
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOvercoming the novel glycan–lectin checkpoints in tumor microenvironments for the success of the cross-presentation-based immunotherapy
Mannat
Jain
a,
Isha M.
Jadhav
a,
Suyash Vinayak
Dangat
a,
Srinivasa Rao
Singuru
a,
Gautam
Sethi
b,
Eiji
Yuba
 *c and
Rajesh Kumar
Gupta
*a
*c and
Rajesh Kumar
Gupta
*a
aProtein Biochemistry Research Centre, Dr. D. Y. Patil Biotechnology and Bioinformatics Institute, Dr. D. Y. Patil Vidyapeeth, Tathawade, Pune-411033, Maharashtra, India. E-mail: jainmannat193@gmail.com; ishajadhav2609@gmail.com; suyashdangat7030@gmail.com; srinus17@gmail.com; rajeshkumar.gupta@dpu.edu.in; guptarajesh76@gmail.com
bDepartment of Pharmacology, Yong Loo Lin School of Medicine, National University of Singapore, 117600, Singapore. E-mail: phcgs@nus.edu.sg
cDepartment of Chemistry & Bioengineering, Graduate School of Engineering, Osaka Metropolitan University, 3-3-138 Sugimoto, Sumiyoshi-ku, Osaka-city, Osaka 558-8585, Japan. E-mail: yuba@omu.ac.jp
First published on 5th May 2025
Abstract
In pursuit of meeting the ever-rising demand for cancer therapies, cross-presentation-based glyconanovaccines (GNVs) targeting C-type lectin receptors (CLRs) on DCs have shown significant potential as cutting-edge cancer immunotherapy. GNVs are an attractive approach to induce anti-cancer cytotoxic T lymphocyte responses. Despite immune checkpoints (ICs) being well established and an obstacle to the success of GNVs, glycan–lectin circuits are emerging as unique checkpoints due to their immunomodulatory functions. Given the role of aberrant tumor glycosylation in promoting immune evasion, mitigating these effects is crucial for the efficacy of GNVs. Lectins, such as siglecs and galectins, are detrimental to the tumor immune landscape as they promote an immunosuppressive TME. From this perspective, this review aims to explore glycan–lectin ICs and their influence on the efficacy of GNVs. We aim to discuss various ICs in the TME followed by drawbacks of immune checkpoint inhibitors (ICIs). We will also emphasize the altered glycosylation profile of tumors, addressing their immunosuppressive nature along with ways in which CLRs, siglecs, and galectins contribute to immune evasion and cancer progression. Considering the resistance towards ICIs, current and prospective approaches for targeting glycan–lectin circuits and future prospects of these endeavors in harnessing the full potential of GNVs will also be highlighted.
1. Introduction
Given the substantial spike in cancer patients globally, the demand for the development of novel cancer therapies is dramatically growing. Current statistics showed around 20 million new cancer cases along with 9.7 million deaths due to cancer in 2022, making it a serious issue.1 Considering this, antigen cross-presentation (XPT), often utilized by dendritic cells (DCs), has been identified as an appealing approach to address and combat the drawbacks of classical anti-cancer therapies. It is known that targeting receptors expressed on DCs with specific ligands can increase the cytotoxic T lymphocyte (CTL) response against cancer cells. Because glycans are recognized as functioning as both tumor-associated antigens (TAA) and ligands for C-type lectin receptors (CLRs) expressed on DCs, they can be used as a therapeutic target for immunotherapy. This knowledge of XPT and glycan-conjugated nanovaccines has been employed to develop ‘glyconanovaccines’ (GNVs) for targeting DCs.2Fig. 1 provides a glance at the XPT pathways and GNVs. | ||
| Fig. 1 Cross-presentation pathways and glyconanovaccines at a glance: (a) two primary mechanisms are involved in the XPT of internalized antigens: the cytosolic pathway and the vacuolar pathway. The internalized antigen is broken down into smaller peptides by endosomal proteases (cathepsin S) in the vacuolar pathway, and these peptides are then loaded onto major histocompatibility complex (MHC) class I molecules. Furthermore, the peptide–MHC complex is transported to the cell surface for recognition by CD8+ T cells. The cytosolic pathway internalizes the exogenous antigen through endocytosis or phagocytosis, and then transports it to the cytosol for further proteosome-mediated degradation to produce shorter antigenic peptides. Furthermore, these antigenic peptides are then loaded onto the MHC class I in the endoplasmic reticulum (ER) after being delivered there by TAP along with the other ER proteins. Furthermore, TAP transports these antigenic peptides to phagosomes, where they get loaded onto MHC class I and are further transferred to the cell surface for their recognition by CD8+ T cells. (b) To enhance antigen presentation and generate a productive T cell response, glycans are multivalently presented on a range of nanocarrier systems. Nanocarriers that are modified with glycans offer glycans in a multivalent form. Furthermore, Toll-like receptor (TLR) ligands can be incorporated into these glyconanocarriers. Examples of glyconanocarriers include glycoliposomes, which can be employed to encapsulate whole tumor antigens as well as adjuvants; glycodendrimers, which can be designed with the appropriate glycan and peptide quantity; and synthetic glycoclusters, and can additionally be created using antigenic peptides from the tumor. (c) DCs efficiently internalise these glycan-modified nanocarrier systems that are laden with tumor antigens in a CLR-specific fashion. Furthermore, these internalised antigens are processed and presented through the MHC class I and MHC class II for their recognition by CD8+ and CD4+ T cells, respectively (created in BioRender. Jain, M. (2025) https://BioRender.com/vs9zk3m). | ||
Numerous cancer immunotherapies have been developed through the years; however, due to the immunosuppressive nature of the tumor microenvironment (TME), they are unable to reach their full potential.3 Immune checkpoints (ICs) are one component of the TME that contributes to the demise of these therapies. Several immune checkpoint inhibitors (ICIs) have been devised to counteract the immune-invasive effects of these checkpoints. ICIs have been revolutionary in the field of immunotherapy due to their ability to mobilize effector T cells to overcome the immunosuppressive niche of regulatory T (Treg) cells and boost the cytotoxic ability of immune cells to further target tumor cells.4,5
ICs are membrane-bound receptors that are located either on the tumor cell surface or immune cells like antigen presenting cells (APCs) and T cells; inhibiting them can be a prominent paragon of an effective immunotherapeutic strategy.4 ICIs may be able to reestablish immune surveillance that tumor cells had managed to evade in order to further boost the activity of T cells, which can also be apt for increasing the efficacy of immunotherapy.6 Several ICs are present at different stages of the cancer immunity cycle, with cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4), programmed death ligand-1 (PD-L1), programmed cell death-1 (PD-1), lymphocyte-activation gene 3 (LAG-3), T cell immunoglobulin and mucin-containing molecule 3 (TIM-3), T cell immunoreceptor with immunoglobulin and immunoreceptor tyrosine-based inhibitory motif domains (TIGIT), and B and T lymphocyte attenuator (BTLA) being the key ones implicated in the development of cancer and the suppression of anti-tumor immunity.7
Despite the pioneering advancements in cancer immunotherapy over the past decade, chemotherapy and radiotherapy continue to be the most frequently utilized cancer treatment modalities. However, the current era of cancer therapeutics has witnessed the rise of anti-checkpoint antibodies as prominent immunotherapeutic agents being directed against immune-suppressing cell surface receptors. Up until now, the Food and Drug Administration (FDA) has approved monoclonal antibody (mAb)-based ICIs for various cancer types, including ipilimumab (CTLA-4 inhibitor), cemiplimab, nivolumab and pembrolizumab (PD-1 inhibitors), as well as durvalumab, atezolimumab, and avelumab (PD-L1 inhibitors).4 However, despite the advancements in ICI therapy, it is benificial in only few cancers due to the resistance posed by the immunosuppressive TME,8 as shown in Fig. 2.
