbogu and D. A. Steinhauer, Influenza hemagglutinin (HA) stem region mutation that stabilizes or destabilizes the structure of multiple HA subtypes, Virology, 2015, 89, 4504–4516 CrossRef CAS.
- Y. Zhang, C. Xu, H. Zhang, G. Dacai Liu, C. Xue and Y. Cao, Targeting hemagglutinin: approaches for broad protection against the influenza A virus, Viruses, 2019, 11, 405 CrossRef CAS PubMed.
- S. J. Gamblin, L. F. Haire, R. J. Russell, D. J. Stevens, B. Xiao, Y. Ha, N. Vasisht, D. A. Steinhauer, R. S. Daniels, A. Elliot, D. C. Wiley and J. J. Skehel, The structure and receptor binding properties of the 1918 influenza hemagglutinin, Science, 2004, 303, 1838–1842 CrossRef CAS.
- M. Lazniewski, W. K. Dawson, T. Szczepinska and D. Plewczynski, The structural variability of the influenza A hemagglutinin receptor-binding site, Briefings Funct. Genomics, 2018, 17, 415–427 CAS.
- S. J. Stray and L. B. Pittman, Subtype- and antigenic site-specific differences in biophysical influences on evolution of influenza virus hemagglutinin, Virol. J., 2012, 9, 91 CrossRef CAS PubMed.
- B. F. Koel, D. F. Burke, T. M. Bestebroer, S. van der Vliet, G. C. Zondag, G. Vervaet, E. Skepner, N. S. Lewis, M. I. Spronken, C. A. Russell, M. Y. Eropkin, A. C. Hurt, I. G. Barr, J. C. de Jong, G. F. Rimmelzwaan, A. D. M. E. Osterhaus, R. A. M. Fouchier and D. J. Smith, Substitutions near the receptor binding site determine major antigenic change during influenza virus evolution, Science, 2013, 342, 976–979 CrossRef CAS PubMed.
- X. Wang, N. A. Ilyushina, V. Y. Lugovtsev, N. V. Bovin, L. K. Couzens, J. Gao, R. P. Donnelly, M. C. Eichelberger and H. Wan, Amino acids in hemagglutinin antigenic site B determine antigenic and receptor binding differences between A(H3N2)v and ancestral seasonal H3N2 influenza viruses, J. Virol., 2017, 91, e01512–e01516 CAS.
- M. E. Cueno, H. Shiotsu, K. Nakano, E. Sugiyama, M. Kikuta, R. Usui, R. Oya and K. Imai, Structural significance of residues 158–160 in the H3N2 hemagglutinin globular head: A computational study with implications in viral evolution and infection, J. Mol. Graphics Modell., 2019, 89, 33–40 CrossRef CAS.
- T. Guarnaccia, L. A. Carolan, S. Maurer-Stroh, R. Lee, E. Job, P. C. Reading, S. Petrie, J. M. McCaw, J. McVernon, A. C. Hurt, A. Kelso, J. Mosse, I. G. Barr and K. L. Laurie, Antigenic drift of the pandemic 2009 A(H1N1) influenza virus in A ferret model, PLoS Pathog., 2013, 9, e1003354 CrossRef CAS PubMed.
- C. Li, M. Hatta, D. F. Burke, J. Ping, Y. Zhang, M. Ozawa, A. S. Taft, S. C. Das, A. P. Hanson, J. Song, M. Imai, P. R. Wilker, T. Watanabe, S. Watanabe, M. Ito, K. Iwatsuki-Horimoto, C. A. Russell, S. L. James, E. Skepner, E. A. Maher, G. Neumann, A. I. Klimov, A. Kelso, J. McCauley, D. Wang, Y. Shu, T. Odagiri, M. Tashiro, X. Xu, D. E. Wentworth, J. M. Katz, N. J. Cox, D. J. Smith and Y. Kawaoka, Selection of antigenically advanced variants of seasonal influenza viruses, Nat. Microbiol., 2016, 1, 16058 CrossRef CAS PubMed.
- M. Igarashi, K. Ito, H. Kida and A. Takada, Genetically destined potentials for N-linked glycosylation of influenza virus hemagglutinin, Virology, 2008, 376, 323–329 CrossRef CAS PubMed.
- J. Du, T. A. Cross and H. Zhou, Recent progress in structure-based anti influenza drug design, Drug Discovery Today, 2012, 17, 1111–1120 CrossRef CAS PubMed.
- R. J. Russell, P. S. Kerry, D. J. Stevens, D. A. Steinhauer, S. R. Martin, S. J. Gamblin and J. J. Skehel, Structure of influenza hemagglutinin in complex with an inhibitor of membrane fusion, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 17736–17741 CrossRef CAS PubMed.
- G. Pezzotti, Raman spectroscopy in cell biology and microbiology, J. Raman Spectrosc., 2021, 52, 2348–2443 CrossRef CAS.
- F. C. Fang, Perspectives series: host/pathogen interactions. Mechanisms of nitric oxide-related antimicrobial activity, J. Clin. Invest., 1997, 99, 2818–2825 CrossRef CAS PubMed.
