
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiochemical characterization and discovery of inhibitors for PfSir2A: new tricks for an old enzyme†
Dickson
Donu
a,
Emily
Boyle
a,
Alyson
Curry
a and
Yana
Cen
 *ab
*ab
aDepartment of Medicinal Chemistry, Virginia Commonwealth University, Richmond, VA 23219, USA. E-mail: ceny2@vcu.edu; Tel: +1 804-828-7405
bCenter for Drug Discovery, Virginia Commonwealth University, Richmond, VA 23219, USA
First published on 23rd January 2025
Abstract
The Sir2 enzyme from Plasmodium falciparum (PfSir2A) is essential for the antigenic variation of this parasite, and its inhibition is expected to have therapeutic effects for malaria. Selective PfSir2A inhibitors are not available yet, partially due to the fact that this enzyme demonstrates extremely weak in vitro deacetylase activity, making the characterization of its inhibitors rather challenging. In the current study, we report the biochemical characterization and inhibitor discovery for this enzyme. PfSir2A exhibits greater enzymatic activity in the presence of DNA for both the peptide and histone protein substrates, suggesting that nucleosomes may be the real substrates of this enzyme. Indeed, it demonstrates robust deacetylase activity against nucleosome substrates, stemming primarily from the tight binding interactions with the nucleosome. In addition to DNA/nucleosome, free fatty acids (FFAs) are also identified as endogenous PfSir2A regulators. Myristic acid, a biologically relevant FFA, shows differential regulation of the two distinct activities of PfSir2A: activates deacetylation, but inhibits defatty-acylation. The structural basis of this differential regulation was further explored. Moreover, synthetic small molecule inhibitors of PfSir2A were discovered through the screening of a library of human sirtuin regulators. The mechanism of inhibition of the lead compounds were investigated. Collectively, the mechanistic insights and inhibitors described in this study will facilitate the future development of small molecule PfSir2A inhibitors as antimalarial agents.
Introduction
It is estimated that over one million people die annually of P. falciparum-related malaria with a majority under the age of five years.1 The development of new therapeutics against malaria is urgent due to increased resistance to current antimalarial drugs due to small changes in parasite DNA. The genome of P. falciparum harbors two sir2 genes, which encode functionally similar but distinct forms of the PfSir2 protein.2,3 Deletion of either of these genes leads to a reduction in overall PfSir2 activity, and substantial de-silencing of a significant portion of the var gene family.3,4 Each var gene encodes a unique variant of the PfEMP1 protein (P. falciparum erythrocyte membrane protein 1), the main virulence factor of P. falciparum. The parasite's genome includes approximately 60 var genes, with all but a single variant of the var genes remaining transcriptionally silent, mainly due to the action of PfSir2.2,5 It is essential for the parasite to express only one var gene at a time, and to switch expression throughout the infection to evade the host's immune response against previously expressed var gene products. This highly regulated gene expression is crucial for sustaining a chronic infection. Disruption of var gene silencing would reveal the full antigenic repertoire to the host prematurely, effectively “vaccinating” the host against all parasite variants. Evidence from analogous gene family in the intestinal parasite Giardia lamblia demonstrate that such silencing disruption can lead to effective vaccination in infected mice, thus supporting the potential efficacy of this approach.6 It is likely that PfSir2 activity will be correspondingly reduced upon inhibitor treatment and that var gene silencing will be disrupted.The current study focuses on one particular PfSir2 isoform, PfSir2A. PfSir2A localizes mainly to the telomeric regions and contributes to the epigenetic regulation of antigenic variation in P. falciparum.7–10PfSir2A, together with its paralogue PfSir2B, regulates the expression of different var gene subsets which are grouped by the promoters including upsA, upsB, upsC, upsD, and upsE.3 Transcriptional profiling reveals that deletion of Pfsir2a induces up-regulation of a subgroup of var genes, particularly affecting members controlled by upsA, upsC, and upsE regions.3,4 Competition is known to exist between the heterochromatic histone mark H3K9me3 (transcription repression) and euchromatic histone mark H3K9Ac (transcription activation) in the 5′-flanking region of the var genes.11 In line with the mutually exclusive relationship between H3K9me3 and H3K9Ac, the lack of PfSir2A activity in knock-out cells lowers the abundance of repressive H3K9me3 marks.12 Additionally, PfSir2A is highly enriched in the nucleolus where it fine-tunes ribosomal RNA gene transcription.13 At the molecular level, PfSir2A exerts various biological functions through its NAD+-dependent protein deacetylase activity. For example, PfSir2A has been shown to remove acetyl marks from the N-terminal tails of histones H3 and H4 in the in vitro setting.14,15 Furthermore, P. falciparum Alba domain-containing protein (PfAlba3) has been identified as the first non-histone substrate of PfSir2A.16PfSir2A-mediated deacetylation of PfAlba3 at K23 enhances its DNA binding affinity, and stimulates its abasic site-driven endonuclease activity, highlighting its importance in DNA damage response.16 It is discovered recently that PfSir2A is also a lysine defatty-acylase.17 It prefers to remove medium to long chain fatty acyl groups from lysine residues, although the physiological significance of this novel activity remains elusive.
The collective biological insight into PfSir2A function suggests that the enzymatic activity is linked to the maintenance of heterochromatin, in sub-telomeric regions in particular. Multiple studies show, however, that PfSir2A deacetylates histone H3 lysine 9 (H3K9Ac) peptides with a slow turnover rate, several orders of magnitude slower than other sirtuins.15,17 The weak in vitro activity cannot reconcile with the biological observations, suggesting that peptide-independent mechanisms promote more robust PfSir2A activity. In the current study, we report that PfSir2A possesses inherent capacity to tightly bind to DNA to perform efficient deacetylation. We further uncover that PfSir2A demonstrates nucleosome-dependent deacetylase activity. It is reasoned that PfSir2A has intrinsic nucleosome binding functions, and the tight and specific interaction with nucleosomes provides improved catalytic efficiency that allows PfSir2A to reduce histone acetylation.
Given its importance in controlling both the virulence and multiplicity of the parasite, PfSir2A is an attractive drug target to develop antimalarial agents. Herein, we report the discovery of endogenous and synthetic small molecule PfSir2A regulators. A free fatty acid (FFA) such as myristic acid exhibits differential regulation of the two distinct activities of PfSir2A: it activates deacetylation but inhibits defatty-acylation. It is proposed that FFA binding-induced conformational change is responsible for the improved deacetylase efficacy. In the meantime, a FFA also competes with fatty-acylated peptide substrates for the same binding site, resulting in reduced defatty-acylase activity. Furthermore, screening of a library of human sirtuin regulators leads to the discovery of two PfSir2A inhibitors: 3-TYP and nicotinamide riboside (NR). They both serve as non-competitive inhibitors to suppress PfSir2A deacetylation of synthetic peptide and physiological substrates. Taken together, our study provides not only the greatly needed mechanistic understanding of PfSir2A regulation, but also novel chemical scaffolds for future inhibitor development.