 | ||
| Fig. 2 Response and resistance to ICIs: (a) a successful anti-tumor immune response following ICIs requires reactivation and proliferation of T cells. Production of tumor-reactive CD8+ T cells requires successful tumor-associated peptide antigen processing and presentation by the APCs. TCRs recognize the tumor antigens that are presented by the MHC, providing a signal for T cell activation. This activation additionally necessitates the interaction of the co-stimulatory CD28 receptor expressed on T cells with B7 present on the APCs. Tumor-specific CD8+ T cells then further differentiate into effector T cells and lead to tumor cell apoptosis through release of granzyme A/B and perforin and decrease the immunosuppressive capacity of the TME; (b) resistance to ICIs originates when the various stages of the cancer immune cycle are disrupted or blocked. In immunosuppressive TMEs, factors such as inefficient antigen presentation, poor infiltration and suppressed activity of CD8+ T cells, and failure of antigen recognition can reduce antitumor immunity. Loss of sufficient neoantigens and their impaired presentation can lead to decreased T cell cytotoxicity. High intratumor heterogeneity can also result in an ineffective recognition of tumor-specific neoantigens and reduced CTL responses. In addition, mutations in the antigen processing and presentation machinery can trigger resistance to ICIs. The loss of β2M expression leads to a decline in MHC class I cell surface expression, which impairs the antigen presentation to CTLs. Immune cells within the TME, such as T cells, neutrophils, and MØs, are reprogrammed to become pro-tumor cells, which prevent T cells from functioning by activating various ICs and secreting cytokines, including IL-10, VEGF, CCL2, and TGF-β (created in BioRender. Jain, M. (2025) https://BioRender.com/j05c775). | ||
Besides these, other different factors also influence the overall effectiveness of the immunotherapy. Within the TME, a number of mechanisms and factors influence the tumor immune surveillance and enable tumor immune escape. Metabolic reprogramming that is a hallmark of cancer occurs as a result of conditions such as hypoxia and nutrient deprivation and tumor cells adapt rapidly to them. The Warburg effect is one particular effect that results from alterations in the metabolic process of cancer cells from mitochondrial oxidation to glycolysis in cancer cells even in an environment rich in oxygen. These changes are conducive to cancer invasion, metastasis, and a poor prognosis by increasing the activity of glycolytic enzymes (hexokinase, PFK-1, and pyruvate kinase). Tumor metastasis has been linked to various glycolysis processes, mostly involving transcription factors, signaling pathways, non-coding RNAs (ncRNAs), and others. Several noncoding RNAs have been shown to possess potential in influencing glucose metabolism related to tumor metastasis, including circular RNAs (circRNAs), microRNAs (miRNAs) and long noncoding RNAs (lncRNAs). In addition, through the sequestration of miRNAs, circRNAs and lncRNAs mainly act as competitive endogenous RNAs (ceRNAs), modifying target gene expression and playing a significant role in the metabolic processes of cancer cells. Moreover, numerous signaling pathways (such as, HIF, ERK, Wnt/β-catenin, and, PI3K/AKT etc.) together with transcription factors are also involved in glycolysis-driven tumor metastasis. The Warburg effect plays a pivotal role in accelerating cancer progression and hence it is essential to focus on the inhibition of aerobic glycolysis as a potential approach for the treatment of cancer. The Warburg effect is responsible for the increased production of lactate by the tumor cells and other cells present in the TME and creates a highly acidic environment. By suppressing the proliferation of T cells, altering chemotaxis along with the migration of the neutrophils and DCs, boosting Tregs, and induction of myeloid-derived suppressor cells (MDSCs) and M2 macrophages (MØs) infiltration, TME acidification confers on cancer cells a growth advantage over immune cells, enhancing the immunosuppressive activity on T cells and the TME. Due to the preferential utilization of lactate by Tregs, an acidic TME increases PD-1 together with other inhibitory molecules on Tregs but dampens the PD-1 expression on effector T cells. Consistent with this, PD-1 blockade can result in increased PD-1+ Treg function that supports immunotherapy resistance. Therefore, glycolysis inhibition has been suggested as a potentially beneficial method of limiting tumor cell growth and inducing the death of cancer cells, highlighting the relevance of glycolysis enzymes as potential therapeutic targets.9,10 The TME plays a vital role in the progression of cancer, metastasis, and response to immunotherapy. Interactions between cancer cells and elements of the TME, such as cancer-associated fibroblasts (CAFs), extracellular matrix (ECM), and immune cells, as well as signaling molecules, are crucial for tumor growth. Understanding these interactions is very important to address the issue of resistance to immunotherapy. Hence, therapies targeted towards the TME have been a notable development with a revolutionary potential for cancer treatment. Epithelial to mesenchymal transition (EMT) is a mechanism during which epithelial cells gain mesenchymal traits. This transformation is regulated by several components of the TME, largely increasing cell motility and invasiveness, thereby advancing metastasis and cancer development. Therefore, disrupting pro-tumor signaling and enhancing treatment efficiency are the objectives of several therapeutic strategies targeting the TME and EMT. In spite of difficulties such as therapy resistance and EMT flexibility, targeting different elements of the TME provides a promising approach for improving cancer treatment outcomes and metastasis suppression. In the recent upsurge in evidence on the function of the TME towards induction of EMT, cancer development and additional therapeutic effects, targeting the primary players of TME has become a promising therapeutic approach to avoid EMT and metastasis. A number of TME-targeting therapeutic modalities have been designed and these modalities are primarily aimed at targeting major immune cells of adoptive immunity such as B and T cells, NK cells, DCs, neutrophils, MDSCs and TAMs, stromal cells such as CAFs and tumor vasculature and ECM. The main concern at present is to increase the functional immune responses of major immune cell populations against tumors. The most prominent immunotherapies for the treatment of cancer are ICIs, adoptive cell transfer techniques like tumor-infiltrating lymphocytes (TILs), T cell receptors (TCRs), and chimeric antigen receptor (CAR) T cell therapy, and cancer vaccines. These therapies, however, have some key challenges. For example, ICIs are only effective in a minority of tumors, and others do not respond at all. Moreover, ICIs may induce immune-related toxicities, which are potentially fatal. CAR-T cell therapy also faces challenges like antigen specificity and can result in complications such as cytokine release syndrome, neurotoxicity, and chronic hypogammaglobulinemia. Furthermore, it is challenging to extend the success of the CAR-T therapy from hematological cancers to solid tumors, mainly because of tumor heterogeneity, poor T cell trafficking, and the immunosuppressive TME. A greater emphasis is being made on the advancement of combination therapies, which combine traditional cancer therapies with agents that target the TME, in order to combat TME-mediated resistance. These innovative treatments are new-age strategies in oncology designed to address the drawbacks associated with monotherapies. By degrading the immune-protective barriers of the TME, such therapies are capable of overcoming the possible resistance pathways, thus augmenting the effectiveness of conventional treatments. In addition, it is also very important to develop reliable biomarkers that will direct TME-targeted therapies in order to be able to produce important clinical efficacy in cancer patients.11 Hypoxia-inducible factor (HIF) is a nuclear protein that controls oxygen homeostasis through transcriptional activity and numerous target gene profiles. The balance between HIF-1 synthesis and degradation is critical for the cellular response to hypoxia, which is primarily controlled by this protein. Because hypoxia is linked to many conditions, understanding HIF could lead to novel strategies for treating a variety of diseases. HIF-1 is mandatory for the survival and growth of solid tumor cells in hypoxic environments, and HIF-1 inhibition suppresses downstream gene-mediated processes like tumor angiogenesis, metastasis, and resistance, as well as overall tumor survival and growth. Targeting HIF-1α prevents the expression of PD-L1 on tumor cells and on tumor-infiltrating myeloid cells; however, it induces PD-L1 expression on normal tissues via an interferon-gamma (IFN-γ)-mediated pathway. Targeting of the HIF-1α/PD-L1 pathway in cancer cells has been reported to reactivate the TILs and induce tumour rejection. Echinomycin, an inhibitor of HIF-1α, augments the effectiveness of CTLA-4 inhibitors, having an efficacy equivalent to the combination of anti-CTLA-4 and anti-PD-1 but without aggravating immune-related adverse effects. By enhancing the tolerance function of PD-L1 in normal tissues while eliminating its function in immune evasion in the TME, HIF-1α inhibition provides a safer and more potent cancer immunotherapy approach.12,13 Another factor that can be considered is tumor necrosis factor-alpha (TNF-α) which is a pleiotropic pro-inflammatory cytokine belonging to the TNF family. TNF-α was originally discovered to have the ability to destroy cancer cells, but it was later found that it could also induce apoptosis, inflammation and cell proliferation. As its mechanism of action differs depending on the cancer, it cannot be employed as a straight anti-cancer drug due to its potent inflammatory and tumour-promoting properties. Rather, understanding its complex signaling can be helpful in finding new therapeutic targets for specific cancers.14 Apart from this, human papilloma-virus (HPV) linked cancers are those that are associated with high-risk HPV infections occurring in multiple locations but have similar biological and immunological characteristics. HPV inhibits the activation of immune cells like NK and cytotoxic T cells by altering the antigen presentation pathway. One of the main causes of cervical cancer is a persistent oncogenic HPV infection. Although cervical carcinomas have an abundance of CD8+ T cells in their stroma, HPV-induced downregulation of chemoattractants renders these cells ineffective in preventing tumor progression. Viral oncoproteins E6 and E7 play significant roles in the initiation and malignant progression. Overexpression of the HPV16E7 oncoprotein has been found to boost the PD-L1 expression while suppressing CTL activity and peripheral blood mononuclear cell (PBMC) proliferation.15,16
Due to their expression in the TME, several lectins and glycans have recently emerged as novel tumor checkpoints.17 Aberrant glycosylation is an eminent trait of tumor cells that has been extensively investigated in several studies.18 Altered glycan expression accelerates tumor growth, and their expression pattern can be utilized as a biomarker to distinguish between various cancers.19 These glycans interact with their respective lectin receptors expressed on either tumor cells or immune cells, dampening the anti-tumor immune response.20 Consequently, it is crucial to uncover the glycan–lectin networks implicated in tumor growth and metastasis since they provide a prospective target for tumor immunotherapeutics. Additionally, blocking these interactions might prove to be a splendid strategy to boost the efficacy of anti-tumor immunotherapies.
In this review, we aim to explore glycan–lectin ICs and their influence on the efficacy of GNVs. We also discuss various ICs in the TME, followed by the drawbacks of immune checkpoint inhibitors (ICIs). We will also emphasize the altered glycosylation profile of tumors, addressing their immunosuppressive nature along with ways in which CLRs, siglecs, and galectins contribute in immune evasion and cancer progression. Considering the resistance towards ICIs, current and prospective approaches for targeting glycan–lectin circuits and future prospects of these endeavors for harnessing the full potential of GNVs will also be highlighted.
