
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Non-enzymatic posttranslational protein modifications in protein aggregation and neurodegenerative diseases
Tim
Baldensperger
 a,
Miriam
Preissler
ab and
Christian F. W.
Becker
*a
a,
Miriam
Preissler
ab and
Christian F. W.
Becker
*a
aUniversity of Vienna, Faculty of Chemistry, Institute of Biological Chemistry, Währinger Str. 38, 1090 Vienna, Austria. E-mail: tim.baldensperger@univie.ac.at; miriam.preissler@univie.ac.at; christian.becker@univie.ac.at
bUniversity of Vienna, Vienna Doctoral School in Chemistry (DoSChem), Währinger Str. 42, 1090 Vienna, Austria
First published on 19th December 2024
Abstract
Highly reactive metabolic intermediates and other small molecules frequently react with amino acid side chains, leading to non-enzymatic posttranslational modifications (nPTMs) of proteins. The abundance of these modifications increases under high metabolic activity or stress conditions and can dramatically impact protein structure and function. Although protein quality control mechanisms typically mitigate the effects of these impaired proteins, in long-lived and degradation-resistant proteins, nPTMs accumulate. In some cases, such as cataract development and diabetes, clear links between nPTMs, aging, and disease progression have been established. In neurodegenerative diseases such as Alzheimer's and Parkinson's disease, a key question is whether accumulation of nPTMs is a cause or consequence of protein aggregation. This review focuses on major nPTMs found on proteins with central roles in neurodegenerative diseases such as α-synuclein, β-amyloid, and tau. We summarize current knowledge on the formation of these modifications and discuss their potential impact on disease onset and progression. Additionally, we examine what is known to date about how nPTMs impair cellular detoxification, repair, and degradation systems. Finally, we critically discuss the available methodologies to systematically investigate nPTMs at the molecular level and outline suitable approaches to study their effects on protein aggregation. We aim to foster more research into the role of nPTMs in neurodegeneration by adapting methodologies that have proven successful in studying enzymatic posttranslational modifications. Specifically, we advocate for site-specific incorporation of these modifications into target proteins using advanced chemical and molecular biology techniques.
 Tim Baldensperger | Dr Tim Baldensperger completed his PhD in the field of aging-associated protein modifications at Martin-Luther-University (Halle, Germany). During the following stay at the Grune lab at the German Institute of Human Nutrition (Potsdam, Germany), he developed a novel isolation method for lipofuscin protein aggregates, facilitating the validation of the mitochondrial–lysosomal axis theory of aging, which earned him a Young Investigator Award by the Society for Free Radical Research Europe. Currently, he is a postdoctoral fellow at the University of Vienna (Vienna, Austria) studying site-specific effects of post-translational modifications on protein aggregation. |
 Miriam Preissler | Miriam Preissler completed her master's degree at the Friedrich-Schiller-University (Jena, Germany) working under Prof. Christian Hertweck at the Leibniz Institute for Natural Product Research and Infection Biology Hans Knöll Institute (Jena, Germany). She was selected for the Honor's student program at the university, during which she worked on the characterization of unusual enzymes catalyzing natural product formation. She is currently a PhD candidate at the University of Vienna (Vienna, Austria) researching the site-specific effects of select modifications on tau protein. |
 Christian F. W. Becker | Christian F. W. Becker studied chemistry at the University of Dortmund (Germany) and obtained his diploma in 1998. After receiving his PhD in 2001 from the same University he became a postdoctoral fellow with Gryphon Therapeutics in So. San Francisco (USA) from 2002 to 2003. He started his independent career as a group leader at the Max-Planck Institute in Dortmund, Germany in 2004 and was appointed as Professor for Protein Chemistry at the Technische Universität München in 2007. In 2011, he became Professor and Head of the Institute of Biological Chemistry at the University of Vienna, in 2020 founding director of the Vienna Doctoral School in Chemistry (DoSChem) and in 2024 Dean of the Faculty of Chemistry. His group develops and uses chemical as well as biochemical means to generate peptides and proteins with otherwise unattainable (posttranslational) modifications to address fundamental biochemical as well as biomedical and biotechnological challenges. |
Introduction
According to the World Health Organization, dementia affects over 55 million people globally, with this number expected to rise to approximately 140 million by 2050. Dementia is the seventh leading cause of death and resulting annual healthcare costs were estimated to be US$ 1.3 trillion in 2019.1 The majority of dementia cases are attributed to neurodegenerative diseases such as Alzheimer's disease (AD) and Parkinson's disease (PD).2 An unifying hallmark of these pathologies is the accumulation of aggregated proteins.3 For the past decades, researchers have intensely debated why functional proteins such as α-synuclein (αSyn), β-amyloid (Aβ), and tau start to aggregate and accumulate during aging and to what extent these aggregates contribute to neurodegeneration.4–7One hypothesis involves the impact of posttranslational modifications (PTMs), which are chemical changes to proteins formed either strictly regulated by enzymes or non-enzymatically due to reactions with metabolites.8 The effects of enzymatic PTMs such as phosphorylation, acetylation, and ubiquitylation on protein aggregation, degradation, and resulting cytotoxicity have been extensively studied and reviewed.9–13 In stark contrast, the role of non-enzymatic PTMs (nPTMs) is much less understood. This is particularly concerning since nPTMs accumulate with age, are prevalent in diseases associated with increased risk of neurodegeneration, and are highly abundant in protein aggregates of neurodegenerative diseases.14–19
Two key processes leading to nPTMs are oxidative and carbonyl stress.20,21 Formation of reactive oxygen species (ROS) is an inevitable part of cellular metabolism. For instance, oxygen consuming processes such as electron transport chains, NADPH oxidases, and cytochrome P450 systems generate superoxide anion radicals. The formed superoxide is detoxified to hydrogen peroxide, leading to formation of hydroxyl radicals via the transition metal catalyzed Fenton reaction.22 Hydroxyl radicals are the most reactive ROS and directly modify proteins as well as other macromolecules leading to secondary ROS formation, e.g., alkoxyl and peroxyl radicals in the course of lipid peroxidation.21 Furthermore, superoxide readily reacts with the cellular messenger molecule nitric oxide and produces reactive nitrogen species (RNS) like peroxynitrite, which spontaneously decomposes into nitrogen dioxide and hydroxyl radicals.23 When cellular antioxidative and repair systems are overwhelmed by these processes, the resulting oxidative stress leads to accumulation of oxidative nPTMs.20 Similarly, the excessive generation of reactive carbonyl species (RCS) is termed carbonyl stress.24 Carbonyl stress has a strong overlap with oxidative stress, e.g., RCS such as glyoxal (GO), malondialdehyde, and 4-hydroxy-2-nonenal (4-HNE) are generated by oxidative degradation of lipids.21 Energy metabolism is another important source of RCS, for example via triosephosphate degradation as the main source of methylglyoxal (MGO)25 or formation of reactive acyl-CoA species (RACS) in the citric acid cycle.26 Moreover, RCS like deoxyglucosones and their cleavage products are formed from carbohydrates in the Maillard reaction.27 These RCS eventually lead to protein modifications known as advanced lipoxidation endproducts (ALEs) and advanced glycation endproducts (AGEs), depending on their metabolic origin.21 Interestingly, carbonyl stress is not merely a consequence of oxidative stress, but vice versa carbonyl stress is an initiator of oxidative stress by damaging mitochondria28 and inducing inflammation.29
The structures and formation mechanisms of the most relevant nPTMs in the context of protein aggregation and neurodegeneration are summarized in Fig. 1. Major targets of protein oxidation include cysteine, methionine, and tyrosine residues. Cysteine is readily oxidized by ROS to cysteine sulfenic and sulfinic acid, which is a reversible process by the enzyme glutaredoxin.30 Further oxidation to cysteine sulfonic acid is considered as irreversible “overoxidation” and typically prevented by protective glutathionylation.31 Alternatively, cysteine is able to form disulfide bonds with a second cysteine moiety under oxidative conditions, which is a fundamentally important mechanism in protein folding and reversible by several enzymes.32 Methionine, the second sulfur containing proteinogenic amino acid, is oxidized to methionine sulfoxide. This process is reversible via methionine sulfoxide reductases.33 In contrast, further oxidation to methionine sulfone is an irreversible step.34 Tyrosine oxidation is a potential source of protein crosslinking through dityrosine formation.35 Moreover, tyrosine is commonly nitrated by RNS leading to 3-nitrotyrosine.36
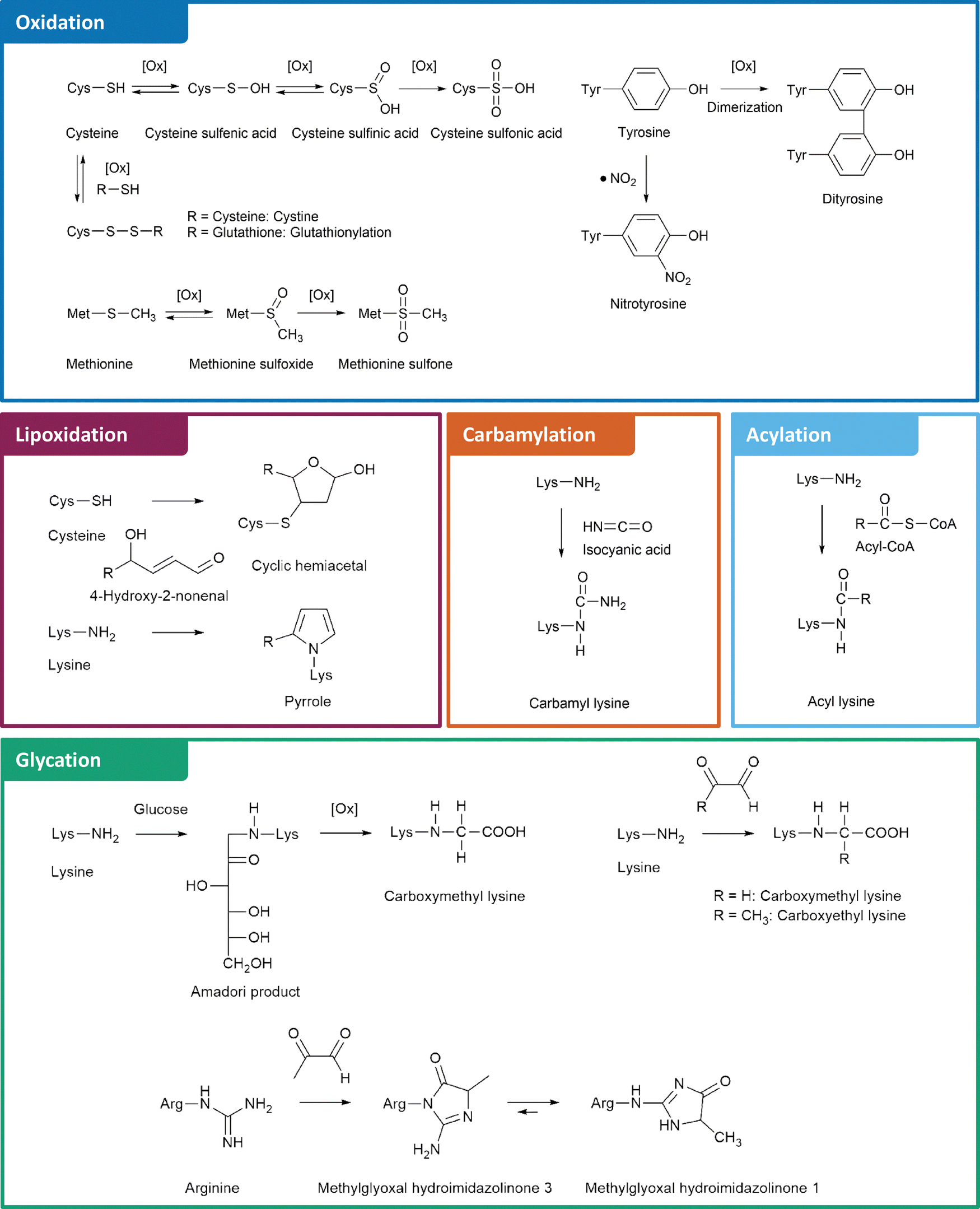 | ||
| Fig. 1 Non-enzymatic posttranslational protein modifications (nPTMs) of protein aggregates. | ||
Beyond direct oxidation of amino acids, several proteins are modified by reactive species originating from oxidative processes. For instance, lipoxidation intermediates such as 4-HNE form various adducts with lysine, cysteine, and histidine residues.21 The myeloperoxidase catalyzed oxidation of thiocyanate or spontaneous urea decomposition results in formation of isocyanic acid,37 which is the precursor of non-enzymatic lysine carbamylation.38 An alternative lysine modification is acylation by RACS.39 Several of these reactions are catalyzed by lysine acyl transferases.40 However, some RACS such as succinyl-CoA form highly reactive intramolecular anhydride structures leading to efficient formation of nPTMs.41
Finally, glycation by the Maillard reaction represents another major pathway of nPTMs, in which a nucleophile, e.g., the amino group of lysine or the guanidino group of arginine reacts with RCS such as deoxyglucosones or MGO. These RCS are central intermediates in glycation cascades and lead to formation of important AGEs, including N6-carboxymethyl lysine (CML), N6-carboxyethyl lysine (CEL), and arginine hydroimidazolinones.42,43
In this review, we aim to comprehensively summarize the complex network of nPTMs reported so far for protein aggregates associated with neurodegenerative diseases. We place particular emphasis on understanding how these nPTMs contribute to the neurotoxicity of protein aggregates, as well as how nPTMs impair cellular defense mechanisms. Last but not least, we critically evaluate the existing literature, describe approaches to generate selectively nPTM-carrying proteins, identify research gaps and propose future directions to enhance both the research quality and to inspire further investigations in this important field.