- W. W. Ackermann, The role of l-methionine in virus propagation, J. Exp. Med., 1951, 93, 337–343 CrossRef CAS PubMed.
- C. C. Valley, A. Cembran, J. D. Perlmutter, A. K. Lewis, N. P. Labello, J. Gao and J. N. Sachs, The methionine-aromatic motif plays a unique role in stabilizing protein structure, J. Biol. Chem., 2012, 287, 34979–34991 CrossRef CAS PubMed.
- R. W. Compans and P. W. Choppin, Reproduction of myxoviruses, in Comprehensive virology, ed. H. Fraenkel-Conrat and R. R. Wagner, Plenum Press, New York, NY, USA, 1975, vol. IV, pp. 179–252 Search PubMed.
- J. Lohmeyer, L. T. Talens and H. D. Klenk, Biosynthesis of the influenza virus envelope in abortive infection, J. Gen. Virol., 1979, 42, 73–88 CrossRef CAS PubMed.
- A. Ali, R. T. Avalos, E. Ponimaskin and D. P. Nayak, Influenza virus assembly: effect of influenza virus glycoproteins on the membrane association of M1 protein, J. Virol., 2000, 74, 8709–8719 CrossRef CAS.
- P. Gomez-Puertas, C. Albo, E. Perez-Pastrana, A. Vivo and A. Portela, Influenza virus matrix protein is the major driving force in virus budding, J. Virol., 2000, 74, 11538–11547 CrossRef CAS PubMed.
- J. Peukes, X. Xiong and J. A. G. Briggs, New structural insight into the multifunctional influenza A matrix protein 1, FEBS Lett., 2021, 595, 2535–2543 CrossRef CAS PubMed.
- T. Shimizu, N. Takizawa, K. Watanabe, K. Nagata and N. Kobayashi, Crucial role of the influenza virus NS2 (NEP) C-terminal domain in M1 binding and nuclear export of vRNP, FEBS Lett., 2010, 585, 41–46 CrossRef.
- R. W. H. Ruigrok, A. Barge, P. Durrer, J. Brunner, K. Ma and G. R. Whittaker, Membrane interaction of influenza virus M1 protein, Virology, 2000, 267, 289–298 CrossRef CAS.
- P. Svancarova and T. Betakova, Conserved methionine 165 of matrix protein contributes to the nuclear import and is essential for influenza A virus replication, Virol. J., 2018, 15, 187 CrossRef PubMed.
- D. P. Nayak, E. K. Hui and S. Barman, Assembly and budding of influenza virus, Virus Res., 2004, 106, 147–165 CrossRef CAS PubMed.
- H. Xie, Z. Lin, P. D. Mosier, U. R. Desai and Y. Gao, The compensatory G88R change is essential in restoring the normal functions of influenza a/WSN/33 virus matrix protein 1 with a disrupted nuclear localization signal, J. Virol., 2013, 87, 345–353 CrossRef CAS PubMed.
- S. Wang, Z. Zhao, Y. Bi, L. Sun, X. Liu and W. Liu, Tyrosine 132 phosphorylation of influenza A virus M1 protein is crucial for virus replication by controlling the nuclear import of M1, J. Virol., 2013, 87, 6182–6191 CrossRef CAS PubMed.
- A. V. Shishkov, V. I. Goldanskii, L. A. Baratova, N. V. Fedorova, A. L. Ksenofontov, O. P. Zhirnov and A. Galkin, The in situ spatial arrangement of the influenza A virus matrix protein M1 assessed by tritium bombardment, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 7827–7830 CrossRef CAS.
- N. Nao, M. Kajihara, R. Manzoor, J. Maruyama, R. Yoshida, M. Muramatsu, H. Miyamoto, M. Igarashi, N. Eguchi, M. Sato, T. Kondoh, M. Okamatsu, Y. Sakoda, H. Kida and A. Takada, A single amino acid in the M1 protein responsible for the different pathogenic potentials of H5N1 highly pathogenic avian influenza virus strains, PLoS One, 2015, 10, e0137989 CrossRef PubMed.
- B. Alvarez and R. Radi, Peroxynitride reactivity with amino acids and proteins, Amino Acids, 2003, 25, 295–311 CrossRef CAS.
- K. Takeuchi and R. A. Lamb, Influenza virus M2 protein ion channel activity stabilizes the native form of fowl plague virus hemagglutinin during intracellular transport, J. Virol., 1994, 68, 911–919 CrossRef CAS PubMed.
- S. Kellokumpu, Golgi pH, ion and redox homeostasis: how much do they really matter?, Front. Cell Dev. Biol., 2019, 7, 93 CrossRef PubMed.
- D. Gravotta, M. Adesnik and D. D. Sabatini, Transport of influenza HA from the trans-Golgi network to the apical surface of MDCK cells permeabilized in their basolateral plasma membranes: energy dependence and involvement of GTP-binding proteins, J. Cell Biol., 1990, 111, 2893–2908 CrossRef CAS.