Methods and materials
Reagents and instruments
All reagents were purchased from Aldrich or Fisher Scientific and were of the highest purity commercially available. HPLC was performed on a Dionex Ultimate 3000 HPLC system equipped with a diode array detector using a Macherey-Nagel C18 reverse-phase column. HRMS spectra were acquired using either a Waters Micromass Q-tof Ultima or a Thermo Scientific Q-Exactive hybrid Quadrupole Orbitrap.Synthetic peptides
Synthetic peptides H3K9Ac: ARTKQTAR(K-Ac)STGGKAPRKQLAS, H3K9Myr: ARTKQTAR(K-Myr)STGGKAPRKQLAS, H4K16Ac: SGRGKGGKGLGKGGA(K-Ac)RHR, and p300K1024Ac: ERSTELKTEI(K-Ac)EEEDQPSTS were synthesized and purified by Genscript. The peptides were purified using HPLC to a purity of >95%.Protein expression and purification
Plasmid of PfSir2A was the generous gift from Dr Hening Lin (University of Chicago). The protein was expressed and purified according to previously published protocol.17 The identity of the protein was confirmed by tryptic digestion followed by LC-MS/MS analysis performed at Vermont Biomedical Research Network (VBRN) Proteomics Facility. Protein concentration was determined via a Bradford assay.Deacetylation/demyristoylation assay
The Km and kcat values of PfSir2A were measured for synthetic peptide substrates.15 A typical reaction was performed in 100 mM phosphate buffer pH 7.5 in a total volume of 250 μL. The reactions contained various concentrations of the peptide substrate and 500 μM NAD+. Reactions were initiated by the addition of 5 μM (deacetylation) or 1 μM (demyristoylation) of PfSir2A and were incubated at 37 °C for 60 min (deacetylation) or 5 min (demyristoylation) before being quenched by 10% TFA. The samples were then injected into an HPLC fitted to a Macherey-Nagel Nucleosil C18 column. Acylated and deacylated peptides were resolved using a gradient of 10–40% acetonitrile in 0.1% TFA. Chromatograms were analyzed at 215 nm. Reactions were quantified by integrating area of peaks corresponding to acylated and deacylated peptides. Rates were plotted as a function of substrate concentration and best fits of points to the Michaelis–Menten equation were performed by using a GraphPad Prism.PfSir2A inhibition assay
A typical reaction contained 500 μM NAD+, 250 μM H3K9Ac or 20 μM H3K9Myr, and varying concentrations of the small molecule inhibitor in 100 mM phosphate buffer pH 7.5. The reactions were initiated by the addition of 5 μM (deacetylation) or 1 μM (demyristoylation) of PfSir2A and were incubated at 37 °C before being quenched by 10% TFA. The samples were then injected into an HPLC fitted to a Macherey-Nagel Nucleosil C18 column. Acylated and deacylated peptides were resolved using a gradient of 10–40% acetonitrile in 0.1% TFA. Chromatograms were analyzed at 215 nm. Reactions were quantified by integrating area of peaks corresponding to acylated and deacylated peptides. Rates were plotted as a function of small molecule inhibitor concentration, and points were fitted to the following equation:| v (%) = v0 (%) − [v0 (%)(10x)/(10x + IC50)] |
Mode of inhibition analysis
Peptide titration reactions containing 0, 10, 100, or 1000 μM inhibitor were incubated with 500 μM NAD+, varying concentrations of H3K9Ac or H3K9Myr in 100 mM phosphate buffer pH 7.5. NAD+ titration reactions containing a 0, 10, 100, or 1000 μM inhibitor were incubated with 250 μM H3K9Ac or 20 μM H3K9Myr, varying concentrations of NAD+ in 100 mM phosphate buffer pH 7.5. Reactions were initiated by the addition of 5 μM (deacetylation) or 1 μM (demyristoylation) of PfSir2A and were incubated at 37 °C before being quenched by 10% TFA. The samples were then injected into an HPLC fitted to a Macherey-Nagel Nucleosil C18 column. Acylated and deacylated peptides were resolved using a gradient of 10–40% acetonitrile in 0.1% TFA. Chromatograms were analyzed at 215 nm. Reactions were quantified by integrating area of peaks corresponding to acylated and deacylated peptides. From the Michaelis–Menten plots, Km and kcat were calculated for different concentrations of the inhibitor.Electrophoretic mobility shift assay (EMSA)
dsDNA was incubated with varying concentrations of PfSir2A (0 to 20 μM) in DNA binding buffer (20 mM Tris–HCl, pH 8.0, 150 mM NaCl, 2 mM DTT, 5% glycerol).18 The samples were incubated on ice for 20 min. The samples were then resolved on a native 4–20% TBE gel at 4 °C. After SYBR safe staining, the visualization and quantification of the gels were carried out using a Biorad ChemiDoc MP imaging system and Image Lab software. Any species migrating at a slower electrophoretic rate than free DNA were considered as a DNA–protein complex. Relative complex formation was plotted as a function of protein concentration and fitted to the equation: f = fmax[PfSir2A]h/(Kd + [PfSir2A]h), where f is the fractional saturation, fmax is maximum complex formation, [PfSir2A] is the protein concentration, h is the Hill slope, and Kd reflects the binding affinity of the proteins to the DNA construct.Microscale thermophoresis (MST) binding assay
The MST experiments were performed using a Monolith NT.115 instrument (NanoTemper Technologies).19 The purified recombinant His-tagged PfSir2A was labeled by the RED-tris-NTA 2nd generation dye (NanoTemper Technologies). The protein concentration was adjusted to 200 nM in PBS-T buffer, while the dye concentration was set to 100 nM. Equal volumes (100 μL) of protein and dye solutions were mixed and incubated at room temperature in the dark for 30 min. Sixteen solutions of ssDNA with decreasing concentrations were prepared in the MST buffer by serial half-log dilution. Each solution was mixed with an equal volume of the labeled PfSir2A. The binding affinity analysis was performed using standard MST capillaries, 40% excitation power (Nano-RED) and medium MST power at room temperature. The Kd value was determined with the MO. Affinity Analysis software (NanoTemper Technologies), using three independent MST measurements.Cell culture
HEK293 cells were cultured in DMEM supplemented with 10% fetal bovine serum (FBS), 100 U mL−1 penicillin and 100 mg mL−1 streptomycin. Cells were maintained in a humidified 37 °C incubator with 5% CO2.Metabolic labeling
Cells were treated with either 20 μM azido myristic acid or DMSO (vehicle control) for 18 h.20 Cells were harvested, re-suspended in PBS, and pelleted at 1000g for 5 min at 4 °C. The cell pellet was then dissolved in RIPA buffer (Thermo Scientific) with halt protease phosphatase inhibitor cocktail (Thermo Scientific). The lysate was centrifuged at 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for 5 min at 4 °C. The supernatant was used for the defatty-acylation analysis.
