
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Advanced reactivity-based sequencing methods for mRNA epitranscriptome profiling†
Zhihe
Cai‡
a,
Peizhe
Song‡
a,
Kemiao
Yu
b and
Guifang
Jia
 *abc
*abc
aSynthetic and Functional Biomolecules Center, Beijing National Laboratory for Molecular Sciences, Key Laboratory of Bioorganic Chemistry and Molecular Engineering of Ministry of Education, College of Chemistry and Molecular Engineering, Peking University, Beijing 100871, China. E-mail: guifangjia@pku.edu.cn
bPeking-Tsinghua Center for Life Sciences, Peking University, Beijing 100871, China
cBeijing Advanced Center of RNA Biology, Peking University, Beijing 100871, China
First published on 10th December 2024
Abstract
Currently, over 170 chemical modifications identified in RNA introduce an additional regulatory attribute to gene expression, known as the epitranscriptome. The development of detection methods to pinpoint the location and quantify these dynamic and reversible modifications has significantly expanded our understanding of their roles. This review goes deep into the latest progress in enzyme- and chemical-assisted sequencing methods, highlighting the opportunities presented by these reactivity-based techniques for detailed characterization of RNA modifications. Our survey provides a deeper understanding of the function and biological roles of RNA modification.
1. Introduction
Since their initial discovery in the mid-20th century, many kinds of RNA modifications have been identified, marking a significant boost in the field of epitranscriptomics and enhancing our understanding of transcriptomics. These modifications, found across various RNA species, participate in complex aspects of gene regulation. Originally, the study of RNA modifications mainly focused on abundant non-coding RNAs such as ribosomal RNAs (rRNAs) and transfer RNAs (tRNAs), uncovering their critical functions in RNA biogenesis, stability, translation, and splicing.1,2 Recent research studies have extended the landscape to include low abundant and highly divergent RNAs like messenger RNAs (mRNAs) and regulatory non-coding RNAs (ncRNAs), which present challenges for studies using traditional biochemical methods.Among all RNA modifications, N6-methyladenosine (m6A) stands out as the most abundant internal modification found in mRNAs, alongside other significant modifications like N6,2′-O-dimethyladenine (m6Am), N1-methyladenine (m1A), 5-methylcytosine (m5C), N4-acetylcytidine (ac4C), N7-methylguanosine (m7G), pseudouridine (Ψ), inosine (I) and 2′-O-methylation (Nm).3–12 These modifications are known to influence key regulatory processes including transcription, translation, RNA stability and other fate determination processes.2,7,9,11–29
The advent of next-generation sequencing (NGS) has enabled the finding of these modifications globally, helping us to understand their biological functions. The goal of ideal sequencing methods is to accurately identify the locations and stoichiometries of RNA modifications. Although antibody-based enrichment followed by sequencing has been a basic and fundamental technique for studying these modifications, it often falls short in terms of accuracy and stoichiometry, which could give us a false sense of their precise location and proportion.30–33 Additionally, antibody-based RNA modification sequencing methods often exhibit biases due to non-specific and off-target binding, which can also introduce sequencing bias.34 In this review, we summarize the current research progress of these reactivity-based next-generation sequencing methods, addressing the challenges they face and discussing their application in functional studies of the epitranscriptome (Table S1, ESI†).
2. mRNA modifications
The central dogma of molecular biology posits that genetic information flows from DNA to RNA, and then to functional proteins. RNA, which exists in various forms such as mRNA, tRNA, rRNA, long non-coding RNA (lncRNA), and small RNA (sRNA), performs diverse roles. Beyond its role in carrying genetic sequences, RNA is subject to many covalent modifications that play critical roles in gene regulation. These modifications introduce an additional layer of RNA function, serving as a dedicated mechanism, with advancements in detection techniques enhancing our exploration of their functional significance.Each chemical modification has a distinct regulatory impact on RNA metabolism and the overall function. For example, m6A, the predominant internal modification in mRNA, modulates various aspects such as transcription, splicing, nuclear export, stability, translation, and even secondary structure.6,35–49 On the other hand, m6Am is found in the first position adjacent to the 5′ cap structure in many mammalian mRNA molecules. In mRNA, m1A distributes mainly in 5′ UTR and regulates translation by altering the RNA structure of translation initiation sites.9,10 The internal m7G modification enhances mRNA translation efficiency, while ac4C in mRNA affects translation.25–27,50–54 Within mRNA, m5C affects RNA export, stability, and also translational regulation.15,55 Moreover, the introduction of Ψ into mRNA not only increases protein production but also modifies translation dynamics.20,56–58 The A-to-I editing events influence many layers of gene regulation, such as amino acid alteration, translation, alternative splicing, nuclear retention and nonsense-mediated mRNA decay (NMD).59–63 Additionally, Nm may play essential roles in translation and RNA splicing.64–66
The evolution of RNA modification detection technologies has significantly contributed to our comprehensive understanding of the epitranscriptome and facilitated downstream functional studies. With the advent of next-generation sequencing, numerous sequencing technologies have been developed to map modified nucleotides across the transcriptome, unlocking their regulatory functions of RNA modifications. The distinction between modified and unmodified nucleotides enables antibody-based recognition and enrichment for modifications like m6A, m6Am, m1A, m5C, ac4C and also m7G.9,10,23,25,26,53,67–75 However, aside from the typically high cost of antibody-related products, challenges also arise with modifications that share similar structures, such as m6A and m6Am, which cannot be differentiated through antibody enrichment.67–69 Even for a single type of RNA modification, antibodies can exhibit non-specific cross-reactivity and batch-to-batch variability. This limitation has driven the development of new strategies for mapping RNA modifications through reactivity-based strategies with specific enzymes and/or chemicals. The following sections include detection methods concentrating on nine important and distinct modifications (m6A, m6Am, m1A, m5C, ac4C, m7G, Ψ, I, and Nm) on mRNAs (Fig. 1 and Table S1, ESI†), which have been highly studied, highlighting the evolution made in this leading field.
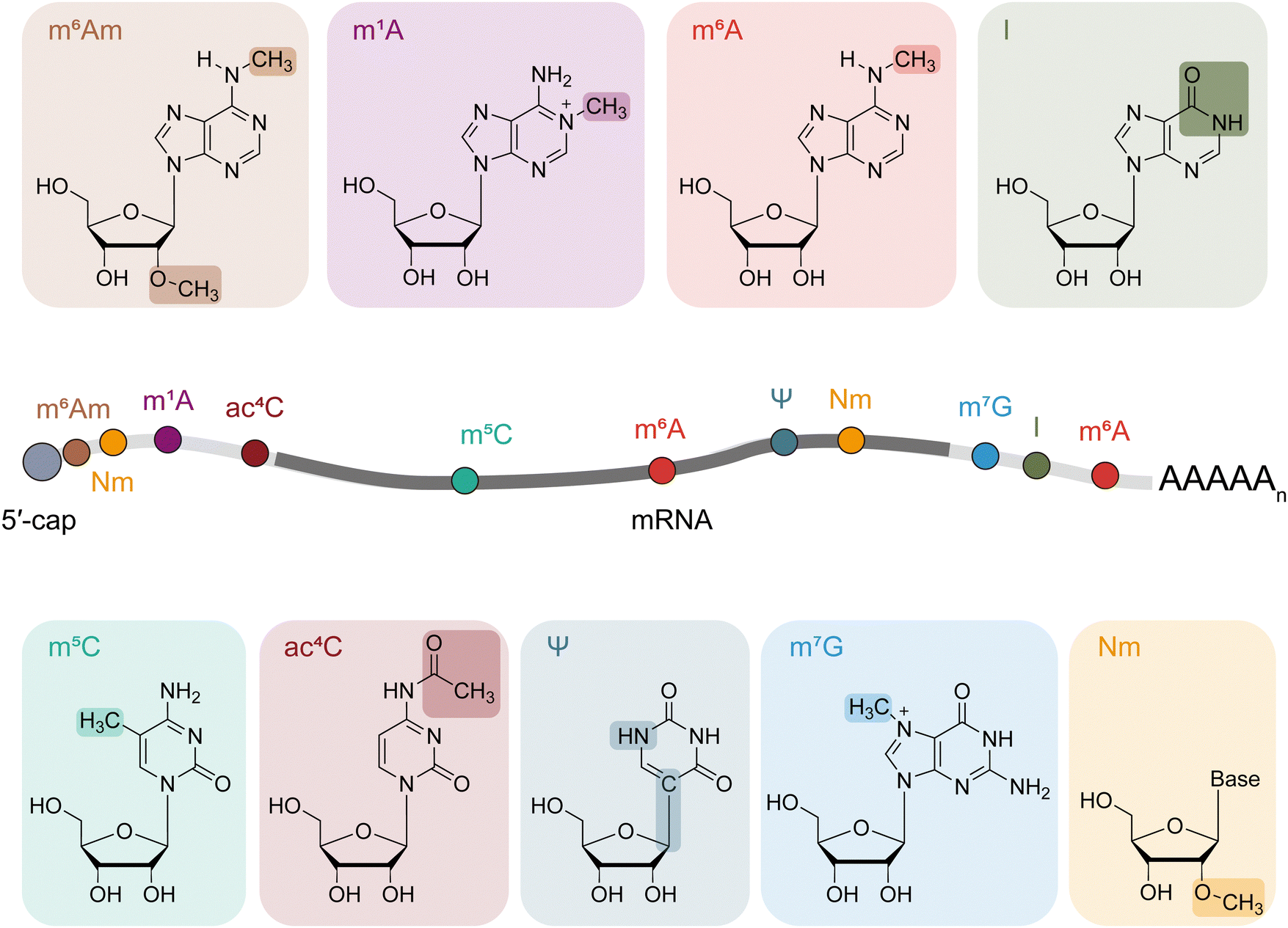 | ||
| Fig. 1 Distribution and chemical structure of mRNA modifications discussed in this review. | ||
3. Sequencing methods
3.1 m6A detection methods
To date, m6A methylation sites have been mapped to the transcriptome, predominantly located in long exons, near stop codons, and mostly in 3′ untranslated regions (3′ UTRs) in mammalian cells.67,68 In this section, we will primarily summarize those reactivity-based methods for locus-specific m6A detection and quantification. | ||
| Fig. 2 Overview of enzyme-assisted sequencing methods for the detection of m6A modifications. (a) DART-seq and scDART-seq. (b) m6A-REF-seq and MAZTER-seq. | ||
DART-seq has the unique function of irreversibly marking m6A sites over several hours allowing APOBEC1-YTH to access and edit structurally hidden sites under physiological conditions. Therefore, it identifies a broader range of sites than antibody-based methods. Moreover, DART-seq has the ability to monitor different mutation sites within individual transcripts, allowing for the determination of m6A sites’ presence within the same transcript by long-read sequencing.83 Potential applications of DART-seq include m6A profiling in various cell types under different physiological states and the detection of m6A within specific cellular compartments by incorporating localization elements into the APOBEC1-YTH fusion. However, it relies on the in vivo overexpression of APOBEC1-YTH, which limits its efficiency for in vitro applications in transfection-challenging materials. Additionally, with only 60% RNA substrates targeted by the YTH domain containing m6A, false negatives present a challenge that may be addressed by enhancing the affinity and specificity of the YTH domain. Besides that, C-to-U base editing can result in unfavorable gene expression, potentially leading to protein dysfunction, which could influence cellular homeostasis.
MazF, identified as a methylation-sensitive endoribonuclease, uniquely cleaves the unmethylated ACA motif but not the methylated (m6A)CA motif. Using this specificity, two similar methods, m6A-REF-seq and MAZTER-seq,86,87 have been developed. These techniques involve treating parallel samples—comparing either control cells to m6A writer knockout cells, where METTL3, as a crucial methyltransferase, is essential for m6A modification, or RNA subjected to FTO demethylation reactions versus untreated RNA—with MazF, followed by RNA sequencing. Ideally, after MazF treatment, RNA fragments should initiate at an ACA site and end just before the next ACA site, allowing reads to span m6A sites (Fig. 2b). The presence of m6A inversely correlates with cleavage efficiency, enabling the identification and quantification of m6A sites using the MAZTER-MINE computational pipeline.86 This pipeline calculates cleavage efficiencies at the 5′ and 3′ ends from RNA-seq data to estimate m6A abundance at specific sites. By analyzing ACA sequences in paired samples, these methods can pinpoint transcriptomic m6A sites within these specific motifs and quantify methylation levels with single-base precision.