2. Tumor checkpoints: current scenario
2.1. Principal tumor checkpoints
ICs have been proved to be crucial in cancer immunotherapy and inflammatory responses. ICs have been observed to be dysregulated in various cancers. ICs, as well as other regulatory cells, including M2 MØs, MDSCs, Treg cells, and cytokines, are often triggered during malignancies and infections. Due to the dysregulation of immune checkpoint signals, cancer cells can subvert the immune response and deviously bypass the anticancer immunity.21 Two types of signal are necessary for T cell activation: the first signal is produced when the antigenic peptide/MHC present on APCs interacts with the TCR along with other signals entailing co-signaling chemicals that are independent of any antigen. It is noteworthy that ICs, which operate as co-stimulators or co-inhibitors, strictly control T cell activation. Engagement of costimulatory receptors, such as CD28, is also essential for MHC/antigenic peptide complex and TCR interaction to result in T cell proliferation and migration toward a particular antigen. Contrarily, if coinhibitory receptors are activated concurrently with MHC/antigenic peptide complex and TCR binding, it will disrupt the T cell activation.22,23CTLA-4 is an inhibitory costimulatory molecule expressed by activated CD4+ and CD8+ T cells, B cells, human muscle cells, placental fibroblasts, monocytes (MO), granulocytes, embryonic stem cells, etc. CTLA-4 downregulates T cell activation by interfering with CD28:B7 binding. Intracellular expression of CTLA-4 on mature DCs (mDCs) leads to downregulation of DC maturation and antigen presentation.24,25
PD-L1 is displayed by B and T cells, NK cells, several APCs, tumor cells, and vascular endothelial cells (ECs). PD-1, which is a receptor for PD-L1, is expressed by B and T cells, NK cells, DCs, MOs, and MØs. PD-L1, upon interacting with PD-1, downregulates T cell activity, which helps PD-L1-expressing tumor cells to escape CTL-mediated cell death.26
TIM-3 is largely expressed on fully differentiated T helper cell type 1 (Th1) and, upon binding to Gal-9, reduces Th1 responses by inducing the death of IFN-producing Th1 cells. The soluble form of TIM-3 suppresses Th1 responses, lowering antigen-specific T cell responses and hence downregulating anti-tumor immunity.27 TIM-3 also serves as a potent inhibitor of CD4+ T helper and CD8+ CTLs.28
LAG-3 is expressed on B cells, NK cells, TILs, and DCs. LAG-3 has a higher affinity for the nonholomorphic region of MHC class II compared with CD4 and is in charge of suppressing the proliferation, activation, and homeostasis of T cells. LAG-3 also exhibited synergistic effects with PD-1 and PD-L1, and further LAG-3 and PD-1 collectively inhibit T cell function along with antitumor immune responses.29,30
TIGIT is found on memory and regulatory CD4+ T cells, NK cells, T cells, follicular helper T cells, as well as follicular Treg cells.31,32 TIGIT and CD226 compete for binding with CD155. CD155 binding to CD226 increases activation of T cells; however, binding of CD155 to TIGIT leads to decreased T cell activation. In melanoma patients, it has been shown that the expression of TIGIT is elevated in TILs and antigen-specific CD8+ T cells. Furthermore, CD155 expression is also increased in melanoma cells and the interaction of TIGIT with CD155 leads to the inhibition of T cell responses. Similarly, an increase in TIGIT+CD8+ T cells has been shown in gastric cancer and these undergo metabolic reprogramming and show functional T cell exhaustion. Additionally, CD155 overexpression in gastric cancer cells and its interaction with TIGIT inhibits glucose uptake by TIGIT+CD8+ T cells, impairing T cell effector functions.33
BTLA is highly expressed by activated T cells; however its expression is low on naive T cells, NK cells, MØs, B cells, and DCs.34 Hematopoietic and parenchymal cells such as breast, esophageal, ovarian, melanoma, and colorectal cancer cells express high amounts of herpesvirus entry mediator (HVEM), which induces BTLA tyrosine phosphorylation and inhibits T cell proliferation upon binding with BTLA.35,36 Elevated expression of BTLA on NK cells competes with CD160 for binding to HVEM (a common ligand for both), to provide inhibitory signals for NK cell cytotoxic activity, therefore impairing the immunosurveillance.37
2.2. Checkpoints’ role in tumor progression
Cancer progression has been aided by various immune subversion mechanisms such as anti-inflammatory cytokine production, induction of Treg cells, and expression of ICs.38 Increased expression of ICs and their role in cancer progression and immune evasion is widely known. Expression of CTLA-4 was significantly elevated in patients with stage IV medullary thyroid cancer, and patients with higher CTLA-4 levels are at high risk of tumor recurrence.39 Another study demonstrated that 30% of cervical malignancies were positive for CTLA-4.40 Overexpression of PD-L1 in early-stage tongue squamous cell carcinomas (SCC) was reported, and lymph node metastasis (LNM)-positive cases had higher PD-L1.41 Increased PD-L1 levels in around 20% of triple negative breast cancer (TNBC) cases were found to be involved in inhibition of proliferation and enhanced apoptosis of T cells.42 Aberrant expression of PD-L1 is also associated with increased risk of cancer progression and cancer-specific death in clear cell renal cell carcinoma (ccRCC).43TIM-3 is overexpressed in cervical cancer, and patients with greater levels of TIM-3 had advanced grades of cancer, higher metastatic potential, and lower overall survival rates.44 Overexpression of TIM-3 has also been observed in glioblastoma, where it is the most prevalent co-inhibitory IC and plays a regulatory role in the malignant behavior of glioma cells. TIM-3 is also responsible for an immunosuppressive TME by promoting anti-inflammatory/pro-tumorigenic MØ activation and T cell exhaustion.45 Increased TIM-3 expression on CD4+ T cells was also observed in non-small cell lung cancer (NSCLC) and was found to be responsible for LNM and advanced cancer stages.46
LAG-3 overexpression on TILs from NSCLC was found to be correlated with PD-1 overexpression on TILs as well as PD-L1 on tumor cells. LAG-3-overexpressing TILs were more prevalent in nonadenocarcinoma than in adenocarcinoma, and the presence of LAG-3 alone or in combination with PD-1/PD-L1 was responsible for early postoperative recurrence.47 LAG-3 expression has also been linked to angiogenesis and poor prognosis in those with hepatocellular carcinoma (HCC).48
A subset of TILs was also shown to overexpress TIGIT. High-affinity ligand of TIGIT, CD155, was shown to be overexpressed on pancreatic ductal adenocarcinoma (PDAC) cells and increased signaling via the TIGIT/CD155 axis was implicated in immune evasion in PDAC.49 A considerable rise in the number of TIGIT+ T cells in gastric cancer patients is engaged in immune dysfunction and tumor growth.33
Overexpression of BTLA in gastric cancer was found to be associated with LNM and cancer progression.35 Patients with lymphatic invasion and advanced tumor stage had increased levels of BTLA expression, which resulted in shorter relapse-free and overall survival, indicating the role of BTLA in cancer progression and poor prognosis in NSCLC.34
2.3. Impact of ICs on GNV-mediated immune responses
XPT-based glycan-conjugated nanovaccines ‘the GNVs’ are emerging as a novel cancer immunotherapy strategy. GNVs are effective for targeting CLRs on APCs because glycans are widely present in the body, and their immunogenicity is relatively low. Therefore, glycans are ideal in the preparation of DC-targeted immunotherapies. However, due to various genetic and epigenetic alterations caused by high mutation rates in cancer, several inhibitory ICs are upregulated, which suppress the anti-tumor immunity and promote the immunosuppressive TME, which can be detrimental to the effectiveness of the GNVs, making it necessary to inhibit these ICs.2 CTLA-4, upon engagement with B7 on APCs, has been implicated in a variety of inhibitory processes, for example, inhibition of cytokine production and T cell proliferation. B7 engagement with CTLA-4+ Tregs induces activation of the enzyme indoleamine 2,3-dioxygenase, which results in the initiation of tryptophan catabolism in DCs, and thus reduces T cell proliferation and survival.50 Downregulation of CD80/CD86 expression on APCs occurs through CTLA-4-dependent Treg trogocytosis. This process disrupts cis-CD80/PD-L1 heterodimers, leading to an increase in the population of APCs that are low in CD80 and high in PD-L1 expression. Together, these effects allow Tregs to have dual suppressive actions: they limit the T cell stimulatory activity of APCs and promote the upregulation of PD-L1, which inhibits PD-1+ effector T cells.51 The presence of CTLA-4 on breast cancer cells is shown to suppress DC maturation and inhibit the capability of DCs to promote the differentiation of naïve CD4+ T cells into IFN-γ+ Th1 effector cells. DCs treated with CTLA-4+ breast cancer cells showed less potency in stimulating CD8+ T cells to produce granzyme B, thus suppressing the functions of CTLs and antitumor immune activation in a CTLA-4-dependent manner.52 Elevated PD-1 expression on TILs as well as MART-1/Melan-A melanoma antigen-specific CD8+ T cells is associated with the exhausted phenotype of TILs with an impaired effector function.53 PD-L1 expression on tumor cells is able to directly inhibit the CD8+ T cell cytotoxicity in the TME, supporting tumor growth by suppressing the anti-tumor immune response aimed towards PD-L1-expressing tumor cells.54 TIM-3 negatively regulates innate and adaptive immunity and plays a significant role in cancer immunity. TIM-3 has been shown to regulate membrane transfer among TIM-3-expressing APCs and CD8+ TILs in a phosphatidylserine-dependent manner in the TME and suppress CD8+ T cell-mediated anti-tumor immunity via T cell trogocytosis and fratricide killing.55 TIM-3 is highly expressed on human DCs, which coordinates with TLRs promoting inflammation, and once the Th1 response is generated, TIM-3, which is expressed on terminally differentiated Th1 cells, induces Gal-9 upregulation, resulting in a termination of the Th1 immune response. Tim-3 also acts in the regulation of pro- and anti-inflammatory innate immune responses. The expression of proinflammatory cytokine IL-12 and anti-inflammatory cytokine IL-10, as well as the activation-associated up-regulation of PD-1, are dependent on Tim-3 expression and signaling. Tim-3 acts as a brake on TLR-driven IL-12 and IL-10 expression during innate immune responses determining the inflammatory outcome.56,57 LAG-3 negatively regulates the activation of T cells and contributes to the exhaustion of CD8+ T cells. Expression of TIGIT is associated with decreased cytokine production, degranulation, and cytotoxicity in human NK cells and can directly inhibit the activation and proliferation of effector T cells.582.4. Existing strategies to tackle checkpoint-mediated immune evasion