000g for 5 min at 4 °C. The supernatant was used for the defatty-acylation analysis.
Defatty-acylation assay
Cell lysate was incubated with 500 μM NAD+, 10 μM PfSir2A with or without calf thymus DNA at 37 °C for 1 h.20 The samples were then incubated with 15 mM iodoacetamide at room temperature for 30 min before TAMRA-DBCO was added. The samples were incubated at room temperature for another 30 min. Subsequently, the samples were heated with NH2OH (60 mM, pH 7.2) at 95 °C for 7 min before being resolved by SDS-PAGE. The destained gel was analyzed with in-gel fluorescence scanning using a Biorad ChemiDoc MP imager (excitation at 532 nm, 580 nm cut-off filter and 30 nm band-pass).Western blot
The samples were resolved on a 10% SDS-PAGE gel and transferred to an Immobilon PVDF transfer membrane (Biorad).21 The blot was blocked with 5% nonfat milk, probed with primary antibody, washed with TBST, followed by incubation with anti-rabbit HRP-conjugated secondary antibody. The signal was then detected by Clarity western ECL substrate (Biorad).Results and discussion
Characterization of PfSir2A activity in vitro
We have expressed and purified PfSir2A with an N-terminal His-tag. It can deacetylate a panel of peptide substrates.15 The kinetic constants for PfSir2A-mediated deacetylation under steady-state conditions at pH 7.5 have been determined (Table 1). The recombinant enzyme is also able to deacetylate H3K9Ac in calf thymus histones (Fig. 1A and B). In an in vitro setting, PfSir2A demonstrates weak deacetylase activity towards the synthetic peptide substrates, consistent with a previous report.17 In contrast, the catalytic efficiency (kcat/Km) of demyristoylation is 4400-fold higher than that of the deacetylation (5140 M−1 s−1vs. 1.15 M−1 s−1, Table 1), confirming the robust defatty-acylase activity of PfSir2A.17| Substratea | Additive | K m (μM) | k cat (10−4 s−1) | M.A.f |
|---|---|---|---|---|
| a At saturating NAD+ concentration. b Sequence: H3(1–22). c Sequence: H4(1–19). d Sequence: p300(1034–1053). e [dsDNA] = 200 ng μL−1. f MA: Maximal activation = kcat(act.) × Km(unact.)/(kcat(unact.) × Km(act.)). | ||||
| H3K9Acb | 556 ± 54 | 6.4 ± 0.8 | 1.0 | |
| H3K9Ac | dsDNA | 175 ± 22 | 8.2 ± 0.5 | 4.1 |
| H4K16Acc | 195 ± 42 | 4.3 ± 0.5 | ||
| p300K1040Acd | 102 ± 21 | 11.4 ± 1.2 | ||
| H3K9Myrb | 4.9 ± 1.1 | 252 ± 17 | 1.0 | |
| H3K9Myr | dsDNA | 0.95 ± 0.3 | 305 ± 28 | 6.2 |
 | ||
| Fig. 1 Analysis of PfSir2A interaction with DNAs. (A) Representative western blots showing the deacetylase activity of PfSir2A on calf thymus histone in the presence or absence of dsDNA; (B) quantification of the western blot results. Statistical significance was determined by a Student's t-test: *p < 0.05 vs. no dsDNA control; (C) representative gel image of the EMSA analysis of the interaction of PfSir2A with a circular plasmid; (D) EMSA analysis of the interaction of PfSir2A with the nucleosome assembly 601 sequence DNA. Top: Representative gel image; bottom: binding affinity analysis; (E) determination of binding affinity of PfSir2A to ssDNA using MST. The sequence of the ssDNA is GAAGTCGGGGATGGCAGAGGCAGTGCT. | ||
PfSir2A activities are activated by DNA
Inspired by the early suggestions that PfSir2A is a telomeric binding factor,7,10,22 we performed a simple PfSir2A-DNA binding experiment. The ability of PfSir2A to bind to DNA was evaluated using the electrophoretic mobility shift assay (EMSA) with a circular plasmid (Fig. 1C): in the samples containing PfSir2A and plasmid (lanes 3 to 11), the formation of a PfSir2A–DNA complex can be readily detected (indicated by the red arrow). Similarly, PfSir2A demonstrates tight binding to the nucleosome assembly 601 sequence DNA23 (a 147 bp dsDNA with high affinity for histone octamers and often used for in vitro nucleosome assembly) with a Kd value of 4.0 ± 0.4 μM (Fig. 1D). PfSir2A also binds to ssDNA (Kd = 138 nM) as determined via a microscale thermophoresis (MST) assay (Fig. 1E). The interaction between PfSir2A and nucleic acids is sequence-independent.These initial findings prompted us to investigate whether the deacetylase activity of PfSir2A can be activated upon binding to DNA. Indeed, in the presence of calf thymus DNA (dsDNA sheared to an average size less than 2000 bp), the catalytic efficiency of deacetylation exhibits a 4-fold improvement towards the peptide substrate (Table 1), owing to a reduced Km and an increased kcat. Furthermore, the effect of DNA on physiological PfSir2A substrate was also assessed. Calf thymus histone was incubated with recombinant PfSir2A and NAD+ with or without dsDNA. The acetylation level of H3K9 was then probed using western blotting. The addition of dsDNA caused a significant reduction of H3K9Ac compared to the no DNA controls (Fig. 1A and B). To our knowledge, these results represent the first direct evidence that the enzymatic activity of PfSir2A can be stimulated upon binding to DNA strands.
Studies from the last two decades have suggested that PfSir2A is a promiscuous enzyme with multiple enzymatic activities. In addition to deacetylation15 and mono-ADP-ribosylation,14PfSir2A also harbors defatty-acylation activity.17 As PfSir2A is poised at the intersection between chromatin modulation and genome maintenance, understanding how this novel activity is regulated is highly relevant. In our preliminary study, a myristoylated synthetic H3K9 peptide (H3K9Myr) was used as the substrate for the characterization of the defatty-acylase activity of PfSir2A. The inclusion of dsDNA caused a marked reduction of Km (from 4.9 to 0.95 μM, Table 1), leading to a 6-fold activation of the demyristoylase activity.