α-Synuclein
α-Synuclein (αSyn) is highly abundant in the central nervous system, making up approximately 0.5 to 1.0% of cytosolic brain proteins.44 It is predominantly located at the presynaptic termini of neurons and colocalized with several proteins responsible for neurotransmitter release and re-uptake of synaptic vesicles.45 Despite extensive research, the exact role of αSyn in neurotransmitter shuttling remains controversial.46 Knock-out and overexpression experiments suggest a regulatory function, as absence of αSyn caused a lack of synaptic vesicles, while overexpression enhanced the number of available vesicles (Fig. 2(A)).47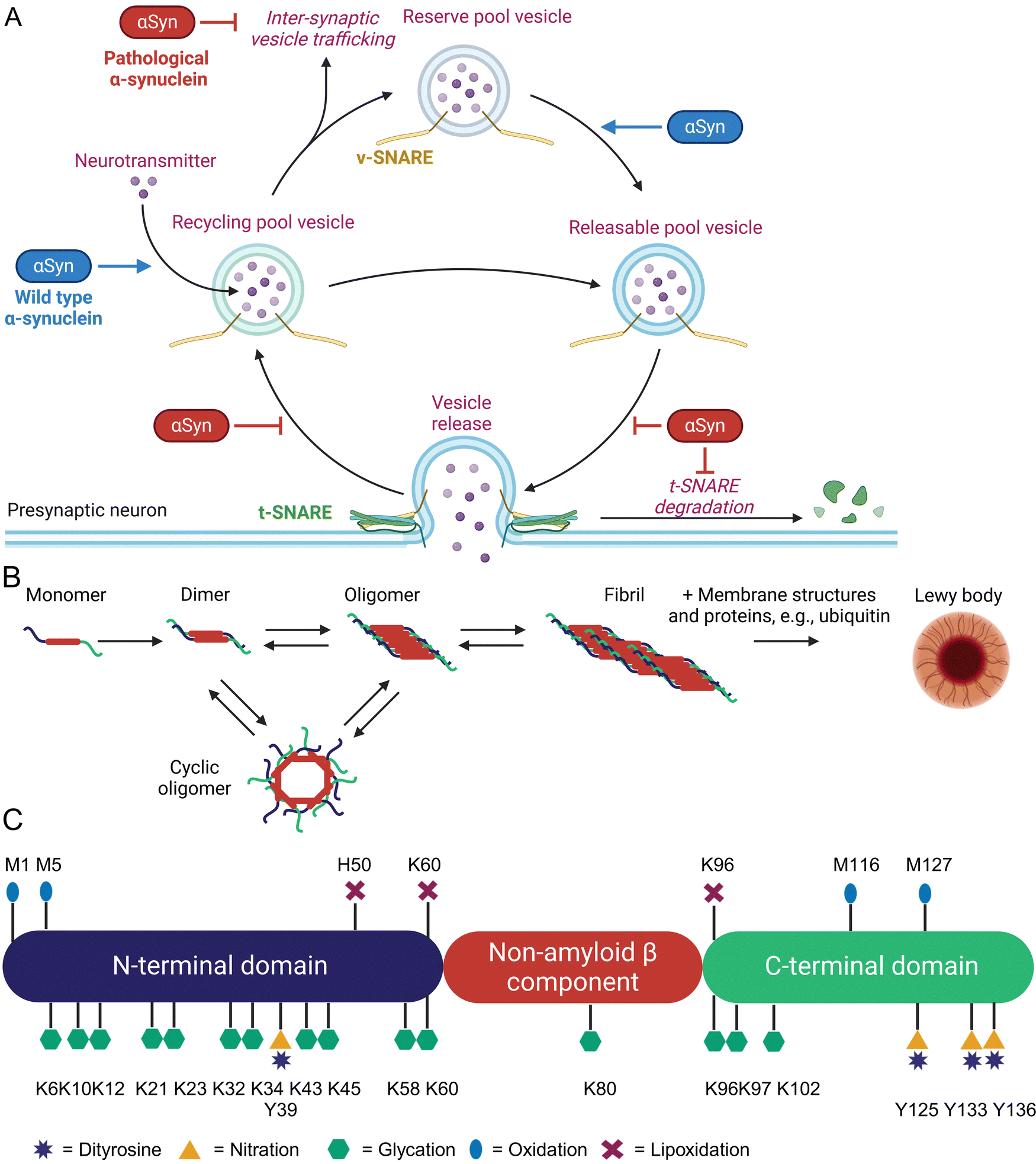 | ||
| Fig. 2 Overview of function and aggregation of α-synuclein, including known non-enzymatic posttranslational modifications. Cellular function and pathogenic effects of α-synuclein (A). Oligomerization, fibrillization, and aggregation of α-synuclein (B). Non-enzymatic posttranslational modifications of α-synuclein (C). | ||
The 14.4 kDa αSyn protein comprises 140 amino acids and lacks cysteine, arginine, as well as tryptophan residues.48 Structurally, αSyn consists of three distinct domains: an amphipathic and lysine-rich amino terminus (residues 1–60), the hydrophobic non-amyloid-β component region (residues 61–95), and a disordered, acidic carboxy-terminal tail (residues 96–140).49 These domains form an intrinsically disordered protein,50 but upon binding to lipid membranes the N-terminus rearranges into an α-helix of 95 amino acids.51 This binding is mediated by repetitive KTKEGV motifs mainly located in the N-terminal region.52 The non-amyloid-β component domain is mandatory for aggregation of αSyn through β-sheet formation.53 Last but not least, the negatively charged C-terminus has been shown to bind Ca2+ and can exhibit chaperone-like activity.54 Initially, αSyn was identified as the non-amyloid-β component in amyloid plaques found in the brains of Alzheimer's disease patients.44 It is also a major component of protein aggregates known as Lewy bodies and Lewy neurites, which are hallmarks of neurodegenerative diseases termed as synucleinopathies, e.g., Parkinson's disease (PD), dementia with Lewy bodies, multiple systems atrophy, and pure autonomic failure.47 Aggregation of αSyn involves several intermediates such as dimers, oligomers, fibrils, and finally Lewy body formation, which additionally contain various proteins (ubiquitin, p62) and lipids (membrane structures) as shown in Fig. 2(B).55 Recent research indicates that oligomeric forms of αSyn, rather than its fibrils or aggregates, are the toxic species contributing to cellular toxicity, similar to other pathologic protein aggregation events.56–58 These oligomers exert their detrimental effects through several mechanisms including increased Ca2+ influx by altered membrane integrity,59 mitochondrial damage,60 lysosomal leakage,61 and disruption of microtubules.62 A critical and still unanswered question is, why a functional important protein such as αSyn starts to form toxic protein aggregates. Approximately 5 – 10% of PD patients have a genetic predisposition due to specific mutations in the SNCA gene, which leads to the expression of harmful αSyn variants.57 For the remaining PD patients, the onset appears to occur spontaneously. Parkinson's disease is predominantly an age-related condition, rarely diagnosed in individuals under 50 years of age, but affecting approximately 1% of people over 60 years old.63 Additional risk factors include exposure to environmental toxins, pesticides, and heavy metals, as well as comorbidities such as head injuries or viral infections. Conversely, lifestyle interventions such as regular physical activity and coffee consumption have been shown to have protective effects.64 Type 2 diabetes has also been identified as a risk factor for PD. An English retrospective study involving 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 190323 individuals demonstrated a 1.3 to 1.5-fold increased risk of PD among diabetics.65 Similarly, a Korean follow-up study with 2362072 participants found that the hazard rate of developing PD increased between 1.1 and 2.8 times, depending on the severity of diabetes mellitus.66
190323 individuals demonstrated a 1.3 to 1.5-fold increased risk of PD among diabetics.65 Similarly, a Korean follow-up study with 2362072 participants found that the hazard rate of developing PD increased between 1.1 and 2.8 times, depending on the severity of diabetes mellitus.66
Based on these studies, a common feature of all risk factors in PD is the generation of inflammation, oxidative and carbonyl stress that also leads to enhanced non-enzymatic protein modifications (Fig. 2(C)) at various αSyn sites.15,20
One of the most abundant nPTMs in αSyn is oxidation of methionine to methionine sulfoxide.33,67 Residue 5 is the most susceptible residue for methionine sulfoxide formation and was already formed by treatment of recombinantly expressed αSyn with UV light. Oxidation of M5 produced fibrilization-incompetent αSyn and favored the formation of neurotoxic oligomers.68 Oxidation of M1 and M5 by hydrogen peroxide drastically inhibited αSyn degradation by the 20S proteasome and this effect was nullified in a M1,5A mutant.69 Dopamine was able to oxidize M116 and M127 leading to soluble and cytotoxic oligomers, which were rendered harmless by M127A mutagenesis in a cellular test system.67 Oxidation of M127 reduces phosphorylation of nearby Y125 and possibly S129, which are important modulators of αSyn aggregation and toxicity.70 Oxidation of all 4 methionine residues in αSyn to the sulfoxides dramatically inhibited fibrillization,71 an effect which was proportional to the amount of oxidized methionine residues,72 and decreased the binding capacity in a model of synaptic vesicles.73 Interestingly, methionine oxidation at position M1 and M5 is reversible by methionine sulfoxide reductases, while positions M116 and M127 are not targeted by this repair system.70 Reversible oxidation of M1 and M5 is considered as a possible ROS scavenger and protects αSyn from oxidation at more harmful positions. Consequently, over-expression of methionine sulfoxide reductase A prevented development of PD-like symptoms in Drosophila melanogaster after ectopic expression of αSyn.74 Tyrosine is another common target of protein oxidation in αSyn, which can form intra- and intermolecular dityrosine crosslinks.75,76 Intramolecular crosslinking of tyrosine residue 39 with 125, 133, or 136 was reported to prevent αSyn fibrilization and aggregation by stabilizing monomers.77 In contrast, intermolecular tyrosine dimerization is a critical and rate limiting step in αSyn aggregation leading to PD.78 Tyrosine is also modified by nitroxidative stress. The group of Lashuel utilized native chemical ligation to generate aSyn variants with site-selective nitrotyrosine residues at positions 39 and 125. These variants formed big amorphous aggregates in contrast to long fibrils formed by wild-type αSyn and the nitrated αSyn had a reduced binding affinity for membrane vesicles.79 The reduced vesicle binding was mainly caused by Y39 nitration and nitrated αSyn was more resistant towards degradation by the 20S proteasome and calpain.80 Despite close proximity, nitration of Y125 had no effect on S129 phosphorylation.79 Lipoxidation byproducts, such as 4-HNE, form adducts with histidine H50, lysine residues K60 and K96, which enhanced formation of αSyn oligomers and toxicity in cultured neurons.81–83 A distinctive characteristic of αSyn is its modification by dopamine and its oxidized metabolites, such as dopamine quinone and 3,4-dihydroxyphenylacetaldehyde.84 Dopaminergic neurons are the primary cell type affected in PD and interactions between αSyn and dopamine have been shown to exacerbate neurotoxicity.85
Deposits of αSyn in both mice and humans accumulate AGEs.86–88 However, whether the accumulation of AGEs is the cause or merely a consequence of protein aggregation remains an unresolved question. Glycation of recombinant αSyn by GO and MGO induced oligomerization, inhibited fibrilization and resulted in small spherical aggregates.89 Glycation by MGO furthermore resulted in loss of binding to anionic lipid membranes.90 Extensive modification of all 14 lysine residues to form CEL led to oligomer formation and prevented fibrillization91 as well as binding to small vesicles.92 Increase of MGO levels by knock-down of glyoxalase 1 or triosephosphate isomerase increased αSyn aggregates and toxicity in yeast, Lund human mesencephalic cells, and Drosophila melanogaster.88 Intracerebroventricular MGO injection in mice exacerbated PD-like symptoms.93 So far, only the group of Fleming-Outeiro has produced selectively CML/CEL-modified αSyn fragments and a full-length version of the PD-associated αSyn variant E46K, with a single CEL modification at position 46. However, the biological evaluation of these modified species remains to be conducted.94
β-Amyloid
β-Amyloid (Aβ) is a 4 kDa fragment originating from proteolytic cleavage of the amyloid precursor protein (APP). This precursor is expressed by brain neurons, astrocytes, vascular and blood cells.95 APP has three isoforms APP695, APP751, and APP770 arising from alternative splicing. APP695 is the predominant isoform in the brain and lacks a 56 amino acid Kunitz Protease Inhibitor domain found in APP751 and both the Kunitz Protease Inhibitor domain and a 19 amino acid OX-2 domain present in APP770.96 APP is a transmembrane protein that spans the extracellular space from the N-terminus to the intracellular lumen at the C-terminus. It is processed by secretases either in a non-amyloidogenic or an amyloidogenic pathway.97 In the non-amyloidogenic pathway, cleavage by α-secretase generates soluble APPα (sAPPα) and the C83α subunit.98 C83α is further processed by γ-secretase to form the APP intracellular domain (AICD) and the p3 fragment.99 Extracellular sAPPα is a neuroprotective protein and vitally important for cognitive function.100 The amyloidogenic pathway involves APP cleavage by β-secretase into soluble APPβ (sAPPβ) and the C99β subunit.101 The latter is further processed by γ-secretase to yield AICD and Aβ peptides, which consist of 37 to 43 amino acids.102 The major product Aβ 1–40 is about 10 times more abundant in cerebrospinal fluid than Aβ 1–42.102 Aβ 1–42 forms intracellular oligomers and is the main component of extracellular amyloid plaques,103 which are a hallmark of Alzheimer's disease (AD).104 Alternatively, β-secretase has a second APP cleavage site leading to a C89β fragment, which is further processed to Aβ 11–42 and lacks amyloidogenic properties (Fig. 3(A)).105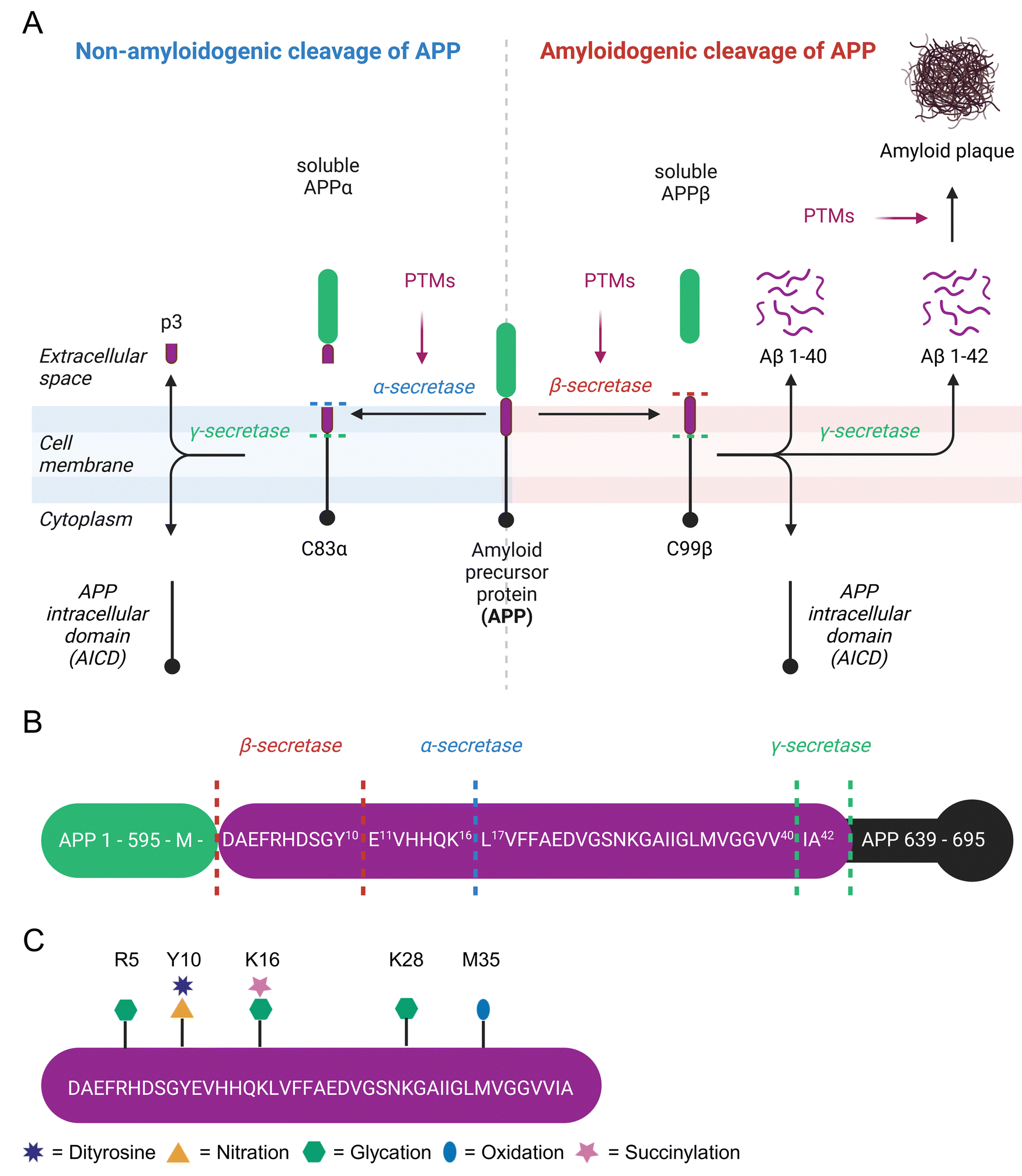 | ||
| Fig. 3 Overview of amyloid-beta, formation of β-amyloid aggregates, and known non-enzymatic posttranslational modification sites. Non-amyloidogenic and amyloidogenic processing of amyloid precursor protein (A). Possible cleavage sites of secretases in the amyloid precursor protein sequence (B). Non-enzymatic posttranslational modifications of β-amyloid 1–42 (C). | ||
A small portion (<5%) of AD cases are early-onset variants caused by mutations in the APP, PSEN1, or PSEN2 genes.106 However, the majority of AD patients is affected by late-onset forms with the disease affecting 1 in 9 individuals over the age of 65 and 1 in 3 individuals over the age of 85, making age the leading risk factor of AD.107 The most significant genetic risk factor for late-onset AD is the polymorphism in the apolipoprotein E (ApoE) gene. The ApoE ε4 allele is present in 60% of Caucasian AD patients. The risk of developing AD is 2–3 times higher with one ε4 allele and about 12 times higher in those with two ε4 alleles compared to ApoE ε3.108 People with ApoE ε4 polymorphism suffer from impaired glucose metabolism, increased oxidative stress, and enhanced neuroinflammation as potential molecular drivers of AD as reviewed previously.109 In contrast, ApoE ε2 is protective against development of AD.110 Beside genetic predisposition, various comorbidities contribute to AD development. For instance, 80% of AD patients have insulin resistance or type 2 diabetes111 and diabetes increases the AD risk 2–5 fold compared to healthy individuals.112 Further diseases facilitating AD are cardiovascular diseases, traumatic brain injury, epilepsy, and depression.113 Again, these risk factors are associated with inflammation, oxidative stress, and carbonyl stress leading to extensive nPTMs.114,115
While modulation of APP cleavage by PTMs has been proposed, it has not been experimentally verified.116,117 Several amino acids next to secretase cleavage sites are prone to posttranslational modification (Fig. 3(B)). Recently, succinylation of APP residue K612 (=K16 in Aβ) was detected in 9 out of 10 brains from AD patients, but not in age-matched controls without dementia.118 This specific residue is the cleavage site of α-secretase and involvement of this PTM in shifting APP cleavage towards amyloidogenic cleavage comes to mind. Furthermore, the N-terminal amino acid in APP at the β-secretase cleavage site located right next to the Aβ 1–42 motif is M696. Oxidation of this site was investigated more than 25 years ago, but unfortunately the exact identity of β-secretase (= beta-site amyloid precursor protein cleaving enzyme 1) was unknown at this time, leading to tests with unsuitable proteases.119 The second β-secretase cleavage site of APP is next to Y10 of Aβ, which is readily nitrated and forms dityrosine crosslinks.120,121 In mice this site is preferred by β-secretase122 and leads to non-amyloidogenic Aβ 11–42.105