The high sensitivity and specificity of MazF, combined with a straightforward experimental procedure without antibody enrichment, make these methods particularly suited for limited samples, including those from pathological tissues or early embryos. However, due to the exclusive recognition of the ACA motif, only partial m6A sites can be detected. Factors such as the secondary structure of RNA, MazF enzyme activity, and sequence preference may influence result accuracy; on the other hand, the quantitation also needs to be challenged by the limited enzymatic efficiency of MazF.34 Nevertheless, the potential to uncover more sites exists, either by finding additional enzymes with different motif specificities or by optimizing current enzymes to recognize a broader set of m6A methylation sites.
 | ||
| Fig. 3 Overview of enzyme- and chemical-assisted sequencing methods for the detection of m6A modifications. (a) m6A-SEAL. (b) m6A-label-seq. (c) m6A-SAC-seq. Rib, ribose. DTT, dithiothreitol. Allyl-SeAM, Se-allyl-adenosyl methionine. Allyl-SAM, S-allyl-adenosyl methionine. | ||
Due to the high specificity of FTO to oxidize the methyl group of m6A, m6A-SEAL minimizes false-positive signals compared to MeRIP-seq, offering an antibody-free method with high sensitivity, specificity, and reliability for mapping m6A transcriptome-wide. Interestingly, both m6Am and m6A serve as FTO substrates, but FTO exhibits greater reactivity with m6Am in vitro. This differential reactivity means the potential to distinguish between m6A and m6Am by fine-tuning the oxidation condition. Moreover, given that FTO can also oxidize DNA 6mA to N6-hydroxymethyldeoxyadenosine (d6mA) under certain conditions, it has the potential for adapting m6A-SEAL for DNA 6mA detection. While m6A-SEAL has a base resolution comparable to MeRIP-seq (about 200 nt), achieving single-base resolution could be possible by optimizing the reverse transcription process to induce truncations or mutations near the dm6A site in order to enhance the accuracy in mapping m6A modifications under the single-base resolution.67,68
In addition to in vitro reaction and labeling strategies, researchers have also developed metabolic labeling methods for single-base m6A detection. The biogenesis of m6A in mRNA involves the transfer of a methyl group from S-adenosyl methionine (SAM) to specific adenosine (A) sites within RNA by the m6A methyltransferase complex, a central process similar to many fundamental biological processes.90 However, the inherent chemical stability of the methyl group on m6A and its consistent base pairing pattern with A present substantial challenges for precise detection by high-throughput sequencing techniques, perplexing the accurate mapping of m6A modifications throughout the transcriptome. To address these challenges, researchers have developed a metabolic labeling approach known as m6A-label-seq (Fig. 3b).91 This method enables transcriptome-wide, single-base resolution detection of m6A by substituting the methyl group with an allyl group. This substitution is facilitated by feeding cells with Se-adenosyl-L-selenomethionine, a small-in-size methionine analog, which leads to the metabolic incorporation of an allyl group into specific adenosine sites, producing a modified nucleotide termed N6-allyladenosine (a6A).92–94 Due to the structural similarity of the isopentenyl and allyl groups, labeled a6A allows for selective enrichment by commercial N6-isopentenyladenosine (i6A) antibodies. The a6A modification then undergoes specific iodination (I2)-induced cyclization to form N1,N6-cyclized adenosine (cyc-A), leading to misincorporations during reverse transcription because of steric hindrance, thereby enabling precise identification of m6A sites.
Despite its precision, m6A-label-seq identifies fewer sites compared to other methods, attributed to low incorporation efficiency and the associated loss of quantitative information. This highlights the necessity for methodological advancements to enhance both incorporation and chemical transformation efficiencies. Future improvements might involve engineering more efficient methionine adenosyl methyltransferases for increased a6A yield or refining reverse transcriptase enzymes to boost mutation efficiency during sequencing. Additionally, the application of Se-adenosyl-L-selenomethionine could induce cellular stress, potentially influencing sequencing results from the bottom up. Thus, further refinement of the method is required to reduce such side effects and improve result reliability.
Similar to m6A-label-seq, m6A-SAC-seq provides a selective and quantitative strategy for high-resolution mapping of m6A across the transcriptome (Fig. 3c).95 Both techniques utilize the chemical reactivity of the allyl group, with m6A-SAC-seq uniquely comprising an in vitro enzymatic reaction by MjDim1 (a homolog of Dim1 in M. jannaschii) for allyl labeling at m6A sites.91,95 This process involves the enzymatic transfer of an allyl group from allyl-SAM to both m6A and A, resulting in the formation of a6m6A and a6A, respectively. Subsequent iodination-induced cyclization causes base misincorporation during reverse transcription, thereby enabling precise m6A identification at the single-nucleotide level without the need for enrichment.
The selectivity of MjDim1-catalyzed allyl transfer from allylic-SAM is approximately tenfold higher for m6A than A, with the human immunodeficiency virus (HIV) reverse transcriptase inducing higher mutation rates at the labeled and cyclized a6m6A sites compared to cyclized a6A sites. One of the key advantages is its minimal RNA input requirement, making it highly adaptable for studies with limited sample availability. Additionally, using RNA samples treated with the m6A demethylase FTO as a background control enhances the specificity of this method, allowing for the differentiation of true m6A modifications from background noise. Another significant benefit of m6A-SAC-seq is its capability for m6A quantification through mutation rate correlation. This feature makes it possible to acquire quantitative data on m6A levels, offering insights into the dynamic modification proportion under various conditions. However, the method does exhibit certain limitations, including a motif preference for GAC over AAC, which may result in the under-detection of some m6A sites, especially for the m6A quantitation. Although it can identify approximately 80% of m6A sites—largely due to the prevalence of the GAC motif among 70–75% of m6A sites—it faces challenges in detecting AAC sites. Moreover, the requirement for higher sequencing depth compared to antibody-based methods may restrict its widespread use in settings with limited sequencing capabilities.
 | ||
| Fig. 4 Overview of sequencing methods for the detection of m6A modifications using nitration and/or deamination reaction. (a) m6A-ORL-seq. (b) NOseq. (c) eTAM-seq. (d) GLORI. Rib, ribose. TDO, thiourea dioxide. TEAA, triethylamine acetate. | ||
Similar to m6A-ORL-seq, NOseq is a method for detecting m6A by exploiting its resistance to chemical deamination by nitrous acid (Fig. 4b).97 This innovative technique includes a deamination process, which converts cytidine to uridine, adenosine to inosine (I), guanine (G) to xanthosine (X), and m6A to N6-methyl-N6-nitrosoadenosine (NO-m6A) while leaving uridine unaltered. The resulting sequence changes are analyzed using a specialized mapping algorithm designed to handle the sequence degeneration caused by deamination, ensuring precise detection of m6A sites. NOseq was experimentally validated by detecting known m6A sites in human rRNA and lncRNA MALAT1, as well as several candidate m6A sites in the Drosophila melanogaster transcriptome. The method proved effective in identifying m6A with partial modification levels around 50%, and this threshold could be lowered to approximately 10% when combined with m6A immunoprecipitation. Although NOseq represents a significant advancement in m6A detection, offering us a powerful tool for exploring RNA modifications and their biological functions, future improvements could further enhance the reaction efficiency and reduce the RNA degradation affected by nitrous acid.
Evolved TadA-assisted N6-methyladenosine sequencing (eTAM-seq) is a technique designed to achieve high-resolution profiling and quantification of m6A across the transcriptome (Fig. 4c). This method uses enzyme-assisted adenosine deamination to detect and quantify m6A modifications with exceptional precision.98 Central to eTAM-seq is the use of a hyperactive variant of the TadA enzyme, TadA8.20, which selectively converts unmethylated A to I, while leaving m6A sites unaltered. During reverse transcription, inosines are recognized as G, allowing for the identification of m6A as persistent adenosine signals. This approach facilitates not only transcriptome-wide m6A mapping but also site-specific quantification with minimal RNA input, making it a powerful tool for epitranscriptomic studies.
One of the primary advantages of eTAM-seq is its ability to provide base-resolution mapping of m6A sites. This high level of precision enables the accurate localization of m6A modifications across the transcriptome, which is critical for understanding the functional roles of these modifications in gene expression regulation. Additionally, eTAM-seq is characterized by its low input requirement, capable of detecting and quantifying m6A modifications with as few as ten cells or 250 picograms (pg) of total RNA. This sensitivity represents a significant improvement over traditional methods, which often necessitate much larger quantities of RNA. Another key benefit of eTAM-seq is its preservation of RNA integrity. Unlike chemical deamination methods, which can degrade RNA and compromise the accuracy of results, eTAM-seq employs an enzymatic approach that maintains the structural integrity of RNA, reducing the risk of sample loss and ensuring more reliable data.96,97 Furthermore, eTAM-seq offers quantitative capabilities, allowing researchers to not only detect the presence of m6A modifications but also quantify the extent of methylation at specific sites. eTAM-seq can be adapted for various applications, including potential single-cell m6A profiling, which could provide unprecedented insights into the heterogeneity of m6A modifications at the individual cell level.
Despite its many advantages, eTAM-seq has certain limitations. The efficiency of the deamination process is partially dependent on the RNA secondary structure. Highly structured RNA regions may obstruct the accession of enzyme, leading to incomplete deamination and potentially resulting in false negatives. Additionally, the method may yield false-positive signals due to other adenine modifications that resist deamination by the enzyme, although this issue can be mitigated by using demethylases like FTO to confirm confident m6A sites. Another limitation of eTAM-seq is its reduced sensitivity to low methylation levels. This method may not accurately detect m6A sites with methylation levels below 25%, which could result in the underrepresentation of certain modifications in the data. Moreover, the accuracy of m6A mapping using eTAM-seq requires the use of control samples to estimate site accessibility, adding an extra layer of complexity to the experimental workflow.
Given that the reaction efficiency of m6A-SAC-seq varies depending on the motif surrounding each m6A site, the lack of quantitative information from m6A-ORL-seq/NOseq, and the potential for false negatives and inefficiency in detecting low methylation level sites using deamination enzyme, significant progress needs to be made in the pursuit of more accurate m6A detection. Existing methods often face challenges such as limited site-specific resolution, motif biases, and the complexities associated with sophisticated experiment and computational analysis.83,86–88,95 However, GLORI represents a breakthrough in m6A detection technology, overcoming these hurdles to achieve precise, single-base identification and quantification of m6A sites (Fig. 4d).99 Using a catalytic system discovered through screening combinations of chemical reactions, nitrite can efficiently deaminate unmethylated adenosine into inosine, achieving an A-to-I conversion rate surpassing 98%. In this system, glyoxal reacts with guanosine in borate buffer to protect the exocyclic amino group forming a glyoxal-guanosine adduct (G*) and with adenosine at the N6 position to generate a N-(hydroxymethylene) hemiaminal derivative, which acts as a catalyst in the deamination process. During reverse transcription, inosine pairs with cytidine and is subsequently read as guanosine in sequencing, resulting in an A-to-G conversion. In contrast, m6A is unaffected and remains identifiable as adenosine. This method allows for the absolute quantification of m6A at the single-base level by assessing the proportion of A in sequencing reads, allowing this chemical reaction to distinguish between methylated and unmethylated adenosines accurately.
GLORI has a strong ability to detect m6A accurately, which makes it a valuable tool in m6A functional research. GLORI sets itself apart with its antibody-free, highly sensitive approach, capable of detecting even low levels of m6A modifications with high technical repeatability. It consistently identifies the canonical DRAC motif (D = G/A/T, R = A/G) in m6A sites and has been applied in studies of dynamic m6A regulation under stress conditions. These findings demonstrate GLORI has the ability to explore the function of m6A in essential biological processes and stress responses. However, the transformation efficiency of the chemical reaction between nitrite and adenosine is largely influenced by the length of the transcripts. Moreover, despite its high A-to-I conversion rate, the treatment with glyoxal and nitrite leads to the degradation of RNA into relatively short chains of nucleic acids, complicating sequencing and data analysis.
3.2 m6Am detection methods
m6Am is a 5′-terminal modification found at the first nucleotide following the mRNA cap. When adenine is the first transcribed nucleotide in mammalian mRNAs, it can undergo a co-transcriptional methylation to form N6,2′-O-dimethyladenosine.77,100,101 The relative content of this modification is approximately one-tenth of that of m6A. Given its specific location at the first base of mRNA, m6Am has a potential role in regulating translation. Notably, this modification is not exclusive to mRNAs; m6Am is also found internally within U2 snRNAs.102High-throughput sequencing methods, combined with the immunoprecipitation of fragmented RNAs using m6A-specific antibodies, have been developed to identify m6Am-containing RNAs.67–69 However, these methods have a significant limitation—anti-m6A antibodies cannot distinguish between m6Am and m6A. This makes sequencing techniques like m6A-seq/MeRIP-seq and methylation iCLIP (miCLIP) less effective for accurate m6Am mapping. To directly identify m6Am, researchers have developed CAPturAM, a novel antibody-free chemical biology approach that directly enriches and probes physiological PCIF1 targets. In this method, cap-m6Am is enzymatically propargylated using PCIF1 with a synthetic AdoMet analog. The propargylated m6Am is then selectively biotinylated and enriched using magnetic streptavidin beads. This strategy is expected to significantly enhance transcriptome-wide studies by identifying PCIF1 targets and m6Am sites. Despite its promising potential, CAPturAM currently faces certain limitations, including residual internal background propargylation, even after stringent optimization of enzymatic modification conditions. It is crucial to address this issue to improve the specificity and accuracy. Future advancements in CAPturAM are expected to incorporate RNA-seq, which would present an antibody-free technique for the direct and comprehensive identification of m6Am sites across the transcriptome.
