
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThiophosphate bioisosteres of inositol hexakisphosphate enhance binding affinity and residence time on bacterial virulence factors†
Rebecca
Cummer
 a,
Garvit
Bhatt
ab,
Lauren M.
Finn
c,
Bettina G.
Keller
c,
Bhushan
Nagar
b and
Bastien
Castagner
*a
a,
Garvit
Bhatt
ab,
Lauren M.
Finn
c,
Bettina G.
Keller
c,
Bhushan
Nagar
b and
Bastien
Castagner
*a
aDepartment of Pharmacology and Therapeutics, McGill University, Montréal, Québec H3G 1Y6, Canada. E-mail: bastien.castagner@mcgill.ca; Fax: +514-398-2045; Tel: +514-398-2181
bDepartment of Biochemistry, McGill University, Montréal, Québec H3G 1Y6, Canada
cDepartment of Biology, Chemistry, Pharmacy, Freie Universität, Arnimallee 22, 14195, Berlin, Germany
First published on 20th March 2025
Abstract
Inositol phosphates are essential for mammalian cell signalling with critical roles in cellular processes. The fully phosphorylated inositol phosphate, myo-inositol hexakisphosphate (IP6), modulates numerous eukaryotic proteins and bacterial virulence factors. It has been suggested that the high charge density of IP6 causes restructuring of virulence factors in mammalian cells, activating their enzymatic activity. IP6 is challenging to study due to its phytase instability and propensity to precipitate. Here we suggest that the thiophosphate bioisostere, myo-inositol hexakisthiophosphate (IT6), will mitigate these issues, as thiophosphate substitution has been found to be phytase resistant and improve solubility. Assessment of the chemical properties of IT6 has indeed validated these characteristics. In addition, we performed biophysical characterization of IT6 binding to the virulence factors Salmonella enterica serovar Typhimurium AvrA, Vibrio parahaemolyticus VopA, and Clostridioides difficile TcdB. Our data show that the higher charge density of IT6 increased its binding affinity and residence time on the proteins, which improved stabilization of the bound-state. IT6 is a valuable tool for structural biology research and the described biophysical characteristics of thiophosphate substitution are of value in medicinal chemistry.
Introduction
Inositol phosphates and their lipidated phosphatidyl inositol congeners are important signaling molecules with crucial roles in cellular processes such as calcium release,1 insulin signaling,2 and metabolic signaling pathways.3 Inositol phosphates are either synthesized from a lipidated inositol core or glucose-6-phosphate via a network of kinases, phosphatases, and hydrolases that modulate the attachments on the myo-inositol core.3 Inositol phosphates can exist with various degrees of phosphorylation, ranging from 1 to 6 phosphates, with each species having different functions in the cell. The fully phosphorylated myo-inositol hexakisphosphate (IP6) modulates numerous eukaryotic proteins and bacterial virulence factors from mammalian and plant pathogens. Examples of mammalian virulence factors include: large clostridial toxins,4,5 multifunctional autoprocessing RTX toxin,6,7 HIV-1 gag-hexamer,8Legionella pneumophila effector kinase,9 and the Salmonella enterica serovar Typhimurium type III effector AvrA.10 The virulence factors enter host cells either via toxin generated pores or a type III secretion system. It is only upon entering their target that the host cytosolic IP6 modulates the target function through allosteric regulation, by promoting protein stabilization due to the high charge density. Therefore, the IP6 binding event can form a target specific catalytically active enzyme by stabilizing the virulence factor.The physicochemical properties of IP6 permit its varied roles in biological processes. IP6 is densely charged,11 a strong chelator of divalent cations,12 and susceptible to hydrolysis. This simultaneously permits the tight regulation of the varied inositol phosphate species while also acting as a source of phosphate energy regulation.13–15 Consequently, the compound is hydrophilic, susceptible to cation precipitation,16 and low in abundance due to its high turnover rate,17 making IP6 challenging to study. Given the importance of IP6 in biology, the development of biomimetics that can tune-out these challenging properties are of value as both structural biology tools and as therapeutics.18,19
In an effort to synthesize an IP6 biomimetic that satisfies the aforementioned properties, thiophosphate and sulfate substitutions have been previously exploited.20,21 Historically, thiophosphate substitution has been used in nucleotide synthesis and with lower phosphorylated myo-inositols to circumvent enzymatic hydrolysis.22–26 Thiophosphate drug mimetics have also been found to have an improved lipophilicity due to a decreased hydrogen bond basicity (HBB) favoring octanol in log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P octanol water partition.27 In high concentrations of divalent cations, like those found in the gastrointestinal (GI) tract, thiophosphate substitution improves compound solubility.19 These results suggest that a fully thiophosphorylated IP6 analog would satisfy the physiochemical properties required to make a promising IP6 biomimetic. In addition, thiophosphate substitution has been found to improve the potency and stability of the target enzyme(s).19,28,29 Here, we show that myo-inositol hexakisthiophosphate (IT6), as previously synthesized by Chen et al.,30 is a useful biomimetic of IP6 with enhanced resistance towards phytase enzymes and solubility in the presence of divalent cations. Notably, IT6 can stabilize the Salmonella enterica serovar Typhimurium effector AvrA and Vibrio parahaemolyticus effector VopA more than their natural co-factor, IP6, which was attributed to better binding affinity and slower dissociation rate.