The Aβ 1–42 peptide itself is heavily modified at various sites (Fig. 3(C)). Aβ extracted from senile plaques is often oxidized at M35.123 Selective oxidation of M35 by H2O2 to methionine sulfoxide results in a threefold lower fibrillization rate compared to native Aβ.124 Neurotoxicity of native Aβ and M35 sulfoxide Aβ in primary cortical neuron cell culture is very similar,125 but replacement of M35 by norleucine significantly decreases toxicity in rat neuronal cells.126 Tyrosine oxidation leads to intermolecular dityrosine crosslinks between Y10 residues of Aβ and is commonly detected in AD brains.120 Dityrosine Aβ dimers are highly efficient in membrane permeabilization and exhibit significantly higher toxicity compared to wild-type Aβ.127,128 Hence, dityrosine formation is considered as a central mechanism in AD development.129 Nitroxidative stress causes nitration of Y10. A study involving in vitro nitration of Aβ found inhibition of fibrillization, increase of oligomerization, and consequently increased neuronal toxicity.130 The inhibition of Aβ aggregation by nitrotyrosine was further verified by dynamic light scattering experiments.121 Conversely, Kummer et al. measured increased aggregation of Aβ after Y10 nitration in vitro and reduced amyloid plaques as well as cognitive decline in an AD mice model after inhibition of nitric oxide synthetase 2.131 Senile plaques of AD patients contain Aβ adducts with lipoxidation product 4-HNE.132 However, 4-HNE only has minor if any effects on aggregation of Aβ1–40. Unfortunately, this study did not check the influence of 4-HNE on Aβ1–42 aggregation.133 Addition of 4-HNE to a neuroblastoma cell line is highly cytotoxic, but conditional expression of a C-terminal APP fragment has not further increased this effect.134 The Aβ peptide contains 3 aspartic acid residues at positions 1, 7, and 23, which are all prone to D-isomerization.135 The formation of D-aspartate is enhancing proteolytic resistance and aggregation of Aβ.136
Protein glycation in AD patients is up to three times higher than in control subjects and even further increased in Aβ plaques.137 MGO treatment decreases Aβ fibrilization kinetics,138 but increases Aβ aggregate size133in vitro and enhances toxicity of C-terminal APP fragment expressed in neuroblastoma cells.134 While the arginine modification MGO-derived hydroimidazolone 1 (MG-H1) is the most abundant AGE in cerebrospinal fluid of AD patients,139 MGO preferentially glycates K16 over R5 in Aβ.140 Synthetic Aβ incubated with MGO is more toxic for hippocampal neurons compared to unmodified Aβ and treatment of Tg2576 mice with dicarbonyl scavenger aminoguanidine ameliorated cognitive decline in this AD model.141 Solid phase peptide synthesis (SPPS) was used to site-specifically incorporate CEL modifications in Aβ at positions K16, K28, and double-mutation of K16 and K28.142 According to this study, K28CEL has no effect on fibrilization, while K16CEL slows fibril formation, and K16,28CEL alters the aggregate morphology. Nevertheless, K16CEL and K28CEL exhibit higher toxicity in differentiated SH-SY5Y cells compared to unmodified Aβ, whereas K16,28CEL completely loses its toxic properties.142 These findings clearly indicate that in vitro and in cell evaluation is required to assess nPTM effects, posing the challenge for all synthetic, modified peptide and protein samples to be transferred into in vivo systems in a sensible manner.
Tau
The microtubule associated protein tau was first purified from porcine brain alongside tubulin.143,144 Further research has placed tau in the central and peripheral nervous system, being most abundant in neuronal axons.145 Tau is typically associated with the promotion of polymerization, assembly and stabilization of microtubules (Fig. 4(A)), thus contributing to the structural integrity of neurons and axonal transport.146 When present in other intracellular compartments or extracellular locations, tau exhibits additional functions such as protecting DNA from peroxidation-induced damage within the nucleus.147 Numerous other potential roles for tau remain under investigation.148–152 | ||
| Fig. 4 Overview of tau protein, physiological function and aggregation process, and known non-enzymatic posttranslational modification sites. Physiological and pathological function of tau in stabilization or disintegration of microtubule (A). Domain structure of tau 4R2N isoform (B). Non-enzymatic posttranslational modifications of tau 4R2N. in vivo nPTMs detected in samples above, in vitro generated nPTMs below (C). | ||
Structurally, tau belongs to the family of intrinsically disordered proteins and exhibits no preferred three-dimensional structure.153 Tau exists in six isoforms of varying length, differing at their N-terminal and microtubule-binding regions. Three varying N-termini derive from alternative splicing of exons 2 and 3, with either no N-terminal extension (0N), 29 (1N) or 58 (2N) additional amino acids present. Furthermore, tau isoforms may contain three or four repeated microtubule-binding domains (3R or 4R) by exclusion or inclusion of exon 10.154,155 Hence, making 4R2N the longest tau isoform, comprising of 441 amino acids. Intrinsically disordered proteins often act as scaffolds for signaling and regulatory functions, allowing for highly promiscuous interactions reflected in the number and diversity of potential tau functions. The 4R2N isoform of tau is characterized by a negatively charged N-terminus (amino acids 1–121), followed by a domain with a high excess of positive charge (122–250), and repeat regions (Fig. 4(B)) with a moderate excess of positive charge (251–390) as well as a negatively charged at the C-terminal domain (391–441). Based on sequence features and the N (N1, N2) and R (R1–R4) domains, 4R2N tau may also be subdivided into an N-terminal domain (NTD; 1–150), proline-rich domain (PRD; 151–243), microtubule binding region (MTBR; 244–368) and C-terminal domain (CTD; 369–441). Containing a large number of residues subject to PTMs, such as phosphorylation, acetylation or ubiquitylation, enables fine-tuning of tau's biological activity.152,156 Aberrant modifications, particularly hyperphosphorylation, are implicated in tau-related diseases, coined tauopathies. By disruption of native tau function, neuronal transport, axonal transport and stability are perturbed (Fig. 4(A)), making tauopathies neurodegenerative diseases, categorized by the disease-associated isoforms 3R, 4R, and 3R + 4R. Some of these include Alzheimer's disease (AD), amyotrophic lateral sclerosis, familial frontotemporal dementia, and Pick's disease.152 It is important to note that tau dysfunction may contribute to disease pathology, though it may not be the primary cause. A defining characteristic of tauopathies, first observed by Alois Alzheimer in the brains of AD patients in 1906, is the presence of insoluble inclusions composed of neurofibrillary tangles (NFTs) formed by tau protein.157 These NFTs consist of paired helical filaments (PHFs) of tau.158,159 Notably, tauopathies are distinguished by unique pathological filament structures, as the conformers vary between different diseases, although they remain consistent among patients with the same condition.160,161
The major focus of previous research was fibrillar tau in AD. Aggregated tau isolated from brain samples carries a specific pattern of phosphorylation different to that of healthy controls and is discussed in detail in several reviews.162–165 Furthermore, modifications such as acetylation and ubiquitylation have garnered increasing interest and have been studied by different approaches allowing controlled installation of PTMs in tau.166–170 However, tau aggregation can also be influenced by a variety of nPTMs, which will be discussed in the following.
Tau protein isolated from AD patients’ brains and in helical filaments is glycated (Fig. 4(C)).171–173 Non-specific in vitro glycation of tau by reducing sugars enhances aggregation of isoforms 4R2N and 3R2N and reduces tubulin binding.174–176 Combining non-specific phosphorylation and glycation of tau enhances aggregation.177–179 Such undefined glycation reactions can lead to tau variants glycated at up to 24 lysine residues (Fig. 4(C)).175 However, such ambiguous and most likely excessive glycation of a target protein does not reflect (patho-) physiological conditions and in turn does not allow understanding the impact of individual modifications. We have previously introduced the site-specific carboxymethylation of K294 into tau via protein semi-synthesis and found an inhibitory effect on tubulin polymerization, without directly impacting tau aggregation.167 In all likelihood, changes in tau properties induced by glycations are modulated by more specific glycation events controlled by concentration of electrophiles and accessibility of reactive side chains, arguably severely reducing the relevance of experiments carried out via unselective glycations. Here, indirect effects of nPTMs, for example on chaperone systems that should prevent protein aggregation, can also come into play as was recently demonstrated for argpyrimidine modifications of heat shock protein 27 (Hsp27). Activity of Hsp27 towards several client proteins was severely impacted by one or more argpyrimidine residues incorporated by protein semi-synthesis.180
Another prevalent nPTM found at elevated levels in brain isolates from AD patients is 3-nitrotyrosine.181Via nitrotyrosine specific antibodies, nitration at Y18, Y29 and Y197 was detected. While nitration of Y18 and Y197 is observed in AD brains and age-matched controls, nitration at Y29 appears to be specific for AD and Pick's disease.182–184 Y197 is also endogenously nitrated in mouse brain samples and PC12 cells as models for AD.185 Single-site nitration of Y18 inhibits tau aggregation in vitro.182 Effects of nitration were also evaluated in vitro through addition of RNS such as peroxynitrite. Based on these results, nitration may occur in hierarchical fashion with Y18 and Y29 being preferred over Y197 and Y394.186 However, the results of such artificial nitration conditions need to be carefully evaluated. Formation of larger tau aggregates is reduced for this variant with multiple nitrations, however, these modifications may induce oligomerization of tau.187
nPTMs can control peptide and protein aggregation as demonstrated for non-specific carbamylation of short peptide fragments of tau containing neighboring lysine residues. Such modified peptides strongly vary in their aggregation behavior with 140KKAKGA145 not aggregating, 148KTKIAT153 strongly aggregating, and 224KKVAVV229 showing medium aggregation levels. Additionally, 254KNVKSK259 aggregates at high concentrations or at low concentrations upon adding nonpolar residues and 368NKKIETHKLTF378 shows a concentration-dependent aggregation.188 Specific carbamylation of the N-terminally acetylated hexapeptides PHF6 (306VQIVYK311) and PHF6* (275VQIINK280) led to rapidly aggregating PHF6 and formation of fibrils that exhibit increased cytotoxicity, whereas PHF6* shows an extended lag-time and a slow, linear aggregation profile when acetylated and carbamylated. In the same study non-specific carbamylation of full-length tau was tested, resulting in a protein or protein mixture that aggregates quickly even in the absence of an external inducer that is commonly used in such in vitro aggregation assay to reduce measurement time and to increase reproducibility.189
Oxidative and reducing conditions modulate the cysteine thiol group. Here, several reports analyzed the impact of disulfide linkages or the oxidation state of cysteine on tau behavior. In summary, these findings implicate the disulfide bridge between C291 and C322 or intermolecular bonds in increased aggregation and polymerization of tau. Tau aggregates accumulate in fly retina and mouse primary cultured neurons under oxidative stress, which the authors link back to intermolecular disulfide bond formation with cysteine-deletions disrupting assembly.190 Aggregation kinetics were also shown to be increased under oxidizing conditions and with all cysteines intact. Alanine replacement of cysteine at position 291 or 322 reduces dimer assembly rate, an effect stronger for 2R and 3R than 4R isoform, assuming dimerization and polymerization of 3R to rely on C322 intermolecular disulfide formation, which in turn acts as polymerization seed. The disulfide linkage between 3R1N and 4R1N accelerates fibrillation kinetics.191 The oxidation state of C322 in particular appears to modulate tau-associated toxicity and dysfunction.181,192 Oxidation of 4R2N induces formation of structurally distinct fibrils, which could be resolubilized through the addition of reducing agents.193
An optional lysine PTM, which has garnered increasing interest, is succinylation. Despite hypotheses on the origin of this PTM conflicting, besides putative succinyl transferases, a non-enzymatic process is considered a likely cause, therefore, being detailed for tau in the following.41,194 Recently, first insights into the succinylation of lysine residue 311 were revealed, demonstrating that succinylated lysine occurs in nine out of ten AD patient's brain samples, however, not in healthy controls.118 Specific succinylation of the PHF6 peptide at K311 accelerates aggregation, whereas modification of K280 in PHF6* does not aggregate under the same conditions. Furthermore, treatment of tau K19 (comprising residues Q244-E372) with succinyl-CoA results in complete loss of function in microtubule binding assays. Lastly, specific succinylation of the tau 296–321 peptide at K311 decreases binding affinity for tubulin.195 Specific modification was achieved by SPPS.