To examine if lysine fatty-acylation of cellular proteins can be modulated by PfSir2A, azido myristic acid (Az-myr) was used for the metabolic labeling of cells. As shown in Fig. 2A, HEK293 cells were treated with Az-myr (20 μM) which can be metabolically incorporated into fatty-acylated, especially N- and S-myristoylated, cellular proteins. The cell lysate was then incubated with PfSir2A in the presence or absence of dsDNA. Subsequently, the labeled proteins were covalently tethered to TAMRA-DBCO via strain-promoted “click” conjugation. NH2OH was used to remove the potential cysteine fatty-acylation. Thus, the in-gel fluorescence signal will mainly represent the lysine fatty-acylation level. Compared to the control cell lysate, Az-myr-treated cell lysate demonstrated significantly increased labeling intensity (Fig. 2B). Treatment with PfSir2A in the presence of NAD+ reduced the fluorescence signals of numerous proteins, confirming the previous hypothesis that PfSir2A can remove lysine fatty-acylation from endogenous protein targets.17 The addition of dsDNA to the sample caused further signal reduction of several highlighted proteins. Although the chemical nature of lysine acylation has been known to regulate the level of PfSir2A catalytic efficiency,17 these substrate preferences have never been studied in combination with additional modulators such as nucleic acids. Our data suggest that DNA-complexed PfSir2A displays enhanced defatty-acylase activity.
 | ||
| Fig. 2 Defatty-acylase activity of PfSir2A can be activated by dsDNA. (A) Schematic representation of the metabolic labeling; (B) defatty-acylation of cellular proteins by PfSir2A. The lysate from Az-myr treated cells showed increased labeling compared to the control cells. Incubation of the lysate with PfSir2A in the presence of dsDNA reduced the labeling intensity of several proteins compared to the no DNA control. Arrows highlight proteins with decreased fluorescence due to dsDNA exposure. | ||
Differential regulation of PfSir2A activity by free fatty acid
A co-crystal structure of PfSir2A with NAD+ and a bound H3H9Myr peptide provides the structural basis of the preferred defatty-acylase activity.17PfSir2A has a long hydrophobic tunnel to accommodate the long chain fatty-acyl groups (Fig. 3A). Structural alignment of PfSir2A-AMP (pdb: 3JWP) and PfSir2A-NAD+-H3K9Myr (pdb: 3U3D) suggests that binding of the H3K9Myr peptide drives the zinc-binding domain to rotate clockwise to the Rossman fold domain, resulting in PfSir2A moving from an inactive open state to a productive closed state.24 It is reasonable to speculate that the hydrophobic tunnel of PfSir2A allows it to bind to free fatty acids (FFAs) for conformational rearrangement and the stimulation of its deacetylase activity.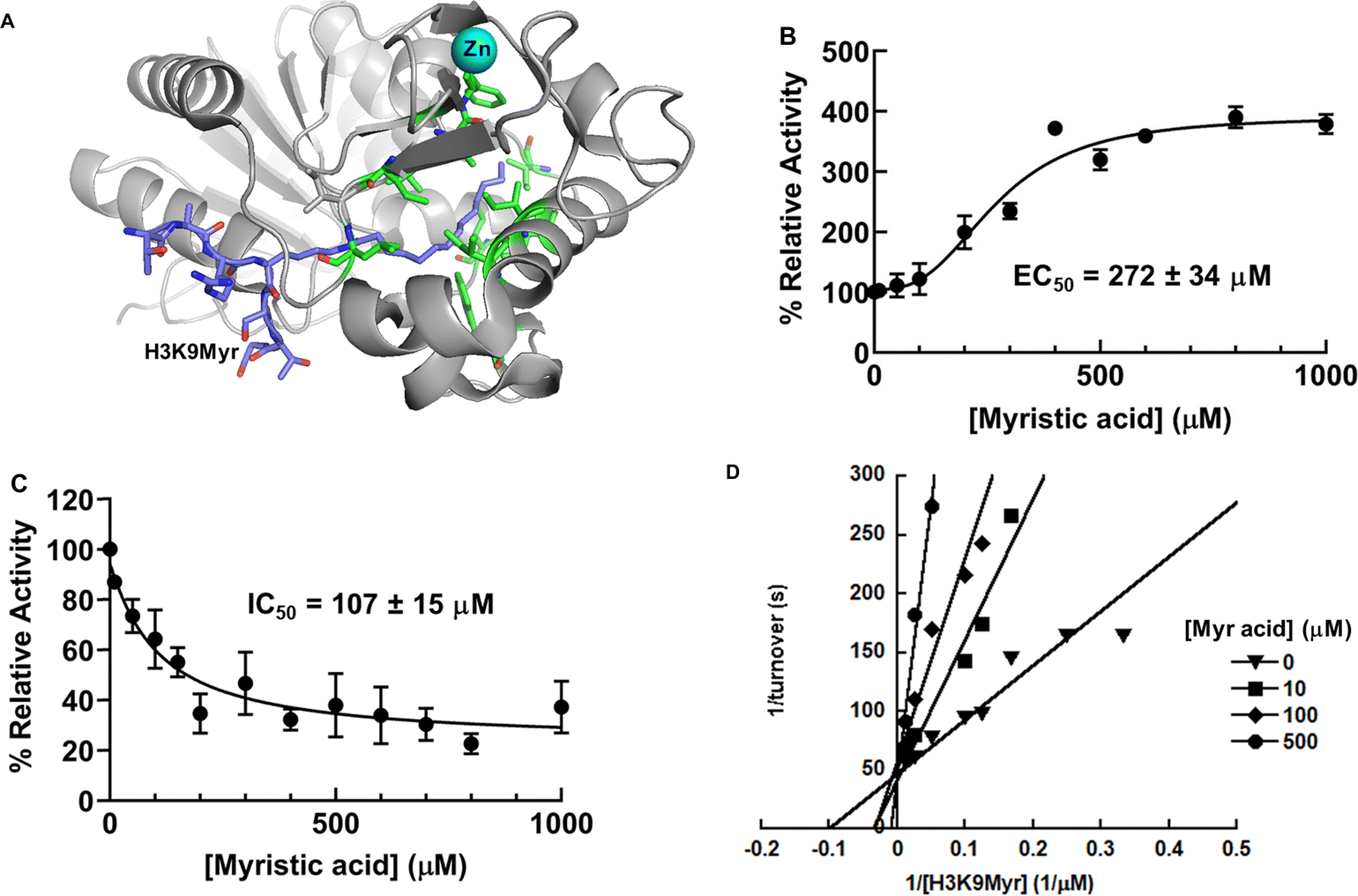 | ||
| Fig. 3 Differential regulation of PfSir2A activity by myristic acid. (A) PfSir2A in complex with H3K9Myr (pdb: 3U3D). H3K9Myr: purple; zinc: cyan; hydrophobic amino acids in the binding tunnel: green; (B) activation of PfSir2A deacetylase activity by myristic acid; (C) inhibition of PfSir2A demyristoylase activity by myristic acid; (D) double-reciprocal plot showing that myristic acid is a competitive inhibitor of PfSir2A's demyristoylase activity. | ||
To assess if FFAs can activate PfSir2A deacetylation, the enzyme was incubated with NAD+ and H3K9Ac peptide in the presence of myristic acid. The concentrations of myristic acid were maintained below its critical micelle concentration (CMC).25 Indeed, the deacetylation was stimulated by myristic acid in a concentration-dependent fashion (Fig. 3B): a maximum of 4-fold activation with an EC50 of 272 ± 34 μM. The EC50 value is well within the physiological concentrations of FFAs in humans.26
The ability of myristic acid to activate PfSir2A deacetylation supports our initial hypothesis that myristic acid occupies the same binding pocket as the myristoylated peptide substrate. To further confirm this, the effect of myristic acid on the demyristoylase activity of PfSir2A was also examined. As shown in Fig. 3C, demyristoylation of H3K9Myr was inhibited by myristic acid in a dose-dependent manner with an IC50 value of 107 ± 15 μM. The mode of inhibition was then investigated by steady-state inhibition analysis. Double reciprocal plot showed that myristic acid was competitive with H3K9Myr peptide as evidenced by a series of lines that intersect at the y-axis (Fig. 3D), consistent with the notion that FFA competes with myristoylated peptide for the same binding site.