3.3 m1A detection methods
The N1 site of the adenine base undergoes methylation to form the m1A modification. Intriguingly, m1A possesses a positive charge under physiological conditions. The presence of the N1 methyl group modifies the structure of the base, influencing its free energy, the manner it pairs with other bases, and the mechanics of pairing. Notably, m1A can engage in Hoogsteen base pairing, potentially leading to mismatches during the reverse transcription process.103 Comprehensive investigation of m1A heavily relies on the development of detection methodologies, which have progressed from traditional chemical analysis to modern high-throughput sequencing techniques. | ||
| Fig. 5 Overview of sequencing methods for the detection of m1A modifications using demethylase AlkB. Rib, ribose. AMV, avian myeloblastosis virus reverse transcriptase. TGIRT, thermostable group II intron reverse transcriptase. | ||
However, the truncated complementary DNA (cDNA) synthesis might lead to the loss of information of the m1A methylation. Like m1A-ID-seq, m1A-MAP-seq and m1A-IP-seq combines enzymatic demethylation and reverse transcription under different conditions using thermostable group II intron reverse transcriptase (TGIRT) and RT-1306, respectively (Fig. 5).23,70 RT-1306, an engineered novel reverse transcriptase, yields a tenfold increase in full-length cDNA production and a higher ratio of reads to truncated products compared to TGIRT.70 Demethylated RNA is subjected to RT to generate cDNA, followed by library preparation for subsequent comparison. Untreated RNA, on the other hand, induces misincorporation by TGIRT or RT-1306. Therefore, the precise location of the m1A modification can be determined, facilitating the identification of m1A modification sites with single-nucleotide resolution. m1A-quant-seq, modified from m1A-IP-seq, incorporates synthetic m1A oligonucleotides for estimating m1A stoichiometry.70 However, reliance on demethylation treatment can lead to false negatives, particularly when RNA methylation abundance is low or when the methylation site is located within complex structures that resist demethylation processes. Moreover, the calibration curve conforms to a nonlinear equation, indicating that RT-1306 may still cause some degree of truncation in biological RNA samples. This truncation reduces sensitivity at certain sites, complicating the accurate detection and mapping of modifications.
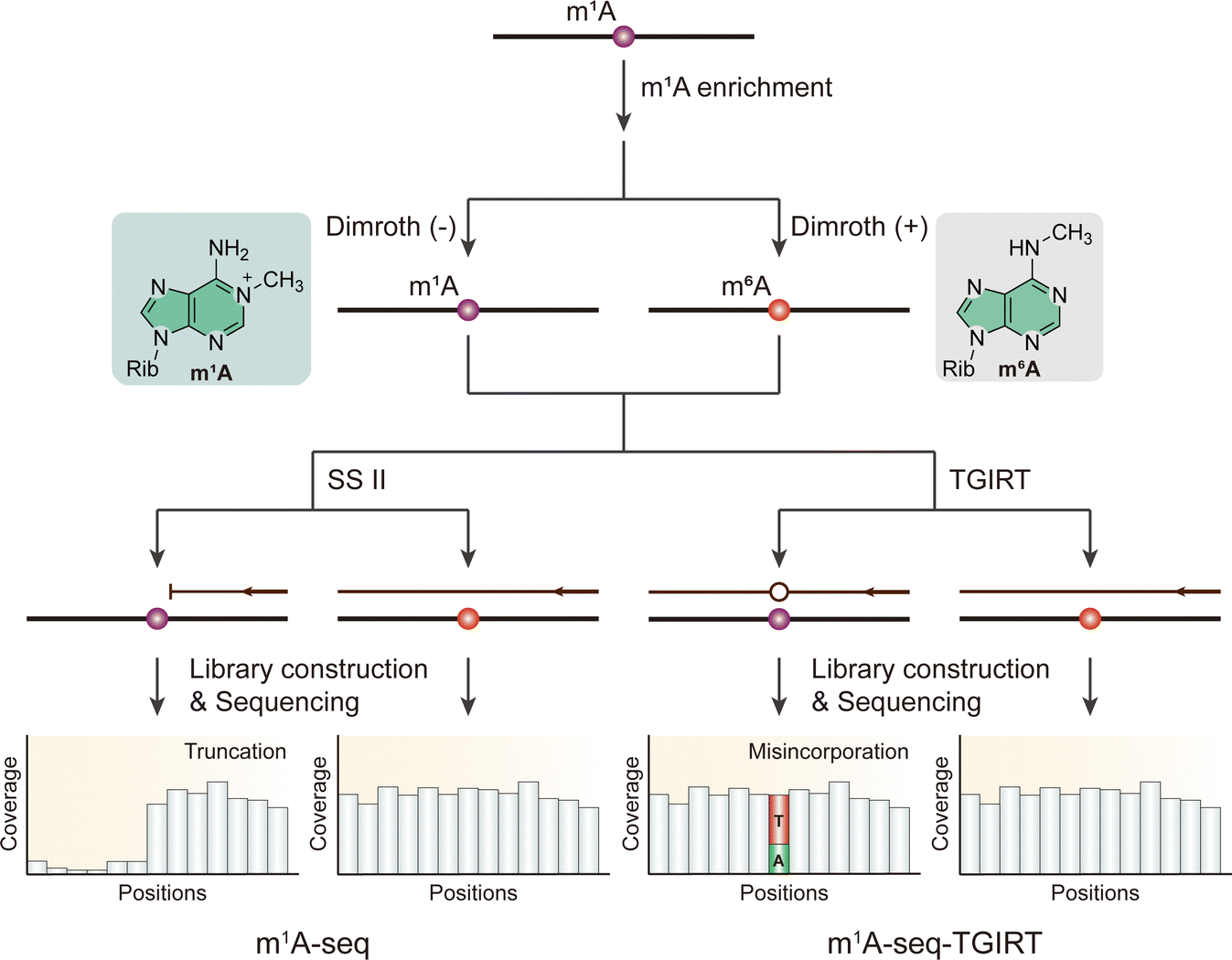 | ||
| Fig. 6 Overview of sequencing methods for the detection of m1A modifications using Dimroth rearrangement. Rib, ribose. SS II, SuperScript II. TGIRT, thermostable group II intron reverse transcriptase. | ||
3.4 m5C detection methods
Although methylated cytosine residues at position 5 (5mC) are very common in DNA, m5C in RNA did not initially gain much attention due to its lower abundance. Previous methods for detecting m5C in RNA, such as high-performance liquid chromatography (HPLC) and mass spectrometry (MS), required exceedingly high amounts of RNA and could only reliably identify methylated sites in highly abundant and stable ncRNAs, such as tRNAs and rRNAs.106,107 However, with the development of high-throughput sequencing approaches, m5C has been found to be widely distributed in mRNA.108 Notably, m5C is often located near argonaute-binding regions within the 3′ UTR or in the vicinity of the translational start site of mRNA.8,16Bisulfite sequencing (BS-seq), a gold standard method for detecting m5C in genomic DNA (gDNA), is based on the chemical deamination of cytosines with sodium bisulfite (NaHSO3) treatment and has been applied to mRNA.109,110 Sodium bisulfite deaminates unmethylated cytosines into uridines in single-stranded DNA or RNA, while methylated cytosines remain unconverted.110,111 During subsequent analysis, unmethylated C is read as T, while methylated C is still read as C. Using bisulfite sequencing, thousands of m5C sites in mRNA have been identified in humans.16 In general, while bisulfite sequencing can achieve single-base-resolution detection of m5C, its limitation in converting cytosines of single-stranded nucleic acids can lead to incomplete conversion in RNA secondary structure regions, resulting in a large number of false-positive m5C sites. To address this, RBS-seq, which uses heating and formamide to denature RNA and improves the C-to-U conversion efficiency in double-stranded regions, has been developed (Fig. 7a).110 This method has identified 486 candidate m5C sites in mammalian mRNA.112 Additionally, achieving a high conversion rate requires prolonged incubation under consecutive acidic and alkaline conditions, which also causes RNA degradation. This degradation can compromise the subsequent reverse transcription and PCR amplification steps. To overcome this issue, an ultrafast bisulfite sequencing method (UBS-seq) has been developed for mapping 5-methylcytosine in both DNA and RNA.113 UBS-seq optimizes the reaction by using ammonium salts of bisulfite and sulfite and performing the reaction at 98 °C for approximately 10 minutes, affording a substantially lower background than previous approaches.
 | ||
| Fig. 7 Overview of sequencing methods for the detection of m5C modifications. (a) RBS-seq. (b) Aza-IP. (c) miCLIP. (d) m5C-TAC-seq. Rib, ribose. TET, ten-eleven translocation methylcytosine dioxygenase. AI, 1,3-indandione. DTT, dithiothreitol. | ||
Two members of the NSUN family, NSUN2 and NSUN6, are responsible for mRNA methylation, with 90% of m5C sites being sensitive to NSUN2 depletion and a small fraction being NSUN6 substrates.113 Considering NSUN2 methylates the majority of m5C sites, two methods have been developed based on the catalytic methylation mechanism. In the 5-azacytidine-mediated RNA immunoprecipitation method (Aza-IP), 5-azacytidine (5-AzaC), a cytidine analog with a nitrogen substitution at carbon 5, is randomly incorporated into nascent RNA by RNA polymerases in cells overexpressing an epitope-tagged m5C RNA methyltransferase (Fig. 7b).114 The incorporation of 5-AzaC affects the release of methyl transferase at carbon 5, forming a stable covalent bond. After immunoprecipitation, specific C-to-G conversion can be observed at targeted C residues, enabling the detection of m5C at single-base resolution. Using Aza-IP, the direct targets of NSUN2 and DNMT2, a tRNA m5C methyltransferase, can be identified, revealing specific methylated cytosines. miCLIP has been developed to successfully identify transcriptome-wide m5C sites methylated by NSUN2.73 This method utilizes the catalytic principle that a cysteine-to-alanine mutation (C271A) in the NSUN2 protein impedes the release of enzymes from the protein–RNA complex. This results in the formation of a stable covalent bond between NSUN2 and its RNA target, causing truncation during reverse transcription and thereby generating single-nucleotide-resolution information (Fig. 7c). While both Aza-IP and miCLIP methodologies are dependent on the formation of covalent bonds between the RNA methylase and its substrate, the accuracy of m5C detection methods is still compromised by challenges such as nonspecific antibody binding and mislocalization of methyltransferases. To address this challenge, a BS-free, base-resolution m5C detection strategy was enabled by TET-assisted chemical labeling (m5C-TAC) (Fig. 7d).115 In m5C-TAC-seq, m5C is first oxidized to f5C, then labeled with an azido derivative of 1,3-indandione (AI). This labeling facilitates the enrichment of m5C-containing RNAs via biotin pull-down and induces C-to-T transitions at m5C sites. Importantly, this method is gentle on RNA and does not affect unmodified Cs, enabling the direct detection of m5C even in RNAs with low abundance or low sequence complexity.
3.5 ac4C detection methods
ac4C was initially discovered during the characterization of eukaryotic rRNAs and tRNAs.116,117 The production of ac4C in eukaryotic RNA is unique, relying exclusively on the enzyme N-acetyltransferase 10 (NAT10).28,117 Through an adenosine triphosphate (ATP)-dependent process, NAT10 executes this function by shifting an acetyl group from acetyl-CoA to the exocyclic N4-amine of cytidine.118It has been noticed that ac4C does not disrupt traditional base pairing. However, when subjected to treatment with two equivalents of sodium cyanoborohydride (NaCNBH3), ac4C transforms into a reduced nucleobase, tetrahydro-N4-acetylcytidine (H4-ac4C), which can be misread as a U during reverse transcription, causing C to T mutations at ac4C sites (Fig. 8a).119 While several other modified nucleobases (such as m7G, dihydropyridine, and N3-methylcytidine) are also susceptible to reduction by hydride donors, the hydrolytic lability of ac4C can be exploited to chemically deacetylate RNA for control experiments.120 In ac4C-seq, one experimental sample is treated with NaCNBH3 under acidic conditions (reduction), while two control samples are subjected to acidic conditions without a reducing agent (mock-treated) and deacetylation followed by NaCNBH3 treatment under alkali situation (deacetylated and reduction-treated).119 Employing ac4C-seq allows for the detection of ac4C modifications distributed within the transcriptome at single-nucleotide resolution. Nevertheless, this method does demand a higher sample input, and low depth continues to be a significant factor limiting the detection of mRNA acetylation in certain circumstances.