With the advent of immunotherapy, therapeutic approaches for the treatment of cancer have undergone a significant transformation recently.59 James Allison and Tasuku Honjo in 2018 shared the Nobel Prize in Physiology & Medicine in recognition of their independent work developing approaches that boost the immune response against tumor cells. ICIs refer to a group of mAb-based therapies that strive to prevent ICs from engaging with their ligands. These treatments mainly target PD-1, PD-L1, and CTLA-4. Due to the longevity of the responses and the impacts on overall survival of the patients, ICIs have received a lot of attention. Although various antibodies are already FDA-approved for various cancers, therapies targeting ICs, such as TIM-3, TIGIT, and BTLA, are still under development.60CTLA-4, being the negative regulator of T cell function, has emerged as a desirable therapeutic target for boosting T cell activity. Ipilimumab, marketed as Yervoy®, was first FDA-approved in 2011 as a human CTLA-4-blocking antibody implicated in the treatment of metastatic melanoma.61 Primary analysis of pooled data from multiple studies, 10 prospective and 2 retrospective, assessing the long-term survival of 1861 patients with advanced melanoma showed that ipilimumab treatment has a 3-year survival rate with the survival time of some patients nearing 10 years.62 Although ipilimumab is presently only licensed as a therapy for melanoma patients, it is being studied for the treatment of several other cancers, including renal cell carcinoma (RCC), NSCLC, HCC, colorectal cancer (CRC) and esophageal cancer (NCT02231749, NCT02477826, NCT01658878, NCT02060188 and NCT03143153). Another CTLA-4-blocking antibody, tremelimumab (brand name Imjudo), in combination with durvalumab, was approved by the FDA for treating adult individuals having unresectable HCC. Furthermore, the combination of tremelimumab with durvalumab and platinum-based chemotherapy was also approved for treating adult patients with metastatic NSCLC.63
Following the findings of KEYNOTE-001 clinical trial (NCT01295827), which examined patients with unresectable or metastatic melanoma, the FDA initially approved pembrolizumab, marketed as Keytruda®, as the first humanized IgG4 antibody against PD-1, in September 2014.64 The FDA approved nivolumab, a completely human IgG4 monoclonal antibody (mAb) sold as Opdivo®, in December 2014. Its approval was based on the outcomes of the CheckMate 037 study, which demonstrated a superior therapeutic efficacy of nivolumab compared with chemotherapy in those with metastatic melanoma where tumors had progressed even after treatment with ipilimumab.65,66 Libtayo (cemiplimab-rwlc) is a PD-1-blocking antibody used for treating patients with cutaneous squamous cell carcinoma (CSCC) (approved in 2018) and basal cell carcinoma (BCC) (approved in 2021).67,68
Three PD-L1-blocking antibodies have so far received FDA approval. Genentech's atezolizumab, marketed as Tecentriq, was approved in 2016 to treat adult patients with metastatic urothelial carcinoma who were ineligible for cisplatin-containing chemotherapy or those who experienced disease progression during or after platinum-containing chemotherapy.69 Additionally, this therapy can be used in conjunction with bevacizumab, paclitaxel, and carboplatin as first-line therapy for patients suffering from metastatic non-squamous NSCLC who do not have mutations of epidermal growth factor receptor (EGFR)/anaplastic lymphoma kinase (ALK).70 Adult patients having unresectable locally advanced or metastatic HCC who have not undergone prior systemic therapy can receive atezolizumab in conjunction with bevacizumab.71 Atezolizumab can be utilized as a first-line therapy for the patients with extensive-stage small cell lung cancer (ES-SCLC) in conjunction with carboplatin and etoposide.72 Furthermore, the combination of the PD-L1 inhibitor atezolizumab with the BRAF pathway-targeting drugs cobimetinib and vemurafenib has been shown to be a safe and effective therapy option for patients with BRAF V600 mutation-positive metastatic melanoma.73 Imfinzi (durvalumab) is another PD-L1-blocking antibody which is implicated in the treatment of patients with locally advanced, unresectable stage III NSCLC.74 Durvalumab can also be used as first-line therapy for adult patients with ES-SCLC in conjunction with etoposide and either carboplatin or cisplatin.75 Avelumab, a fully human IgG1 anti-PD-L1 antibody marketed as Bavencio®, was authorized in 2017 for treating patients of 12 years of age and above with metastatic Merkel cell carcinoma (MCC).76 In 2022, the FDA-approved Opdualag™, which combines nivolumab targeting PD-1 along with the LAG-3-blocking antibody relatlimab for the treatment of adult and pediatric patients aged 12 years and older having unresectable or metastatic melanoma.77
Despite the astounding performance of ICIs in enhancing the rate of outcome in a particular patient population, only 20–30% of patients having RCC, NSCLC, and melanoma have benefitted through this therapy. This unresponsiveness towards ICIs may be characterized into two classes of patients: those who did not react whatsoever (primary resistance) and those who relapsed following incomplete response to treatment with ICIs (acquired resistance). These non-responder patients are subjected to exorbitant therapy expenditures and side effects, with little advantage from these therapies. The key reason for patients not efficiently benefiting from ICIs is associated with the defect in the behavior of the T cells within the TME. Tumor intrinsic resistance is generally due to the loss or gain of mutations in the tumor suppressor genes and oncogenes, respectively. As for the extrinsic factors of resistance, an immunosuppressive TME evades the immune-mediated eradication of cancer cells by depleting the required nutrition or by producing materials that are deleterious for the immune cells. Even though the ICIs potentiate T cell activation, other essential steps are required for the immune-mediated killing of the tumor to precisely control the tumor growth.78 Apart from this, various genetic, epigenetic, and metabolic changes prevent trafficking of T cells to the tumor site trigger dysfunction in immune cells, hinder antigen presentation, and promote tumor survival. Additionally, lifestyle factors, such as obesity and microbiome composition, also affects the resistance towards ICIs.79 It has been shown that TROP2 overexpression is recognized as a major driver of primary resistance to PD-L1 blockade in advanced NSCLC, resulting in a significantly lower progression-free survival (PFS) and worse overall survival (OS) in atezolizumab-treated patients. Mechanistically, overexpression of TROP2 suppresses anti-tumor immunity by reducing T cell infiltration. It has also been shown that the overexpression of TROP2 is also linked with decreased gamma delta T cells and Th1 cells within the TME.80 In another study, it was shown that there are 3 key mechanisms that lead to resistance to ICIs in melanoma, namely antigen loss via melanoma de-differentiation, impaired antigen presentation, and immune cell exclusion through PTEN loss. The de-differentiation is driven by the sustained tumor-intrinsic IFNγ signaling which is associated with the altered melanoma secretome and promotes an immunosuppressive TME that is enriched with exhausted CD8+ T cells. In addition, defects in MHC class I and MHC class II expression, often induced by mutations in β2M, and CIITA, were also found to disrupt antigen presentation. Additionally, PTEN loss is linked to immune exclusion, particularly in brain metastases, preventing effective T cell infiltration.81 Furthermore, resistance to ICIs in melanoma develops through distinct mechanisms depending on the type of therapy. Anti-CTLA-4-resistant tumors have sustained immune response with expanded TCR clones. However, these tumors contained high numbers of FOXP3+ T cell whose immunosuppressive activity is TCR signaling-dependent indicating that the augmented TCR clonality displays a heightened immunosuppressive milieu mediated by FOXP3+ Treg cells. In contrast, anti-PD1-resistant tumors are immune-depleted, bearing significantly reduced CD8+ T-cell infiltration, unexpanded TCR clones, and loss of MHC class I expression. Such an immune-poor TME is associated with melanoma cells with a de-differentiated phenotype that lacks expression of MHC class I molecules.82 Apart from this, it has been shown that one of the major obstacles to the success of PD-L1 blockade in NSCLC is acquired resistance, in over 60% of initial responders. Acquired resistance has been proved to be associated with persistent IFNγ signaling, which causes immune dysfunction and CD8+ T cell exhaustion.83 Additionally, tumor-associated fibrosis in NSCLC is found to contribute resistance to ICB. Large amounts of fibrosis have been shown to be associated with reduced T cell infiltration, impaired DCs, and altered MØ phenotypes, favoring immunosuppression. CAFs expressing Col13a1 also promote immune resistance by recruiting MØs and Tregs while restricting DC and T cell recruitment.84 Furthermore, gastric cancer peritoneal metastases (GCPM) have been shown to develop an immunosuppressive TME governed by the stroma-myeloid niche which consists of SPP1+TAMs along with thrombospondin 2 (THBS2)+ matrix CAFs (mCAFs), which promote resistance to ICB therapy in GCPM. This resistance is mediated by CAF–MØ crosstalk through the C3–C3a receptor 1 (C3AR1) axis, where aberrantly accumulated THBS2+mCAFs facilitate the peritoneum-specific tissue-resident MØ recruitment as well as their conversion into SPP1+TAMs through C3 and C3AR1, establishing a tumor-promoting stroma-myeloid niche in GCPM.85 In another study it has been revealed that chronic IFN-γ stimulation leads to epigenetic changes in cancer cells, creating inflammatory memory domains (IFN-IMDs) that sustain immune suppression. STAT1 controls active enhancers for IFN-IMDs, while interferon regulatory factor 3 (IRF3) maintains chromatin accessibility even after IFN signaling diminishes. The primed enhancers upregulate IFN-stimulated resistance signature (ISG.RS) genes like OAS1, amplifying the IFN-I pathway and promoting the expression of immune inhibitory genes. This prevents IFN production by immune cells, disrupts T cell–DC interactions, and induces CD8+ T cell exhaustion. Eventually, resistant cancer cells acquire IMDs to develop a self-sustaining IFN-I-mediated feedback loop that reinforces immune dysfunction and ICB resistance.86
Given the resistance to ICIs, it is crucial to look for other checkpoints implicated in immunosuppressive TMEs and ways to block them. Glycan–lectin interactions are one such focal point that can be studied for their reverberations in immunosuppressed TMEs and the fashion in which they can be blocked.
3. Glycans as new emerging checkpoint in TMEs
Nearly all essential molecular processes, such as intracellular signaling, cell–cell communication, cell–matrix adhesion, and various signal transduction cascades, involve glycan expression on the cell surface and ECM, which can be present as free form or as glycoconjugates. Glycosylation is a post-transcriptional modification that involves the enzymatic activity of glycosyltransferases, which give rise to glycosidic linkages between saccharides and other saccharides, proteins, or lipids. Glycoconjugates are mainly described on the basis of the nature and type of linkage with their non-glycosyl part. Glycoproteins can consist of various glycan structures which are covalently bound to polypeptide backbones. N- and O-linked glycosylation are major mechanisms involved in the addition of glycans to polypeptide backbones. Glycosylation, being a non-template process, has scope for a range of modifications in their machinery induced by various internal or external stimuli, which can be blamed for the development of various diseases.19,87 Therefore, aberrant glycosylation is considered as hallmark of various cancers.3.1. Aberrant glycosylation in TMEs