Another commonly observed irreversible, non-enzymatic modification occurring during protein aging is deamidation of asparagine. A similar deamidation can be observed as a consequence of asparagine isomerization (Fig. 5), which may occur enzyme catalyzed or enzyme-free and which has also been observed for tau.196,197 This observation was triggered by broad bands in Western blots of tau PHFs.198 This notable feature was further investigated by analysis of the protein samples in these bands and revealed the presence of significant amounts of D-aspartate in tau.196 This finding has been confirmed by another study focusing on smeared bands found for PHF. Here deamidation and isomerization are found on residues N381 and N387 giving rise to isoaspartates.199 Another study detected isoaspartate at D193, N381 and N387 in PHF tau, supporting these findings.200 Employing antibodies specific for D387 and isoD387, the PHF smear was immunostained to a higher degree than PHFs, which led to the conclusion that the modifications mainly occur after fibril formation.201 This observation touches on a critical point when studying nPTMs as we still struggle with determining the sequence of events in forming nPTMs.
 | ||
| Fig. 5 Asparagine deamidation and aspartate isomerization reaction proceeding via the succinimide intermediate. | ||
In contrast to this, deamidation can also lead to functional consequence as described for asparagine residue 279. In this case, Asn to Asp conversion negatively affects microtubule assembly.202 Intriguingly, extensive deamidation of 3R and 4R tau is exclusively observed in AD, yet neither progressive nuclear palsy or corticobasal degeneration. In order to study the molecular basis site-specific D-isomerization in tau R2 and R3, the main components of tau PHF core, peptide with D-Asp were generated by SPPS. These model peptides exhibit reduced time-dependent transition from random coil to β-sheet following heparin treatment compared to a wild-type R2 peptide.203 Another study employing synthetic R2 and R3 peptides respectively, demonstrates notable attenuation of the inhibitory effect of cyanidin on fibrillation for peptides containing D-isomerized aspartate. Additionally, peptides of this series show an altered fibril morphology.204 No synergistic effects for the double-isomerized peptide compared to the single isomerized variants are observed. Additionally, isomerization of serine in tau R3 affect β-sheet transition or fibril formation compared to the WT R3 peptide.203
Prevention of protein aggregation by avoiding or repairing nPTMs
In the previous sections, we highlighted the importance of non-enzymatic posttranslational modifications (nPTMs) in the process of protein aggregation. Cells employ several enzymatic defense mechanisms to prevent accumulation of nPTMs (Fig. 6). The first line of defense involves the detoxification of potentially harmful electrophiles,205 which can arise from elevated levels of oxidative and carbonyl stress. The second line of defense focuses on the repair of mildly damaged proteins. Last but not least, severely damaged proteins are degraded via different pathways.205 Although these systems are crucial for preventing accumulation of proteins damaged by nPTMs, the enzymes involved in these processes are themselves susceptible to modifications. The following sections will summarize the known nPTMs in these enzyme systems and the resulting effects as a contributing factor in protein aggregation.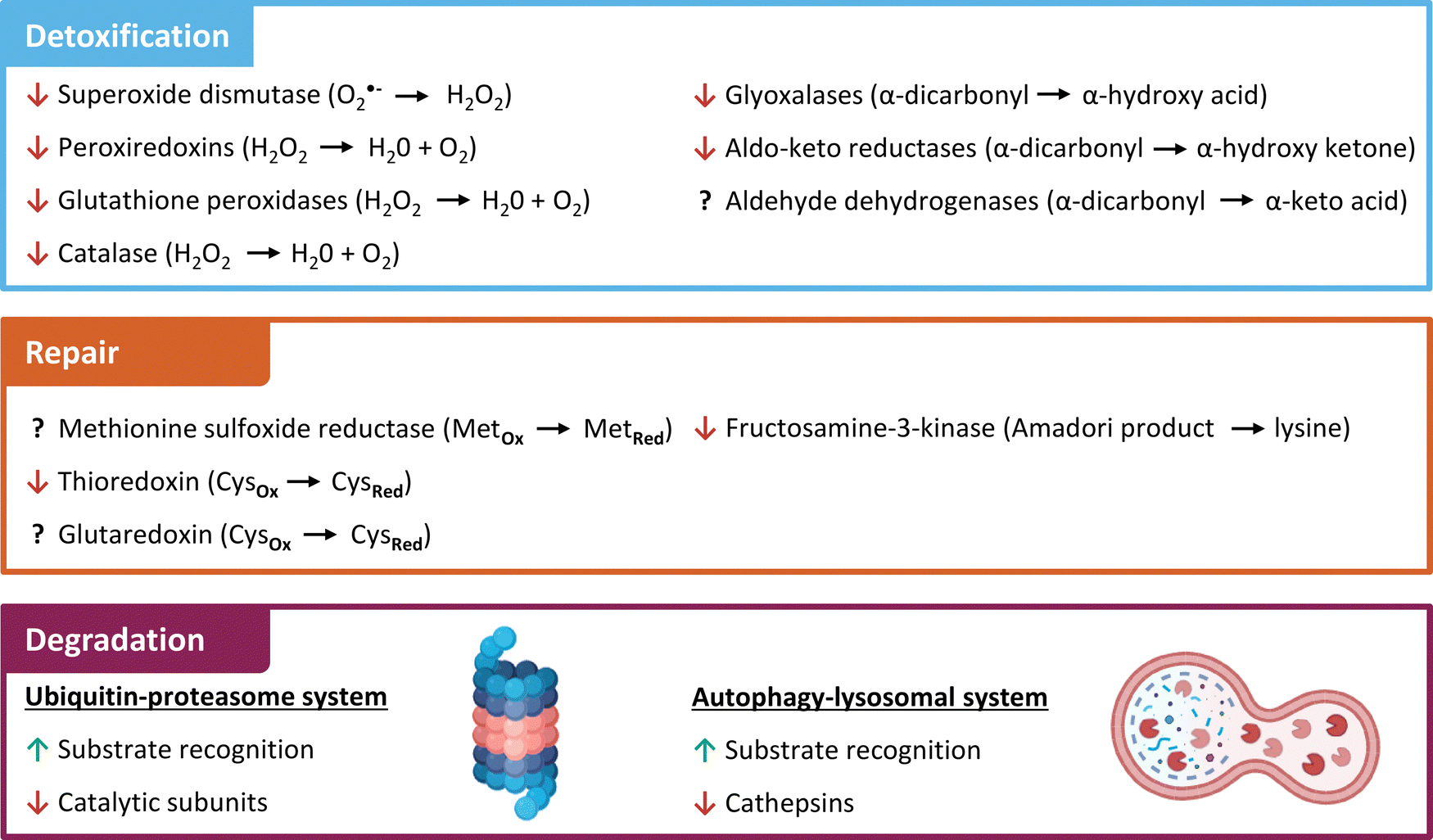 | ||
| Fig. 6 Effects of non-enzymatic posttranslational protein modification on cellular defense systems against protein aggregation. Effects of non-enzymatic posttranslational protein modifications on cellular detoxification, repair, and degradation systems are indicated by red arrows(inhibition), green arrows (activation), and question marks (unknown). | ||
The predominant enzymes responsible for elimination of hydrogen peroxide in cells are peroxiredoxins (Prx), glutathione peroxidases (GPx), and catalase. Their activities are regulated by a plethora of enzymatic modifications,218 but studies about the influence of nPTMs are rather scarce. Nevertheless, a shift from peroxidase to chaperone activity was reported after hyperoxidation of cysteine residues in Prx.219 Non-specific glycation by hyperglycemic conditions inhibits catalase220 as well as Gpx221 activity in vitro.
Short-chained dicarbonyls such as GO and MGO are mainly detoxified by the glutathione dependent glyoxalase system.222 The activity of this fundamentally important system is decreased in aged brains.223 Glyoxalase 1 is inactivated by nitration in cell culture224 and glutathionylation of C139 inhibits the enzyme.225 In tissues with low glyoxalase expression levels and in case of complex carbonyls, the detoxification is catalyzed by NADPH dependent aldo-keto reductases or aldehyde dehydrogenases.226 Elevated ROS levels are reported to cause cysteine oxidation and decrease enzymatic activity of aldo-keto reductases.25,226 Another example is the Parkinson disease protein 7 (DJ-1) that has various cellular functions such as transcription regulation, antioxidative modulation, and chaperone activity.227 DJ-1 scavenges glycated aSyn228 and was postulated as a potential deglycase.229 However, DJ-1 primarily counteracts glycation through its glyoxalase activity, reducing free MGO levels and shifting the equilibrium away from reversibly protein-bound MGO.230 DJ-1 is closely related to development of PD and point mutations231 or oxidative modification of C106 promote aSyn aggregation in PD patients.232
Oxidized cysteine residues and unnatural disulfide bonds are repaired by the NADPH dependent thioredoxin–thioredoxin reductase system (Trx–TrxR system). Known target proteins are Msr and Prx.234 Trx itself is inactivated by oxidation of active site C33 and C35, which can be reactivated by the selenoprotein TrxR.235 However, oxidation of Trx at positions C62, C69, and C73 is not reversible by TrxR and resulted in a loss of activity.236 Alternatively, glutathione dependent glutaredoxin (Grx) reduces cysteine oxidation and removes glutathionylation.237 While the treatment of epithelial cells with MGO results in loss of Trx activity, the Grx is unaffected in contrast.238
Early intermediates in the glycation reaction cascade such as the Amadori product between lysine residues and glucose can potentially be repaired by fructosamine-3-kinase (FN3K). The enzymatic phosphorylation at the 3-hydroxy position of the Amadori product destabilizes the ketoamine leading to non-enzymatic breakdown into the native lysine and 3-deoxyglucosone.239 This protein deglycase is inactivated by oxidative dimerization at C32.240 Altogether, the presence of repair mechanisms for nPTMs emphasize the relevance and potential threat of nPTMs for protein homeostasis. In case repair of modified proteins fails, nPTM carrying proteins that have been rendered non- or malfunctional must be degraded.
The UPS is unable to degrade cross-linked and small protein aggregates.243 Hence, the ALS is the last clearing pathway for these early species of protein aggregation.248 Similar to degradation by the UPS, moderate substrate unfolding by nPTMs increases accessibility for higher degradation levels and the ALS pathway is upregulated in stressed cells, but at some point key enzymes in ALS are also affected by oxidative damage.249 As an example autophagy-related protein 4 (ATG4), which is an important initiator of the autophagy cascade, is reversely inhibited by cysteine oxidation.250 Heat-shock cognate 70 (Hsc70), which is required for substrate detection in chaperone mediated autophagy, loses its chaperone activity by glycation.251 Finally, central proteases of the ALS such as cathepsins are reportedly inhibited by GO/MGO glycation in cell lysates.252
Discussion
We have summarized here the effects of nPTMs on key proteins involved in neurodegeneration as described to date. A significant lack of molecular details becomes apparent when comparing the known facts on such nPTMs with “regular” enzyme-mediated PTMs. Therefore, many studies are merely descriptive and rely on identifying (and quantifying) nPTMs with antibodies against such modification and more recently on proteomics studies. The latter generate large datasets that need to be rigorously analyzed to identify physiologically relevant nPTMs but do not contribute to deciphering the molecular basis of how nPTMs effect protein function. A still unanswered question is the extent to which nPTMs and enzymatic modifications, such as lysine glycation versus enzymatic ubiquitylation, compete for the same modification sites and influence each other. The absence of precise model systems and analytical probes has significantly limited our ability to establish robust connections between oxidative stress, elevated levels of reactive metabolites, and protein aggregation. Hence, addressing these critical points is vitally important in future studies.Many of the “mechanistic” studies on proteins carrying nPTMs cited here generate samples under highly artificial in vitro conditions based on incubation with glycating agent or oxidants at elevated temperature, with excessive reactant concentration in non-physiological solvents. In turn they mostly rely on in vitro measurement for example of protein aggregation via thioflavin T assay but do not aim at toxicological evaluations. Furthermore, most studies on nPTMs focus exclusively on in vitro systems, with limited efforts to validate these modifications or their effects in vivo. For example, the pathological relevance of nPTMs has rarely been verified using brain samples from patients with neurodegenerative diseases. Bridging this gap will require developing innovative tools to model nPTMs under physiologically relevant conditions and combining them with in vivo studies to evaluate their pathological impact.
Cell culture and in vivo approaches based on gene knock-outs of detoxification systems and/or genetic modifications inducing oxidative stress that both lead to an increase in nPTMs on proteins also suffer from a lack of molecular resolution and often complex reactions cascades. As an example, knock-down of glyoxalase 1 is a frequently used method to increase intracellular MGO concentrations. However, cells adapt their metabolism leading to mixed results, which makes it even more challenging to distinguish causes and effects of nPTMs.253,254
Obvious solutions to this dilemma can be found in approaches used to study enzyme-based PTMs such as depicted in Fig. 7. Chemical synthesis of peptides and proteins represents a direct way of addressing these questions by producing peptides and even proteins via chemoselective ligation approaches with nPTMs almost without any restrictions and with atomic precision.255–257 Currently this approach still suffers from the limited availability of many of the known nPTMs as useful building blocks for SPPS. Here, only few examples exist with robust synthetic access routes that either are long-known non-proteinogenic amino acids (e.g. citrulline) or have been more recently added to the available repertoire such as pentosinane and argpyrimidine.258,259
 | ||
| Fig. 7 Methods for site-selective incorporation of non-enzymatic posttranslational protein modifications (indicated as X). Chemical synthesis via solid phase peptide synthesis in combination with chemoselective ligations reactions is the most flexible approach. Challenges related to protein size can be overcome by combing expressed protein segments with synthetic segments in protein semi-synthesis. To produce modified proteins in cells, exchange of one proteinogenic amino acid by supplementation of a structurally related non-proteinogenic amino acid is a viable method. This approach is exceeded by amber codon suppression in selectivity and flexibility (works in eukaryotic cells and expands the set of proteinogenic amino acids). | ||
Another classic but often misleading approach has been copied from enzyme-based PTMs and is related to mutational exchange of proteinogenic amino acids, e.g. to suppress nPTMs such as nitrosylation on tyrosine by replacing it with phenyalanine.260 Such mutations have proven to be of limited use for PTMs (e.g. to mimic phosphoserine with glutamic acid) and we expect similar challenges when expanding this approach towards nPTMs. But we need to stress that mutational analysis by replacing reactive with unreactive amino acids (e.g. lysine to alanine mutations) remains a valuable tool.