P octanol water partition.27 In high concentrations of divalent cations, like those found in the gastrointestinal (GI) tract, thiophosphate substitution improves compound solubility.19 These results suggest that a fully thiophosphorylated IP6 analog would satisfy the physiochemical properties required to make a promising IP6 biomimetic. In addition, thiophosphate substitution has been found to improve the potency and stability of the target enzyme(s).19,28,29 Here, we show that myo-inositol hexakisthiophosphate (IT6), as previously synthesized by Chen et al.,30 is a useful biomimetic of IP6 with enhanced resistance towards phytase enzymes and solubility in the presence of divalent cations. Notably, IT6 can stabilize the Salmonella enterica serovar Typhimurium effector AvrA and Vibrio parahaemolyticus effector VopA more than their natural co-factor, IP6, which was attributed to better binding affinity and slower dissociation rate.
Results and discussion
Synthesis and characterization of IT6
The thiophosphorylated IP6 analog, IT6, was synthesized in a two-step process as described previously (Fig. 1).30 First, myo-inositol was thiophosphorylated by a PIII method, followed by sulfur oxidation, generating myo-inositol hexakis-dibenzylthiophosphate (IT6-Bn). Next, the benzyl protected thiophosphate groups were removed with sodium in liquid ammonia and the compound was purified via size-exclusion chromatography. | ||
| Fig. 1 Scheme of the synthesis of hexakis-thiophosphate (IT6)30 and hexakis-dibenzylphosphate (IP6-Bn). mCPBA, 3-chloroperbenzoic acid, DCM, dichloromethane; DMF, dimethylformamide; O/N, over night; RT, room temperature; THF, tetrahydrofuran. | ||
Interestingly, upon purifying IT6-Bn we noticed a sizeable decrease in compound polarity compared to the phosphorylated equivalent, myo-inositol hexakis-dibenzylphosphate (IP6-Bn, Fig. S1A, ESI†). To elaborate on this, we determined the calculated logP (clogP) of both compounds and confirmed that IT6-Bn was more lipophilic than IP6-Bn (20.77 vs. 10.69, respectively). To explain these differences, we determined the Hückel charge on each element for both compounds (Fig. S1B, ESI†) and the corresponding compound polar surface area (IP6-Bn = 286.56 Å2vs.IT6-Bn = 166.14 Å2). These results corroborate data from Columbus et al. that suggests that thiophosphate substitution of aprotically substituted compounds reduces the hydrogen bond basicity (pKHB) value, improving lipophilicity.27 It should be noted that in respect to IP6 and IT6, this property is not observed as the phosphate and thiophosphate functional groups on IP6 and IT6 have different pK values, and the lipophilicities of both compounds are pH dependent. For example, IT6 has a net charge of −12 at pH 7.4, whereas IP6 has a net charge of −8, making IT6 less lipophilic than IP6 at a physiological pH.19
Stability of IT6
First, we evaluated whether IT6 was resistant to enzymatic hydrolysis by a commercial native wheat phytase. Phytases are a class of enzymes that catalyze the hydrolysis of IP6 into lower phosphorylated myo-inositols and inorganic phosphate (Fig. 2B).31 Dietary wheat phytase is of physiological importance as it significantly contributes to IP6 dephosphorylation in mammals.32 To test whether IT6 was resistant to enzymatic hydrolysis, we quantified the stability of IP6 and IT6 in the presence of phytase-4 over 32 h via31P NMR with the internal standard trimethyl phosphate (TMP). 31P NMR revealed that IT6 remained completely intact over 32 h, whereas IP6 was hydrolysed within the first hour of phytase exposure (Fig. 2C). The apparent thiophosphate phytase resistance is likely a result of the thiophosphate phosphoryl being less positively charged than on the phosphate, due to the thiophosphate sulfur being less electronegative than the oxygen. Since phytase catalysis is initiated by a nucleophilic attack on the phosphorus, the decrease in positive charge reduces the likelihood of said nucleophilic attack. As a result, the thiophosphate esters exhibit greater stability toward neutral and base-catalyzed hydrolysis than the corresponding phosphate esters. These results suggest that IT6 is more resistant to phytase hydrolysis than IP6.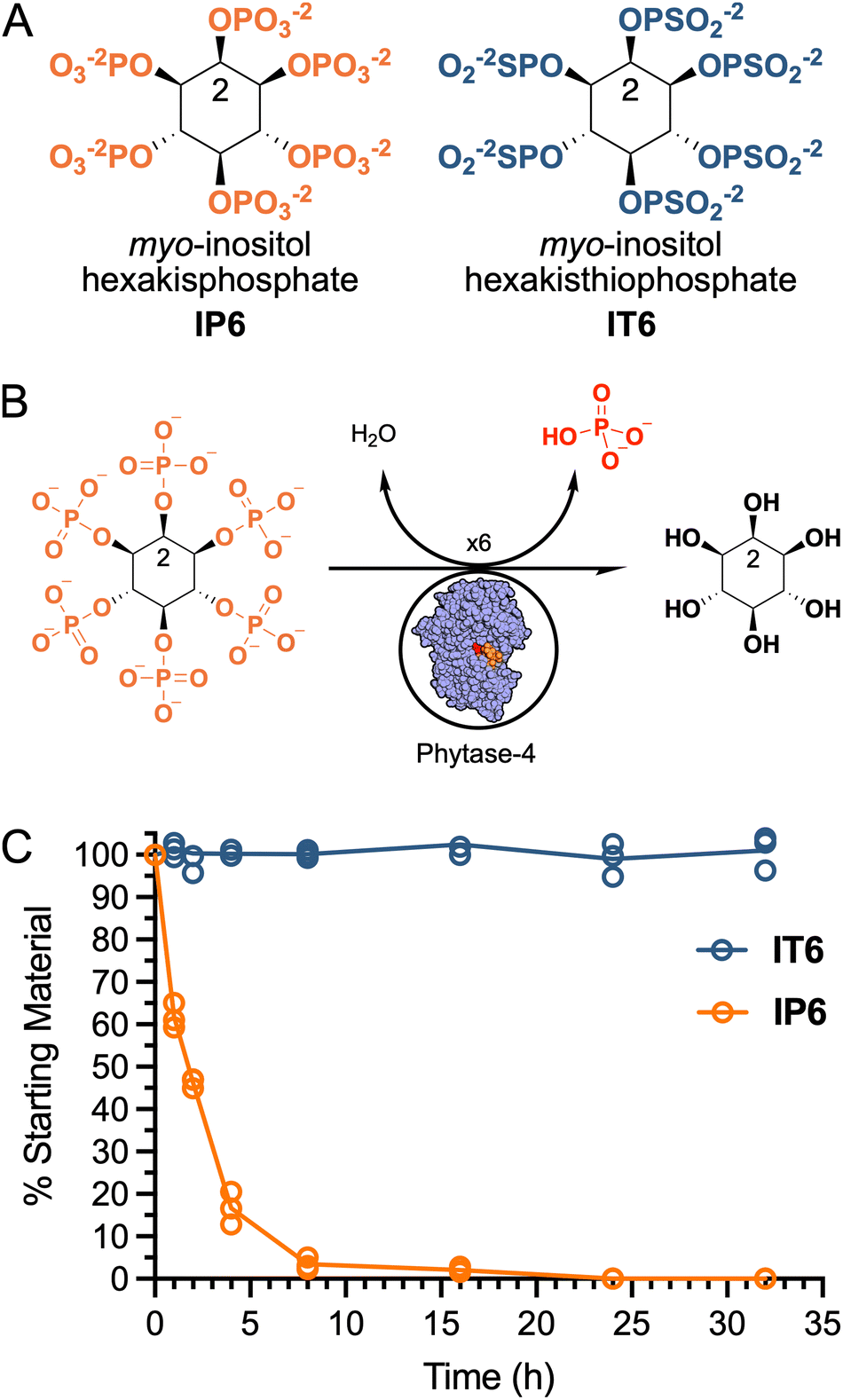 | ||