Altered glycosylation is a well-recognized trait of cancer cells that contributes to the development of tumor-associated carbohydrates. Tumor cells display a vast array of glycosylation modifications compared with normal cells. As these glycan modifications are protein-, site-, and cell-specific, they encourage molecular heterogeneity and functional variation within diverse cell populations. Apart from protein glycosylation, each cell also displays a specific glycolipid profile, and with the transformation of normal cells, there is a change in glycolipid profile as a result of certain developmental arrests or regressions. Incomplete synthesis and neo-synthesis are the key mechanisms vital for the tumor-associated alterations in glycans. Incomplete synthesis is usually observed in the initial stages of cancer development, which is the repercussion of the hindered development of glycans on normal epithelial cells due to the partial impairment of transcription of glycogenes involved in the production of complex glycans leading to the formation of truncated glycans such as sialyl Tn (STn) on malignant cells. The development of particular antigens, like sialyl Lewis a (SLea) and sialyl Lewis x (SLex), which are expressed de novo in various cancers, is a common example of neo-synthesis, frequently found in advanced stages of cancer.87–90 Altered glycan expression in cancer cells occurs due to a number of reasons, which include altered expression (under/over) of glycosyltransferases as a consequence of dysregulation at transcriptional levels,91 impaired chaperone function92 and alteration in glycosidase activity.93 A change in the tertiary conformation of the peptide backbone is another factor contributing to altered glycan expression. Altered glycosylation can also be attributed to the variability of diverse acceptor substrates, along with the availability and abundance of sugar nucleotide donors and cofactors.94 The expression as well as the placement of pertinent glycosyltransferases in the Golgi apparatus can also influence glycan expression.95The process of cancer development, tumor progression and subsequent metastatic spread has been shown to be linked to a loss of cell–cell adhesion, and acquisition of the ability to migrate and invade the surrounding healthy tissues. These processes have also been linked to EMT. Changes in the glycan profile of cancer cells have been shown to influence migratory and invasive processes, which contribute to the development of metastatic cancer.96,97
E-cadherin, a vital transmembrane glycoprotein, serves as a key cell–cell adhesion molecule in epithelial cells, playing a crucial role in EMT.97 The interplay between O-mannosylation and N-glycosylation is responsible for the dysregulation of E-cadherins.98 E-cadherin mediates cell–cell adhesion that functions in the formation of a multiprotein complex that is anchored to the actin cytoskeleton, known as adherens junctions (AJs). E-cadherin-mediated AJs are prone to changes in their composition and stability because of their dynamic nature. Changes in the composition and amount of the N-linked glycan status of E-cadherins affects the stability of AJs by directly affecting E-cadherin-related protein complexes and their links to the cytoskeleton as described in Fig. 3.99 Overexpression of the gene DPAGT1 is associated with excessive N-glycosylation of E-cadherin, which is implicated in the production of complex N-glycan modified E-cadherins linked to impaired maturation of AJs, demonstrating reduced connection with γ-catenin, α-catenin, and vinculin.100 In diffuse gastric carcinoma, N-acetylglucosaminyltransferases V have shown detrimental effects on E-cadherin as they catalyze its modification through the production of β1,6 GlcNAc branched structures, which leads to disruption of the cadherin/catenin complex, thereby impairing cell aggregation and further resulting in tumor cell invasion and progression.101 Furthermore, elevated O-GlcNAcylation in fibroblast cell lines increases the transcriptional activity of β-catenin and elevates the expression of β-catenin and E-cadherin, which is associated with increased cell migration. In vivo studies in a murine orthotropic CRC model also showed that elevated O-GlcNAcylation is responsible for increased tumor and metastatic progression, and a high mortality rate.102 On the other hand, in ovarian cancer, elevated O-GlcNAcylation facilitates cancer cell mobility and resonates with a decrease in E-cadherin levels along with inhibition of E-cadherin/catenin complex formation, thereby lowering the intercellular adhesion.103
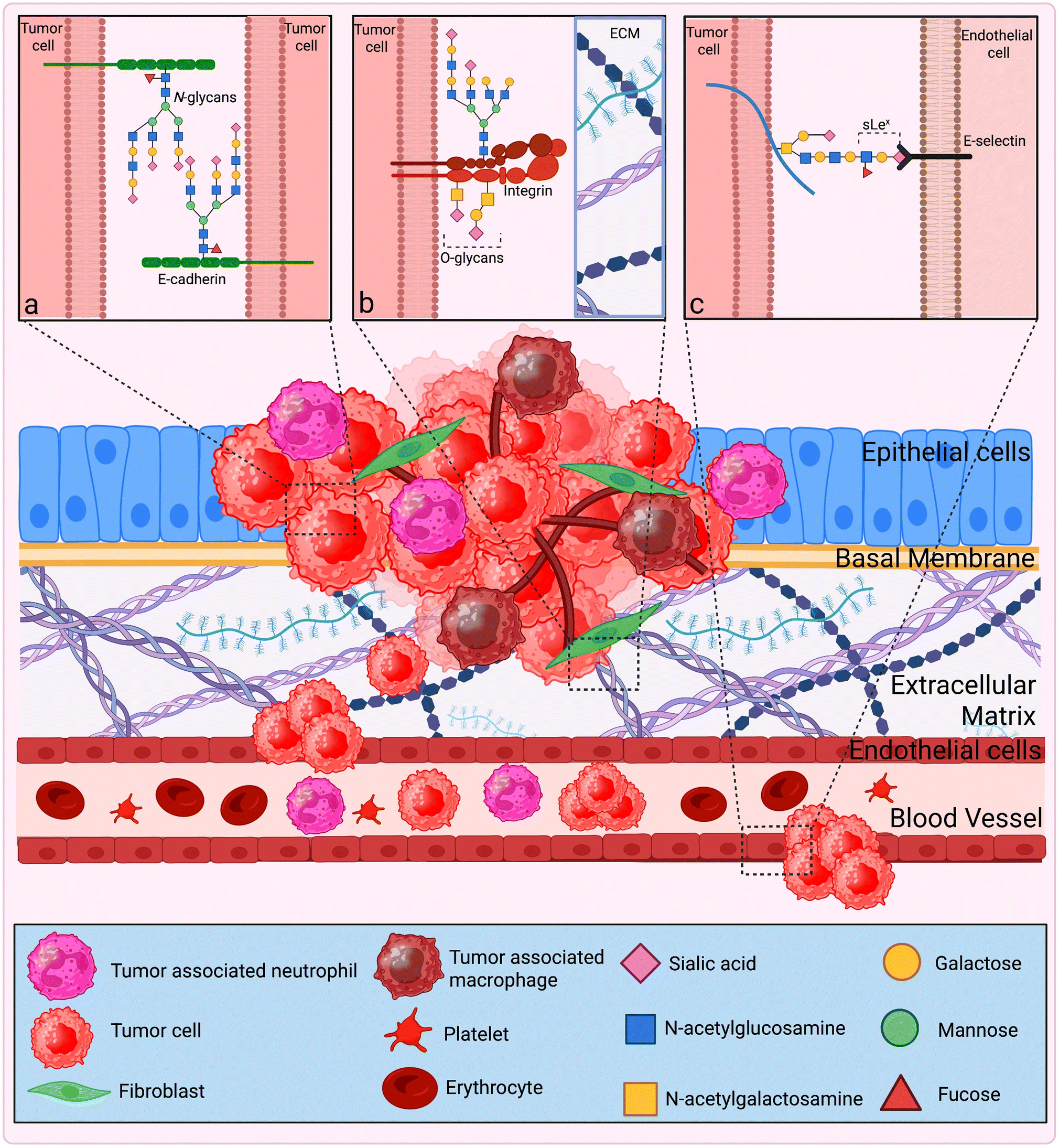 | ||
| Fig. 3 Glycosylation of adhesion molecules and their role in cancer progression: loss of cell–cell contact and cell adhesion has been seen during cancer growth, with implications for immune evasion and metastatic spread. The increased tendency of cancer cells for migration, which is followed by EMT, is also connected to a loss of cell adhesion. These mechanisms have been demonstrated to be influenced by the glycosylation of the adhesion molecules present in the TME. (a) The alteration of E-cadherin through the addition of β1,6-N-acetylglucosamine (β1,6GlcNAc)-branched N-glycan structures via increased activity of N-acetylglucosaminyltransferase V weakens cell adhesion and further increases tumor cell invasion. Alterations in O-GlcNAcylation are linked to the development of cancer; (b) integrins exhibit differential glycosylation in both O- and N-linked glycans during the migration of tumor cells. Interactions between cells and the ECM are hampered by terminal sialylation, which encourages a more migratory and invasive phenotype; (c) cancer cell adhesion and metastasis are aided during extravasation by the tumor-associated carbohydrates SLea and SLex, which act as ligands for receptors involved in adhesion, such as E-selectin, P-selectin, and L-selectin (created in BioRender. Jain, M. (2025) https://BioRender.com/k82s187). | ||
Aberrant O-glycosylation is a common occurrence on the tumor cell surface and is often associated with poor prognosis and adverse outcomes in cancer patients. In Golgi bodies, T, Tn, and STn antigens can be synthesized with the help of glycosyltransferases like T-synthase and ST6GalNAc-I as shown in Table 1. Core 1 synthase-specific molecular chaperone (Cosmc) is a distinct molecular chaperone for T-synthase, facilitating its proper folding within the ER. Dysregulation of glycosyltransferases, molecular chaperones, or the environment can all contribute to O-glycan dysregulation. Tn, STn, and T antigens are frequently neo- or over-expressed in various cancers, including gastric, prostate, colon, endometrial, breast, esophageal, and lung cancer.104
| Glycosyltransferase | Type of glycosylation | Implication in cancer | Ref. |
|---|---|---|---|
| Polypeptide N-acetylgalactosamine-transferase 1 (GALNT1) | O-Glycosylation | In gastric cancer, GALNT1 overexpression enhances malignancy via promoting the Wnt/β-catenin signaling pathway through aberrant O-glycosylation of CD44 | 105 |
| Polypeptide N-acetylgalactosamine-transferase 2 (GALNT2) | O-Glycosylation | By altering O-glycosylation and EGFR activity, GALNT2, which is overexpressed along the invasive edge of OSCC, enhances the invasive capabilities of OSCC cells | 106 |
| Polypeptide N-acetylgalactosamine-transferase 3 (GalNAc-T3) | O-Glycosylation | GalNAc-T3 affects the growth and survival of pancreatic cancer cells by supporting the activity of O-glycosylated proteins | 107 |
| Polypeptide N-acetylgalactosamine-transferase 4 (GalNAc-T4) | O-Glycosylation | In CRC, GalNAc-T4 is increased in stages I and II (non-metastatic stages) and enhances the capacity for colony and sphere formation, two critical aspects of cell tumorigenicity | 108 |
| Polypeptide N-acetylgalactosamine-transferase 6 (GALNT6) | O-Glycosylation | By directly interacting with and O-glycosylating the chaperone protein GRP78, GALNT6 increases MEK1/2/ERK1/2 signaling in lung cancer cells, hence promoting EMT | 109 |
| Polypeptide N-acetylgalactosamine-transferase 7 (GALNT7) | O-Glycosylation | GALNT7 promotes the growth of prostate tumors via altering the O-glycosylation of prostate cancer cells | 110 |
| Polypeptide N-acetylgalactosamine-transferase 10 (GALNT10) | O-Glycosylation | Ability of gastric cancer cells to proliferate and migrate could be regulated by GALNT10 | 111 |
| Polypeptide N-acetylgalactosamine-transferase 12 (GALNT12) | O-Glycosylation | GALNT12 affects the PI3K/Akt/mTOR axis, which promotes the malignant features of glioblastoma multiforme | 112 |
| Polypeptide N-acetylgalactosamine-transferase 14 (GALNT14) | O-Glycosylation | GALNT14 promotes the first stage of mucin-type O-glycosylation and changes cell motility, proliferation, EMT gene expression, and MMP-2 activity, all of which lead to breast cancer invasion | 113 |
| Core 1 β1,3-galactosyltransferase (C1GALT1) | O-Glycosylation | By altering the O-glycosylation and activity of FGFR2, C1GALT1 overexpression increases the invasive potential as well as stem-like cell characteristics of colon cancer cells | 114 |
| N-Acetylgalactosaminide α-2,6-sialyltransferase 1 (ST6GalNAc-I) | Sialic acid addition to initiating GalNAc residues forming STn on glycoproteins | By altering the sialylation of MUC5AC, ST6GalNAc-I facilitates lung cancer metastasis | 115 |
| ST3 β-galactoside α-2,3-sialyltransferase 1 (ST3GAL1) | Addition of α2,3-linked sialic acid to Gal β1,3 GalNAc | The sialylation of Tn to STn increases when ST3GAL1 is overexpressed. In ovarian cancer, overexpression of ST3GAL1 promotes cell invasion, migration, and proliferation | 116 |
| ST6 β-galactoside α2,6-sialyltransferase 1 (ST6GAL1) | Addition of α2,6-linked sialic acids to N-glycosylated proteins | ST6GAL1 promotes tumor cell migration, invasion, apoptosis resistance, and EMT | 117 |
| Core2 β-1,6-N-acetylglucosaminyltransferase (C2GnT) | Forms N-acetylglucosamine branches in the O-glycans (core2 O-glycans) of cell surface glycoproteins | C2GnT encourages prostate cancer cells to survive longer in the host bloodstream, which will promote prostate cancer metastasis | 118 |
| N-Acetylglucosaminyltransferases V (GnT-V) | Addition of β1,6 GlcNAc branching N-glycans | E-cadherin is destabilized by GnT-V, which results in its mislocalization and unstable adherens junctions, which hinder cell–cell adhesion, tumor cell invasion, and tumor cell progression | 101 |
| Fucosyltransferase IV (FUT 4) | Fucosylate oligosaccharides by transferring GDP fucose to the terminal N-acetylglucosamine with the α1,3-linkage | Encourages cell proliferation and cell cycle progression by overexpressing Ley | 119 |
| Fucosyltransferase VI (FUT 6) | Fucosylate sialylated acceptors and produce sialyl Lewis antigens | Boosts the tumor growth by altering the expression of p21 and the PI3K/Akt signaling pathway | 120 |
| Fucosyltransferase VII (FUT 7) | Fucosylate sialylated acceptors and produce sialyl Lewis antigens | Promotes the EMT and immune infiltration in bladder urothelial carcinoma | 121 |
| Fucosyltransferase VIII (FUT 8) | Core fucosylation | Promotes invasion and metastasis of pancreatic ductal adenocarcinoma | 122 |