Similarly, the exchange of sensitive residues such as methionine by norleucine (to exclude any oxidative damage) can be advantageous but requires the use of methionine-auxotrophic bacterial expression strains together with supplementation of norleucine (Fig. 7) and therefore additional levels of engineering.261 In a more flexible approach, amber stop-codon suppression (or more generally termed genetic code expansion) has been successfully used to site-selectively incorporate PTMs into a variety of proteins by relying on orthogonal pairs of tRNAs and tRNA synthetases that process specific non-proteinogenic amino acids and allow their incorporation via ribosomal protein synthesis.262,263 This approach could be quite easily expanded towards nPTMs if the modified amino acids are available together with suitable orthogonal pairs of tRNA and tRNA synthetases. It would be even more intriguing to combine such a code expansion approach with protein semi-synthesis as this allows modifying all parts of the protein of interest though with different flexibility.
In summary, more molecular details are needed to determine how nPTMs influence protein structure and function. Increasing insights into these mechanisms will demonstrate if intervention into processes leading to nPTMs can be exploited on a therapeutic level.
Abbreviations
| Aβ | Amyloid beta |
| AD | Alzheimer's disease |
| AGE(s) | Advanced glycation endproduct(s) |
| ALE(s) | Advanced lipoxidation endproduct(s) |
| ALS | Autophagy lysosomal system |
| APP | Amyloid precursor protein |
| ApoE | Apolipoprotein E |
| ATG4 | Autophagy-related protein 4 |
| CEL | N 6-Carboxyethyl lysine |
| CML | N 6-Carboxymethyl lysine |
| CTD | C-terminal domain |
| FN3K | Fructosamine-3-kinase |
| GO | Glyoxal |
| Gpx | Glutathione peroxidase |
| Grx | Glutaredoxin |
| 4-HNE | 4-Hydroxy-2-nonenal |
| Hsc70 | Heat-shock cognate 70 protein |
| Hsp | Heat-shock protein |
| MGO | Methylglyoxal |
| Msr | Methionine sulfoxide reductase |
| MTBR | Microtubule binding region |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| NFTs | Neurofibrillary tangles |
| NTD | N-Terminal domain |
| nPTM(s) | Non-enzymatic posttranslational protein modification(s) |
| PD | Parkinson's disease |
| PHF | Paired helical filaments |
| PRD | Proline-rich domain |
| Prx | Peroxiredoxin |
| PTM(s) | Posttranslational protein modification(s) |
| RACS | Reactive acyl-CoA species |
| RCS | Reactive carbonyl species |
| RNS | Reactive nitrogen species |
| ROS | Reactive oxygen species |
| sAPPα | soluble amyloid precursor protein α |
| SPPS | Solid phase peptide synthesis |
| SOD1 | Copper/zinc superoxide dismutase |
| SOD2 | Manganese superoxide dismutase |
| αSyn | α-Synuclein |
| Trx | Thioredoxin |
| TrxR | Thioredoxin reductase |
| UPS | Ubiquitin proteasomal system |
Data availability
No primary research results, software or code have been included and no new data were generated or analyzed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Fig. 2, 3, and 6 were created in https://www.BioRender.com. Open access funding provided by University of Vienna.References
- G. Livingston, J. Huntley, K. Y. Liu, S. G. Costafreda, G. Selbæk, S. Alladi, D. Ames, S. Banerjee, A. Burns and C. Brayne, Lancet, 2024, 404, 572–628 CrossRef PubMed.
- O. Hansson, Nat. Med., 2021, 27, 954–963 CrossRef PubMed.
- D. M. Wilson, M. R. Cookson, L. Van Den Bosch, H. Zetterberg, D. M. Holtzman and I. Dewachter, Cell, 2023, 186, 693–714 CrossRef PubMed.
- B. N. Dugger and D. W. Dickson, Cold Spring Harbor Perspect. Biol., 2017, 9, a028035 CrossRef PubMed.
- P. T. Lansbury and H. A. Lashuel, Nature, 2006, 443, 774–779 CrossRef PubMed.
- H. A. Lashuel, Curr. Opin. Chem. Biol., 2021, 64, 67–75 CrossRef PubMed.
- C. A. Ross and M. A. Poirier, Nat. Rev. Mol. Cell Biol., 2005, 6, 891–898 CrossRef PubMed.
- L.-N. Schaffert and W. G. Carter, Brain Sci., 2020, 10, 232 CrossRef PubMed.
- P. Magalhães and H. A. Lashuel, NPJ Parkinsons Dis., 2022, 8, 93 CrossRef PubMed.
- S. P. Moon, A. T. Balana and M. R. Pratt, Curr. Opin. Chem. Biol., 2021, 64, 76–89 CrossRef CAS PubMed.
- H. Olzscha, Biol. Chem., 2019, 400, 895–915 CrossRef CAS PubMed.
- S. Park, J. H. Lee, J. H. Jeon and M. J. Lee, BMB Rep., 2018, 51, 265 CrossRef CAS PubMed.
- M. R. Pratt, T. Abeywardana and N. P. Marotta, Biomolecules, 2015, 5, 1210–1227 CrossRef CAS PubMed.
- N. Ahmed and P. J. Thornalley, Diabetes, Obes. Metab., 2007, 9, 233–245 CrossRef CAS PubMed.
- T. Baldensperger, M. Eggen, J. Kappen, P. R. Winterhalter, T. Pfirrmann and M. A. Glomb, Sci. Rep., 2020, 10, 7596 CrossRef PubMed.
- M. J. Finelli, Front. Aging Neurosci., 2020, 12, 254 CrossRef PubMed.
- V. N. Gladyshev, S. B. Kritchevsky, S. G. Clarke, A. M. Cuervo, O. Fiehn, J. P. de Magalhães, T. Mau, M. Maes, R. L. Moritz, L. J. Niedernhofer, E. Van Schaftingen, G. J. Tranah, K. Walsh, Y. Yura, B. Zhang and S. R. Cummings, Nat. Aging, 2021, 1, 1096–1106 CrossRef PubMed.
- J. Li, D. Liu, L. Sun, Y. Lu and Z. Zhang, J. Neurol. Sci., 2012, 317, 1–5 CrossRef PubMed.
- A. L. Santos and A. B. Lindner, Oxid. Med. Cell. Longevity, 2017, 2017, 5716409 CrossRef PubMed.
- R. Kehm, T. Baldensperger, J. Raupbach and A. Höhn, Redox Biol., 2021, 42, 101901 CrossRef PubMed.
- G. Vistoli, D. De Maddis, A. Cipak, N. Zarkovic, M. Carini and G. Aldini, Free Radical Res., 2013, 47, 3–27 CrossRef PubMed.
- C. M. C. Andrés, J. M. Pérez de la Lastra, C. Andrés Juan, F. J. Plou and E. Pérez-Lebeña, Int. J. Mol. Sci., 2023, 24, 1841 CrossRef PubMed.
- L. Adams, M. C. Franco and A. G. Estevez, Exp. Biol. Med., 2015, 240, 711–717 CrossRef PubMed.
- S. Menini, C. Iacobini, M. Vitale, C. Pesce and G. Pugliese, Cancers, 2021, 13, 313 CrossRef PubMed.
- N. Rabbani and P. J. Thornalley, Biochem. Biophys. Res. Commun., 2015, 458, 221–226 CrossRef PubMed.
- A. G. Trub and M. D. Hirschey, Trends Biochem. Sci., 2018, 43, 369–379 CrossRef PubMed.
- M. Smuda and M. A. Glomb, J. Agric. Food Chem., 2013, 61, 10198–10208 CrossRef PubMed.
- N. Rabbani and P. J. Thornalley, Biochem. Soc. Trans., 2008, 36, 1045–1050 CrossRef PubMed.
- T. Teissier and É. Boulanger, Biogerontology, 2019, 20, 279–301 CrossRef PubMed.
- M. Trujillo, B. Alvarez and R. Radi, Free Radical Res., 2016, 50, 150–171 CrossRef.
- C. L. Grek, J. Zhang, Y. Manevich, D. M. Townsend and K. D. Tew, J. Biol. Chem., 2013, 288, 26497–26504 CrossRef PubMed.
- B. P. Tu and J. S. Weissman, J. Cell Biol., 2004, 164, 341–346 CrossRef PubMed.
- W. Vogt, Free Radical Biol. Med., 1995, 18, 93–105 CrossRef PubMed.
- D. A. Butterfield, T. Reed, S. F. Newman and R. Sultana, Free Radical Biol. Med., 2007, 43, 658–677 CrossRef PubMed.
- J. W. Heinecke, W. Li, G. A. Francis and J. A. Goldstein, J. Clin. Invest., 1993, 91, 2866–2872 CrossRef PubMed.
- P. J. White, A. Charbonneau, G. J. Cooney and A. Marette, Am. J. Physiol.: Endocrinol. Metab., 2010, 299, E868–E878 CrossRef PubMed.
- A. Badar, Z. Arif and K. Alam, IUBMB Life, 2018, 70, 267–275 CrossRef PubMed.
- L. Gorisse, C. Pietrement, V. Vuiblet, C. E. Schmelzer, M. Köhler, L. Duca, L. Debelle, P. Fornès, S. Jaisson and P. Gillery, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 1191–1196 CrossRef PubMed.
- T. Baldensperger and M. A. Glomb, Front. Cell Dev. Biol., 2021, 9, 664553 CrossRef PubMed.
- Y. Xu, Z. Shi and L. Bao, Mol. Cell. Proteomics, 2022, 21, 100193 CrossRef PubMed.
- G. R. Wagner, D. P. Bhatt, T. M. O'Connell, J. W. Thompson, L. G. Dubois, D. S. Backos, H. Yang, G. A. Mitchell, O. R. Ilkayeva, R. D. Stevens, P. A. Grimsrud and M. D. Hirschey, Cell Metab., 2017, 25, 823–837 CrossRef PubMed.
- T. Baldensperger, T. Jost, A. Zipprich and M. A. Glomb, J. Agric. Food Chem., 2018, 66, 1898–1906 CrossRef PubMed.
- A. Klöpfer, R. Spanneberg and M. A. Glomb, J. Agric. Food Chem., 2011, 59, 394–401 CrossRef PubMed.
- A. Iwai, E. Masliah, M. Yoshimoto, N. Ge, L. Flanagan, H. A. de Silva, A. Kittel and T. Saitoh, Neuron, 1995, 14, 467–475 CrossRef PubMed.
- J. Burré, M. Sharma, T. Tsetsenis, V. Buchman, M. R. Etherton and T. C. Südhof, Science, 2010, 329, 1663–1667 CrossRef PubMed.
- K. Makasewicz, S. Linse and E. Sparr, JACS Au, 2024, 4, 1250–1262 CrossRef PubMed.
- P. Calabresi, A. Mechelli, G. Natale, L. Volpicelli-Daley, G. Di Lazzaro and V. Ghiglieri, Cell Death Dis., 2023, 14, 176 CrossRef PubMed.
- K. Uéda, H. Fukushima, E. Masliah, Y. Xia, A. Iwai, M. Yoshimoto, D. A. Otero, J. Kondo, Y. Ihara and T. Saitoh, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 11282–11286 CrossRef PubMed.
- H. A. Lashuel, C. R. Overk, A. Oueslati and E. Masliah, Nat. Rev. Neurosci., 2013, 14, 38–48 CrossRef CAS PubMed.
- Y. Tong, P. Zhang, X. Yang, X. Liu, J. Zhang, M. Grudniewska, I. Jung, D. Abegg, J. Liu and J. L. Childs-Disney, Proc. Natl. Acad. Sci. U. S. A., 2024, 121, e2306682120 CrossRef PubMed.
- W. S. Davidson, A. Jonas, D. F. Clayton and J. M. George, J. Biol. Chem., 1998, 273, 9443–9449 CrossRef PubMed.
- G. Fusco, A. De Simone, T. Gopinath, V. Vostrikov, M. Vendruscolo, C. M. Dobson and G. Veglia, Nat. Commun., 2014, 5, 3827 CrossRef PubMed.
- M. Periquet, T. Fulga, L. Myllykangas, M. G. Schlossmacher and M. B. Feany, J. Neurosci., 2007, 27, 3338–3346 CrossRef PubMed.
- M. S. Nielsen, H. Vorum, E. Lindersson and P. H. Jensen, J. Biol. Chem., 2001, 276, 22680–22684 CrossRef PubMed.
- A.-L. Mahul-Mellier, J. Burtscher, N. Maharjan, L. Weerens, M. Croisier, F. Kuttler, M. Leleu, G. W. Knott and H. A. Lashuel, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 4971–4982 CrossRef PubMed.
- D. Emin, Y. P. Zhang, E. Lobanova, A. Miller, X. Li, Z. Xia, H. Dakin, D. I. Sideris, J. Y. L. Lam, R. T. Ranasinghe, A. Kouli, Y. Zhao, S. De, T. P. J. Knowles, M. Vendruscolo, F. S. Ruggeri, F. I. Aigbirhio, C. H. Williams-Gray and D. Klenerman, Nat. Commun., 2022, 13, 5512 CrossRef PubMed.
- D. Ottolini, T. Calí, I. Szabò and M. Brini, Biol. Chem., 2017, 398, 77–100 Search PubMed.
- D. J. Rinauro, F. Chiti, M. Vendruscolo and R. Limbocker, Mol. Neurodegener., 2024, 19, 20 CrossRef PubMed.
- K. M. Danzer, D. Haasen, A. R. Karow, S. Moussaud, M. Habeck, A. Giese, H. Kretzschmar, B. Hengerer and M. Kostka, J. Neurosci., 2007, 27, 9220–9232 CrossRef PubMed.
- L. J. Hsu, Y. Sagara, A. Arroyo, E. Rockenstein, A. Sisk, M. Mallory, J. Wong, T. Takenouchi, M. Hashimoto and E. Masliah, Am. J. Pathol., 2000, 157, 401–410 CrossRef CAS PubMed.
- D. Freeman, R. Cedillos, S. Choyke, Z. Lukic, K. McGuire, S. Marvin, A. M. Burrage, S. Sudholt, A. Rana, C. O'Connor, C. M. Wiethoff and E. M. Campbell, PLoS One, 2013, 8, e62143 CrossRef CAS PubMed.
- M. A. Alim, Q.-L. Ma, K. Takeda, T. Aizawa, M. Matsubara, M. Nakamura, A. Asada, T. Saito, M. xKaji, M. Yoshii, S. Hisanaga and K. Uéda, J. Alzheimer's Dis., 2004, 6, 435–442 Search PubMed.
- O. B. Tysnes and A. Storstein, J. Neural Transm., 2017, 124, 901–905 CrossRef PubMed.
- D. Belvisi, R. Pellicciari, A. Fabbrini, M. Costanzo, S. Pietracupa, M. De Lucia, N. Modugno, F. Magrinelli, C. Dallocchio, T. Ercoli, C. Terravecchia, A. Nicoletti, P. Solla, G. Fabbrini, M. Tinazzi, A. Berardelli and G. Defazio, Neurology, 2020, 95, e2500–e2508 CrossRef CAS PubMed.