| Fig. 2 Stability of IP6 and IT6 against phytase-4. (A) Structure of IP6 and IT6. (B) Phytase-4 enzymes remove the phosphate groups from IP6via hydrolysis (phytase-4 PDB 6GJ2).31 (C) Percent of 4 mM of IP6 or IT6 detectable via31P NMR over 32 h in the presence of phytase-4. n =3 data points are shown. | ||
Solubility of IT6
It was previously observed that IT6 was soluble in a simulated GI tract environment whereas IP6 precipitated in the presence of the concentrations of MgCl2 and CaCl2 found in the intestinal lumen.19 Cummer et al. found that the thiophosphate groups on IT6 have a lower pKa than the phosphates on IP6, resulting in IT6 having a higher charge density at pH 7.4. They suggested that the greater charge density of IT6 gave the compound a higher electroneutrality threshold point, thus enabling solubility at physiologically relevant concentrations of divalent cations. To explore this theory, we wanted to determine whether IT6 was soluble in all concentrations of CaCl2, or whether a critical point for precipitation existed for IT6 that was previously untested. We determined the concentration of soluble IP6 and IT6 when 4 mM of each ligand was combined with a serial dilution of CaCl2 at the physiological pH 7.4 in the presence of TMP. Phosphorus NMR with proton coupling (31P–{1H} NMR) was run on the supernatant to determine the concentration of soluble ligand, and these values were plotted against the corresponding CaCl2 concentration (Fig. 3A and B). We found that IT6 does precipitate out of solution in the presence of CaCl2, although it requires a much higher concentration than IP6 (32 mM vs. 4 mM CaCl2). Therefore, IT6 is soluble in physiologically relevant concentrations of CaCl2, as its precipitation threshold is outside the physiologically relevant range (1–10 mM).33 | ||
| Fig. 3 Solubility differences between IP6 and IT6. (A) Amount of soluble IP6 and (B) IT6 at pH 7.4 in the presence of an increasing concentration of CaCl2. The amount of soluble ligand was calculated via31P–{1H} NMR with TMP as an internal standard. Data are curve fit with a plateau followed by one phase decay in Prism 10. X0 is noted with the gray line, and the corresponding ratio of CaCl2 to ligand that induces precipitation is reported. (C) Stacked 31P–{1H} NMR spectra of IT6 (blue/red) and IP6 (orange) with varied concentrations of CaCl2. The presence of CaCl2 elicits the formation of an IT6-Ca complex (IT6·xCa2+, red) with a unique 31P–{1H} NMR profile. The percentage of IT6 and IT6·xCa2+ was calculated for each 31P–{1H} NMR spectrum. The average particle diameter (nm) of 4 mM IP6 (D) and IT6 (E) at pH 7.4 was calculated at various concentrations of CaCl2via DLS. | ||
While performing the quantification of soluble IT6via31P–{1H} NMR an unexpected observation was made. Two species of IT6 were detected, one that corresponded with the 31P–{1H} NMR spectrum of deprotonated IT6,19 and another that only appeared in the presence of CaCl2. The second species increased in quantity as CaCl2 was added to the sample, and this change was inversely proportional to that of the deprotonated IT6 species, indicating that the second species was a by-product of calcium chelating to IT6 (IT6·xCa2+, Fig. 3C). This observation was not apparent for IP6 bound to Ca2+. These results suggest that Ca2+ first chelates to IT6, forming a complexed species, which then precipitates out of solution, as the concentration of soluble IT6 only decreased after all of the deprotonated IT6 was converted into the complexed form as observed by 31P–{1H} NMR. Of note, IP6 has been found to undergo a ring-flip with five axial substituents in the presence of certain environmental factors, such as metal cation composition.34 However, 13C NMR of the IT6 samples indicated that the putative IT6·xCa2+ species was not a by-product of a “flipped” conformation (Fig. S4, ESI†).