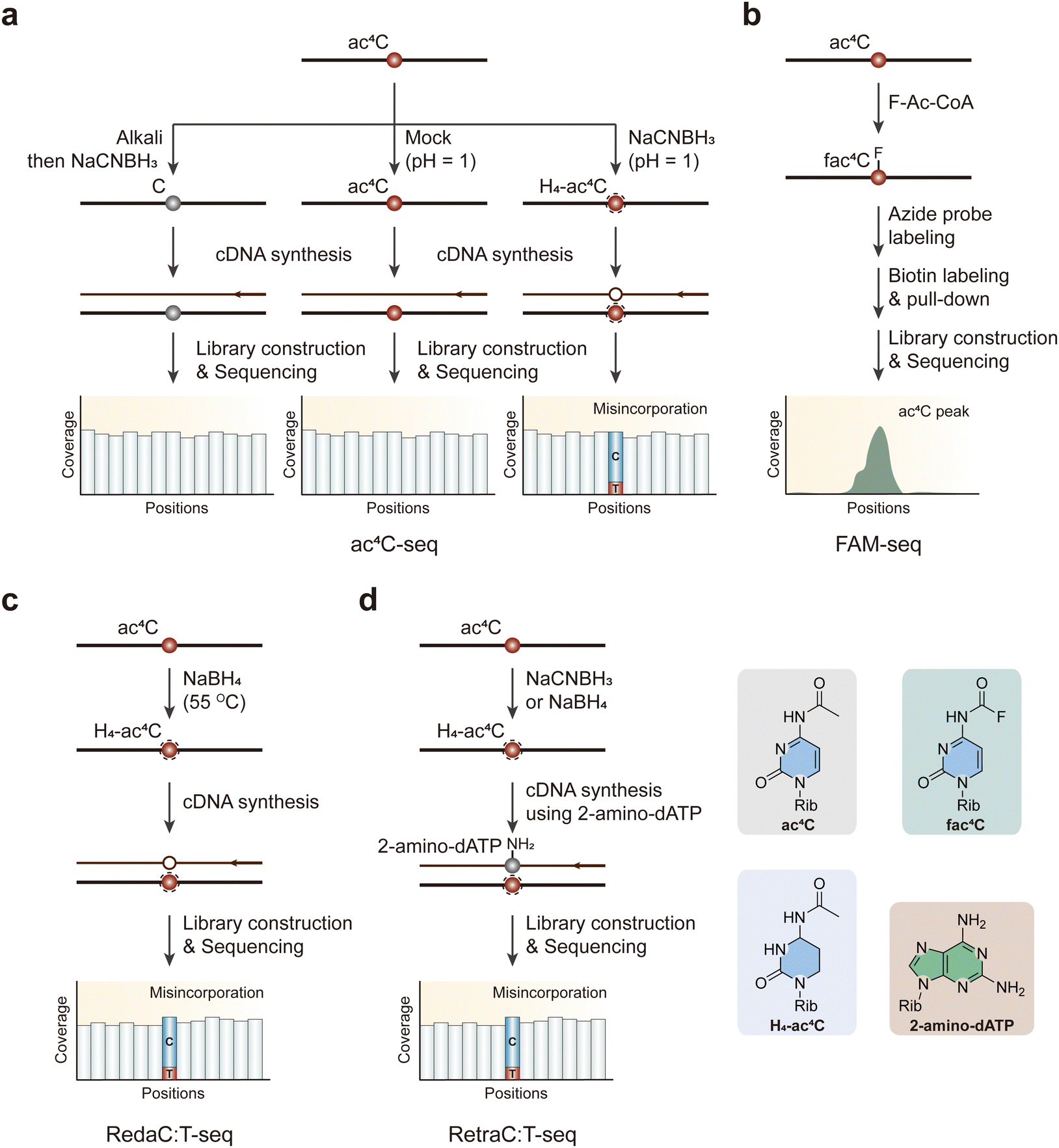 | ||
| Fig. 8 Overview of sequencing methods for the detection of ac4C modifications. (a) ac4C-seq. (b) FAM-seq. (c) RedaC:T-seq. (d) RetraC:T-seq. Rib, ribose. H4-ac4C, tetrahydro-N4-acetylcytidine. | ||
An antibody-free FAM-seq method utilizes antibody-free fluorine-assisted metabolic sequencing to detect ac4C within RNA (Fig. 8b).121 The acetyl group donor, acetyl coenzyme A (Ac-CoA), serves as the substrate for the ac4C methyltransferase NAT10. This enzyme transfers an acetyl group to the N4-position of cytidines, marking the site of acetylation. To pinpoint cytidine acetylation throughout the transcriptome, researchers fed cells with sodium fluoroacetate. This pro-metabolite compound can be ligated to CoA by acetyl-CoA synthetase within the cell to produce F-Ac-CoA. Subsequently, the acetyltransferase NAT10 transfers the fluoroacetate group from F-Ac-CoA to the N4 position of a target cytidine in RNA. Previous research has evidenced that fluoroacetamide can be effectively converted to biotin or fluorophore tags through the fluorine-thiol displacement reaction (FTDR) with high selectivity and yield.122 Capitalizing on this, the researchers employed an azide probe containing a benzenethiol structure to react with N4-fluoroacetylcytidine (fac4C). Following a click reaction with dibenzocyclooctyne-biotin, the biotin-labeled modified RNAs were enriched for library construction. This innovative approach allows for a more precise identification and analysis of ac4C modifications in the RNA, contributing to our understanding of their role in various cellular processes. However, adding sodium fluoroacetate to cell cultures may lead to inevitable false signals in sequencing data.
Avoiding the side effects caused by metabolic labeling, RedaC:T-seq is an advanced sequencing method designed to identify ac4C in RNA with high precision.123 This technique involves chemically reducing ac4C to tetrahydro-ac4C using NaBH4, which induces C-to-T mismatches during reverse transcription (Fig. 8c). These mismatches are detectable by sequencing and serve as markers for ac4C sites. Key features of RedaC:T-seq include its high sensitivity and specificity, achieved by comparing treated samples to untreated and NAT10-knockout controls, ensuring accurate identification of ac4C sites. The comprehensive coverage allows for detailed mapping of ac4C across various RNA regions even in low-abundance transcripts. However, some researchers have wondered whether the ac4C sites provided are not reproducible because of irreproducibility of the mismatch pattern, technical biases and low complexity reads in the sequencing data.124 Thus, avoiding the inefficient reduction reaction and low mismatch rate, which results in the inability of RedaC:T-seq to detect ac4C modification on mRNAs, researchers have developed an improved method called RetraC:T-seq (Fig. 8d).125 This method utilizes NaBH4 or NaCNBH3 to reduce ac4C to tetrahydro-ac4C, which leads to C-to-T mismatches during cDNA synthesis with modified dNTPs, such as 2-NH2-dATP. These mismatches are then detected via sequencing, allowing for precise mapping of ac4C sites. This technique offers improved sensitivity and specificity over previous methods, facilitating better understanding of its role in RNA biology.
3.6 m7G detection methods
m7G, a positively charged, crucial modification mainly found at the cap of eukaryotic mRNA, participates in regulating mRNA export, translation, and splicing.75,126–128 m7G also appears internally within tRNA, rRNA, and eukaryotic mRNA.75,129–136 The primary techniques for detecting internal m7G in mRNA include antibody-based and chemical-assisted sequencing methods. The m7G-MeRIP-seq method, which employs an m7G-specific antibody, has successfully identified over 3000 internal m7G peaks in mammalian cell lines.25 Nevertheless, this approach, dependent entirely on an anti-m7G antibody to enrich RNA fragments containing m7G, is hindered by several drawbacks, such as a lack of single-base resolution, low detection sensitivity, and high background noise. To address these issues, chemical-assisted sequencing methods that leverage the unique chemical reactivity of m7G have been developed, enabling the detection of m7G methylome distribution features in human cells at base resolution.To achieve base resolution mapping of m7G with an orthogonal approach, several chemical-assisted sequencing methods, which are referred to as m7G-seq, borohydride reduction sequencing (BoRed-seq), mutational profiling sequencing (m7G-MaP-seq), tRNA reduction and cleavage sequencing (TRAC-seq) and m7G-seq with stoichiometry information (m7G-quant-seq) were developed by using the unique chemical reactivity of m7G in a reduction-induced depurination reaction (Fig. 9).25,53,137–139 However, m7G-seq and m7G-MaP-seq are two of the methods capable of mapping m7G sites on mRNAs, as demonstrated by their results, particularly after incorporating a decapping step into the protocol to generate the necessary substrate for sequencing.25,137 In m7G-seq, the positive charge on the five-membered ring makes m7G particularly susceptible to NaBH4-mediated reduction, which eliminates the aromaticity of the five-membered ring attached to the ribose without affecting unmodified G. Reduced m7G forms an apurinic/apyrimidinic site (AP site), also known as an abasic site, after heating in an acidic solution, generating an RNA abasic site that can be captured by biotin-ligated hydrazide in a one-pot reaction, resulting in biotinylated RNA. The biotinylated sites are predominantly mutated to T, as well as other bases, during HIV-1 reverse transcriptase (RT)-mediated reverse transcription, enabling the detection of m7G sites at single-base resolution.25 Similarly, BoRed-seq, TRAC-seq and m7G-quant-seq also employ the NaBH4- or KBH4-mediated reduction process, followed by depurination under mild conditions to generate abasic sites.25,53 In TRAC-seq, an additional aniline treatment is employed to facilitate the cleavage of the RNA backbone specifically at m7G-modified sites. In m7G-MaP-seq, NaBH4-meidated RNA abasic sites were misincorporated with moloney murine leukemia virus reverse transcriptase (MMLV) reverse transcriptase to record the positions of m7G modifications during reverse transcription.137 All these chemical-assisted high throughput sequencing methods for m7G could provide precise location of m7G at the nucleotide level in various RNA types, while the treatment of NaBH4 might affect other types of RNA modification and a high sequencing depth is needed because of RNA degradation.
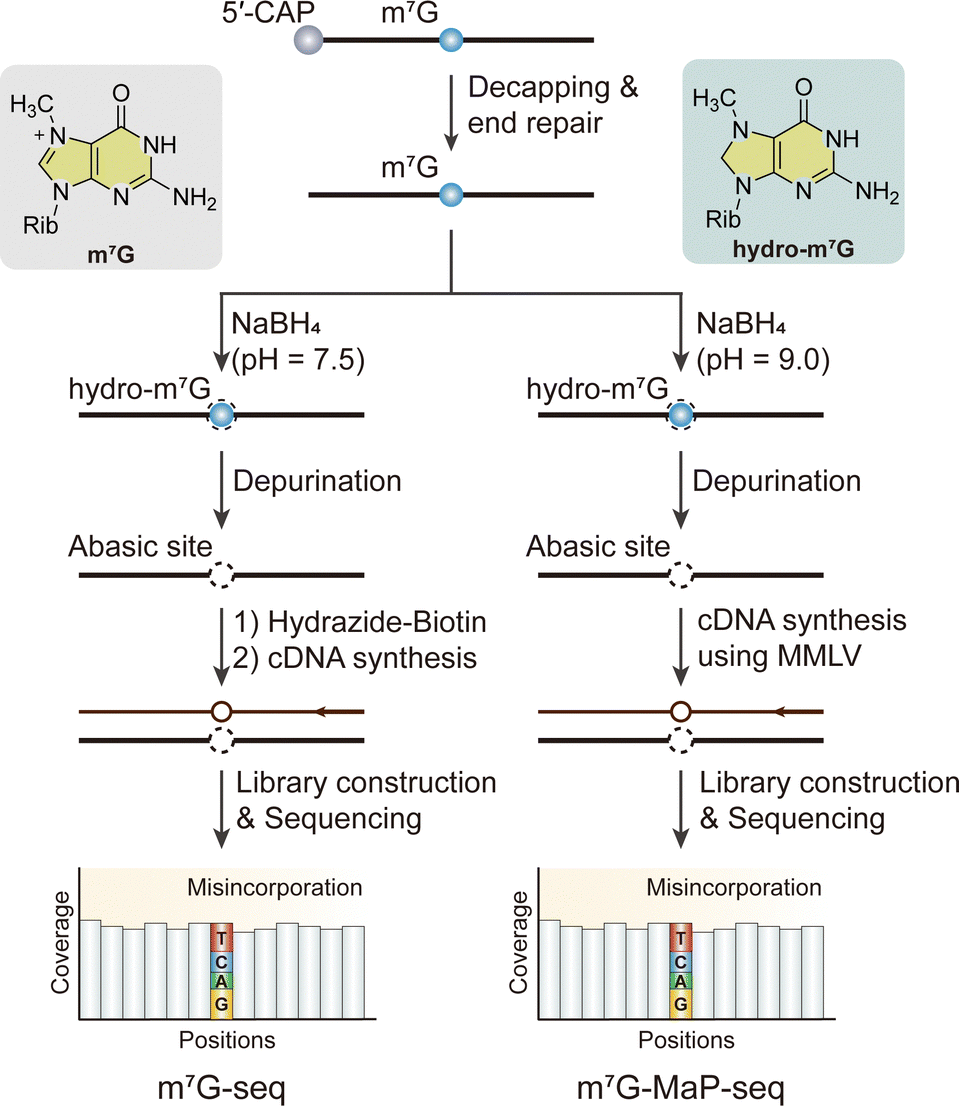 | ||
| Fig. 9 Overview of sequencing methods for the detection of internal m7G modifications. Rib, ribose. MMLV, moloney murine leukemia virus reverse transcriptase. | ||
3.7 Ψ detection methods
Ψ is one of the most abundant RNA modifications in eukaryotes, earning it the title of the “fifth nucleoside”.112 In yeast, Ψ is present at many positions in tRNAs, at 46 positions across the four rRNAs (25S, 18S, 5.8S, and 5S), and at six positions in snRNA U1, U2, and U5.140–146 Although base pairing of Ψ is similar to uridine, isomerization allows the potential formation of an extra hydrogen bond.147 This additional bond could lend greater structural stability to RNA molecules, underlining the functional importance of Ψ beyond mere structural decoration. Moreover, Ψ has also been identified in mRNA, highlighting its broader role in the RNA world beyond its traditional confines within rRNA and tRNA. Mass spectrometry studies have quantified the ratio of Ψ to uridine (Ψ/U) in human cell lines, finding it to be on the order of approximately 0.2–0.6%, which is comparable to the ratio of m6A to adenosine (m6A/A) in these cells.148 This similarity in abundance with m6A suggests that Ψ might also have important functions in RNA biology.The challenge of Ψ detection in RNA sequences stems from the fact that Ψ is mass-silent and indistinguishable from regular uridine bases during reverse transcription. This difficulty has led to the development of chemical-assisted sequencing methods that rely on the specificity of N-cyclohexyl-N′-(4-methylmorpholinium)ethylcarbodiimide (CMC) for labeling and distinguishing Ψ from U.149 The mechanism of CMC is based on its covalent binding to the N3 position of U, G, and Ψ residues, resulting in the formation of CMC-U, CMC-G, and CMC-Ψ adducts, respectively. Upon alkaline treatment, the unique chemical stability of the CMC-Ψ adduct under these conditions means it remains intact while the CMC moieties linked to U and G are removed. This stability is harnessed in sequencing methodologies; the presence of a CMC-Ψ adduct causes reverse transcription to terminate, thus facilitating the detection of Ψ at single-base resolution. Using this strategy, three CMC-based profiling methods, including pseudo-seq, Ψ-seq, and PSI-seq, have been successful in mapping Ψ modifications at single-base resolution, particularly in yeast and human mRNA (Fig. 10a).57,150,151 However, existing profiling methods do not pre-enrich Ψ-containing RNAs, potentially missing low-abundance pseudouridylation events. To address this, CeU-seq employs a CMC derivative, N3-CMC, which forms Ψ-CMC-N3 adducts that can be further labeled with biotin for pull-down assays, allowing pre-enrichment of Ψ-containing RNA fragments (Fig. 10b).148 Remarkably, CeU-seq has successfully identified a significant number of Ψ sites across various samples, including 1889 sites in human mRNA from HEK293T cells, 1543 sites in mouse liver, and 1741 sites in mouse brain. Furthermore, the detection capabilities of Ψ-CMC-induced mutation/deletion patterns can also be combined with highly sensitive qPCR analysis,152 allowing for the detection of locus-specific Ψ modifications across different RNA types, thus broadening the scope and application of pseudouridylation detection in RNA biology.