var gene transcription is epigenetically regulated by PfSir2A,3,8 a NAD+-dependent histone deacetylase. Given recent evidence supporting that FFAs can reduce histone acetylation levels at specific loci,27,28 we hypothesize that metabolism and epigenetic regulation of gene transcription may be controlled by FFA availability. The parasites acquire FFA through either de novo synthesis or scavenging from the host cells.29 Some recent studies reveal that P. falciparum senses nutrient availability to induce epigenetic reprogramming.30,31 When FFAs are abundant, the deacetylase activity of PfSir2A can be stimulated for targeted histone deacetylation and var gene silencing. FFAs may also serve as an intrinsic regulatory factor allowing PfSir2A to switch from a defatty-acylase to a deacetylase, although the biological significance of PfSir2A-catalyzed defatty-acylation remains elusive.
PfSir2A demonstrates nucleosome-dependent activity
Despite a plethora of reported in vivo functions, the biochemical aspects of PfSir2A activity and regulation remain poorly understood. The recombinant PfSir2A is found to be rather inactive, but activatable toward acetylated substrates by complexation with DNAs. This suggests the existence of a more potent, but previously uncharacterized, PfSir2A activation mechanism. Considering the chromatin association and function,7,8 it is reasonable to hypothesize nucleosomes as potential PfSir2A activators.To study PfSir2A-nucleosome interactions, a highly purified recombinant human 601-positioned nucleosome (EpiCypher) was used. In this nucleosome, histone H3 contains an acetyllysine at position 9 (H3K9Ac dNuc). The binding interaction was assessed using the EMSA. The titration of recombinant PfSir2A into nucleosome resulted in the formation of a higher molecular weight band (Fig. 4A). H3K9Ac dNuc effectively interacts with PfSir2A with a KD value of 1.4 ± 0.2 μM (Fig. 4B). To further characterize if binding to nucleosome promotes more efficient histone deacetylation, the ability of PfSir2A to deacetylate H3K9Ac dNuc was evaluated using western blotting. The enzyme showed enhanced deacetylation of nucleosome substrate as compared to free histone proteins (Fig. 4C and D). Our data suggest that PfSir2A harbors the intrinsic capacity to bind tightly and remove acetyl groups efficiently from nucleosomes. Currently, we are using a unique combination of HPLC-based enzyme activity assays, EMSA, as well as site-directed mutagenesis and truncation analysis to investigate the molecular features required for the intimate PfSir2A-nucleosome interactions, and how these interactions contribute to the efficient removal of acetyl marks from histone lysines.
 | ||
| Fig. 4 PfSir2A demonstrates nucleosome-dependent deacetylase activity. (A) EMSA analysis of PfSir2A interactions with H3K9Ac dNuc; (B) binding affinity analysis. The KD was determined to be 1.4 ± 0.2 μM; (C) western blot analysis of PfSir2A deacetylation of H3k9Ac dNuc; (D) quantification of the western blot results in (C). The H3K9Ac level was normalized by the amount of histone H2B protein. | ||
Repurposing human sirtuin regulators as PfSir2A inhibitors
Although combination therapies have succeeded in reducing the global burden of malaria, multidrug resistance is emerging worldwide. Innovative antimalarial drugs that kill all-life-cycle stages of parasites are urgently needed. PfSir2A controls both the virulence and multiplicity of the parasite, and disruption of this gene results in the loss of mono-allelic expression of var genes. Thus, this epigenetic regulator is an attractive drug target for overcoming multidrug resistance.Currently, there are no PfSir2A-selective inhibitors. Nicotinamide (NAM) is the physiological Sir2 inhibitor.32,33 A recent study indicates that NAM inhibits PfSir2A deacetylase activity in vivo.34 However, this compound can be directly incorporated into metabolic pathways and shows broad-spectrum inhibition against Sir2s. A lysine-based tripeptide analog has been shown to inhibit recombinant PfSir2A activity, and inhibit the intra-erythrocytic parasite growth.35 However, this compound exhibits comparable potency against human SIRT1. An ideal PfSir2A inhibitor should have a novel scaffold that allows it to target PfSir2A in a selective manner.
The development of human sirtuin regulators has grown exponentially during the last two decades, with several compounds in clinical trials for various diseases.36 These prior studies not only provide chemical entities for drug discovery, but also insight into sirtuin structure, selectivity, and biological functions. All of these can be harnessed for the development of PfSir2A inhibitors. To this end, we screened a commercially available library of 28 human sirtuin regulators (Selleckchem) at 10, 100, and 1000 μM against recombinant PfSir2A. Using the HPLC-based assay described in “Methods and Materials”, the effect of these compounds of PfSir2A-catalyzed deacetylation was evaluated. 3-TYP and nicotinamide riboside (NR) demonstrated concentration-dependent inhibition of PfSir2A deacetylase activity with more than 50% inhibition at 1 mM concentration (Fig. 5A). The IC50 value of NR, a water soluble compound, was determined to be 155.2 ± 14.9 μM (Fig. 5B). Due to its limited solubility, the IC50 of 3-TYP was estimated to be 59.6 ± 3.4 μM (Fig. 5C).
 | ||
| Fig. 5 Discovery of PfSir2A inhibitor from a human sirtuin regulator library. (A) Screening of human sirtuin regulators (from Selleckchem) on PfSir2A-catalyzed peptide deacetylation at 10, 100, and 1000 μM concentrations; (B) and (C) concentration-dependent inhibition of PfSir2A-mediated deacetylation by NR (B) or 3-TYP (C); (D) representative western blots showing that 3-TYP and NR inhibit PfSir2A deacetylation of H3K9Ac in calf thymus histone; (E) quantification of the western blotting results. | ||
3-TYP is a SIRT3-selective inhibitor with an IC50 value of 38 ± 5 μM.37 This compound also exhibits cellular activity: incubation of the cells with 3-TYP leads to increased mitochondrial acetylation, significantly reduced ATP levels, and markedly increased superoxide, consistent with the phenotypes observed in sirt3-deficient mice.37 Due to its structural similarity to nicotinamide (NAM), it is proposed that 3-TYP and NAM inhibit sirtuins via the same mode of action. Indeed, increasing 3-TYP concentration caused a significant reduction of the Vmax value, but negligible changes to the Km (Fig. S1, ESI†), suggesting that this compound is non-competitive with an acetyllysine peptide substrate.