Finally, the presence of precipitate formation was confirmed by dynamic light scattering (DLS, Fig. 3D and E). The mean diameter of IP6 and IT6 in solution at pH 7.4 increased as the amount of soluble ligand decreased. The size of the precipitate positively correlated with increased concentrations of CaCl2. Collectively, these results support the theory that the increased charge density of IT6 requires a greater concentration of CaCl2 to cause charge neutralization of the compound, which initiates precipitation.
Characterizing the binding interaction between IT6 and AvrA
Cummer et al. previously found that IT6 had a higher binding affinity for the Clostridioides difficile toxin B (TcdB) cysteine protease domain (CPD) allosteric binding site than its natural co-factor IP6. They suggested that this was due to the increased net charge density of IT6 as the target binding site was electropositive, thus enhancing the electrochemical interaction.19 Here we aimed to determine whether this property could be ascertained for other IP6-dependent virulence factors.AvrA is an S. typhimurium effector protein, which suppresses c-JUN N-terminal kinase (JNK) signalling in mammals through acetylation of mitogen activated receptor kinases 4 and 7 (MKK4/7). Acetylation of MKK4/7 is suspected to reduce the upregulation of the host inflammatory response upon bacterial infection.35 The acetyltransferase has two closely associated domains: a regulatory region and a catalytic region. The regulatory region mediates binding of the cofactors IP6 and acetyl CoA (AcCoA), and the catalytic region acetylates the bound substrate (Fig. 4A).10 Binding of IP6 induces structural changes that form the substrate binding sites.36 The IP6 allosteric binding site contains many basic amino acids that are positively charged at a neutral pH and are stabilized by the negatively charged co-factor, as observed for TcdB CPD.
 | ||
| Fig. 4 Characterization of the binding interaction between bacterial virulence factors and IP6versusIT6. (A) Crystal structure of AvrAΔL140 bound to IP6 and AcCoA (PDB 6BE0).10IP6 binds to an allosteric binding site in the regulatory region of AvrA that controls the formation of the AcCoA active site. IP6 binds to the very positively charged allosteric binding site. Inset: Docking experiments show that IT6 binds to the same positively charged pocket.37,38 The binding interaction between AvrA and TcdB CPD with IP6 and IT6 was determined via ITC. The resultant thermograms (B) and (E) and the equilibration time curves (C) and (D) are shown for IP6/IT6 with AvrA and IP6/IT6 with TcdB CPD (F) and (G). | ||
We wanted to determine whether IT6 had a stronger affinity to AvrA than IP6. The equilibrium dissociation constants (Kd) of IP6 and IT6 were determined via isothermal calorimetry (ITC) (Fig. 4B and E). The Kd of IT6 was 2.5-fold lower than that of IP6 for AvrA, which was the same fold-change observed for TcdB CPD (Table 1). Of note, the reported Kd of AvrA with IP6 was significantly lower than previously described (139 nM versus 5.18 μM).10 We suspect that this discrepancy arose from the use of different sources of IP6. IP6 needs to be stored in dry conditions, because if water of crystallization is present in the industrial preparation, IP6 is susceptible to hydrolysis (Fig. S6, ESI†). If IP6 was in fact a lower phosphorylated species, the resultant decreased net charge would explain the discrepancy with the previously reported Kd.