 | ||
| Fig. 10 Overview of sequencing methods for the detection of Ψ modifications. (a) Ψ-seq, PSI-seq and pseudo-seq. (b) CeU-seq. (c) HydraPsiSeq. (d) RBS-seq, BID-seq and PRAISE. CMC, N-cyclohexyl-N′-(4-methylmorpholinium) ethylcarbodiimide. Rib, ribose. | ||
Since CMC-based profiling methods are prone to variation, making this approach only semi-quantitative, the lack of a quantitative method hinders our ability to comprehensively understand the prevalence of pseudouridylation in the transcriptome and to evaluate its dynamics. To address this issue, a novel quantitative Ψ mapping technique, HydraPsiSeq, was developed. This method relies on specific protection from hydrazine/aniline cleavage of Ψ (Fig. 10c).153 In principle, hydrazine cleaves uridine residues, forming abasic sites that are then treated with aniline to fragment the RNA strands. Since Ψ is unaffected by hydrazine, intact RNA fragments are retained. Ψ sites can be precisely located and quantitatively analyzed by comparing them to the reference genome. Although this strategy does not allow enrichment, HydraPsiSeq provides a systematic approach for mapping and accurately quantifying pseudouridines in RNAs, with potential applications in disease, development, and stress response.
Additionally, Ψ can undergo irreversible labeling through a bisulfite reaction combined with hydroquinone,154 leading to the formation of a ribose ring-opening adduct. This Ψ-bisulfite adduct was later found to induce reverse transcriptase bypass, allowing its detection as 1–2 nucleotide deletion signatures during sequencing (Fig. 10d).110 However, traditional bisulfite conditions, such as RBS-seq, while capable of detecting m1A simultaneously, suffer from limited labeling efficiency, which can result in incomplete conversion and an increased deletion rate. This limitation restricts the accuracy of methylation detection and can obscure subtle epigenetic modifications. To this end, two independent studies introduced approaches for the absolute quantification of transcriptome-wide Ψ, namely BID-seq and PRAISE.155,156 BID-seq employs an adjusted pH with sodium hydroxide (NaOH), while PRAISE enhances effective ion concentrations during the bisulfite reaction without adding hydroquinone. Both methods inhibit C-to-T conversion and significantly improve reaction efficiency towards Ψ. In the context of mRNA modification, BID-seq and PRAISE identified thousands of Ψ sites along with their modification stoichiometry, highlighting the absolute quantitative capability of the optimized bisulfite chemistry.
3.8 I detection methods
Recent studies on inosine and A-to-I (adenosine-to-inosine) RNA editing reveal that it's a common feature across various transcripts, indicating its significant biological function.157 This A-to-I editing is enzymatically facilitated by the family of adenosine deaminases acting on RNA (ADAR) enzymes.157 ADAR-mediated A-to-I base editing, particularly in dsRNA regions, results in the conversion of A to I, which is then interpreted as G during translation. To achieve accurate detection of inosine modifications on a transcriptome-wide level, a variety of high-throughput sequencing methods have been established, each employing unique strategies such as direct sequencing, chemical-assisted sequencing, and enzyme-based enrichment techniques.The most conventional method to identify A-to-I editing sites is direct sequencing by comparing cDNA sequences with their corresponding genomic DNA sequences. This approach relies on the reverse transcription step, where inosines are read as guanosines. Thus, the appearance of A-to-G mismatches between the cDNA and the genomic sequence is indicative of A-to-I editing. However, this method can be limited by the difficulty in distinguishing true editing events from sequencing errors or PCR artifacts, particularly in regions with high noise or pseudogene regions.
To overcome the limitations of direct sequencing, chemical-assisted approaches like inosine chemical erasing (ICE) have been developed (Fig. 11a).158 Combined with high-throughput sequencing, ICE-seq is based on the cyanoethylation of inosines, which blocks reverse transcription at modified sites, thereby allowing direct identification of inosines in sequencing reads. This method is highly specific and may not require genomic DNA as a reference, making it a reliable technique for detecting editing events without the confused effects of SNP (single nucleotide polymorphism) or sequencing errors.
 | ||
| Fig. 11 Overview of sequencing methods for the detection of I modifications. (a) ICE-seq. (b) EndoVIPER-seq. eEndoV, Escherichia coli endonuclease V. Rib, ribose. | ||
Another important method for identifying inosine modifications involves enzyme-based enrichment, called endonuclease V inosine precipitation enrichment sequencing (EndoVIPER-seq) (Fig. 11b).159 It uses specific endonuclease eEndoV (Escherichia coli endonuclease V) to recognize fragmented inosine-containing RNA, enriching for edited regions before sequencing. This approach can improve the sensitivity of inosine detection, particularly in samples with low editing frequencies or in non-repetitive regions where editing is not abundant.
These diverse sequencing methods have collectively advanced our understanding of A-to-I RNA editing, revealing its widespread occurrence across the transcriptome and its involvement in numerous biological processes. As new techniques continue to be developed, the resolution and accuracy of inosine detection are expected to improve, providing deeper insights into the function of inosine across the transcriptome.
3.9 Nm detection methods
The post-transcriptional modification 2′-O-methylation (Nm), found on the ribose of all four ribonucleosides, is a significant RNA modification commonly recognized in noncoding RNAs. This includes the 5′ RNA cap in viruses and higher eukaryotes, as well as within small nucleolar RNA (snoRNA) and rRNA.Recent advancements have led to the development of techniques for transcriptome-wide mapping of Nm using high-throughput sequencing, capable of confirming established Nm sites on abundant rRNA. RiboMeth-Seq employs alkaline RNA cleavage followed by high-throughput sequencing; because 2′-O-methylation protects nucleotides from alkaline fragmentation, modified residues are excluded from the RNA library.160,161 Similarly, RibOxi-seq also utilizes alkaline RNA cleavage but is enriched for methylated fragments by preventing the ligation of unmethylated fragments to adaptors.162 Other methods that exploit the steric properties of Nm include 2OMe-seq and MeTH-seq, which sequence a cDNA library prepared through reverse transcription under restrictive conditions, such as low dNTP or low magnesium concentrations, causing reverse transcription to terminate at Nm sites (Fig. 12a).163,164 However, only MeTH-seq is used to map Nm sites on mRNAs.
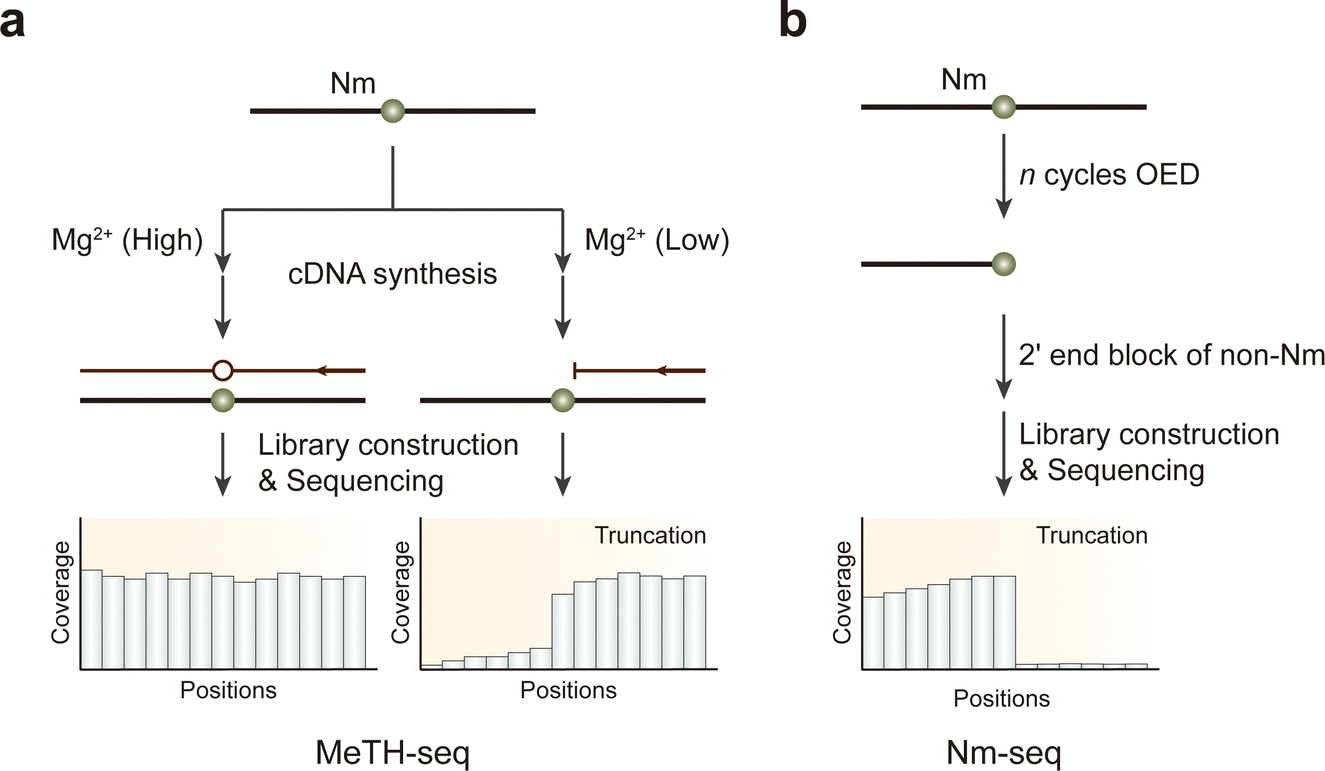 | ||
| Fig. 12 Overview of sequencing methods for the detection of Nm modifications. (a) MeTH-seq. (b) Nm-seq. OED, oxidation-elimination-dephosphorylation. | ||
Although these approaches effectively detect Nm modifications on abundant RNAs, they encounter challenges when applied to less abundant RNAs, such as mRNA. These methods often lack stoichiometric information and can lead to RNA sample degradation due to chemical treatments. To achieve single-nucleotide precision profiling in mRNA species, Nm-seq was developed to map thousands of Nm sites in human mRNA (Fig. 12b).165 Nm-seq employs multiple rounds of oxidation-elimination-dephosphorylation (OED) to iteratively remove 2′-unmodified nucleotides from the 3′ end of fragmented RNA. 2′-O-Methylated nucleotides resist OED, resulting in their enrichment at the 3′ end of the fragments. After several rounds of OED, a final oxidation-elimination reaction is performed without dephosphorylation, creating unligatable 3′ monophosphate ends on fragments ending with unmodified nucleotides. In contrast, 3′ adaptors are ligated onto fragments with 2′-O-methylated ends that retain a ligatable 3′ OH. Consequently, the 3′ end of each RNA fragment corresponds to the 2′-O-methylated nucleotide, which is then mapped using high-throughput sequencing.
4. Conclusions and outlook
In the last decade, the field of RNA epigenetics has advanced remarkably, starting with the discovery of the reversible m6A modification.166 Initially, detection methods required large amounts of material and were costly, but newer techniques now provide single-nucleotide resolution with lower input requirements and costs. This progress has been driven by the development of reactivity-based sequencing methods, which have significantly enhanced our ability to profile RNA modifications with high precision and sensitivity.These methods are notable for their high sensitivity and specificity, allowing for the detection of low-abundance modifications and offering comprehensive coverage of RNA modification landscapes. Their ability to provide single-base resolution is essential for understanding the precise roles of these modifications in RNA biology. Additionally, these techniques are antibody-free, which reduces the risk of cross-reactivity and false positives, enhancing the reliability of the results.
Despite these advancements, challenges remain. The complexity of these procedures and the need for specialized equipment and expertise can limit accessibility. High sequencing depth requirements can be resource-intensive, and potential technical biases necessitate careful experimental design and data analysis. Some modifications, such as hm5C, are still challenging to precisely detect with current methods. Continuous refinement and the development of orthogonal validation methods are essential for improving accuracy and expanding applicability.