- E. De Pablo-Fernandez, R. Goldacre, J. Pakpoor, A. J. Noyce and T. T. Warner, Neurology, 2018, 91, e139–e142 CrossRef PubMed.
- K. Han, B. Kim, S. H. Lee and M. K. Kim, NPJ Parkinsons Dis., 2023, 9, 11 CrossRef PubMed.
- K. Nakaso, N. Tajima, S. Ito, M. Teraoka, A. Yamashita, Y. Horikoshi, D. Kikuchi, S. Mochida, K. Nakashima and T. Matsura, PLoS One, 2013, 8, e55068 CrossRef CAS PubMed.
- P. Carmo-Gonçalves, A. S. Pinheiro, L. Romão, J. Cortines and C. Follmer, Amyloid, 2014, 21, 163–174 CrossRef PubMed.
- B. Alvarez-Castelao, M. Goethals, J. Vandekerckhove and J. G. Castaño, Biochim. Biophys. Acta, 2014, 1843, 352–365 CrossRef PubMed.
- A. Binolfi, A. Limatola, S. Verzini, J. Kosten, F. X. Theillet, H. M. Rose, B. Bekei, M. Stuiver, M. van Rossum and P. Selenko, Nat. Commun., 2016, 7, 10251 CrossRef PubMed.
- V. N. Uversky, G. Yamin, P. O. Souillac, J. Goers, C. B. Glaser and A. L. Fink, FEBS Lett., 2002, 517, 239–244 CrossRef PubMed.
- M. J. Hokenson, V. N. Uversky, J. Goers, G. Yamin, L. A. Munishkina and A. L. Fink, Biochemistry, 2004, 43, 4621–4633 CrossRef PubMed.
- A. B. Uceda, J. Frau, B. Vilanova and M. Adrover, Int. J. Biol. Macromol., 2023, 229, 92–104 CrossRef PubMed.
- R. Wassef, R. Haenold, A. Hansel, N. Brot, S. H. Heinemann and T. Hoshi, J. Neurosci., 2007, 27, 12808–12816 CrossRef PubMed.
- M. K. Tiwari, P. M. Hägglund, I. M. Møller, M. J. Davies and M. J. Bjerrum, Redox Biol., 2019, 26, 101262 CrossRef PubMed.
- Y. K. Al-Hilaly, L. Biasetti, B. J. Blakeman, S. J. Pollack, S. Zibaee, A. Abdul-Sada, J. R. Thorpe, W. F. Xue and L. C. Serpell, Sci. Rep., 2016, 6, 39171 CrossRef PubMed.
- C. Sahin, E. C. Østerlund, N. Österlund, J. Costeira-Paulo, J. N. Pedersen, G. Christiansen, J. Nielsen, A. L. Grønnemose, S. K. Amstrup, M. K. Tiwari, R. S. P. Rao, M. J. Bjerrum, L. L. Ilag, M. J. Davies, E. G. Marklund, J. S. Pedersen, M. Landreh, I. M. Møller, T. J. D. Jørgensen and D. E. Otzen, J. Am. Chem. Soc., 2022, 144, 11949–11954 CrossRef PubMed.
- S. Krishnan, E. Y. Chi, S. J. Wood, B. S. Kendrick, C. Li, W. Garzon-Rodriguez, J. Wypych, T. W. Randolph, L. O. Narhi, A. L. Biere, M. Citron and J. F. Carpenter, Biochemistry, 2003, 42, 829–837 CrossRef PubMed.
- R. Burai, N. Ait-Bouziad, A. Chiki and H. A. Lashuel, J. Am. Chem. Soc., 2015, 137, 5041–5052 CrossRef PubMed.
- R. Hodara, E. H. Norris, B. I. Giasson, A. J. Mishizen-Eberz, D. R. Lynch, V. M. Y. Lee and H. Ischiropoulos, J. Biol. Chem., 2004, 279, 47746–47753 CrossRef PubMed.
- E. J. Bae, D. H. Ho, E. Park, J. W. Jung, K. Cho, J. H. Hong, H. J. Lee, K. P. Kim and S. J. Lee, Antioxid. Redox Signaling, 2013, 18, 770–783 CrossRef PubMed.
- A. Trostchansky, S. Lind, R. Hodara, T. Oe, I. A. Blair, H. Ischiropoulos, H. Rubbo and J. M. Souza, Biochem. J., 2006, 393, 343–349 CrossRef PubMed.
- W. Xiang, J. C. Schlachetzki, S. Helling, J. C. Bussmann, M. Berlinghof, T. E. Schäffer, K. Marcus, J. Winkler, J. Klucken and C. M. Becker, Mol. Cell. Neurosci., 2013, 54, 71–83 CrossRef PubMed.
- J. W. Werner-Allen, J. F. DuMond, R. L. Levine and A. Bax, Angew. Chem., Int. Ed., 2016, 55, 7374–7378 CrossRef PubMed.
- D. E. Mor, M. J. Daniels and H. Ischiropoulos, Mov. Disord., 2019, 34, 167–179 CrossRef PubMed.
- R. Castellani, M. A. Smith, P. L. Richey and G. Perry, Brain Res., 1996, 737, 195–200 CrossRef PubMed.
- Y. G. Choi and S. Lim, Biochimie, 2010, 92, 1379–1386 CrossRef PubMed.
- H. Vicente Miranda, É. M. Szegő, L. M. A. Oliveira, C. Breda, E. Darendelioglu, R. M. de Oliveira, D. G. Ferreira, M. A. Gomes, R. Rott, M. Oliveira, F. Munari, F. J. Enguita, T. Simões, E. F. Rodrigues, M. Heinrich, I. C. Martins, I. Zamolo, O. Riess, C. Cordeiro, A. Ponces-Freire, H. A. Lashuel, N. C. Santos, L. V. Lopes, W. Xiang, T. M. Jovin, D. Penque, S. Engelender, M. Zweckstetter, J. Klucken, F. Giorgini, A. Quintas and T. F. Outeiro, Brain, 2017, 140, 1399–1419 CrossRef PubMed.
- D. Lee, C. W. Park, S. R. Paik and K. Y. Choi, Biochim. Biophys. Acta, 2009, 1794, 421–430 CrossRef PubMed.
- A. Farzadfard, A. König, S. V. Petersen, J. Nielsen, E. Vasili, A. Dominguez-Meijide, A. K. Buell, T. F. Outeiro and D. E. Otzen, J. Biol. Chem., 2022, 298, 101848 CrossRef PubMed.
- L. Mariño, R. Ramis, R. Casasnovas, J. Ortega-Castro, B. Vilanova, J. Frau and M. Adrover, Chem. Sci., 2020, 11, 3332–3344 RSC.
- A. B. Uceda, J. Frau, B. Vilanova and M. Adrover, Cell. Mol. Life Sci., 2022, 79, 342 CrossRef PubMed.
- A. Chegão, M. Guarda, B. M. Alexandre, L. Shvachiy, M. Temido-Ferreira, I. Marques-Morgado, B. Fernandes Gomes, R. Matthiesen, L. V. Lopes and P. R. Florindo, NPJ Parkinsons Dis., 2022, 8, 51 CrossRef PubMed.
- C. Bosbach, L. M. Gatzemeier, K. I. Bloch von Blottnitz, A. König, U. Diederichsen, C. Steinem and T. F. Outeiro, Org. Biomol. Chem., 2024, 22, 2670–2676 RSC.
- H. Hampel, J. Hardy, K. Blennow, C. Chen, G. Perry, S. H. Kim, V. L. Villemagne, P. Aisen, M. Vendruscolo, T. Iwatsubo, C. L. Masters, M. Cho, L. Lannfelt, J. L. Cummings and A. Vergallo, Mol. Psychiatry, 2021, 26, 5481–5503 CrossRef PubMed.
- N. D. Belyaev, K. A. Kellett, C. Beckett, N. Z. Makova, T. J. Revett, N. N. Nalivaeva, N. M. Hooper and A. J. Turner, J. Biol. Chem., 2010, 285, 41443–41454 CrossRef PubMed.
- P. Sharma, P. Srivastava, A. Seth, P. N. Tripathi, A. G. Banerjee and S. K. Shrivastava, Prog. Neurobiol., 2019, 174, 53–89 CrossRef PubMed.
- E. Kojro and F. Fahrenholz, in Alzheimer's Disease: Cellular and Molecular Aspects of Amyloid β, ed. J. R. Harris and F. Fahrenholz, Springer, US, Boston, MA, 2005, pp. 105–127 DOI:10.1007/0-387-23226-5_5.
- S. Chandran and D. Binninger, Antioxidants, 2024, 13, 21 CrossRef PubMed.
- S. M. Ohline, C. Chan, L. Schoderboeck, H. E. Wicky, W. P. Tate, S. M. Hughes and W. C. Abraham, Mol. Brain, 2022, 15, 5 CrossRef PubMed.
- X. Lin, G. Koelsch, S. Wu, D. Downs, A. Dashti and J. Tang, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 1456–1460 CrossRef PubMed.
- J. Y. Hur, Exp. Mol. Med., 2022, 54, 433–446 CrossRef PubMed.
- K. W. Menting and J. A. Claassen, Front. Aging Neurosci., 2014, 6, 165 Search PubMed.
- E. Karran and B. De Strooper, Nat. Rev. Drug Discovery, 2022, 21, 306–318 CrossRef PubMed.
- X. Zhang and W. Song, Alzheimer's Res. Ther., 2013, 5, 46 CrossRef PubMed.
- S. Murakami and P. Lacayo, Front. Aging Neurosci., 2022, 14, 996030 CrossRef PubMed.
- C. Reitz, E. Rogaeva and G. W. Beecham, Neurol.:Genet., 2020, 6, e512 Search PubMed.
- D. M. Michaelson, Alzheimer's Dementia, 2014, 10, 861–868 CrossRef PubMed.
- D. A. Butterfield and M. P. Mattson, Neurobiol. Dis., 2020, 138, 104795 CrossRef PubMed.
- E. M. Reiman, J. F. Arboleda-Velasquez, Y. T. Quiroz, M. J. Huentelman, T. G. Beach, R. J. Caselli, Y. Chen, Y. Su, A. J. Myers, J. Hardy, J. Paul Vonsattel, S. G. Younkin, D. A. Bennett, P. L. De Jager, E. B. Larson, P. K. Crane, C. D. Keene, M. I. Kamboh, J. K. Kofler, L. Duque, J. R. Gilbert, H. E. Gwirtsman, J. D. Buxbaum, D. W. Dickson, M. P. Frosch, B. F. Ghetti, K. L. Lunetta, L.-S. Wang, B. T. Hyman, W. A. Kukull, T. Foroud, J. L. Haines, R. P. Mayeux, M. A. Pericak-Vance, J. A. Schneider, J. Q. Trojanowski, L. A. Farrer, G. D. Schellenberg, G. W. Beecham, T. J. Montine, G. R. Jun, E. Abner, P. M. Adams, M. S. Albert, R. L. Albin, L. G. Apostolova, S. E. Arnold, S. Asthana, C. S. Atwood, C. T. Baldwin, R. C. Barber, L. L. Barnes, S. Barral, J. T. Becker, D. Beekly, E. H. Bigio, T. D. Bird, D. Blacker, B. F. Boeve, J. D. Bowen, A. Boxer, J. R. Burke, J. M. Burns, N. J. Cairns, L. B. Cantwell, C. Cao, C. S. Carlson, C. M. Carlsson, R. M. Carney, M. M. Carrasquillo, H. C. Chui, D. H. Cribbs, E. A. Crocco, C. Cruchaga, C. DeCarli, M. Dick, R. S. Doody, R. Duara, N. Ertekin-Taner, D. A. Evans, K. M. Faber, T. J. Fairchild, K. B. Fallon, D. W. Fardo, M. R. Farlow, S. Ferris, D. R. Galasko, M. Gearing, D. H. Geschwind, V. Ghisays, A. M. Goate, N. R. Graff-Radford, R. C. Green, J. H. Growdon, H. Hakonarson, R. L. Hamilton, K. L. Hamilton-Nelson, L. E. Harrell, L. S. Honig, R. M. Huebinger, C. M. Hulette, G. P. Jarvik, L.-W. Jin, A. Karydas, M. J. Katz, J. S. K. Kauwe, J. A. Kaye, R. Kim, N. W. Kowall, J. H. Kramer, B. W. Kunkle, A. P. Kuzma, F. M. LaFerla, J. J. Lah, Y. Y. Leung, J. B. Leverenz, A. I. Levey, G. Li, A. P. Lieberman, R. B. Lipton, O. L. Lopez, C. G. Lyketsos, J. Malamon, D. C. Marson, E. R. Martin, F. Martiniuk, D. C. Mash, E. Masliah, W. C. McCormick, S. M. McCurry, A. N. McDavid, S. McDonough, A. C. McKee, M. Mesulam, B. L. Miller, C. A. Miller, J. W. Miller, J. C. Morris, S. Mukherjee, A. C. Naj, S. O’Bryant, J. M. Olichney, J. E. Parisi, H. L. Paulson, E. Peskind, R. C. Petersen, A. Pierce, W. W. Poon, H. Potter, L. Qu, J. F. Quinn, A. Raj, M. Raskind, B. Reisberg, J. S. Reisch, C. Reitz, J. M. Ringman, E. D. Roberson, E. Rogaeva, H. J. Rosen, R. N. Rosenberg, D. R. Royall, M. A. Sager, M. Sano, A. J. Saykin, L. S. Schneider, W. W. Seeley, A. G. Smith, J. A. Sonnen, S. Spina, P. S. George-Hyslop, R. A. Stern, R. H. Swerdlow, R. E. Tanzi, J. C. Troncoso, D. W. Tsuang, O. Valladares, V. M. Van Deerlin, L. J. Van Eldik, B. N. Vardarajan, H. V. Vinters, S. Weintraub, K. A. Welsh-Bohmer, K. C. Wilhelmsen, J. Williamson, T. S. Wingo, R. L. Woltjer, C. B. Wright, C.-K. Wu, C.-E. Yu, L. Yu, Y. Zhao and Alzheimer's Disease Genetics Consortium, Nat. Commun., 2020, 11, 667 CrossRef PubMed.
- B. Kim and E. L. Feldman, Exp. Mol. Med., 2015, 47, e149–e149 CrossRef PubMed.
- H. V. Miranda and T. F. Outeiro, J. Pathol., 2010, 221, 13–25 CrossRef PubMed.
- G. A. Edwards III, N. Gamez, G. Escobedo Jr, O. Calderon and I. Moreno-Gonzalez, Front. Aging Neurosci., 2019, 11, 146 CrossRef PubMed.
- J. W. Kinney, S. M. Bemiller, A. S. Murtishaw, A. M. Leisgang, A. M. Salazar and B. T. Lamb, Alzheimer's Dementia, 2018, 4, 575–590 CrossRef PubMed.
- A. Tramutola, C. Lanzillotta, M. Perluigi and D. A. Butterfield, Brain Res. Bull., 2017, 133, 88–96 CrossRef PubMed.