| Protein | Ligand | ΔH (kcal mol−1) | K d (nM) | K off (s−1) | χ 2 |
|---|---|---|---|---|---|
| AvrA | IP6 | −21.09 (0.12) | 139.4 (16.4) | 1.711 × 10−3 (1.71 × 10−4) | 0.751 |
| IT6 | −35.56 (0.32) | 55.39 (15.4) | 1.156 × 10−3 (1.91 × 10−4) | 1.300 | |
| TcdB CPD | IP6 | −22.94 (0.05) | 1918 (34.6) | 4.086 × 10−2 (5.52 × 10−3) | 0.173 |
| IT6 | −9.386 (0.14) | 413.3 (57.8) | 6.912 × 10−3 (7.22 × 10−4) | 1.623 |
To look beyond the strength of interaction we can also investigate the durability of the binary ligand–protein complex. The improved electrostatic interaction between IT6 and AvrA/TcdB CPD is a molecular determinant of the dissociation rate constant (koff), and this dissociation process defines the drug-target residence time (τR).39 We determined the koff of the interactions via the kinetic ITC (kinITC) methodology utilized by the AFFINImeter software.40,41 The equilibration time curves (ETC, Fig. 4C, D, F and G) were curve fit with a one-site model, and the goodness of fit for each measurement had a χ2 below 2, indicating a strong fit. The ETC's gave a koff measurement from the ITC raw data (Table 1). The lower koff of IT6 indicated that it had a longer-lived ligand-target complex with both AvrA and TcdB CPD than IP6 (AvrA τR = 14.4, 9.7 min; TcdB CPD τR = 2.4, 0.4 min). Therefore, the increased charge density of IT6 has an effect on the duration of target activation.
Stabilization of virulence factor effector proteins AvrA and VopA
Molecular dynamics simulations have been performed with both TcdB CPD and AvrA to show the structural modifications associated with IP6 binding.36,42 Finn et al. previously found that the conformational changes that accompany allosteric binding of IP6 to the CPD on TcdB and toxin A (TcdA) were stabilized by a network of residue interactions, complementary to the unbound state residue network. The network involves electrostatic interactions between IP6 and arginine and lysine residues in the allosteric binding site. In the IP6-bound conformation, an α-loop formed a ‘lid’ over the drug binding pocket, which acted to block the escape of the ligand, and a β-flap rotated ∼90°, which resulted in the formation of the cysteine protease active site.42 Similarly, Zhang et al. showed that binding of IP6 to the allosteric binding site of AvrA induced structural changes that constrained two α-helices around IP6, which caused two β-strands in the regulatory domain to form the AcCoA and substrate binding sites.36The simple one-step binding/dissociation model that was used to describe the binding interaction between IT6/IP6 and AvrA/TcdB-CPD may be insufficient to describe the interaction as the ligands induce conformational changes to the proteins. The conformational changes result in greater steric and electronic complementarity between the drug molecule and its binding site on the target and often greater occlusion from the solvent. The conformational changes associated with the transition from unbound to bound strengthen the binding of the ligand to its target and simultaneously decrease the rate of drug dissociation. Consequently, the τR of the drug–protein complex is considerably augmented. Therefore, understanding these changes by other biophysical methods can be of importance.
Here, we determined whether IT6 was more capable than IP6 at stabilizing AvrA and VopA, an ortholog of AvrA which is found in other enteric pathogens like V. parahaemolyticus.43 To test this, we performed a differential scanning fluorimetry (DSF) experiment to determine the melting temperature (TM) of AvrA and VopA alone and in the presence of serial dilutions of IP6 and IT6. We then determined the change in TM (ΔTM) for each ligand concentration when compared to the TM of AvrA or VopA alone (Fig. 5A and C, respectively). The sizable leftward shift in the dose response curve corresponding to IT6 suggests that IT6 may have an improved binding affinity when compared to IP6, and these results are corroborated by the ITC results for AvrA. However, when attempting to assess the binding affinity for VopA via ITC, we found that the protein was unstable in the absence of the ligand. VopA rapidly lost functionality or misfolded within hours after purification, leading to inconsistent ITC results.