Looking ahead, future efforts should focus on developing absolute quantitative methods, single-cell level analyses, and time-resolved studies of RNA modification dynamics. Nanopore sequencing, which has shown potential for simultaneous detection of multiple modifications at single-base resolution, requires improvements in cost and accuracy.167–169 Combining enzyme- and chemical-assisted methods could enhance detection signals.
Moreover, many RNA modifications with significant biological functions remain undetected by current high-throughput methods. Optimizing existing technologies could help identify these modifications. The discovery of new modifications and sequencing techniques will likely accelerate the use of RNA modifications as biomarkers for disease diagnosis and treatment.170
In summary, advancing RNA modification detection technologies will likely focus on increasing sensitivity and specificity, reducing complexity, and minimizing technical biases. These improvements will broaden the application of these methods in diverse biological contexts, including gene regulation, cellular processes, and disease mechanisms. Understanding RNA modifications and their roles will be crucial for developing targeted therapies, underscoring the importance of continued research in this field. Because of the complex roles of RNA modifications, this requires advanced mapping and quantification methods as existing methods can hardly identify many kinds of modifications across all RNA types simultaneously, which might by an important point for medical diagnosis of diseases. This is further complicated by the lack of type-specific modification patterns and varying abundance across RNAs. Understanding RNA modifications and their roles will be crucial for developing targeted therapies, underscoring the importance of continued research in this field, such as pseudouridine and N1-methylpseudouridine (m1Ψ) in mRNA vaccine immunogenicity and effective half-life.171–173 In a larger sense, it is clear that the function of RNA modifications makes them an excellent therapeutic target for further investigation.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Biological Breeding-National Science and Technology Major Project (023ZD04073), the National Natural Science Foundation of China (no. 22225704, 22321005, and 92053109), and the National Basic Research Program of China (2019YFA0802201).References
- X. Song and R. N. Nazar, FEBS Lett., 2002, 523, 182–186 CrossRef PubMed.
- J. Karijolich, C. Yi and Y. T. Yu, Nat. Rev. Mol. Cell Biol., 2015, 16, 581–585 CrossRef.
- Y. Wang and G. Jia, Essays Biochem., 2020, 64, 967–979 CrossRef PubMed.
- Y. Wang, X. Zhang, H. Liu and X. Zhou, Chem. Soc. Rev., 2021, 50, 13481–13497 RSC.
- M. C. Owens, C. Zhang and K. F. Liu, Mol. Cell, 2021, 81, 4116–4136 CrossRef CAS PubMed.
- D. Wiener and S. Schwartz, Nat. Rev. Genet., 2021, 22, 119–131 CrossRef CAS.
- X. Li, X. Xiong and C. Yi, Nat. Methods, 2016, 14, 23–31 CrossRef PubMed.
- J. E. Squires, H. R. Patel, M. Nousch, T. Sibbritt, D. T. Humphreys, B. J. Parker, C. M. Suter and T. Preiss, Nucleic Acids Res., 2012, 40, 5023–5033 CrossRef CAS PubMed.
- D. Dominissini, S. Nachtergaele, S. Moshitch-Moshkovitz, E. Peer, N. Kol, M. S. Ben-Haim, Q. Dai, A. Di Segni, M. Salmon-Divon, W. C. Clark, G. Zheng, T. Pan, O. Solomon, E. Eyal, V. Hershkovitz, D. Han, L. C. Doré, N. Amariglio, G. Rechavi and C. He, Nature, 2016, 530, 441–446 CrossRef CAS.
- X. Li, X. Xiong, K. Wang, L. Wang, X. Shu, S. Ma and C. Yi, Nat. Chem. Biol., 2016, 12, 311–316 CrossRef CAS.
- K. I. Zhou, C. V. Pecot and C. L. Holley, RNA, 2024, 30, 570–582 CrossRef CAS PubMed.
- M. Sakurai, H. Ueda, T. Yano, S. Okada, H. Terajima, T. Mitsuyama, A. Toyoda, A. Fujiyama, H. Kawabata and T. Suzuki, Genome Res., 2014, 24, 522–534 CrossRef CAS PubMed.
- I. Barbieri and T. Kouzarides, Nat. Rev. Cancer, 2020, 20, 303–322 CrossRef CAS PubMed.
- S. Delaunay and M. Frye, Nat. Cell Biol., 2019, 21, 552–559 CrossRef CAS.
- Y. S. Chen, W. L. Yang, Y. L. Zhao and Y. G. Yang, Wiley Interdiscip. Rev.: RNA, 2021, 12, e1639 CrossRef CAS PubMed.
- X. Yang, Y. Yang, B. F. Sun, Y. S. Chen, J. W. Xu, W. Y. Lai, A. Li, X. Wang, D. P. Bhattarai, W. Xiao, H. Y. Sun, Q. Zhu, H. L. Ma, S. Adhikari, M. Sun, Y. J. Hao, B. Zhang, C. M. Huang, N. Huang, G. B. Jiang, Y. L. Zhao, H. L. Wang, Y. P. Sun and Y. G. Yang, Cell Res., 2017, 27, 606–625 CrossRef CAS.
- X. Chen, A. Li, B. F. Sun, Y. Yang, Y. N. Han, X. Yuan, R. X. Chen, W. S. Wei, Y. Liu, C. C. Gao, Y. S. Chen, M. Zhang, X. D. Ma, Z. W. Liu, J. H. Luo, C. Lyu, H. L. Wang, J. Ma, Y. L. Zhao, F. J. Zhou, Y. Huang, D. Xie and Y. G. Yang, Nat. Cell Biol., 2019, 21, 978–990 CrossRef CAS PubMed.
- Y. Yang, L. Wang, X. Han, W. L. Yang, M. Zhang, H. L. Ma, B. F. Sun, A. Li, J. Xia, J. Chen, J. Heng, B. Wu, Y. S. Chen, J. W. Xu, X. Yang, H. Yao, J. Sun, C. Lyu, H. L. Wang, Y. Huang, Y. P. Sun, Y. L. Zhao, A. Meng, J. Ma, F. Liu and Y. G. Yang, Mol. Cell, 2019, 75, 1188–1202.e1111 CrossRef CAS.
- X. Li, S. Ma and C. Yi, Curr. Opin. Chem. Biol., 2016, 33, 108–116 CrossRef CAS PubMed.
- K. Karikó, H. Muramatsu, F. A. Welsh, J. Ludwig, H. Kato, S. Akira and D. Weissman, Mol. Ther., 2008, 16, 1833–1840 CrossRef PubMed.
- D. E. Eyler, M. K. Franco, Z. Batool, M. Z. Wu, M. L. Dubuke, M. Dobosz-Bartoszek, J. D. Jones, Y. S. Polikanov, B. Roy and K. S. Koutmou, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 23068–23074 Search PubMed.
- J. Karijolich and Y. T. Yu, Nature, 2011, 474, 395–398 Search PubMed.
- X. Li, X. Xiong, M. Zhang, K. Wang, Y. Chen, J. Zhou, Y. Mao, J. Lv, D. Yi, X. W. Chen, C. Wang, S. B. Qian and C. Yi, Mol. Cell, 2017, 68, 993–1005.e1009 CrossRef.
- X. Xiong, X. Li and C. Yi, Curr. Opin. Chem. Biol., 2018, 45, 179–186 CrossRef PubMed.
- L. S. Zhang, C. Liu, H. Ma, Q. Dai, H. L. Sun, G. Luo, Z. Zhang, L. Zhang, L. Hu, X. Dong and C. He, Mol. Cell, 2019, 74, 1304–1316.e1308 CrossRef PubMed.
- D. Arango, D. Sturgill, N. Alhusaini, A. A. Dillman, T. J. Sweet, G. Hanson, M. Hosogane, W. R. Sinclair, K. K. Nanan, M. D. Mandler, S. D. Fox, T. T. Zengeya, T. Andresson, J. L. Meier, J. Coller and S. Oberdoerffer, Cell, 2018, 175, 1872–1886.e1824 CrossRef PubMed.
- G. Jin, M. Xu, M. Zou and S. Duan, Mol. Ther.–Nucleic Acids, 2020, 20, 13–24 CrossRef.
- D. Dominissini and G. Rechavi, Cell, 2018, 175, 1725–1727 CrossRef.
- P. Kudrin, A. Singh, D. Meierhofer, A. Kuśnierczyk and U. A. V. Ørom, EMBO Rep., 2024, 25, 1814–1834 CrossRef.
- K. Li, J. Peng and C. Yi, Fundam. Res., 2023, 3, 738–748 CrossRef.
- M. Helm, F. Lyko and Y. Motorin, Nat. Commun., 2019, 10, 5669 CrossRef PubMed.
- Y. Zhang, L. Lu and X. Li, Exp. Mol. Med., 2022, 54, 1601–1616 CrossRef.
- Y. Zhang, L. Lu and X. Li, Exp. Mol. Med., 2022, 54, 1601–1616 CrossRef CAS PubMed.
- Z. Zhang, T. Chen, H. X. Chen, Y. Y. Xie, L. Q. Chen, Y. L. Zhao, B. D. Liu, L. Jin, W. Zhang, C. Liu, D. Z. Ma, G. S. Chai, Y. Zhang, W. S. Zhao, W. H. Ng, J. Chen, G. Jia, J. Yang and G. Z. Luo, Nat. Methods, 2021, 18, 1213–1222 CrossRef CAS PubMed.
- S. Höfler and O. Duss, Life Sci. Alliance, 2024, 7, e202302240 CrossRef.
- J. Zhou, J. Wan, X. Gao, X. Zhang, S. R. Jaffrey and S. B. Qian, Nature, 2015, 526, 591–594 CrossRef CAS PubMed.
- A. Li, Y. S. Chen, X. L. Ping, X. Yang, W. Xiao, Y. Yang, H. Y. Sun, Q. Zhu, P. Baidya, X. Wang, D. P. Bhattarai, Y. L. Zhao, B. F. Sun and Y. G. Yang, Cell Res., 2017, 27, 444–447 CrossRef CAS PubMed.
- H. Du, Y. Zhao, J. He, Y. Zhang, H. Xi, M. Liu, J. Ma and L. Wu, Nat. Commun., 2016, 7, 12626 CrossRef.
- X. Wang, B. S. Zhao, I. A. Roundtree, Z. Lu, D. Han, H. Ma, X. Weng, K. Chen, H. Shi and C. He, Cell, 2015, 161, 1388–1399 CrossRef.
- Y. Fu, D. Dominissini, G. Rechavi and C. He, Nat. Rev. Genet., 2014, 15, 293–306 CrossRef PubMed.
- I. A. Roundtree, G. Z. Luo, Z. Zhang, X. Wang, T. Zhou, Y. Cui, J. Sha, X. Huang, L. Guerrero, P. Xie, E. He, B. Shen and C. He, eLife, 2017, 6, e31311 CrossRef PubMed.
- C. R. Alarcón, H. Goodarzi, H. Lee, X. Liu, S. Tavazoie and S. F. Tavazoie, Cell, 2015, 162, 1299–1308 CrossRef PubMed.
- L. J. Deng, W. Q. Deng, S. R. Fan, M. F. Chen, M. Qi, W. Y. Lyu, Q. Qi, A. K. Tiwari, J. X. Chen, D. M. Zhang and Z. S. Chen, Mol. Cancer, 2022, 21, 52 CrossRef PubMed.
- W. Huang, T. Q. Chen, K. Fang, Z. C. Zeng, H. Ye and Y. Q. Chen, J. Hematol. Oncol., 2021, 14, 117 CrossRef PubMed.
- X. Wang, R. Ma, X. Zhang, L. Cui, Y. Ding, W. Shi, C. Guo and Y. Shi, Mol. Cancer, 2021, 20, 121 CrossRef PubMed.
- P. C. He and C. He, EMBO J., 2021, 40, e105977 CrossRef.
- S. Zaccara, R. J. Ries and S. R. Jaffrey, Nat. Rev. Mol. Cell Biol., 2019, 20, 608–624 CrossRef PubMed.
- F. Xiong, R. Wang, J. H. Lee, S. Li, S. F. Chen, Z. Liao, L. A. Hasani, P. T. Nguyen, X. Zhu, J. Krakowiak, D. F. Lee, L. Han, K. L. Tsai, Y. Liu and W. Li, Cell Res., 2021, 31, 861–885 CrossRef.
- J. Wei, X. Yu, L. Yang, X. Liu, B. Gao, B. Huang, X. Dou, J. Liu, Z. Zou, X. L. Cui, L. S. Zhang, X. Zhao, Q. Liu, P. C. He, C. Sepich-Poore, N. Zhong, W. Liu, Y. Li, X. Kou, Y. Zhao, Y. Wu, X. Cheng, C. Chen, Y. An, X. Dong, H. Wang, Q. Shu, Z. Hao, T. Duan, Y. Y. He, X. Li, S. Gao, Y. Gao and C. He, Science, 2022, 376, 968–973 CrossRef.
- A. Ramanathan, G. B. Robb and S. H. Chan, Nucleic Acids Res., 2016, 44, 7511–7526 CrossRef.
- Y. Luo, Y. Yao, P. Wu, X. Zi, N. Sun and J. He, J. Hematol. Oncol., 2022, 15, 63 CrossRef PubMed.