NR is a recently discovered NAD+ precursor.38 It is metabolically transformed by NR kinases (NRK1 and NRK2) to nicotinamide mononucleotide (NMN).38–40 Subsequently, NMN can be incorporated into NAD+via the Preiss–Handler pathway.41 We and others have demonstrated that NR treatment increases cellular NAD+ concentrations significantly.42–44 And sirtuin activity is known to be activated with elevated intracellular NAD+ contents.45,46 Most recently, our group also discovered that NR is a direct activator of human SIRT5.21 It selectively activates SIRT5 deacetylation on both the synthetic peptide and physiological substrates.21 In the current study, NR is identified as a non-competitive PfSir2A inhibitor, as evidenced by the Michaelis–Menten kinetic analysis (Fig. S2, ESI†).
3-TYP and NR not only inhibit PfSir2A deacetylation of synthetic peptides, but also suppress its deacetylation of histone proteins. As shown in Fig. 5D and E, 3-TYP or NR treatment increases the acetylation level of H3K9 in calf thymus histone in a concentration-dependent manner. At the highest concentration tested, both compounds restore the level of H3K9Ac as compared to the no enzyme controls. The structures of 3-TYP and NR will be further optimized for improved potency and selectivity.
Conclusions and perspectives
P. falciparum is the causative agent responsible for the most severe form of human malaria. The parasite uses antigenic variation to avoid host antibody recognition, leading to greatly extended periods of infection.47 This antigenic variation is mediated by the differential control of a family of var genes that encode the surface protein PfEMP1.48 Each individual parasite expresses only a single var gene at a time. All the other family members are maintained in a transcriptionally silent state, a process controlled by epigenetic mechanisms.49Recent studies suggest the key role of P. falciparum silent information regulator 2A (PfSir2A) in regulating var gene transcription.3,8 Enzymatically, PfSir2A is a NAD+-dependent histone deacetylase, catalyzing the removal of acetyl groups from histone tails.15,50PfSir2A has been shown to associate with silenced var genes and localizes to chromosome-end clusters and telomere repeats within the nucleus.3 Furthermore, genetic ablation of PfSir2A results in the de-repression of a subset of var genes.7 All of this evidence points to the idea that heterochromatic silencing mediated by PfSir2A could directly control the expression of var virulence genes. PfSir2A thus serves as a novel therapeutic target for the development of antimalarial drugs.
The weak in vitro deacetylase activity of PfSir2A makes the characterization of its biochemical properties and the development of selective inhibitors extremely challenging. We hypothesize, with strong preliminary data, PfSir2A is a highly active histone deacetylase, and this activity is nucleosome dependent. We use a combination of biochemical, chemical biology, and biophysical approaches to uncover the underlying principles leading to the assembly of PfSir2A-DNA or PfSir2A-nucleosome complex, and how this binding is coupled to efficient histone deacetylation.
In addition to DNA and nucleosome, cellular FFAs have also been identified as endogenous PfSir2A regulators. Myristic acid, a C14 fatty acid, is able to stimulate the deacetylase activity, but suppresses the defatty-acylase activity. The binding of myristic acid in the large, hydrophobic binding pocket of PfSir2A causes a conformational rearrangement for enhanced deacetylation. The same binding event also prevents fatty-acylated lysine substrate from binding, leading to the inhibition of defatty-acylation. The data are consistent with the hypothesis that the parasite can sense nutrient availability and respond with antigenic switching.
The biological relevance of PfSir2A inspires us to conduct inhibitor repurposing for the development of PfSir2A inhibitors. Screening of a commercial library of human sirtuin regulators led to the discovery of two lead compounds: 3-TYP and NR. Both compounds inhibit the deacetylase activity of PfSir2A with synthetic peptide substrates and histone substrates. The mode of inhibition of the two molecules were further analyzed. Based on these two lead compounds, potent and PfSir2A-selective inhibitors will be developed using a suite of enzyme kinetic analysis, structure–activity relationship study, and structure-based drug design.
The current study reveals new understandings on the regulation of PfSir2A activity, and new scaffolds for PfSir2A inhibitor. It also provides proof of concept to gain mechanistic and chemical insights into complex enzyme reactions involved in epigenetic control and protein modifications.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported in part by 1R01GM143176-01A1 from NIH/NIGMS (to Y. C.), and Jeffress Trust Award in Interdisciplinary Research from Thomas F. and Kate Miller Jeffress Memorial Trust and Bank of America (to Y. C.). MS analysis reported in this manuscript was performed at the Vermont Biomedical Research Network (VBRN) proteomics facility supported by P20GM103449 (NIH/NIGMS).References
- B. Rieke, Overview of the worldwide Handling with the Disease in Prevention, Diagnostics and Therapy WHO World Malaria Report 2017, Flugmedizin Tropenmedizin Reisemedizin, 2018, 25, 5 CrossRef.
- L. Cui and J. Miao, Chromatin-mediated epigenetic regulation in the malaria parasite Plasmodium falciparum, Eukaryot. Cell, 2010, 9(8), 1138–1149 CrossRef CAS PubMed.
- C. J. Tonkin, C. K. Carret, M. T. Duraisingh, T. S. Voss, S. A. Ralph, M. Hommel, M. F. Duffy, L. M. Silva, A. Scherf, A. Ivens, T. P. Speed, J. G. Beeson and A. F. Cowman, Sir2 paralogues cooperate to regulate virulence genes and antigenic variation in Plasmodium falciparum, PLoS Biol., 2009, 7(4), e84 CrossRef PubMed.
- C. J. Merrick, R. Dzikowski, H. Imamura, J. Chuang, K. Deitsch and M. T. Duraisingh, The effect of Plasmodium falciparum Sir2a histone deacetylase on clonal and longitudinal variation in expression of the var family of virulence genes, Int. J. Parasitol., 2010, 40(1), 35–43 CrossRef CAS PubMed.
- L. Swamy, B. Amulic and K. W. Deitsch, Plasmodium falciparum var gene silencing is determined by cis DNA elements that form stable and heritable interactions, Eukaryot. Cell, 2011, 10(4), 530–539 CrossRef CAS PubMed.
- F. D. Rivero, A. Saura, C. G. Prucca, P. G. Carranza, A. Torri and H. D. Lujan, Disruption of antigenic variation is crucial for effective parasite vaccine, Nat. Med., 2010, 16(5), 551–557 CrossRef CAS PubMed.