 | ||
| Fig. 5 Stabilization of the effector proteins AvrA and VopA. Dose–response curves for binding of IP6 and IT6 with AvrA (A) and VopA (C) and the corresponding temperature stabilization. Stabilization of AvrA and VopA was determined via DSF. The melting temperature (TM) was determined for the proteins alone and in the presence of a serial dilution of IP6 or IT6. The difference in melting temperature (ΔTM) was then plotted and the data were fit with a nonlinear curve fit. Mean ± SD, n = 5. Maximal ΔTM (°C) for IP6 and IT6 with AvrA (B) and VopA (D), as determined in (A) and (C), respectively. Mean ± SD, n = 5; independent t-test ****p < 0.0001. | ||
To quantify the stabilizing effect of each ligand, we performed a nonlinear curve fit on the data to determine the maximal ΔTM for IP6 and IT6 on AvrA and VopA (Fig. 5B and D, respectively). We found that IT6 had a significantly higher ΔTM than IP6 when bound to both AvrA (3.2 °C vs. 4.4 °C) and VopA (12.4 °C vs. 13.5 °C), which reflected increased protein stabilization. Additionally, the enhanced stabilization of IT6 over IP6 was previously observed for TcdB CPD.19 These results add further support to the theory that the improved charge density of the thiophosphate biomimetic, IT6, strengthens the electrostatic interaction with the IP6-dependent protein stabilizing the bound state, as observed here and previously with TcdB.19
Conclusions
Here, we propose IT6 as an interesting biomimetic of IP6 due to its suggested phytase stability, improved lipophilicity, solubility, and lower Kd and increased stabilization of the IP6-dependent proteins AvrA and TcdB-CPD. First, we found that IT6 is resistant to phytase-4 degradation due to the thiophosphate phosphoryl group being less positively charged, making a nucleophilic attack unlikely, and circumventing the hydrolysis reaction. Second, we found that aprotic thiophosphates are less polar than phosphates, while protic thiophosphates have a greater charge density; these apparent contradictory properties have manifested themselves in multiple interesting capabilities. The decreased polarity of IT6-Bn in comparison with IP6-Bn has resulted in an improved lipophilicity of the functional group. Although this does not put IT6-Bn, or IT6, within the cell permeability range according to Lipinski's rules, other strategies have been developed to optimize the delivery of highly anionic compounds.44 The increased charge density of thiophosphates at pH 7.4 has resulted in IT6 solubility in physiological concentrations of divalent cations due to a higher concentration threshold for charge neutralization. The higher charge density has also improved the Kd, τR and stabilization of IT6 for TcdB CPD, AvrA, and VopA in comparison with IP6. The unique properties of thiophosphate analogs suggest several potential practical applications that warrant further exploration. First, the drug-target τR model predicts that durable pharmacodynamics can be achieved by developing drug molecules with a long τR on their target.39 These results suggest that further research should explore whether thiophosphate substitution of functional groups involved in electrostatic interactions improves the pharmacodynamic properties of other pharmaceuticals. Second, stabilization of IP6-dependent proteins by IT6 might facilitate the formation of tractable crystal/ligand complexes for X-ray structural analysis.44Abbreviations
| AcCoA | Acetyl CoA |
| K a | Association constant |
| clogP | Calculated logP |
| ΔTM | Change in melting temperature |
| JNK | c-JUN N-terminal kinase |
| CPD | Cysteine protease domain |
| k off | Dissociation rate constant |
| DLS | Dynamic light scattering |
| K d | Equilibrium dissociation constant |
| GI | Gastrointestinal |
| IP6-Bn | myo-Inositol hexakis-dibenzylphosphate |
| IT6-Bn | myo-Inositol hexakis-dibenzylthiophosphate |
| HBB | Hydrogen bond basicity |
| ITC | Isothermal calorimetry |
| kinITC | Kinetic ITC |
| MKK4/7 | Mitogen activated receptor kinases 4 and 7 |
| T M | Melting temperature |
| IP6 | myo-Inositol hexakisphosphate |
| IT6 | myo-Inositol hexakisthiophosphate |
| τ R | Residence time |
| TcdB | Toxin B |
| TMP | Trimethyl phosphate. |
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
We would like to acknowledge Dr Kim Munro and the Centre for Structural Biology Research (CRBS), supported by the Fond de Recherche du Québec–Santé (FRQS), for use of the core facilities. NMR experiments were recorded at the Québec/Eastern Canada High Field NMR Facility, supported by the Canada Foundation for Innovation, McGill University Faculty of Science and Department of Chemistry. Mass Spectrometry and DLS experiments were performed by the McGill Chemistry Characterization facility. Funding for this project was provided by a Canadian Institutes of Health Research (CIHR) project grant (PJT-173262) to Dr Bastien Castagner and a Natural Science and Engineering Research council of Canada (NSERC) discovery grant (RGPIN-2020-04908) to Dr Castagner. Dr Castagner is a tier 2 Canada Research Chair (CRC) in Therapeutic Chemistry.References
- K. A. Woll and F. Van Petegem, Physiol. Rev., 2022, 102, 209–268 CAS.
- A. J. López-Gambero, C. Sanjuan, P. J. Serrano-Castro, J. Suárez and F. Rodríguez de Fonseca, Biomedicines, 2020, 8, 295 CrossRef PubMed.
- B. Tu-Sekine and S. F. Kim, Int. J. Mol. Sci., 2022, 23, 6747 CrossRef CAS PubMed.