Under normal circumstances, Tn antigen is modified through O-glycosylation to form elongated and complex O-glycans through T-synthase in the Golgi bodies of the cell. This process makes Tn antigen undetectable in healthy tissues. T-synthase and Cosmc play a crucial role in normal O-glycosylation processes. However, dysfunction in Cosmc due to mutations, deletions, or hypermethylation can cause Tn antigen expression in various cancers.123 Furthermore, unique adhesive interactions between cancer cells and endothelium mediated by altered glycosylation, especially GalNAc-glycans like Tn antigen, are often associated with metastatic dissemination and poor prognosis in a number of malignancies.124 Additionally, Tn antigen has also been shown to be implicated in activation of the EMT pathway in CRC, which was indicated by a decrease in expression of epithelial cell marker expression like E-cadherin and a consequent increase in the expression of mesenchymal markers like N-cadherin, vimentin, and Snail owing to upregulated expression of H-Ras in Tn+ CRC cells.123 Interaction of Tn+ lung cancer cells with macrophage galactose-type lectin 2 (MGL2)+CD11c+F4/80+ cells is what contributes to the recruitment of IL-10-producing T cells and tumor angiogenesis. Additionally, the ability of Tn+ lung cancer cells to secrete vascular endothelial growth factor (VEGF) is linked to CD31 expression on these cells, which most likely results in the development of highly vascularized lung tumors.125 In addition to these, Tn antigen in TNBCs promotes lung metastasis in conjugation with local and systemic immunosuppression at the site of the primary tumor and the metastasis.126
STn antigen is another truncated O-glycan that is produced by sialyltransferase ST6GalNAcI. Not only does ST6GalNAc I produce STn antigen, but it is also capable of transforming the whole glycosylation pattern of various glycoproteins in a variety of malignancies. In breast cancer, concomitant with these alterations in glycosylation pattern, STn-expressing tumors exhibit reduced cell adherence and enhanced mobility which is responsible for the increased tumorigenic potential of breast cancer cells.127 A contribution of STn antigen to the metastatic spread of gastric cancer is also reported. Expression of STn antigen on the cancer cell surface leads to heightened intraperitoneal metastasis and tumor growth along with a shortened survival time, which may be caused due to accelerated cell proliferation, enhanced migration, modified adherence to target matrices or cells, and/or decreased apoptotic activity. Also, MUC1 and CD44 are the major carrier proteins of STn antigen, indicating their possible involvement in acquisition of the metastatic phenotype by gastric cancer cells.128 Both Tn and STn antigens are upregulated in ovarian cancer cells found in serous effusions, suggesting that this is a transient phenotypic shift that promotes metastasis.129 Aberrant expression of STn in pancreatic cancer boosts the tumor growth and metastasis as well as decreases the survival by inducing EMT and stemness features in PDAC cells.130 It is likely that in carcinomas, STn expression reduces homotypic aggregation, thereby facilitating the escape of individual cells from the primary tumor. Consequently, migration of STn-expressing single cells makes the underlying mucosa more amenable to invasion, which eventually allows these cells to reach lymph or blood vessels. But for metastatic cells to spread and invade the target region, they need to have specific adhesion properties. Thus, these characteristics of the STn antigen aid in the explanation of how it alters the phenotype of malignant cancer by promoting more aggressive cell activity, such as a decrease in cell–cell aggregation, increase in ECM adherence, migration, and further invasion.131
Overexpression of fucosylated epitopes is often observed on the cancer cell surface (Fig. 4) and is mainly ascribed to elevated expression of relevant fucosyltransferases (FUTs). Lewis (Le) antigens belong to the human histo-blood group antigen system and are made up of terminal fucosylated carbohydrate epitopes. Numerous cancers such as lung, breast, colorectal, and pancreatic cancer have been documented to overexpress Lewis antigens and associated FUT proteins.132 As seen in lung cancer tissues, adenocarcinoma and squamous cell carcinoma showed substantially higher levels of Lex expression than small cell carcinoma. Contrarily, SLex expression was more prevalent in adenocarcinomas compared with squamous cell or small cell carcinomas. Additionally, Lex and SLex are also implicated in the reduced survival span of patients.133 SLex is expressed more frequently in primary lung cancer than SLea and plays a crucial role in hematogenous metastasis.134 Apart from Lex and SLex, Ley is also expressed in NSCLC tissues and is also considered as a prognostic marker related to the grade of cancer differentiation.135 In young patients (<50 years) having TNBC, Lex functions as a poor prognostic marker for recurrence free-survival and is associated with reduced overall survival.136 Additionally, Ley antigen expression in individuals with malignant breast cancer may be a reliable indication of the level of malignancy. Leb and SLex antigen expression in breast cancer can also predict LNM.137 However, in the case of lymph node negative breast carcinomas, increased Leb/y expression is associated with reduced overall survival of the patients.138 Higher expression of SLea in highly metastatic CRC cells has been attributed to effective extravasation, in contrast to lower levels of SLea expression in non-metastatic cells.139 In addition to being connected with disease aggressiveness, such as undifferentiated histologic type, manner of severe invasion, and LNM, expression of SLex is also associated with tumor relapse.140
 | ||
| Fig. 4 Changes in glycosylation pattern with transformation of healthy cells to malignant cells: changes in glycosylation are common during cellular transformation and can happen early or late in the course of cancer. Among other essential biological functions, glycans support the recognition of immune cells, cell–cell adhesion, and cell–ECM adhesion. Malignant cells often exhibit complex and branched oversialylated and/or fucosylated N-glycans compared with healthy cells. Elongated LacNAc chains on N-glycans are also overexpressed on the cancer cells. On the other hand, less complex and truncated O-glycans like T, Tn, and STn antigens, are expressed by malignant cells. Furthermore, a number of sialic acid-expressing glycosphingolipids known as gangliosides, including trisialogangliosides like GT1b, disialogangliosides like GD1a, GD2, and GD1b, and monosialogangliosides like GM3 and GM1a, have been linked to malignancy. Overexpression of “core” fucosylation, which attaches to the innermost GlcNAc of N-glycans, is also considered crucial for cancer development and progression. Selectin ligands SLea/x, which are terminal structures present on both N- and O-glycans, may also be expressed by some cancer cells. These surface structural changes promote invasion, lymphatic and hematogenous dispersion, immune evasion, and the development of motile and plastic cell forms. Cancer-associated glycans disrupt normal intracellular signaling transduction pathways that lead to the activation of carcinogenic characteristics by disrupting the normal functioning of cell–surface receptors (created in BioRender. Jain, M. (2025) https://BioRender.com/s28p848). | ||
3.2. Immune cell-specific glycan coat
Glycosylation occurs in all secreted or cell surface proteins, as well as in lipids, making it continuously accompany the contacts between molecules on the cell surface or in serum. The immune response stems from contacts between cells and molecules, such as the formation of immunological synapses between T cells and specialized cells, as well as interactions between antigens and antibodies. Moreover, it is highly probable that glycans contribute to and influence these immune encounters. DCs are crucial for antitumoral immunity, in which they can stimulate specific CTLs to target tumor cells.141 DCs, being the primary APCs, hold promise for various anti-cancer immunotherapies. However, one of the significant challenges is approaches in enhancement of the maturation profile of DCs and, specifically, antigen presentation to CTLs.142 DC maturation leads to significant changes in the expression of genes related to glycosylation such as galactosyltransferases, fucosyltransferases, and sialyltransferases. This results in increased expression of LacNAc, sialylated glycans, and Lewis structures.143 Sialic acids, abundant on DCs, are shown to be crucial in the regulation of DC maturation and their interaction with other lymphocytes, such as T cells.144 The expression of sialic acids on DCs has the capacity to suppress both the maturation of DCs and their co-stimulatory functions. Human monocyte-derived DCs (moDCs) are also heavily sialylated and elimination of these sialic acids by sialidases like neuraminidase accelerates DC maturation. Sialidase treatment of DCs facilitates the induction of various phenotypic and functional aspects of maturation, including enhanced antigen presentation, co-stimulatory molecules, and generation of Th1 cytokines.142,145 Increased sialic acid expression on tDCs is responsible for their tolerogenic state. However, withdrawal of sialic acids results in improved stimulatory activity of iDCs, which in turn encourages T cell activation and proliferation.146 Sialic acid-containing glycans expressed on T cell surfaces and APCs can serve as alternative ligands for CD28. These glycans compete with well-established activatory ligand CD80 on the APCs, leading to a weakened co-stimulation. Furthermore, sialic acid removal resulted in increased activation of the naive T cells during antigen presentation, as well as enhanced revival of effector T cells.1473.3. Immune regulation by glycans