- L. H. Freitas-Junior, R. Hernandez-Rivas, S. A. Ralph, D. Montiel-Condado, O. K. Ruvalcaba-Salazar, A. P. Rojas-Meza, L. Mancio-Silva, R. J. Leal-Silvestre, A. M. Gontijo, S. Shorte and A. Scherf, Telomeric heterochromatin propagation and histone acetylation control mutually exclusive expression of antigenic variation genes in malaria parasites, Cell, 2005, 121(1), 25–36 CrossRef CAS PubMed.
- M. T. Duraisingh, T. S. Voss, A. J. Marty, M. F. Duffy, R. T. Good, J. K. Thompson, L. H. Freitas-Junior, A. Scherf, B. S. Crabb and A. F. Cowman, Heterochromatin silencing and locus repositioning linked to regulation of virulence genes in Plasmodium falciparum, Cell, 2005, 121(1), 13–24 CrossRef CAS PubMed.
- A. Scherf, J. J. Lopez-Rubio and L. Riviere, Antigenic variation in Plasmodium falciparum, Annu. Rev. Microbiol., 2008, 62, 445–470 CrossRef CAS PubMed.
- L. Mancio-Silva, A. P. Rojas-Meza, M. Vargas, A. Scherf and R. Hernandez-Rivas, Differential association of Orc1 and Sir2 proteins to telomeric domains in Plasmodium falciparum, J. Cell Sci., 2008, 121(Pt 12), 2046–2053 CrossRef CAS PubMed.
- J. J. Lopez-Rubio, A. M. Gontijo, M. C. Nunes, N. Issar, R. Hernandez Rivas and A. Scherf, 5' flanking region of var genes nucleate histone modification patterns linked to phenotypic inheritance of virulence traits in malaria parasites, Mol. Microbiol., 2007, 66(6), 1296–1305 CrossRef CAS PubMed.
- J. J. Lopez-Rubio, L. Mancio-Silva and A. Scherf, Genome-wide analysis of heterochromatin associates clonally variant gene regulation with perinuclear repressive centers in malaria parasites, Cell Host Microbe, 2009, 5(2), 179–190 CrossRef CAS PubMed.
- L. Mancio-Silva, J. J. Lopez-Rubio, A. Claes and A. Scherf, Sir2a regulates rDNA transcription and multiplication rate in the human malaria parasite Plasmodium falciparum, Nat. Commun., 2013, 4, 1530 CrossRef PubMed.
- C. J. Merrick and M. T. Duraisingh, Plasmodium falciparum Sir2: an unusual sirtuin with dual histone deacetylase and ADP-ribosyltransferase activity, Eukaryot. Cell, 2007, 6(11), 2081–2091 CrossRef CAS PubMed.
- J. B. French, Y. Cen and A. A. Sauve, Plasmodium falciparum Sir2 is an NAD + -dependent deacetylase and an acetyllysine-dependent and acetyllysine-independent NAD+ glycohydrolase, Biochemistry, 2008, 47(38), 10227–10239 CrossRef CAS PubMed.
- C. Banerjee, S. Nag, M. Goyal, D. Saha, A. A. Siddiqui, S. Mazumder, S. Debsharma, S. Pramanik and U. Bandyopadhyay, Nuclease activity of Plasmodium falciparum Alba family protein PfAlba3, Cell Rep., 2023, 42(4), 112292 CrossRef CAS PubMed.
- A. Y. Zhu, Y. Zhou, S. Khan, K. W. Deitsch, Q. Hao and H. Lin, Plasmodium falciparum Sir2A preferentially hydrolyzes medium and long chain fatty acyl lysine, ACS Chem. Biol., 2012, 7(1), 155–159 CrossRef CAS PubMed.
- Z. A. Wang, J. W. Markert, S. D. Whedon, M. Yapa Abeywardana, K. Lee, H. Jiang, C. Suarez, H. Lin, L. Farnung and P. A. Cole, Structural Basis of Sirtuin 6-Catalyzed Nucleosome Deacetylation, J. Am. Chem. Soc., 2023, 145(12), 6811–6822 CrossRef CAS PubMed.
- B. Suenkel, S. Valente, C. Zwergel, S. Weiss, E. Di Bello, R. Fioravanti, M. Aventaggiato, J. A. Amorim, N. Garg, S. Kumar, D. B. Lombard, T. Hu, P. K. Singh, M. Tafani, C. M. Palmeira, D. Sinclair, A. Mai and C. Steegborn, Potent and Specific Activators for Mitochondrial Sirtuins Sirt3 and Sirt5, J. Med. Chem., 2022, 65(20), 14015–14031 CrossRef CAS PubMed.
- W. Kang, A. Hamza, A. M. Curry, E. Korade, D. Donu and Y. Cen, Activation of SIRT6 Deacetylation by DNA Strand Breaks, ACS Omega, 2023, 8(44), 41310–41320 CrossRef CAS PubMed.
- A. M. Curry, S. Rymarchyk, N. B. Herrington, D. Donu, G. E. Kellogg and Y. Cen, Nicotinamide riboside activates SIRT5 deacetylation, FEBS J., 2023, 290(19), 4762–4776 CrossRef CAS PubMed.
- M. C. Dalmasso, S. J. Carmona, S. O. Angel and F. Aguero, Characterization of Toxoplasma gondii subtelomeric-like regions: identification of a long-range compositional bias that is also associated with gene-poor regions, BMC Genomics, 2014, 15(1), 21 CrossRef PubMed.
- P. T. Lowary and J. Widom, New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning, J. Mol. Biol., 1998, 276(1), 19–42 CrossRef CAS PubMed.
- K. G. Hoff, J. L. Avalos, K. Sens and C. Wolberger, Insights into the sirtuin mechanism from ternary complexes containing NAD+ and acetylated peptide, Structure, 2006, 14(8), 1231–1240 CrossRef CAS PubMed.
- A. L. Fameau, J. Ventureira, B. Novales and J. P. Douliez, Foaming and emulsifying properties of fatty acids neutralized by tetrabutylammonium hydroxide, Colloids Surf., A, 2012, 403, 87–95 CrossRef CAS.
- T. Tholstrup, B. Sandstrom, A. Bysted and G. Holmer, Effect of 6 dietary fatty acids on the postprandial lipid profile, plasma fatty acids, lipoprotein lipase, and cholesterol ester transfer activities in healthy young men, Am. J. Clin. Nutr., 2001, 73(2), 198–208 CrossRef CAS PubMed.
- J. Xu, B. Christian and D. B. Jump, Regulation of rat hepatic L-pyruvate kinase promoter composition and activity by glucose, n-3 polyunsaturated fatty acids, and peroxisome proliferator-activated receptor-alpha agonist, J. Biol. Chem., 2006, 281(27), 18351–18362 CrossRef CAS PubMed.
- D. B. Jump, S. D. Clarke, A. Thelen and M. Liimatta, Coordinate regulation of glycolytic and lipogenic gene expression by polyunsaturated fatty acids, J. Lipid Res., 1994, 35(6), 1076–1084 CrossRef CAS.