- J. Reineke, S. Tenzer, M. Rupnik, A. Koschinski, O. Hasselmayer, A. Schrattenholz, H. Schild and C. Von Eichel-Streiber, Nature, 2007, 446, 415–419 CrossRef CAS PubMed.
- K. E. Orrell and R. A. Melnyk, Microbiol. Mol. Biol. Rev., 2021, 85, e0006421 CrossRef PubMed.
- K. Prochazkova and K. J. F. Satchell, J. Biol. Chem., 2008, 283, 23656–23664 CrossRef CAS PubMed.
- A. Shen, P. J. Lupardus, V. E. Albrow, A. Guzzetta, J. C. Powers, K. C. Garcia and M. Bogyo, Nat. Chem. Biol., 2009, 5, 469–478 CrossRef CAS PubMed.
- R. A. Dick, K. K. Zadrozny, C. Xu, F. K. M. Schur, T. D. Lyddon, C. L. Ricana, J. M. Wagner, J. R. Perilla, B. K. Ganser-Pornillos, M. C. Johnson, O. Pornillos and V. M. Vogt, Nature, 2018, 560, 509–512 CrossRef CAS PubMed.
- A. Sreelatha, C. Nolan, B. C. Park, K. Pawłowski, D. R. Tomchick and V. S. Tagliabracci, J. Biol. Chem., 2020, 295, 6214–6224 CrossRef CAS PubMed.
- J. M. Labriola, Y. Zhou and B. Nagar, Biochemistry, 2018, 57, 4985–4996 CrossRef CAS PubMed.
- A. J. R. Costello, T. Glonek and T. C. Myers, Carbohydr. Res., 1976, 46, 159–171 CrossRef CAS PubMed.
- J. Torres, S. Domínguez, M. F. Cerdá, G. Obal, A. Mederos, R. F. Irvine, A. Díaz and C. Kremer, J. Inorg. Biochem., 2005, 99, 828–840 CrossRef CAS PubMed.
- S. R. Hull and R. Montgomery, J. Agric. Food Chem., 1995, 43, 1516–1523 CrossRef CAS.
- Y. Moritoh, S. Abe, H. Akiyama, A. Kobayashi, R. Koyama, R. Hara, S. Kasai and M. Watanabe, Nat. Commun., 2021, 12, 4847 CrossRef CAS PubMed.
- M. Nguyen Trung, S. Kieninger, Z. Fandi, D. Qiu, G. Liu, N. K. Mehendale, A. Saiardi, H. Jessen, B. Keller and D. Fiedler, ACS Cent. Sci., 2022, 8, 1683–1694 CrossRef CAS PubMed.
- A. S. M. G. M. Akond, H. Crawford, J. Berthold, Z. I. Talukder and K. Hossain, Am. J. Food Technol., 2011, 6, 235–243 CrossRef CAS PubMed.
- X. Li, Q. Wei, K. Zhao, W. Wang, B. Liu, W. Li and J. Wang, ACS Sens., 2023, 8, 4484–4493 CrossRef CAS PubMed.
- M. E. Ivarsson, E. Durantie, C. Huberli, S. Huwiler, C. Hegde, J. Friedman, F. Altamura, J. Lu, E. F. Verdu, P. Bercik, S. M. Logan, W. Chen, J.-C. Leroux and B. Castagner, Cell Chem. Biol., 2019, 26, 17–26.e13 CrossRef CAS PubMed.
- R. Cummer, F. Grosjean, R. Bolteau, S. E. Vasegh, S. Veyron, L. Keogh, J.-F. Trempe and B. Castagner, J. Med. Chem., 2024, acs.jmedchem.4c01408 Search PubMed.
- A. Dostálková, F. Kaufman, I. Křížová, B. Vokatá, T. Ruml and M. Rumlová, J. Virol., 2020, 94, 10–1128 CrossRef PubMed.
- A. Dostálková, B. Vokatá, F. Kaufman, P. Ulbrich, T. Ruml and M. Rumlová, Viruses, 2021, 13, 129 CrossRef PubMed.
- G. M. Blackbum, G. E. Taylor, G. R. J. Thatcher, M. Prescott and A. G. McLennan, Nucleic Acids Res., 1987, 15, 6991–7004 CrossRef PubMed.
- G. M. Blackburn, P. R. Ashton, M.-J. Guo, M. Rogers, G. Taylor, A. Guranowski and D. Watts, Heteroat. Chem., 1991, 2, 163–170 CrossRef CAS.
- D. Lampe, C. Liu and B. V. L. Potter, J. Med. Chem., 1994, 37, 907–912 CrossRef CAS PubMed.
- A. M. Cooke, S. R. Nahorski and B. V. L. Potter, FEBS Lett., 1989, 242, 373–377 CrossRef CAS PubMed.
- Y. Xu, X. Liu and G. D. Prestwich, Tetrahedron Lett., 2005, 46, 8311–8314 CrossRef CAS.