- R. A. Lamb, S. L. Zebedee and C. D. Richardson, Influenza virus M2 protein is an integral membrane protein expressed on the infected-cell surface, Cell, 1985, 40, 627–633 CrossRef CAS PubMed.
- O. Roetzschke, J. M. Lau, M. Hofstaetter, K. Falk and J. L. Strominger, A pH-sensitive histidine residue as control element for ligand release from HLA-DR molecules, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 16946–16950 CrossRef PubMed.
- L. H. Pinto, L. J. Holsinger and R. A. Lamb, Influenza virus M2 protein has ion channel activity, Cell, 1992, 69, 517–528 CrossRef CAS PubMed.
- C. Wang, R. A. Lamb and L. H. Pinto, Activation of the M2 ion channel of influenza virus: a role for the transmembrane domain histidine residue, Biophys. J., 1995, 69, 1363–1371 CrossRef CAS.
- K. J. Schweighofer and A. Pohorille, Computer simulation of ion channel gating: the M2 channel of influenza A virus in a lipid bilayer, Biophys. J., 2000, 78, 150–163 CrossRef CAS.
- M. S. P. Sansom, I. D. Kerr, G. R. Smith and H. S. Son, The influenza A virus M2 channel: A molecular modeling and simulation study, Virology, 1997, 233, 163–173 CrossRef CAS.
- J. Mould, H. C. Li, C. S. Dudlak, J. D. Lear, A. Pekosz, R. A. Lamb and L. H. Pinto, Mechanism for proton conduction of the M(2) ion channel of influenza A virus, J. Biol. Chem., 2000, 275, 8592–8599 CrossRef CAS.
- V. S. Mandala, S.-Y. Liao, M. D. Gelenter and M. Hong, The transmembrane conformation of the influenza B virus M2 protein in lipid bilayers, Sci. Rep., 2019, 9, 3725 CrossRef PubMed.
- L. H. Pinto and R. A. Lamb, The M2 proton channels of influenza A and B viruses, J. Biol. Chem., 2006, 281, 8997–9000 CrossRef CAS PubMed.
- V. S. Mandala, S.-Y. Liao, B. Kwon and M. Hong, Structural basis for asymmetric conductance of the influenza M2 proton channel investigated by solid-state NMR spectroscopy, J. Mol. Biol., 2017, 429, 2192–2210 CrossRef CAS PubMed.
- T. Betakova and A. J. Hay, Comparison of the activities of BM2 protein and its H19 and W23 mutants of influenza B virus with activities of M2 protein and its H37 and W41 mutants of influenza A virus, Arch. Virol., 2009, 154, 1619–1624 CrossRef CAS.
- L. H. Pinto, L. J. Holsinger and R. A. Lamb, Influenza virus M2 protein has ion channel activity, Cell, 1992, 69, 517–528 CrossRef CAS PubMed.
- C. Wang, R. A. Lamb and L. H. Pinto, Activation of the M2 channel of influenza virus: a role for the transmembrane domain histidine residue, Biophys. J., 1995, 69, 1363–1371 CrossRef CAS.
- V. Balannik, V. Carnevale, G. Fiorin, B. G. Levine, R. A. Lamb, M. L. Klein, W. F. Degrado and L. H. Pinto, Functional studies and modeling of pore-lining residue mutants of the influenza A virus M2 ion channel, Biochemistry, 2010, 49, 696–708 CrossRef CAS.
- J. Wang, R. M. Pielak, M. A. McClintock and J. J. Chou, Solution structure and functional analysis of the influenza B proton channel, Nat. Struct. Mol. Biol., 2009, 16, 1267–1271 CrossRef CAS PubMed.
- J. A. Mould, R. G. Paterson, M. Takeda, Y. Ohigashi, P. Venkataraman, R. A. Lamb and L. H. Pinto, Influenza B virus BM2 protein has ion channel activity that conducts protons across membranes, Dev. Cell, 2003, 5, 175–184 CrossRef CAS.
- J. K. Williams, A. A. Shcherbakov, J. Wang and M. Hong, Protonation equilibria and pore-opening structure of the dual-histidine influenza B virus M2 transmembrane proton channel from solid-state NMR, J. Biol. Chem., 2017, 292, 17876–17884 CrossRef CAS.
- G. Pezzotti, E. Ohgitani, M. Shin-Ya, T. Adachi, E. Marin, F. Boschetto, W. Zhu and O. Mazda, Instantaneous “catch-and-kill” inactivation of SARS-CoV-2 by nitride ceramics, Clin. Transl. Med., 2020, 10, e212 CrossRef CAS PubMed.
- G. Pezzotti, R. M. Bock, B. J. McEntire, T. Adachi, E. Marin, F. Boschetto, W. Zhu, O. Mazda and S. B. Bal, In vitro antibacterial activity of oxide and non-oxide bioceramics for arthroplastic devices: I. In situ time-lapse Raman spectroscopy, Analyst, 2018, 143, 3708–3721 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cb00237g |
| This journal is © The Royal Society of Chemistry 2025 |