- G. V. Patil, R. S. Joshi, R. S. Kazi, S. E. Kulsange and M. J. Kulkarni, Med. Hypotheses, 2020, 142, 109799 CrossRef PubMed.
- H. P. Schmitt, Med. Hypotheses, 2006, 66, 898–906 CrossRef PubMed.
- Y. Yang, V. Tapias, D. Acosta, H. Xu, H. Chen, R. Bhawal, E. T. Anderson, E. Ivanova, H. Lin, B. T. Sagdullaev, J. Chen, W. L. Klein, K. L. Viola, S. Gandy, V. Haroutunian, M. F. Beal, D. Eliezer, S. Zhang and G. E. Gibson, Nat. Commun., 2022, 13, 159 CrossRef PubMed.
- M. Rapała-Kozik, A. Kozik and J. Travis, J. Pept. Res., 1998, 52, 315–320 CrossRef PubMed.
- A. Vázquez de la Torre, M. Gay, S. Vilaprinyó-Pascual, R. Mazzucato, M. Serra-Batiste, M. Vilaseca and N. Carulla, Anal. Chem., 2018, 90, 4552–4560 CrossRef PubMed.
- J. Zhao, Q. Shi, Y. Zheng, Q. Liu, Z. He, Z. Gao and Q. Liu, Front. Mol. Neurosci., 2021, 14, 619836 CrossRef PubMed.
- A. Kimura, S. Hata and T. Suzuki, J. Biol. Chem., 2016, 291, 24041–24053 CrossRef PubMed.
- A. M. Boutte, R. L. Woltjer, L. J. Zimmerman, S. L. Stamer, K. S. Montine, M. V. Manno, P. J. Cimino, D. C. Liebler and T. J. Montine, FASEB J., 2006, 20, 1473–1483 CrossRef PubMed.
- M. Friedemann, E. Helk, A. Tiiman, K. Zovo, P. Palumaa and V. Tõugu, Biochem. Biophys. Rep., 2015, 3, 94–99 Search PubMed.
- K. J. Barnham, G. D. Ciccotosto, A. K. Tickler, F. E. Ali, D. G. Smith, N. A. Williamson, Y.-H. Lam, D. Carrington, D. Tew, G. Kocak, I. Volitakis, F. Separovic, C. J. Barrow, J. D. Wade, C. L. Masters, R. A. Cherny, C. C. Curtain, A. I. Bush and R. Cappai, J. Biol. Chem., 2003, 278, 42959–42965 CrossRef PubMed.
- S. M. Yatin, S. Varadarajan, C. D. Link and D. A. Butterfield, Neurobiol. Aging, 1999, 20, 325–330 CrossRef PubMed ; discussion 339.
- T. L. Williams, L. C. Serpell and B. Urbanc, Biochim. Biophys. Acta, 2016, 1864, 249–259 CrossRef PubMed.
- M. B. Maina, G. Burra, Y. K. Al-Hilaly, K. Mengham, K. Fennell and L. C. Serpell, iScience, 2020, 23, 167785 CrossRef PubMed.
- Y. K. Al-Hilaly, T. L. Williams, M. Stewart-Parker, L. Ford, E. Skaria, M. Cole, W. G. Bucher, K. L. Morris, A. A. Sada, J. R. Thorpe and L. C. Serpell, Acta Neuropathol. Commun., 2013, 1, 83 CrossRef PubMed.
- B. Guivernau, J. Bonet, V. Valls-Comamala, M. Bosch-Morató, J. A. Godoy, N. C. Inestrosa, A. Perálvarez-Marín, X. Fernández-Busquets, D. Andreu, B. Oliva and F. J. Muñoz, J. Neurosci., 2016, 36, 11693–11703 CrossRef PubMed.
- M. P. Kummer, M. Hermes, A. Delekarte, T. Hammerschmidt, S. Kumar, D. Terwel, J. Walter, H.-C. Pape, S. König, S. Roeber, F. Jessen, T. Klockgether, M. Korte and M. T. Heneka, Neuron, 2011, 71, 833–844 CrossRef PubMed.
- Y. Ando, T. Brännström, K. Uchida, N. Nyhlin, B. Näsman, O. Suhr, T. Yamashita, T. Olsson, M. E. L. Salhy, M. Uchino and M. Ando, J. Neurol. Sci., 1998, 156, 172–176 CrossRef PubMed.
- K. Chen, J. Maley and P. H. Yu, J. Neurochem., 2006, 99, 1413–1424 CrossRef PubMed.
- R. L. Woltjer, I. Maezawa, J. J. Ou, K. S. Montine and T. J. Montine, J. Alzheimer's Dis., 2003, 5, 467–476 Search PubMed.
- N. P. Sargaeva, C. Lin and P. B. O'Connor, Anal. Chem., 2009, 81, 9778–9786 CrossRef PubMed.
- E. R. Readel, M. Wey and D. W. Armstrong, Anal. Chim. Acta, 2021, 1163, 338506 CrossRef PubMed.
- M. P. Vitek, K. Bhattacharya, J. M. Glendening, E. Stopa, H. Vlassara, R. Bucala, K. Manogue and A. Cerami, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 4766–4770 CrossRef PubMed.
- A. Emendato, G. Milordini, E. Zacco, A. Sicorello, F. Dal Piaz, R. Guerrini, R. Thorogate, D. Picone and A. Pastore, J. Biol. Chem., 2018, 293, 13100–13111 CrossRef PubMed.
- N. Ahmed, U. Ahmed, P. J. Thornalley, K. Hager, G. Fleischer and G. Münch, J. Neurochem., 2005, 92, 255–263 Search PubMed.
- S. M. Fica-Contreras, S. O. Shuster, N. D. Durfee, G. J. K. Bowe, N. J. Henning, S. A. Hill, G. D. Vrla, D. R. Stillman, K. M. Suralik, R. K. Sandwick and S. Choi, J. Biol. Inorg. Chem., 2017, 22, 1211–1222 CrossRef PubMed.
- X. Li, L. Du, X. Cheng, X. Jiang, Y. Zhang, B. Lv, R. Liu, J. Wang and X. Zhou, Cell Death Dis., 2013, 4, e673–e673 CrossRef PubMed.
- J. Ng, H. Kaur, T. Collier, K. Chang, A. E. S. Brooks, J. R. Allison, M. A. Brimble, A. Hickey and N. P. Birch, J. Biol. Chem., 2019, 294, 8806–8818 CrossRef PubMed.
- M. D. Weingarten, A. H. Lockwood, S. Y. Hwo and M. W. Kirschner, Proc. Natl. Acad. Sci. U. S. A., 1975, 72, 1858–1862 Search PubMed.
- D. W. Cleveland, S.-Y. Hwo and M. W. Kirschner, J. Mol. Biol., 1977, 116, 227–247 Search PubMed.
- D. Drubin, S. Kobayashi and M. Kirschner, Ann. N. Y. Acad. Sci., 1986, 466, 257–268 CrossRef PubMed.
- R. Brandt, N. I. Trushina and L. Bakota, Front. Neurol., 2020, 11, 590059 Search PubMed.
- Y. Wei, M.-H. Qu, X.-S. Wang, L. Chen, D.-L. Wang, Y. Liu, Q. Hua and R.-Q. He, PLoS One, 2008, 3, e2600 CrossRef PubMed.
- I. Sotiropoulos, M.-C. Galas, J. M. Silva, E. Skoulakis, S. Wegmann, M. B. Maina, D. Blum, C. L. Sayas, E.-M. Mandelkow, E. Mandelkow, M. G. Spillantini, N. Sousa, J. Avila, M. Medina, A. Mudher and L. Buee, Acta Neuropathol. Commun., 2017, 5, 91 CrossRef PubMed.
- M. Bukar Maina, Y. K. Al-Hilaly and L. C. Serpell, Biomolecules, 2016, 6, 9 CrossRef PubMed.
- P. LoPresti, S. Szuchet, S. C. Papasozomenos, R. P. Zinkowski and L. I. Binder, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 10369–10373 CrossRef PubMed.
- E. Marciniak, A. Leboucher, E. Caron, T. Ahmed, A. Tailleux, J. Dumont, T. Issad, E. Gerhardt, P. Pagesy, M. Vileno, C. Bournonville, M. Hamdane, K. Bantubungi, S. Lancel, D. Demeyer, S. Eddarkaoui, E. Vallez, D. Vieau, S. Humez, E. Faivre, B. Grenier-Boley, T. F. Outeiro, B. Staels, P. Amouyel, D. Balschun, L. Buee and D. Blum, J. Experiment. Med., 2017, 214, 2257–2269 CrossRef PubMed.
- Y. Wang and E. Mandelkow, Nat. Rev. Neurosci., 2016, 17, 22–35 CrossRef CAS PubMed.
- P. Tompa, Trends Biochem. Sci., 2002, 27, 527–533 CrossRef PubMed.
- M. Goedert, M. G. Spillantini, R. Jakes, D. Rutherford and R. A. Crowther, Neuron, 1989, 3, 519–526 CrossRef PubMed.
- M. Goedert and R. Jakes, EMBO J., 1990, 9, 4225–4230 CrossRef PubMed.
- C. Alquezar, S. Arya and A. W. Kao, Front. Neurol., 2021, 11, 595532 CrossRef PubMed.
- A. Alzheimer, Allg. Z. Psychiatr. Psych.-Gerichtl. Med., 1907, 64, 146–148 Search PubMed.
- M. Kidd, Nature, 1963, 197, 192–193 CrossRef PubMed.
- R. D. Terry, J. Neuropathol. Exp. Neurol., 1963, 22, 629–642 CrossRef PubMed.
- S.-G. Kang, G. Eskandari-Sedighi, L. Hromadkova, J. G. Safar and D. Westaway, Front. Neurol., 2020, 11, 590199 CrossRef PubMed.
- D.-e C. Chung, S. Roemer, L. Petrucelli and D. W. Dickson, Mol. Neurodegener., 2021, 16, 57 CrossRef PubMed.
- A. Schneeweis and D. T. S. Pak, Int. J. Biochem. Cell Biol., 2023, 164, 106475 CrossRef PubMed.
- G. Šimić, M. Babić Leko, S. Wray, C. Harrington, I. Delalle, N. Jovanov-Milošević, D. Bažadona, L. Buée, R. De Silva, G. Di Giovanni, C. Wischik and P. R. Hof, Biomolecules, 2016, 6, 6 CrossRef PubMed.
- W. Hu, X. Zhang, Y. C. Tung, S. Xie, F. Liu and K. Iqbal, Alzheimer's Dementia, 2016, 12, 1066–1077 CrossRef PubMed.
- J.-Z. Wang, Z.-H. Wang and Q. Tian, Neurosci. Bull., 2014, 30, 359–366 CrossRef PubMed.
- P. Chakraborty, G. Rivière, A. Hebestreit, A. I. de Opakua, I. M. Vorberg, L. B. Andreas and M. Zweckstetter, Nat. Commun., 2023, 14, 5919 CrossRef PubMed.
- D. Ellmer, M. Brehs, M. Haj-Yahya, H. A. Lashuel and C. F. W. Becker, Angew. Chem., Int. Ed., 2019, 58, 1616–1620 CrossRef PubMed.
- F. Parolini, E. Ataie Kachoie, G. Leo, L. Civiero, L. Bubacco, G. Arrigoni, F. Munari, M. Assfalg, M. D'Onofrio and S. Capaldi, Angew. Chem., Int. Ed., 2023, 62, e202310230 CrossRef PubMed.
- F. Munari, C. G. Barracchia, C. Franchin, F. Parolini, S. Capaldi, A. Romeo, L. Bubacco, M. Assfalg, G. Arrigoni and M. D'Onofrio, Angew. Chem., Int. Ed., 2020, 59, 6607–6611 CrossRef PubMed.
- D. Trivellato, F. Floriani, C. G. Barracchia, F. Munari, M. D'Onofrio and M. Assfalg, Bioorg. Chem., 2023, 132, 106347 CrossRef PubMed.
- M. D. Ledesma, P. Bonay, C. Colaço and J. Avila, J. Biol. Chem., 1994, 269, 21614–21619 CrossRef PubMed.
- M. A. Smith, S. Taneda, P. L. Richey, S. Miyata, S. D. Yan, D. Stern, L. M. Sayre, V. M. Monnier and G. Perry, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 5710–5714 CrossRef PubMed.
- N. Sasaki, R. Fukatsu, K. Tsuzuki, Y. Hayashi, T. Yoshida, N. Fujii, T. Koike, I. Wakayama, R. Yanagihara, R. Garruto, N. Amano and Z. Makita, Am. J. Pathol., 1998, 153, 1149–1155 CrossRef PubMed.
- P. Nacharaju, L.-w Ko and S.-H. C. Yen, J. Neurochem., 1997, 69, 1709–1719 CrossRef PubMed.
- K. Liu, Y. Liu, L. Li, P. Qin, J. Iqbal, Y. Deng and H. Qing, Biochim. Biophys. Acta, 2016, 1862, 192–201 CrossRef PubMed.
- S. D. Yan, S. F. Yan, X. Chen, J. Fu, M. Chen, P. Kuppusamy, M. A. Smith, G. Perry, G. C. Godman, P. Nawroth, J. L. Zweier and D. Stern, Nat. Med., 1995, 1, 693–699 CrossRef PubMed.
- M. D. Ledesma, M. Medina and J. Avila, Mol. Chem. Neuropathol., 1996, 27, 249–258 CrossRef PubMed.
- K. Batkulwar, R. Godbole, R. Banarjee, O. Kassaar, R. J. Williams and M. J. Kulkarni, ACS Chem. Neurosci., 2018, 9, 988–1000 CrossRef PubMed.
- M. Necula and J. Kuret, J. Biol. Chem., 2004, 279, 49694–49703 CrossRef PubMed.
- S. Mukherjee, D. P. Vogl and C. F. W. Becker, ACS Chem. Biol., 2023, 18, 1760–1771 CrossRef PubMed.
- M. R. Reynolds, R. W. Berry and L. I. Binder, Biochemistry, 2005, 44, 1690–1700 Search PubMed.
- J. F. Reyes, C. Geula, L. Vana and L. I. Binder, Acta Neuropathol., 2012, 123, 119–132 CrossRef PubMed.
- M. R. Reynolds, J. F. Reyes, Y. Fu, E. H. Bigio, A. L. Guillozet-Bongaarts, R. W. Berry and L. I. Binder, J. Neurosci., 2006, 26, 10636 CrossRef PubMed.
- J. F. Reyes, Y. Fu, L. Vana, N. M. Kanaan and L. I. Binder, Am. J. Pathol., 2011, 178, 2275–2285 CrossRef PubMed.
- S. Nonnis, G. Cappelletti, F. Taverna, C. Ronchi, S. Ronchi, A. Negri, E. Grassi and G. Tedeschi, Neurochem. Res., 2008, 33, 518–525 CrossRef PubMed.
- M. R. Reynolds, R. W. Berry and L. I. Binder, Biochemistry, 2005, 44, 13997–14009 CrossRef PubMed.
- Y. J. Zhang, Y. F. Xu, X. Q. Chen, X. C. Wang and J.-Z. Wang, FEBS Lett., 2005, 579, 2421–2427 CrossRef PubMed.