- I. Columbus, L. Ghindes-Azaria, R. Chen, L. Yehezkel, O. Redy-Keisar, G. Fridkin, D. Amir, D. Marciano, E. Drug, E. Gershonov, Z. Klausner, S. Saphier, S. Elias, A. Pevzner, Y. Eichen, G. Parvari, B. Smolkin and Y. Zafrani, J. Med. Chem., 2022, 65, 8511–8524 CrossRef CAS PubMed.
- H.-J. Zhang, M. Ociepa, M. Nassir, B. Zheng, S. A. Lewicki, V. Salmaso, H. Baburi, J. Nagel, S. Mirza, B. Bueschbell, H. Al-Hroub, O. Perzanowska, Z. Lin, M. A. Schmidt, M. D. Eastgate, K. A. Jacobson, C. E. Müller, J. Kowalska, J. Jemielity and P. S. Baran, Nat. Chem., 2024, 16, 249–258 CrossRef PubMed.
- B. Novotná, L. Vaneková, M. Zavřel, M. Buděšínský, M. Dejmek, M. Smola, O. Gutten, Z. A. Tehrani, M. Pimková Polidarová, A. Brázdová, R. Liboska, I. Štěpánek, Z. Vavřina, T. Jandušík, R. Nencka, L. Rulíšek, E. Bouřa, J. Brynda, O. Páv and G. Birkuš, J. Med. Chem., 2019, 62, 10676–10690 CrossRef PubMed.
- D. Chen, N. Oezguen, P. Urvil, C. Ferguson, S. M. Dann and T. C. Savidge, Sci. Adv., 2016, 2, e1501240 CrossRef PubMed.
- Degradation of Phytate by the 6-Phytase from Hafnia alvei: A Combined Structural and Solution Study|PLOS ONE, https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0065062, accessed 11 July 2024.
- P. H. Selle and V. Ravindran, Anim. Feed Sci. Technol., 2007, 135, 1–41 CrossRef CAS.
- J. G. J. Hoenderop, B. Nilius and R. J. M. Bindels, Physiol. Rev., 2005, 85, 373–422 CrossRef CAS PubMed.
- L. Kurz, P. Schmieder, N. Veiga and D. Fiedler, Biomolecules, 2023, 13, 645 CrossRef CAS PubMed.
- R. M. Jones, H. Wu, C. Wentworth, L. Luo, L. Collier-Hyams and A. S. Neish, Cell Host Microbe, 2008, 3, 233–244 CrossRef CAS PubMed.
- Z.-M. Zhang, K.-W. Ma, S. Yuan, Y. Luo, S. Jiang, E. Hawara, S. Pan, W. Ma and J. Song, Nat. Struct. Mol. Biol., 2016, 23, 847–852 CrossRef CAS PubMed.
- R. A. Friesner, J. L. Banks, R. B. Murphy, T. A. Halgren, J. J. Klicic, D. T. Mainz, M. P. Repasky, E. H. Knoll, M. Shelley, J. K. Perry, D. E. Shaw, P. Francis and P. S. Shenkin, J. Med. Chem., 2004, 47, 1739–1749 CrossRef CAS PubMed.
- R. A. Friesner, R. B. Murphy, M. P. Repasky, L. L. Frye, J. R. Greenwood, T. A. Halgren, P. C. Sanschagrin and D. T. Mainz, J. Med. Chem., 2006, 49, 6177–6196 CrossRef CAS PubMed.
- R. A. Copeland, D. L. Pompliano and T. D. Meek, Nat. Rev. Drug Discovery, 2006, 5, 730–739 CrossRef CAS PubMed.
- D. Burnouf, E. Ennifar, S. Guedich, B. Puffer, G. Hoffmann, G. Bec, F. Disdier, M. Baltzinger and P. Dumas, J. Am. Chem. Soc., 2012, 134, 559–565 CrossRef CAS PubMed.
- Á. Piñeiro, E. Muñoz, J. Sabín, M. Costas, M. Bastos, A. Velázquez-Campoy, P. F. Garrido, P. Dumas, E. Ennifar, L. García-Río, J. Rial, D. Pérez, P. Fraga, A. Rodríguez and C. Cotelo, Anal. Biochem., 2019, 577, 117–134 CrossRef PubMed.
- L. M. Finn, R. Cummer, B. Castagner and B. G. Keller, Proc. Natl. Acad. Sci. U. S. A., 2025, 122, e2419263122 CrossRef PubMed.
- J. E. Trosky, S. Mukherjee, D. L. Burdette, M. Roberts, L. McCarter, R. M. Siegel and K. Orth, J. Biol. Chem., 2004, 279, 51953–51957 CrossRef CAS PubMed.
- I. Pavlovic, D. T. Thakor, J. R. Vargas, C. J. McKinlay, S. Hauke, P. Anstaett, R. C. Camuña, L. Bigler, G. Gasser, C. Schultz, P. A. Wender and H. J. Jessen, Nat. Commun., 2016, 7, 10622 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cb00228h |
| This journal is © The Royal Society of Chemistry 2025 |