One of the attributes of cancer is its ability to elude host immune surveillance. The innate immune system functions by identifying molecular patterns, such as damage-associated molecular patterns (DAMPs) and pathogen-associated molecular patterns (PAMPs), triggering the secretion of pro-inflammatory cytokines and activation of immune cells.148 However, in order to circumvent the antitumor response, cancer cells employ a range of strategies including downregulation of antigen presentation, production of cytokines that induce an immunosuppressive TME, and expression of ICs.149 The presentation of self-associated molecular patterns on host cells dampens pro-inflammatory signals through a mechanism mediated via the expression of specific glycan epitopes. Immune cells employ glycan-binding proteins (lectins) to recognize these glycan epitopes expressed on the surface of target cells, which attenuates the immune cell activation by inhibiting the activating signaling cascades. In cancer, glycosylation of tumor cells aids in immune tolerance by preventing an effective immune response, frequently by upregulating SAMPs.148 In TNBC tumors, B7 homolog 3 protein (B7H3) which is implicated in tumor cell migration, proliferation, invasion, and angiogenesis, is aberrantly N-glycosylated at NXT motif sites, which gives rise to its stability and immunosuppression capability. To maintain its high expression, FUT8 catalyzes B7H3 core fucosylation at N-glycans. This aberrant glycosylation of B7H3 protein inhibited the 26S proteasome-mediated protein degradation and is responsible for maintaining B7H3 protein stability. Additionally, tumor cells having glycosylated B7H3 show reduced in vivo trafficking of tumor-reactive T and NK cells to tumors. These findings shows that glycosylated B7H3 is a critical immunosuppressive factor that regulates the immune response in TNBC.150 GalNAc glycosylation has also been shown to improve uptake of antigen, MHC class II presentation, and CD4+ T cell activation, all of which led to more robust antibody responses. However, GalNAc glycosylation may arrest MHC class I antigen presentation and activation of CD8+ T cells.151 It has been shown that bladder cancer cells expressing the O-glycan branching enzyme C2GnT are highly metastatic because of their increased ability to evade NK cell immunity.152 T antigen, Tn, and STn are recognized as valuable markers for poorly differentiated adenocarcinomas and mucinous carcinomas, which are often linked to poor clinical outcomes.153 Another study has shown that Tn glycosylation of MUC6 protein significantly alters its B cell and T cell immunogenicity, promoting tumor escape.154 Increased expression of truncated O-glycans, like Tn antigen, has been observed in glioblastoma cell lines, in tissues derived from patients, and lower levels are observed in lower-grade gliomas. Additionally, an increased infiltration of immunosuppressive MGL+CD163+ TAMs that bind to Tn antigen was found in patient-derived glioblastoma tissues. Furthermore, overexpression of O-linked glycans was linked to a higher frequency of immunosuppressive PD-L1+ MØs in murine MGL-Lhi tumors and alterations in immune cell frequencies in the bone marrow. These findings suggest that Tn antigen expression influences both local and systemic immune responses and therefore should be considered for possible therapeutic and diagnostic avenues.1554. Lectin expression and cancer immunity
The recognition of glycans expressed specifically in tumor cells has been shown to influence the TME, significantly contributing to the biology of tumor cells and processes of immune escape and immunomodulation. Lectins expressed on the surface of immune cells are integral participants in glycan recognition. Lectins are defined as proteins that identify glycans in a specific, typically multivalent fashion and translate this recognition and structural details into functional cellular responses. As a result, the biological functions of lectins are widely acknowledged in a range of different cellular processes.156,157 These glycan-binding receptors are either secreted or present on immune cell surfaces and recognize the glycan structures present on protein backbones or on lipid structures. Aberrant glycosylation in the TME leads to selective recognition by lectins and modulation of immune responses. Several galectins and CLRs such as MGL and dendritic cell-specific ICAM-grabbing non-integrin (DC-SIGN) are capable of detecting the altered glycosylation. Importantly, these glycosylation changes have been frequently employed as prognostic markers of disease development.1584.1. Lectin pool and their role in the TME
A diverse range of glycans found in cells and tissues (known as the glycome) encodes the vital biological information that plays an important role in reprogramming the cellular fate and function and, as a result, has a significant impact on the balance among health and illness. One of the common characteristics of the tumorigenic process is changes in the glycosylation signature of immune cells, ECs and tumors. These signatures can affect cell adhesion, EMT, angiogenesis, immunoediting and metastasis. Therefore, by controlling the exposure or masking of particular glycoepitopes, an aberrant glycome may modify cellular activities. The conversion of information encoded in the glycome into biological programs relies on the participation of endogenous lectins. Three major families of lectins that significantly contribute to shaping the inflammatory and tumor microenvironment are C-type lectins (including selectins), galectins, and siglecs.159 Galectins and C-type lectins have received much attention in cancer biology in recent times. I-type lectins, such as siglecs, are also increasingly being recognized as important players in the TME due to their widespread presence. Galectins have gained prominence because of their participation in cancer, prognostic significance, and potential as therapeutic targets.160 Galectins might play a role in transitioning from healthy to neoplastic or inflammatory tissues and may prolong these pathological states through both extracellular and intracellular processes. They influence the hallmarks of tumor progression and resistance to a variety of anticancer treatments, such as chemotherapy, radiotherapy, immunotherapy, targeted therapies, and anti-angiogenic therapy.159 Galectin overexpression in various cancers is associated with the aggressiveness of the tumors and subsequent development into metastatic phenotypes.161 Galectins play a role in immunosuppression, including T cell exhaustion, limiting their ability to survive, encouraging the growth of Tregs, inactivating NK cells, and polarizing myeloid cells to differentiate into cells with an immunosuppressive phenotypic form.162 C-type lectins are a family of lectins expressed as CLRs on various APCs, facilitating the identification and capture of several glycosylated antigens in a calcium (Ca2+)-dependent manner, which perform a plethora of actions as uptake receptors and mediators of cell-to-cell interaction.20,163 Various C-type lectins, including DC-SIGN, CD93, CLEC2, CLEC5A, CLEC14A, LOX-1, MMR, MGL and selectins, can promote interactions of cancer cells and platelets, leukocytes, and ECs, hence encouraging tumor invasion, metastasis, and immune suppression.164–166 Selectins, a family of Ca2+-dependent lectins, are well known for their function in mediating adhesion of immune cells to the endothelium to enable their entry into secondary lymphoid organs and sites of inflammation. L-, E- and P-selectin, which are expressed on leukocytes, ECs, and platelets, respectively, are members of the selectin family.167 All primary physiological roles of selectins are to facilitate the leukocyte recruitment to inflammatory locations or lymphoid tissues. Establishment of immune invasion, dissemination, extravasation and formation of a metastatic niche are all facilitated by leukocyte recruitment to tumor locations in a selectin-dependent manner. Selectins primarily promote tumor development through hijacking inflammatory pathways and therefore play a key role in the development and maintenance of the TME.168 Siglecs are the most well-characterized I-type lectins, are widely expressed in the hematopoietic and immune systems and can facilitate cell to cell interactions along with signaling functions.169 The siglec/sialic acid axis helps the immune cells discriminate between self and non-self during homeostasis. However, by upregulating sialic acids on their surface and/or siglecs on immune cells, tumor cells may utilize this characteristic to evade anti-tumor immunosurveillance.170,171 Annexins, recognized as a novel lectin family, were initially described as Ca2+-dependent membrane phospholipid-binding proteins. Annexins influence tumor formation via dynamic and significant changes in gene expression associated with cancer progression. Annexin expression during carcinogenesis also leads to chemotherapeutic drug resistance.172 A higher expression of annexin A1 promotes resistance to oxaliplatin via autophagy which relies on inhibition of the PI3K/protein kinase B (AKT)/mammalian target of rapamycin (mTOR) signaling pathway.173 In pediatric neuroblastoma, higher expression of annexin A2 was found to be associated with advanced cancer stage, higher number of chemotherapy cycles, tumor metastasis, and poor prognosis.174 Annexin A3 was found to be overexpressed in HCC and was linked with poor prognosis in patients with HCC and enhanced cell proliferation, migration, invasion, and chemotherapeutic drug resistance.1754.2. Cross-talk between lectins and glycans in immune evasion and suppression
Glycans that are associated with cancer, including sialic acid-containing structures, Tn antigen, and Lewis antigen, are often attached to membrane-bound or secreted tumor proteins, like CD43, carcinoembryonic antigen, and mucin 1 (MUC1). They can also be bound to membrane lipids as in gangliosides, disialoganglioside 1 (GD1), and monosialic ganglioside 2 (GM2) and GM3. Furthermore, hijacking glycan responses might aid in immune evasion by influencing the activities of APCs, stimulating the differentiation of tumor-associated or anti-inflammatory M2 MØs, and changing T cell differentiation and NK cell activity.18 Therefore, it is essential to understand the tumor glyco-code and the manner in which glycan–immune lectin interactions can lead to immunosuppression in the TME.2Numerous studies have demonstrated that immune cells express a diversity of lectin receptors, especially DC-SIGN, MGL, and siglecs, which regulate immune suppression by binding to tumor glycans.171,176,177 Examples include the hypersialylation of tumor cells, which boosted the surface expression of the sialic acid which binds to siglec. Strong inhibitory activity is induced by siglecs binding to sialic acids, which suppresses the immune system.2 Melanoma cell hypersialylation was found to be linked to increased tumor development in vivo, which was accompanied by increased Treg cells, decreased effector T cells and NK cell activity.178 Due to the expression of siglec-7 and -9 on NK cells, sialylated glycans expressed on tumor cells can engage directly with NK cells and inhibit their function, protecting tumor cells from the innate immune response.179 Siglec-15 is also linked to a decreased antigen-specific T cell response and is also reported to be highly overexpressed in human cancer cells and in tumor-infiltrating myeloid cells.180 Galectins are novel regulatory checkpoint molecules that have potential as therapeutic targets in various cancers as they are widely expressed in the TME and have been shown to regulate transformation, angiogenesis, cell adhesion, and tumor-immune escape.181 Galectins are known to promote tumor growth by hindering immune surveillance via various mechanisms, such as a boost in T cell apoptosis, suppression of the activation of T cells, promotion of an anti-inflammatory Th2 profile, enhanced production of Foxp3+ Tregs, differentiation of IL-27+IL-10+ tolerogenic DCs, inhibition of NK cell cytotoxicity, and also polarization of MØs towards an M2 phenotype.182
5. CLR-mediated tumor progression
CLRs expressed on APCs are mannose/fucose-specific CLRs which include DC-SIGN and mannose receptor (MR) along with galactose-specific CLRs such as MGL.183 It is interesting to note that CLRs have affinity for tumor-associated glycan structures in addition to their primary binding to a variety of pathogens.155,1765.1. MGL