- I. N. Shatsky, I. M. Terenin, V. V. Smirnova and D. E. Andreev, Trends Biochem. Sci., 2018, 43, 882–895 CrossRef PubMed.
- L. Pandolfini, I. Barbieri, A. J. Bannister, A. Hendrick, B. Andrews, N. Webster, P. Murat, P. Mach, R. Brandi, S. C. Robson, V. Migliori, A. Alendar, M. d'Onofrio, S. Balasubramanian and T. Kouzarides, Mol. Cell, 2019, 74, 1278–1290.e1279 CrossRef.
- K. E. Sloan, A. S. Warda, S. Sharma, K. D. Entian, D. L. J. Lafontaine and M. T. Bohnsack, RNA Biol., 2017, 14, 1138–1152 CrossRef.
- G. Guo, K. Pan, S. Fang, L. Ye, X. Tong, Z. Wang, X. Xue and H. Zhang, Mol. Ther.–Nucleic Acids, 2021, 26, 575–593 CrossRef PubMed.
- J. Cerneckis, Q. Cui, C. He, C. Yi and Y. Shi, Trends Pharmacol. Sci., 2022, 43, 522–535 CrossRef.
- T. M. Carlile, M. F. Rojas-Duran, B. Zinshteyn, H. Shin, K. M. Bartoli and W. V. Gilbert, Nature, 2014, 515, 143–146 CrossRef PubMed.
- N. M. Martinez, A. Su, M. C. Burns, J. K. Nussbacher, C. Schaening, S. Sathe, G. W. Yeo and W. V. Gilbert, Mol. Cell, 2022, 82, 645–659.e649 CrossRef PubMed.
- E. Y. Levanon, M. Hallegger, Y. Kinar, R. Shemesh, K. Djinovic-Carugo, G. Rechavi, M. F. Jantsch and E. Eisenberg, Nucleic Acids Res., 2005, 33, 1162–1168 CrossRef PubMed.
- S. M. Rueter, T. R. Dawson and R. B. Emeson, Nature, 1999, 399, 75–80 CrossRef CAS.
- L. L. Chen, J. N. DeCerbo and G. G. Carmichael, EMBO J., 2008, 27, 1694–1705 CrossRef.
- H. A. Hundley, A. A. Krauchuk and B. L. Bass, RNA, 2008, 14, 2050–2060 CrossRef PubMed.
- L. Agranat, O. Raitskin, J. Sperling and R. Sperling, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 5028–5033 CrossRef.
- M. T. Bohnsack and K. E. Sloan, Biol. Chem., 2018, 399, 1265–1276 CrossRef.
- J. L. Hyde and M. S. Diamond, Virology, 2015, 479–480, 66–74 CrossRef PubMed.
- P. L. Monaco, V. Marcel, J. J. Diaz and F. Catez, Biomolecules, 2018, 8, 106 CrossRef PubMed.
- D. Dominissini, S. Moshitch-Moshkovitz, S. Schwartz, M. Salmon-Divon, L. Ungar, S. Osenberg, K. Cesarkas, J. Jacob-Hirsch, N. Amariglio, M. Kupiec, R. Sorek and G. Rechavi, Nature, 2012, 485, 201–206 CrossRef PubMed.
- K. D. Meyer, Y. Saletore, P. Zumbo, O. Elemento, C. E. Mason and S. R. Jaffrey, Cell, 2012, 149, 1635–1646 CrossRef PubMed.
- H. Sun, K. Li, X. Zhang, J. Liu, M. Zhang, H. Meng and C. Yi, Nat. Commun., 2021, 12, 4778 CrossRef PubMed.
- H. Zhou, S. Rauch, Q. Dai, X. Cui, Z. Zhang, S. Nachtergaele, C. Sepich, C. He and B. C. Dickinson, Nat. Methods, 2019, 16, 1281–1288 CrossRef PubMed.
- M. Safra, A. Sas-Chen, R. Nir, R. Winkler, A. Nachshon, D. Bar-Yaacov, M. Erlacher, W. Rossmanith, N. Stern-Ginossar and S. Schwartz, Nature, 2017, 551, 251–255 CrossRef PubMed.
- E. Saplaoura, V. Perrera, V. Colot and F. Kragler, J. Visualized Exp., 2020, 14, 159 Search PubMed.
- S. Hussain, A. A. Sajini, S. Blanco, S. Dietmann, P. Lombard, Y. Sugimoto, M. Paramor, J. G. Gleeson, D. T. Odom, J. Ule and M. Frye, Cell Rep., 2013, 4, 255–261 CrossRef CAS.
- S. Lin, Q. Liu, V. S. Lelyveld, J. Choe, J. W. Szostak and R. I. Gregory, Mol. Cell, 2018, 71, 244–255.e245 CrossRef CAS PubMed.
- L. Malbec, T. Zhang, Y. S. Chen, Y. Zhang, B. F. Sun, B. Y. Shi, Y. L. Zhao, Y. Yang and Y. G. Yang, Cell Res., 2019, 29, 927–941 CrossRef CAS.
- K. Chen, G. Z. Luo and C. He, Methods Enzymol., 2015, 560, 161–185 Search PubMed.
- B. Linder, A. V. Grozhik, A. O. Olarerin-George, C. Meydan, C. E. Mason and S. R. Jaffrey, Nat. Methods, 2015, 12, 767–772 CrossRef PubMed.
- M. Ren, H. Pan, X. Zhou, M. Yu and F. Ji, J. Transl. Med., 2024, 22, 584 CrossRef PubMed.
- X. Wang, Z. Lu, A. Gomez, G. C. Hon, Y. Yue, D. Han, Y. Fu, M. Parisien, Q. Dai, G. Jia, B. Ren, T. Pan and C. He, Nature, 2014, 505, 117–120 CrossRef.
- H. Shi, X. Wang, Z. Lu, B. S. Zhao, H. Ma, P. J. Hsu, C. Liu and C. He, Cell Res., 2017, 27, 315–328 CrossRef.
- M. N. Wojtas, R. R. Pandey, M. Mendel, D. Homolka, R. Sachidanandam and R. S. Pillai, Mol. Cell, 2017, 68, 374–387.e312 CrossRef.
- C. Xu, X. Wang, K. Liu, I. A. Roundtree, W. Tempel, Y. Li, Z. Lu, C. He and J. Min, Nat. Chem. Biol., 2014, 10, 927–929 CrossRef PubMed.
- K. D. Meyer, Nat. Methods, 2019, 16, 1275–1280 CrossRef PubMed.
- M. Tegowski, M. N. Flamand and K. D. Meyer, Mol. Cell, 2022, 82, 868–878.e810 CrossRef PubMed.
- M. Tegowski and K. D. Meyer, STAR Protoc., 2022, 3, 101646 CrossRef.
- M. A. Garcia-Campos, S. Edelheit, U. Toth, M. Safra, R. Shachar, S. Viukov, R. Winkler, R. Nir, L. Lasman, A. Brandis, J. H. Hanna, W. Rossmanith and S. Schwartz, Cell, 2019, 178, 731–747.e716 CrossRef.
- Z. Zhang, L. Q. Chen, Y. L. Zhao, C. G. Yang, I. A. Roundtree, Z. Zhang, J. Ren, W. Xie, C. He and G. Z. Luo, Sci. Adv., 2019, 5, eaax0250 CrossRef.
- Y. Wang, Y. Xiao, S. Dong, Q. Yu and G. Jia, Nat. Chem. Biol., 2020, 16, 896–903 CrossRef PubMed.
- Y. Fu, G. Jia, X. Pang, R. N. Wang, X. Wang, C. J. Li, S. Smemo, Q. Dai, K. A. Bailey, M. A. Nobrega, K. L. Han, Q. Cui and C. He, Nat. Commun., 2013, 4, 1798 CrossRef.
- J. Liu, Y. Yue, D. Han, X. Wang, Y. Fu, L. Zhang, G. Jia, M. Yu, Z. Lu, X. Deng, Q. Dai, W. Chen and C. He, Nat. Chem. Biol., 2014, 10, 93–95 CrossRef.
- X. Shu, J. Cao, M. Cheng, S. Xiang, M. Gao, T. Li, X. Ying, F. Wang, Y. Yue, Z. Lu, Q. Dai, X. Cui, L. Ma, Y. Wang, C. He, X. Feng and J. Liu, Nat. Chem. Biol., 2020, 16, 887–895 CrossRef.
- X. Shu, Q. Dai, T. Wu, I. R. Bothwell, Y. Yue, Z. Zhang, J. Cao, Q. Fei, M. Luo, C. He and J. Liu, J. Am. Chem. Soc., 2017, 139, 17213–17216 CrossRef PubMed.
- M. Tomkuvienė, M. Mickutė, G. Vilkaitis and S. Klimašauskas, Curr. Opin. Biotechnol, 2019, 55, 114–123 CrossRef PubMed.
- R. Wang, K. Islam, Y. Liu, W. Zheng, H. Tang, N. Lailler, G. Blum, H. Deng and M. Luo, J. Am. Chem. Soc., 2013, 135, 1048–1056 CrossRef.
- L. Hu, S. Liu, Y. Peng, R. Ge, R. Su, C. Senevirathne, B. T. Harada, Q. Dai, J. Wei, L. Zhang, Z. Hao, L. Luo, H. Wang, Y. Wang, M. Luo, M. Chen, J. Chen and C. He, Nat. Biotechnol., 2022, 40, 1210–1219 CrossRef PubMed.
- Y. Xie, S. Han, Q. Li, Z. Fang, W. Yang, Q. Wei, Y. Wang, Y. Zhou, X. Weng and X. Zhou, Chem. Sci., 2022, 13, 12149–12157 RSC.
- S. Werner, A. Galliot, F. Pichot, T. Kemmer, V. Marchand, M. V. Sednev, T. Lence, J. Y. Roignant, J. König, C. Höbartner, Y. Motorin, A. Hildebrandt and M. Helm, Nucleic Acids Res., 2021, 49, e23 CrossRef PubMed.
- Y. L. Xiao, S. Liu, R. Ge, Y. Wu, C. He, M. Chen and W. Tang, Nat. Biotechnol., 2023, 41, 993–1003 CrossRef PubMed.
- C. Liu, H. Sun, Y. Yi, W. Shen, K. Li, Y. Xiao, F. Li, Y. Li, Y. Hou, B. Lu, W. Liu, H. Meng, J. Peng, C. Yi and J. Wang, Nat. Biotechnol., 2023, 41, 355–366 CrossRef.
- K. Kramer, T. Sachsenberg, B. M. Beckmann, S. Qamar, K. L. Boon, M. W. Hentze, O. Kohlbacher and H. Urlaub, Nat. Methods, 2014, 11, 1064–1070 CrossRef PubMed.
- S. Schwartz, M. R. Mumbach, M. Jovanovic, T. Wang, K. Maciag, G. G. Bushkin, P. Mertins, D. Ter-Ovanesyan, N. Habib, D. Cacchiarelli, N. E. Sanjana, E. Freinkman, M. E. Pacold, R. Satija, T. S. Mikkelsen, N. Hacohen, F. Zhang, S. A. Carr, E. S. Lander and A. Regev, Cell Rep., 2014, 8, 284–296 CrossRef.
- Y. T. Goh, C. W. Q. Koh, D. Y. Sim, X. Roca and W. S. S. Goh, Nucleic Acids Res., 2020, 48, 9250–9261 CrossRef PubMed.
- H. Zhou, I. J. Kimsey, E. N. Nikolova, B. Sathyamoorthy, G. Grazioli, J. McSally, T. Bai, C. H. Wunderlich, C. Kreutz, I. Andricioaei and H. M. Al-Hashimi, Nat. Struct. Mol. Biol., 2016, 23, 803–810 CrossRef.
- X. Li, J. Peng and C. Yi, Methods Mol. Biol., 2017, 1562, 245–255 CrossRef.
- H. Liu, T. Zeng, C. He, V. H. Rawal, H. Zhou and B. C. Dickinson, ACS Chem. Biol., 2022, 17, 1334–1342 CrossRef PubMed.
- B. E. Maden, J. Mol. Biol., 1988, 201, 289–314 CrossRef PubMed.
- G. M. Veldman, J. Klootwijk, V. C. de Regt, R. J. Planta, C. Branlant, A. Krol and J. P. Ebel, Nucleic Acids Res., 1981, 9, 6935–6952 CrossRef CAS PubMed.
- K. E. Bohnsack, C. Höbartner and M. T. Bohnsack, Genes, 2019, 10, 102 CrossRef.
- Y. Li and T. O. Tollefsbol, Methods Mol. Biol., 2011, 791, 11–21 CrossRef PubMed.
- V. Khoddami, A. Yerra, T. L. Mosbruger, A. M. Fleming, C. J. Burrows and B. R. Cairns, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 6784–6789 CrossRef PubMed.
- M. Schaefer, T. Pollex, K. Hanna and F. Lyko, Nucleic Acids Res., 2009, 37, e12 CrossRef PubMed.
- C. Legrand, F. Tuorto, M. Hartmann, R. Liebers, D. Jacob, M. Helm and F. Lyko, Genome Res., 2017, 27, 1589–1596 CrossRef PubMed.
- Q. Dai, C. Ye, I. Irkliyenko, Y. Wang, H. L. Sun, Y. Gao, Y. Liu, A. Beadell, J. Perea, A. Goel and C. He, Nat. Biotechnol., 2024, 42, 1559–1570 CrossRef PubMed.