- A. Lahree, J. Mello-Vieira and M. M. Mota, The nutrient games - Plasmodium metabolism during hepatic development, Trends Parasitol., 2023, 39(6), 445–460 CrossRef CAS PubMed.
- S. Amiar, N. J. Katris, L. Berry, S. Dass, S. Duley, C. S. Arnold, M. J. Shears, C. Brunet, B. Touquet, G. I. McFadden, Y. Yamaryo-Botte and C. Y. Botte, Division and Adaptation to Host Environment of Apicomplexan Parasites Depend on Apicoplast Lipid Metabolic Plasticity and Host Organelle Remodeling, Cell Rep., 2020, 30(11), 3778–3792 CrossRef CAS PubMed.
- V. M. Schneider, J. E. Visone, C. T. Harris, F. Florini, E. Hadjimichael, X. Zhang, M. R. Gross, K. Y. Rhee, C. Ben Mamoun, B. F. C. Kafsack and K. W. Deitsch, The human malaria parasite Plasmodium falciparum can sense environmental changes and respond by antigenic switching, Proc. Natl. Acad. Sci. U. S. A., 2023, 120(17), e2302152120 CrossRef CAS PubMed.
- A. A. Sauve and V. L. Schramm, Sir2 regulation by nicotinamide results from switching between base exchange and deacetylation chemistry, Biochemistry, 2003, 42(31), 9249–9256 CrossRef CAS PubMed.
- J. L. Avalos, K. M. Bever and C. Wolberger, Mechanism of sirtuin inhibition by nicotinamide: altering the NAD(+) cosubstrate specificity of a Sir2 enzyme, Mol. Cell, 2005, 17(6), 855–868 CrossRef CAS PubMed.
- D. Prusty, P. Mehra, S. Srivastava, A. V. Shivange, A. Gupta, N. Roy and S. K. Dhar, Nicotinamide inhibits Plasmodium falciparum Sir2 activity in vitro and parasite growth, FEMS Microbiol. Lett., 2008, 282(2), 266–272 CrossRef CAS PubMed.
- S. P. Chakrabarty, R. Ramapanicker, R. Mishra, S. Chandrasekaran and H. Balaram, Development and characterization of lysine based tripeptide analogues as inhibitors of Sir2 activity, Bioorg. Med. Chem., 2009, 17(23), 8060–8072 CrossRef CAS PubMed.
- A. M. Curry, D. S. White, D. Donu and Y. Cen, Human Sirtuin Regulators: The “Success” Stories, Front. Physiol., 2021, 12, 752117 CrossRef PubMed.
- U. Galli, O. Mesenzani, C. Coppo, G. Sorba, P. L. Canonico, G. C. Tron and A. A. Genazzani, Identification of a sirtuin 3 inhibitor that displays selectivity over sirtuin 1 and 2, Eur. J. Med. Chem., 2012, 55, 58–66 CrossRef CAS PubMed.
- W. Tempel, W. M. Rabeh, K. L. Bogan, P. Belenky, M. Wojcik, H. F. Seidle, L. Nedyalkova, T. Yang, A. A. Sauve, H. W. Park and C. Brenner, Nicotinamide riboside kinase structures reveal new pathways to NAD+, PLoS Biol., 2007, 5(10), e263 CrossRef PubMed.
- K. L. Bogan and C. Brenner, Nicotinic acid, nicotinamide, and nicotinamide riboside: a molecular evaluation of NAD+ precursor vitamins in human nutrition, Annu. Rev. Nutr., 2008, 28, 115–130 CrossRef CAS PubMed.
- R. S. Fletcher, J. Ratajczak, C. L. Doig, L. A. Oakey, R. Callingham, G. Da Silva Xavier, A. Garten, Y. S. Elhassan, P. Redpath, M. E. Migaud, A. Philp, C. Brenner, C. Canto and G. G. Lavery, Nicotinamide riboside kinases display redundancy in mediating nicotinamide mononucleotide and nicotinamide riboside metabolism in skeletal muscle cells, Mol. Metab., 2017, 6(8), 819–832 CrossRef CAS PubMed.
- J. Preiss and P. Handler, Biosynthesis of diphosphopyridine nucleotide. I. Identification of intermediates, J. Biol. Chem., 1958, 233(2), 488–492 CrossRef CAS PubMed.
- A. Tran, R. Yokose and Y. Cen, Chemo-enzymatic synthesis of isotopically labeled nicotinamide riboside, Org. Biomol. Chem., 2018, 16(19), 3662–3671 RSC.
- S. A. Trammell, M. S. Schmidt, B. J. Weidemann, P. Redpath, F. Jaksch, R. W. Dellinger, Z. Li, E. D. Abel, M. E. Migaud and C. Brenner, Nicotinamide riboside is uniquely and orally bioavailable in mice and humans, Nat. Commun., 2016, 7, 12948 CrossRef CAS PubMed.
- C. R. Martens, B. A. Denman, M. R. Mazzo, M. L. Armstrong, N. Reisdorph, M. B. McQueen, M. Chonchol and D. R. Seals, Chronic nicotinamide riboside supplementation is well-tolerated and elevates NAD(+) in healthy middle-aged and older adults, Nat. Commun., 2018, 9(1), 1286 CrossRef PubMed.
- T. Nakagawa, D. J. Lomb, M. C. Haigis and L. Guarente, SIRT5 Deacetylates carbamoyl phosphate synthetase 1 and regulates the urea cycle, Cell, 2009, 137(3), 560–570 CrossRef CAS PubMed.
- P. Belenky, F. G. Racette, K. L. Bogan, J. M. McClure, J. S. Smith and C. Brenner, Nicotinamide riboside promotes Sir2 silencing and extends lifespan via Nrk and Urh1/Pnp1/Meu1 pathways to NAD, Cell, 2007, 129(3), 473–484 CrossRef CAS PubMed.
- G. Geleta and T. Ketema, Severe Malaria Associated with Plasmodium falciparum and P. vivax among Children in Pawe Hospital, Northwest Ethiopia, Malar. Res. Treat., 2016, 2016, 1240962 Search PubMed.
- K. W. Deitsch and R. Dzikowski, Variant Gene Expression and Antigenic Variation by Malaria Parasites, Annu. Rev. Microbiol., 2017, 71, 625–641 CrossRef CAS PubMed.
- A. Cortes and K. W. Deitsch, Malaria Epigenetics, Cold Spring Harbor Perspect. Med., 2017, 7, a025528 CrossRef PubMed.
- S. P. Chakrabarty, Y. K. Saikumari, M. P. Bopanna and H. Balaram, Biochemical characterization of Plasmodium falciparum Sir2, a NAD+ -dependent deacetylase, Mol. Biochem. Parasitol., 2008, 158(2), 139–151 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cb00206g |
| This journal is © The Royal Society of Chemistry 2025 |