- J. Gadhavi, S. Shah, T. Sinha, A. Jain and S. Gupta, FEBS J., 2022, 289, 2562–2577 CrossRef PubMed.
- V. Guru KrishnaKumar, L. Baweja, K. Ralhan and S. Gupta, Biochim. Biophys. Acta, 2018, 1862, 2590–2604 CrossRef PubMed.
- T. Saito, T. Chiku, M. Oka, S. Wada-Kakuda, M. Nobuhara, T. Oba, K. Shinno, S. Abe, A. Asada, A. Sumioka, A. Takashima, T. Miyasaka and K. Ando, Hum. Mol. Genet., 2021, 30, 1955–1967 CrossRef PubMed.
- N. Sahara, S. Maeda, M. Murayama, T. Suzuki, N. Dohmae, S.-H. Yen and A. Takashima, Eur. J. Neurosci., 2007, 25, 3020–3029 CrossRef PubMed.
- E. Prifti, E. N. Tsakiri, E. Vourkou, G. Stamatakis, M. Samiotaki, E. M. C. Skoulakis and K. Papanikolopoulou, Acta Neuropathol. Commun., 2022, 10, 44 CrossRef PubMed.
- D. M. Hatters, J. Biol. Chem., 2021, 297, 101309 CrossRef PubMed.
- Y. Yang and G. E. Gibson, Neurochem. Res., 2019, 44, 2346–2359 CrossRef PubMed.
- Y. Yang, V. Tapias, D. Acosta, H. Xu, H. Chen, R. Bhawal, E. Anderson, E. Ivanova, H. Lin, B. T. Sagdullaev, W. L. Klein, K. L. Viola, S. Gandy, V. Haroutunian, M. F. Beal, D. Eliezer, S. Zhang and G. E. Gibson, bioRxiv, 2019, 10.1101/764837, 764837.
- A. Kenessey, S.-H. Yen, W.-K. Liu, X.-R. Yang and D. S. Dunlop, Brain Res., 1995, 675, 183–189 CrossRef PubMed.
- R. Shapira, G. E. Austin and S. S. Mirra, J. Neurochem., 1988, 50, 69–74 CrossRef PubMed.
- M. Morishima-Kawashima, M. Hasegawa, K. Takio, M. Suzuki, K. Titani and Y. Ihara, Neuron, 1993, 10, 1151–1160 CrossRef PubMed.
- A. Watanabe, K. Takio and Y. Ihara, J. Biol. Chem., 1999, 274, 7368–7378 CrossRef PubMed.
- T. Shimizu, A. Watanabe, M. Ogawara, H. Mori and T. Shirasawa, Arch. Biochem. Biophys., 2000, 381, 225–234 CrossRef PubMed.
- T. Miyasaka, A. Watanabe, Y. Saito, S. Murayama, D. M. A. Mann, M. Yamazaki, R. Ravid, M. Morishima-Kawashima, K. Nagashima and Y. Ihara, J. Neuropath. Exp. Neurol., 2005, 64, 665–674 CrossRef PubMed.
- A. Dan, M. Takahashi, M. Masuda-Suzukake, F. Kametani, T. Nonaka, H. Kondo, H. Akiyama, T. Arai, D. M. A. Mann, Y. Saito, H. Hatsuta, S. Murayama and M. Hasegawa, Acta Neuropathol. Commun., 2013, 1, 54 CrossRef PubMed.
- N. Tochio, T. Murata and N. Utsunomiya-Tate, Biochem. Biophys. Res. Commun., 2019, 508, 184–190 CrossRef PubMed.
- T. Murata, G. Ito and N. Utsunomiya-Tate, Biochem. Biophys. Res. Commun., 2023, 654, 18–25 CrossRef PubMed.
- T. Jung, B. Catalgol and T. Grune, Mol. Aspects Med., 2009, 30, 191–296 CrossRef PubMed.
- E. L. Robb, A. R. Hall, T. A. Prime, S. Eaton, M. Szibor, C. Viscomi, A. M. James and M. P. Murphy, J. Biol. Chem., 2018, 293, 9869–9879 CrossRef PubMed.
- L. MacMillan-Crow, J. P. Crow, J. D. Kerby, J. S. Beckman and J. A. Thompson, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 11853–11858 CrossRef PubMed.
- H. Neumann, J. L. Hazen, J. Weinstein, R. A. Mehl and J. W. Chin, J. Am. Chem. Soc., 2008, 130, 4028–4033 CrossRef PubMed.
- C. J. Banks and J. L. Andersen, Redox Biol., 2019, 26, 101270 CrossRef CAS PubMed.
- F. Yamakura and H. Kawasaki, Biochim. Biophys. Acta, 2010, 1804, 318–325 CrossRef CAS PubMed.
- H. Zhang, C. Andrekopoulos, J. Joseph, K. Chandran, H. Karoui, J. P. Crow and B. Kalyanaraman, J. Biol. Chem., 2003, 278, 24078–24089 CrossRef PubMed.
- H. Yan and J. J. Harding, Biochem. J., 1997, 328(Pt 2), 599–605 CrossRef PubMed.
- P. Polykretis, E. Luchinat, F. Boscaro and L. Banci, Redox Biol., 2020, 30, 101421 CrossRef PubMed.
- J. Fujii, T. Myint, A. Okado, H. Kaneto and N. Taniguchi, Nephrol., Dial., Transplant., 1996, 11, 34–40 CrossRef PubMed.
- J. R. Monteiro Neto, G. D. Ribeiro, R. S. S. Magalhães, C. Follmer, T. F. Outeiro and E. C. A. Eleutherio, Biochim. Biophys. Acta, 2023, 1869, 166835 CrossRef PubMed.
- C. J. Banks, N. W. Rodriguez, K. R. Gashler, R. R. Pandya, J. B. Mortenson, M. D. Whited, E. J. Soderblom, J. W. Thompson, M. A. Moseley, A. R. Reddi, J. S. Tessem, M. P. Torres, B. T. Bikman and J. L. Andersen, Mol. Cell. Biol., 2017, 37, e00354–17 CrossRef PubMed.
- S. Rasouli, A. Abdolvahabi, C. M. Croom, D. L. Plewman, Y. Shi, J. I. Ayers and B. F. Shaw, J. Biol. Chem., 2017, 292, 19366–19380 CrossRef PubMed.
- S. G. Rhee, K. S. Yang, S. W. Kang, H. A. Woo and T. S. Chang, Antioxid. Redox Signaling, 2005, 7, 619–626 CrossRef PubMed.
- J. Bolduc, K. Koruza, T. Luo, J. Malo Pueyo, T. N. Vo, D. Ezeriņa and J. Messens, Redox Biol., 2021, 42, 101959 CrossRef PubMed.
- F. M. Najjar, F. Taghavi, R. Ghadari, N. Sheibani and A. A. Moosavi-Movahedi, Arch. Biochem. Biophys., 2017, 630, 81–90 CrossRef PubMed.
- J. S. Baldwin, L. Lee, T. K. Leung, A. Muruganandam and B. Mutus, Biochim. Biophys. Acta, 1995, 1247, 60–64 CrossRef PubMed.
- N. Rabbani, M. Xue and P. J. Thornalley, in Oxidative Stress, ed. H. Sies, Academic Press, 2020, pp. 759–777 DOI:10.1016/B978-0-12-818606-0.00036-5.
- B. Kuhla, K. Boeck, H.-J. Lüth, A. Schmidt, B. Weigle, M. Schmitz, V. Ogunlade, G. Münch and T. Arendt, Neurobiol. Aging, 2006, 27, 815–822 CrossRef PubMed.
- A. Mitsumoto, K.-R. Kim, G. Oshima, M. Kunimoto, K. Okawa, A. Iwamatsu and Y. Nakagawa, J. Biochem., 2000, 128, 647–654 CrossRef PubMed.
- G. Birkenmeier, C. Stegemann, R. Hoffmann, R. Günther, K. Huse and C. Birkemeyer, PLoS One, 2010, 5, e10399 CrossRef PubMed.
- S. P. Baba, O. A. Barski, Y. Ahmed, T. E. O'Toole, D. J. Conklin, A. Bhatnagar and S. Srivastava, Diabetes, 2009, 58, 2486–2497 CrossRef PubMed.
- L. D. Skou, S. K. Johansen, J. Okarmus and M. Meyer, Cells, 2024, 13, 296 CrossRef PubMed.
- T. B. Atieh, J. Roth, X. Yang, C. L. Hoop and J. Baum, Biomolecules, 2021, 11, 1466 CrossRef PubMed.
- Q. Zheng, N. D. Omans, R. Leicher, A. Osunsade, A. S. Agustinus, E. Finkin-Groner, H. D’Ambrosio, B. Liu, S. Chandarlapaty, S. Liu and Y. David, Nat. Commun., 2019, 10, 1289 CrossRef PubMed.
- M. C. Mazza, S. C. Shuck, J. Lin, M. A. Moxley, J. Termini, M. R. Cookson and M. A. Wilson, J. Neurochem., 2022, 162, 245–261 CrossRef PubMed.
- H. Ariga, K. Takahashi-Niki, I. Kato, H. Maita, T. Niki and S. M. M. Iguchi-Ariga, Oxid. Med. Cell. Longevity, 2013, 2013, 683920 Search PubMed.
- S. Schmidt, D. M. Vogt Weisenhorn and W. Wurst, Mol. Aspects Med., 2022, 86, 101096 CrossRef PubMed.
- H. Weissbach, L. Resnick and N. Brot, Biochim. Biophys. Acta, 2005, 1703, 203–212 CrossRef PubMed.
- M. Dagnell, E. E. Schmidt and E. S. J. Arnér, Free Radical Biol. Med., 2018, 115, 484–496 CrossRef PubMed.
- S. Lee, S. M. Kim and R. T. Lee, Antioxid. Redox Signaling, 2013, 18, 1165–1207 CrossRef PubMed.
- J. Haendeler, Antioxid. Redox Signaling, 2006, 8, 1723–1728 CrossRef PubMed.
- F. T. Ogata, V. Branco, F. F. Vale and L. Coppo, Redox Biol., 2021, 43, 101975 CrossRef PubMed.
- R. Tatsunami, T. Oba, K. Takahashi and Y. Tampo, J. Pharm. Sci., 2009, 111, 426–432 CrossRef PubMed.
- E. Van Schaftingen, F. Collard, E. Wiame and M. Veiga-da-Cunha, Amino Acids, 2012, 42, 1143–1150 CrossRef PubMed.
- S. Shrestha, S. Katiyar, C. E. Sanz-Rodriguez, N. R. Kemppinen, H. W. Kim, R. Kadirvelraj, C. Panagos, N. Keyhaninejad, M. Colonna, P. Chopra, D. P. Byrne, G. J. Boons, E. van der Knaap, P. A. Eyers, A. S. Edison, Z. A. Wood and N. Kannan, Sci. Signaling, 2020, 13, eaax6313 CrossRef PubMed.
- H. A. Bustamante, A. E. González, C. Cerda-Troncoso, R. Shaughnessy, C. Otth, A. Soza and P. V. Burgos, Front. Cell. Neurosci., 2018, 12, 126 CrossRef PubMed.
- T. Jung, A. Höhn and T. Grune, Redox Biol., 2014, 2, 388–394 CrossRef PubMed.
- T. Jung, A. Höhn and T. Grune, Redox Biol., 2014, 2, 99–104 CrossRef PubMed.
- T. Reinheckel, N. Sitte, O. Ullrich, U. Kuckelkorn, K. J. Davies and T. Grune, Biochem. J., 1998, 335, 637–642 CrossRef PubMed.
- J. W. Zmijewski, S. Banerjee and E. Abraham, J. Biol. Chem., 2009, 284, 22213–22221 CrossRef PubMed.
- K. Okada, C. Wangpoengtrakul, T. Osawa, K. Uchida, S. Toyokuni and K. Tanaka, J. Biol. Chem., 1999, 274, 23787–23793 CrossRef PubMed.
- M. A. Queisser, D. Yao, S. Geisler, H.-P. Hammes, G. Lochnit, E. D. Schleicher, M. Brownlee and K. T. Preissner, Diabetes, 2010, 59, 670–678 CrossRef PubMed.
- S. Finkbeiner, Cold Spring Harbor Perspect. Biol., 2020, 12, a033993 CrossRef PubMed.
- M. Pajares, A. Cuadrado, N. Engedal, Z. Jirsova and M. Cahova, Oxid. Med. Cell. Longevity, 2018, 2018, 2450748 CrossRef PubMed.
- R. Scherz-Shouval, E. Shvets, E. Fass, H. Shorer, L. Gil and Z. Elazar, EMBO J., 2007, 26, 1749–1760 CrossRef PubMed.
- J.-i Takino, Y. Kobayashi and M. Takeuchi, J. Gastroenterol., 2010, 45, 646–655 CrossRef PubMed.
- J. M. Zeng, R. A. Dunlop, K. J. Rodgers and M. J. Davies, Biochem. J., 2006, 398, 197–206 CrossRef PubMed.
- J. Morgenstern, T. Fleming, D. Schumacher, V. Eckstein, M. Freichel, S. Herzig and P. Nawroth, J. Biol. Chem., 2017, 292, 3224–3238 CrossRef PubMed.
- B. Stratmann, B. Engelbrecht, B. C. Espelage, N. Klusmeier, J. Tiemann, T. Gawlowski, Y. Mattern, M. Eisenacher, H. E. Meyer, N. Rabbani, P. J. Thornalley, D. Tschoepe, G. Poschmann and K. Stühler, Sci. Rep., 2016, 6, 37737 CrossRef PubMed.
- A. C. Conibear, E. E. Watson, R. J. Payne and C. F. W. Becker, Chem. Soc. Rev., 2018, 47, 9046–9068 RSC.
- Y. Tan, H. Wu, T. Wei and X. Li, J. Am. Chem. Soc., 2020, 142, 20288–20298 CrossRef PubMed.
- R. E. Thompson and T. W. Muir, Chem. Rev., 2020, 120, 3051–3126 CrossRef PubMed.
- E. A. deRamon, V. R. Sabbasani, M. D. Streeter, Y. Liu, T. R. Newhouse, D. M. McDonald and D. A. Spiegel, J. Am. Chem. Soc., 2022, 144, 21843–21847 CrossRef PubMed.
- M. Matveenko, E. Cichero, P. Fossa and C. F. Becker, Angew. Chem., Int. Ed., 2016, 128, 11569–11574 CrossRef.
- C. Chavarría, R. Ivagnes, A. Zeida, M. D. Piñeyro and J. M. Souza, Arch. Biochem. Biophys., 2024, 752, 109858 CrossRef PubMed.
- P. C. Cirino, Y. Tang, K. Takahashi, D. A. Tirrell and F. H. Arnold, Biotechnol. Bioeng., 2003, 83, 729–734 CrossRef PubMed.
- D. de la Torre and J. W. Chin, Nat. Rev. Genet., 2021, 22, 169–184 CrossRef PubMed.
- M. A. Shandell, Z. Tan and V. W. Cornish, Biochemistry, 2021, 60, 3455–3469 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2025 |