- V. Khoddami and B. R. Cairns, Nat. Biotechnol., 2013, 31, 458–464 CrossRef PubMed.
- L. Lu, X. Zhang, Y. Zhou, Z. Shi, X. Xie, X. Zhang, L. Gao, A. Fu, C. Liu, B. He, X. Xiong, Y. Yin, Q. Wang, C. Yi and X. Li, Mol. Cell, 2024, 84, 2984–3000.e2988 CrossRef PubMed.
- S. Ito, Y. Akamatsu, A. Noma, S. Kimura, K. Miyauchi, Y. Ikeuchi, T. Suzuki and T. Suzuki, J. Biol. Chem., 2014, 289, 26201–26212 CrossRef PubMed.
- S. Sharma, J. L. Langhendries, P. Watzinger, P. Kötter, K. D. Entian and D. L. Lafontaine, Nucleic Acids Res., 2015, 43, 2242–2258 CrossRef.
- J. M. Thomas, C. A. Briney, K. D. Nance, J. E. Lopez, A. L. Thorpe, S. D. Fox, M. L. Bortolin-Cavaille, A. Sas-Chen, D. Arango, S. Oberdoerffer, J. Cavaille, T. Andresson and J. L. Meier, J. Am. Chem. Soc., 2018, 140, 12667–12670 CrossRef.
- S. Thalalla Gamage, A. Sas-Chen, S. Schwartz and J. L. Meier, Nat. Protoc., 2021, 16, 2286–2307 CrossRef CAS PubMed.
- W. R. Sinclair, D. Arango, J. H. Shrimp, T. T. Zengeya, J. M. Thomas, D. C. Montgomery, S. D. Fox, T. Andresson, S. Oberdoerffer and J. L. Meier, ACS Chem. Biol., 2017, 12, 2922–2926 CrossRef CAS.
- S. Yan, Z. Lu, W. Yang, J. Xu, Y. Wang, W. Xiong, R. Zhu, L. Ren, Z. Chen, Q. Wei, S. M. Liu, T. Feng, B. Yuan, X. Weng, Y. Du and X. Zhou, J. Am. Chem. Soc., 2023, 145, 22232–22242 CrossRef CAS.
- Z. Lyu, Y. Zhao, Z. Y. Buuh, N. Gorman, A. R. Goldman, M. S. Islam, H. Y. Tang and R. E. Wang, J. Am. Chem. Soc., 2021, 143, 1341–1347 CrossRef CAS PubMed.
- D. Arango, D. Sturgill, R. Yang, T. Kanai, P. Bauer, J. Roy, Z. Wang, M. Hosogane, S. Schiffers and S. Oberdoerffer, Mol. Cell, 2022, 82, 2797–2814.e2711 CrossRef CAS PubMed.
- J. Georgeson and S. Schwartz, Mol. Cell, 2024, 84, 1601–1610.e1602 CrossRef CAS.
- S. Relier, S. Schiffers, H. Beiki and S. Oberdoerffer, RNA, 2024, 30, 938–953 CAS.
- J. D. Lewis and E. Izaurralde, Eur. J. Biochem., 1997, 247, 461–469 CrossRef CAS.
- D. L. Lindstrom, S. L. Squazzo, N. Muster, T. A. Burckin, K. C. Wachter, C. A. Emigh, J. A. McCleery, J. R. Yates, 3rd and G. A. Hartzog, Mol. Cell. Biol., 2003, 23, 1368–1378 CrossRef CAS PubMed.
- S. Muthukrishnan, G. W. Both, Y. Furuichi and A. J. Shatkin, Nature, 1975, 255, 33–37 CrossRef CAS.
- Z. Chen, W. Zhu, S. Zhu, K. Sun, J. Liao, H. Liu, Z. Dai, H. Han, X. Ren, Q. Yang, S. Zheng, B. Peng, S. Peng, M. Kuang and S. Lin, Clin. Transl. Med., 2021, 11, e661 CrossRef CAS PubMed.
- R. Shaheen, G. M. Abdel-Salam, M. P. Guy, R. Alomar, M. S. Abdel-Hamid, H. H. Afifi, S. I. Ismail, B. A. Emam, E. M. Phizicky and F. S. Alkuraya, Genome Biol., 2015, 16, 210 CrossRef PubMed.
- E. A. Orellana, Q. Liu, E. Yankova, M. Pirouz, E. De Braekeleer, W. Zhang, J. Lim, D. Aspris, E. Sendinc, D. A. Garyfallos, M. Gu, R. Ali, A. Gutierrez, S. Mikutis, G. J. L. Bernardes, E. S. Fischer, A. Bradley, G. S. Vassiliou, F. J. Slack, K. Tzelepis and R. I. Gregory, Mol. Cell, 2021, 81, 3323–3338.e3314 CrossRef CAS PubMed.
- J. White, Z. Li, R. Sardana, J. M. Bujnicki, E. M. Marcotte and A. W. Johnson, Mol. Cell. Biol., 2008, 28, 3151–3161 CrossRef CAS PubMed.
- J. Létoquart, E. Huvelle, L. Wacheul, G. Bourgeois, C. Zorbas, M. Graille, V. Heurgué-Hamard and D. L. Lafontaine, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, E5518–E5526 CrossRef.
- N. Husain, K. L. Tkaczuk, S. R. Tulsidas, K. H. Kaminska, S. Cubrilo, G. Maravić-Vlahovicek, J. M. Bujnicki and J. Sivaraman, Nucleic Acids Res., 2010, 38, 4120–4132 CrossRef CAS PubMed.
- S. Haag, J. Kretschmer and M. T. Bohnsack, RNA, 2015, 21, 180–187 CrossRef PubMed.
- Z. Dai, H. Liu, J. Liao, C. Huang, X. Ren, W. Zhu, S. Zhu, B. Peng, S. Li, J. Lai, L. Liang, L. Xu, S. Peng, S. Lin and M. Kuang, Mol. Cell, 2021, 81, 3339–3355.e3338 CrossRef CAS PubMed.
- C. Enroth, L. D. Poulsen, S. Iversen, F. Kirpekar, A. Albrechtsen and J. Vinther, Nucleic Acids Res., 2019, 47, e126 CrossRef CAS.
- S. Lin, Q. Liu, Y. Z. Jiang and R. I. Gregory, Nat. Protoc., 2019, 14, 3220–3242 CrossRef PubMed.
- L.-S. Zhang, C.-W. Ju, C. Liu, J. Wei, Q. Dai, L. Chen, C. Ye and C. He, ACS Chem. Biol., 2022, 17, 3306–3312 CrossRef PubMed.
- M. Charette and M. W. Gray, IUBMB Life, 2000, 49, 341–351 CrossRef PubMed.
- J. Ofengand, FEBS Lett., 2002, 514, 17–25 CrossRef PubMed.
- P. Morais, H. Adachi and Y. T. Yu, Front. Genet., 2021, 12, 652129 CrossRef PubMed.
- M. Penzo and L. Montanaro, Biomolecules, 2018, 8, 38 CrossRef PubMed.
- J. Jiang, D. N. Kharel and C. S. Chow, Biophys. Chem., 2015, 200–201, 48–55 CrossRef PubMed.
- J. Ge and Y. T. Yu, Trends Biochem. Sci., 2013, 38, 210–218 CrossRef PubMed.
- K. Jack, C. Bellodi, D. M. Landry, R. O. Niederer, A. Meskauskas, S. Musalgaonkar, N. Kopmar, O. Krasnykh, A. M. Dean, S. R. Thompson, D. Ruggero and J. D. Dinman, Mol. Cell, 2011, 44, 660–666 CrossRef.
- E. Kierzek, M. Malgowska, J. Lisowiec, D. H. Turner, Z. Gdaniec and R. Kierzek, Nucleic Acids Res., 2014, 42, 3492–3501 CrossRef PubMed.
- X. Li, P. Zhu, S. Ma, J. Song, J. Bai, F. Sun and C. Yi, Nat. Chem. Biol., 2015, 11, 592–597 CrossRef.
- A. Bakin and J. Ofengand, Biochemistry, 1993, 32, 9754–9762 CrossRef CAS.
- S. Schwartz, D. A. Bernstein, M. R. Mumbach, M. Jovanovic, R. H. Herbst, B. X. León-Ricardo, J. M. Engreitz, M. Guttman, R. Satija, E. S. Lander, G. Fink and A. Regev, Cell, 2014, 159, 148–162 CrossRef CAS.
- A. F. Lovejoy, D. P. Riordan and P. O. Brown, PLoS One, 2014, 9, e110799 CrossRef PubMed.
- Z. Lei and C. Yi, Angew. Chem., Int. Ed., 2017, 56, 14878–14882 CrossRef CAS.
- V. Marchand, F. Pichot, P. Neybecker, L. Ayadi, V. Bourguignon-Igel, L. Wacheul, D. L. J. Lafontaine, A. Pinzano, M. Helm and Y. Motorin, Nucleic Acids Res., 2020, 48, e110 CrossRef CAS PubMed.
- R. P. Singhal, Biochemistry, 1974, 13, 2924–2932 CrossRef CAS.
- M. Zhang, Z. Jiang, Y. Ma, W. Liu, Y. Zhuang, B. Lu, K. Li, J. Peng and C. Yi, Nat. Chem. Biol., 2023, 19, 1185–1195 CrossRef CAS.
- Q. Dai, L. S. Zhang, H. L. Sun, K. Pajdzik, L. Yang, C. Ye, C. W. Ju, S. Liu, Y. Wang, Z. Zheng, L. Zhang, B. T. Harada, X. Dou, I. Irkliyenko, X. Feng, W. Zhang, T. Pan and C. He, Nat. Biotechnol., 2023, 41, 344–354 CrossRef CAS.
- Y. Wang, Y. Zheng and P. A. Beal, Enzymes, 2017, 41, 215–268 CAS.
- M. Sakurai, T. Yano, H. Kawabata, H. Ueda and T. Suzuki, Nat. Chem. Biol., 2010, 6, 733–740 CrossRef CAS.
- S. D. Knutson, R. A. Arthur, H. R. Johnston and J. M. Heemstra, J. Am. Chem. Soc., 2020, 142, 5241–5251 CrossRef CAS PubMed.
- U. Birkedal, M. Christensen-Dalsgaard, N. Krogh, R. Sabarinathan, J. Gorodkin and H. Nielsen, Angew. Chem., Int. Ed., 2015, 54, 451–455 CrossRef CAS PubMed.
- V. Marchand, F. Blanloeil-Oillo, M. Helm and Y. Motorin, Nucleic Acids Res., 2016, 44, e135 CrossRef.
- Y. Zhu, S. P. Pirnie and G. G. Carmichael, RNA, 2017, 23, 1303–1314 CrossRef CAS PubMed.
- D. Incarnato, F. Anselmi, E. Morandi, F. Neri, M. Maldotti, S. Rapelli, C. Parlato, G. Basile and S. Oliviero, Nucleic Acids Res., 2017, 45, 1433–1441 CrossRef CAS.
- K. M. Bartoli, C. Schaening, T. M. Carlile and W. V. Gilbert, bioRxiv, 2018, preprint, arXiv:271916 DOI:10.1101/271916.
- Q. Dai, S. Moshitch-Moshkovitz, D. Han, N. Kol, N. Amariglio, G. Rechavi, D. Dominissini and C. He, Nat. Methods, 2017, 14, 695–698 CrossRef CAS PubMed.
- G. Jia, Y. Fu, X. Zhao, Q. Dai, G. Zheng, Y. Yang, C. Yi, T. Lindahl, T. Pan, Y. G. Yang and C. He, Nat. Chem. Biol., 2011, 7, 885–887 CrossRef CAS PubMed.
- S. Ramasamy, V. J. Sahayasheela, S. Sharma, Z. Yu, T. Hidaka, L. Cai, V. Thangavel, H. Sugiyama and G. N. Pandian, ACS Chem. Biol., 2022, 17, 2704–2709 CrossRef.
- A. M. Fleming, J. Zhu, V. K. Done and C. J. Burrows, RSC Chem. Biol., 2023, 4, 952–964 RSC.
- O. Begik, M. C. Lucas, L. P. Pryszcz, J. M. Ramirez, R. Medina, I. Milenkovic, S. Cruciani, H. Liu, H. G. S. Vieira, A. Sas-Chen, J. S. Mattick, S. Schwartz and E. M. Novoa, Nat. Biotechnol., 2021, 39, 1278–1291 CrossRef PubMed.
- L. Qiu, Q. Jing, Y. Li and J. Han, Mol. Biomed., 2023, 4, 25 CrossRef PubMed.
- M. Vadovics, H. Muramatsu, A. Sárközy and N. Pardi, Methods Mol. Biol., 2024, 2786, 167–181 CrossRef PubMed.
- K. D. Nance and J. L. Meier, ACS Cent. Sci., 2021, 7, 748–756 CrossRef PubMed.
- Y. Zhang, H. Yan, Z. Wei, H. Hong, D. Huang, G. Liu, Q. Qin, R. Rong, P. Gao, J. Meng and B. Ying, Int. J. Biol. Macromol., 2024, 270, 132433 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cb00215f |
| ‡ These authors contributed equally: Zhihe Cai and Peizhe Song. |
| This journal is © The Royal Society of Chemistry 2025 |