MGL is a unique member of the CLR family in the human immune system that specifically identifies terminal GalNAc residues, such as LacdiNAc epitope, Tn and STn antigens. Tn antigen is enhanced during oncogenic transitions and cancer development, making it quite prevalent in malignancies, especially those of epithelial origin. Due to the preferential recognition of tumor-associated Tn and STn antigens, MGL effectively distinguishes between TACAs and healthy tissue. MGL signaling aligns with TLR-induced pathways upon ligand binding, leading to an enhanced release of IL-10 by DCs. This process fosters the differentiation of functional human Tr1 cells. Furthermore, the T cell-dependent cytokine response and TCR-mediated signaling are negatively regulated by MGL-mediated binding to effector T cell CD45.This results in decreased T cell proliferation and an increase in T cell apoptosis. Tumor-associated Tn antigens may thereby suppress adaptive immune responses via MGL on several levels, eventually promoting tumor progression.177,184
5.2. DC-SIGN
DC-SIGN, a type II transmembrane CLR that is expressed on myeloid, dermal, interstitial, and moDCs, exhibits an affinity for high mannose-containing glycans and fucose-containing glycans and Lewis antigens. DC-SIGN facilitates the interaction between myeloid cells and tumor cells through the recognition of Lex antigen, and its ligation on the cell surface enhances the production of IL-10 and impairs the production of proinflammatory cytokines. It is worth mentioning that glycan-binding and triggering of DC-SIGN can effectively modify MØ maturation by enhancing IL-10 production and lowering IL-6 production, which may assist in establishing a tolerogenic milieu. It has also been shown that the DC-SIGN–fucose axis also governs early interactions with epithelial cells before they undergo EMT and lose their ability to bind DC-SIGN.20,185,1866. Siglecs and sialic acids
Siglecs, type I transmembrane proteins, belongs to the immunoglobulin superfamily of proteins. Extracellular domains of siglecs contain sialoglycan-recognizing V-set domains strikingly similar to the variable domain of immunoglobulins. The V-set domain contains CRD of siglecs, followed by a different number of C2-set Ig-like domains.187 Depending on certain patterns found in each siglec molecule, such as the immunoreceptor tyrosine-based activation motif (ITAM) and ITIM, siglec members can have either activating or inhibiting effects.188,189 Upon activation of siglecs, their ITIM domain is phosphorylated by Src family kinases, which eventually recruits the SH2-domain, having phosphatases SHP-1 and/or SHP-2, which leads to dephosphorylation of downstream components of immune stimulatory pathways, hence preventing further cellular responses. However, some siglecs (-14, -15, and -16) lack ITIM domains, and consist of positively charged residues present in their transmembrane domain, which allows them to form a complex with ITAM having adaptor proteins, such as DAP10 or DAP12. This results in the recruitment of protein kinases which are responsible for phosphorylating downstream targets, eventually triggering downstream signaling pathways.190Sialic acids are negatively charged nine-carbon carboxylated monosaccharides that are often cover glycans of glycosylated proteins and lipids on the cell surface, making it ideal for their interaction with siglecs, which are essential for immune homeostasis.191 Sialic acids on their exposed terminal positions in the glycan chains also allow them to function as a protective shield for the sub-terminal part of the molecule (e.g. preventing the protease-mediated degradation of glycoproteins) or the cell (e.g. mucous layer of respiratory epithelium). Sialic acid also participates in a variety of recognition mechanisms and the immune system may differentiate among both self and non-self-structures based on the presence of the sialic acid pattern.192 Glycan chains in vertebrate and many invertebrate cells are terminated by sialic acids that facilitate protein stability and trafficking, along with cell–cell and cell–ECM interactions. Many cells have specialized mechanisms for the synthesis of different sialic acids from precursor sugars present in the cytoplasm. After transfer, sialyltransferases in the Golgi incorporate sialic acids into the glycan part of glycoproteins and glycolipids. Around 20 different sialyltransferases have been discovered so far, each one adding sialic acid to fundamental sugars through distinct glycosidic bonds (α2,3, α2,6 or α2,8).193
6.1. Immunosuppressive nature of sialoglycans and siglecs
Sialoglycans on tumor cells can be implicated in the interaction between tumor cells and the ECM, as well as cell–cell interactions within the TME, hence protecting tumor cells from immune recognition. The immunomodulatory properties of sialoglycans are attributed to their specific interactions with siglecs. Siglecs, which are highly expressed in immune cells and detect aberrant sialoglycans expression on tumor cells, frequently suppress the immune system, limiting the immune system from eliminating tumor cells, and further hampering the cancer immunotherapy.194 Therefore, overcoming the immunosuppressive nature of aberrant sialoglycans and their interactions with siglecs is essential for developing successful cancer therapies.Due to their widespread expression on NK cells in the TME, siglec-7 and -9 have been the focus of attention in recent years. In contrast to siglec-7, siglec-9 has been recently found to be preferentially expressed in a fraction of CD56dim NK cells, whereas siglec-7 is recognized as a pan-NK cell marker.179 Siglec-15 has also been in the public eye as an immune suppressor widely expressed on human cancer cells, tumor-infiltrating myeloid cells and TAMs.180
Overexpression of sialoglycans and their engagement with siglecs on NK cells guards tumor cells against innate immunity. Sialic acid-containing ligands of siglec-7 and -9 on NK cells are expressed on tumor cells of varied histological origins and shield malignant cells from NK cell attack.179
During tumor progression, myeloid cells are linked to the promotion of tumor angiogenesis, resistance against antiangiogenic therapies, and suppression of anti-cancer immune responses.195 Siglec-9 on myeloid cells, upon interacting with a cancer-specific MUC1 glycoform (sialylated O-linked glycans MUC1-ST), instructs myeloid cells to release factors involved in TME establishment and disease progression.196 Tumor-derived sialic acids control MO to MØ differentiation by signaling through siglec-7 and -9.197 Siglec-15 is also shown to be involved in suppression of T cell responses.180
The expression and roles of siglecs on cancer-associated DCs have not been extensively defined; however, the expression of siglec-7 and -9 on conventional DCs have been shown to affect T cell activation, antigen presentation, and DC activation.198
Expression of various siglecs and their role in immunosuppression and cancer progression is shown in Table 2.
| Siglecs & their synonyms | Structure | Target cancer types | Recognition of glycan linkages | Role in immune escape and tumor progression | Ref. |
|---|---|---|---|---|---|
| Siglec-1/CD169 |

|
Melanoma, breast cancer, CRC | α-2,3 and α-2,6 linked sialic acid | Siglec-1-expressing subcapsular sinus (SCS) MØs provide anchorage to pioneer metastatic cells and enable efficient metastatic colonization; siglec-1+ MØs play a pro-tumor role by inhibiting CD8+ T cells; siglec-1+ monocytes and TILs serve as biomarkers for pathogenic degrees of CRC | 199–201 |
| Siglec-2/CD22 |

|
B-cell-related lymphomas | α-2,6 linked sialic acid | Siglec-2 is involved in inhibition of B-cell receptor-induced signaling by inhibiting Ca2+ mobilization and cellular activation | 202 |
| Siglec-3/CD33 |

|
Acute myeloid leukemia (AML) | α-2,3 and α-2,6 linked sialic acid | Siglec-3 acts as an inhibitory receptor regulating the NKG2D/DAP10 cytotoxic signaling pathway, which is engaged in self-tolerance and tumor | 203 and 204 |
| Siglec-6/CD327 |

|
Acute myeloid leukemia, chronic lymphocytic leukemia, bladder cancer, CRC | α-2,6 linked sialic acid | Higher expression of siglec-6 is significantly associated with poor overall survival | 205–207 |
| Siglec-7/CD328 |

|
CRC, bladder cancer, AML, head and neck squamous cell carcinoma (HNSCC) | α-2,3, α-2,6 and α-2,8 linked sialic acid | Siglec-7 attenuated NK cell cytotoxicity | 208–212 |
| Siglec-8 |

|
Breast and gastric cancer, ccRCC | α-2,3 and α-2,6 linked sialic acid | Siglec-8 acts as a marker for poor prognosis | 213–215 |
| Siglec-9/CD329 |

|
NSCLC, AML, melanoma, CRC, breast, ovarian and pancreatic cancer | α-2,3 and α-2,6 linked sialic acid | Siglec-9 is responsible for attenuation of CD8+ T cell effector function and NK cell cytotoxicity | 204, 212 and 216–218 |
| Siglec-10 (CD24) |

|
HCC, mantle cell carcinoma, ovarian and breast cancer | α-2,3 and α-2,6 linked sialic acid | Siglec-10 triggers immunosuppression by inhibiting TCR and T cell activation; siglec-10 on NK cells mediates functional damage of NK cells in HCC; siglec-10 on MØs also suppresses T cell activity | 212 and 219–221 |
| Siglec-15 |

|
HNSCC, breast, lung, bladder, liver, renal, pancreatic, colon, endometrioid, and thyroid cancer | α-2,3 and α-2,6 linked sialic acid | Siglec-15 suppress antigen-specific T cell responses | 180, 212 and 222 |
7. Galectins: modulators of cancer progression
Galectins are another class of animal lectins defined by their binding affinity towards β-galactose. The conserved CRDs of roughly 130 amino acids found in all galectins are in charge of the binding of carbohydrates.161 Based on their structure, galectins are categorized into three subfamilies: ‘prototype’, having only one CRD which can dimerize (Gal-1, -2, -5, -7, -10, -13, -14 and -15); chimera-type, having a unique CRD fused with another non-lectin N-terminal domain responsible for oligomerization (Gal-3); and tandem-repeat type, with two CRDs having distinct specificity joined via a flexible peptide linker (Gal-4, -6, -8, -9 and -12).223 Although galectins display biological activity both within and outside the cell they serve as glycan-binding proteins primarily when they are released into the ECM. Galectins govern various extracellular activities such as cell–cell and cell–ECM interactions, cell activation, apoptosis and cytokine secretion.224 Cancer cells and cancer-associated stromal cells often overexpress galectins, especially those cells that do not express certain galectins under normal circumstances. Galectins broadly influence tumor progression via a variety of mechanisms that can be glycosylation-dependent or independent. This altered expression of galectins correlates with aggressiveness of cancer and contributes to cancer proliferation. Galectins interact with LacNAc structures on immune cells such as DCs, T cells or NK cells, and facilitate TH cell differentiation and formation of tolerogenic DCs, TAMs and MDSCs, while also the reducing anti-tumor activity of NK cells. Galectins can also lead to the evasion of tumor growth suppressors, resistance to apoptosis, angiogenesis, and metastasis. They have the ability to alter immune responses and play a crucial role in assisting tumors in evading immune surveillance.18,159,1617.1. Galectin expression in the TME
Cancer cells do not exist in isolation, and to develop and thrive, they need other accessory cells and a favorable environment. The TME may be viewed as a complex tissue made up of a wide variety of different kinds of cell that are engaged in heterotypic interactions with one another which influence the growth and expression of malignant cell phenotypes.225 Galectins engage with certain specific glycosylated ligands to form multivalent interactions that fine-tune the signaling of multiple cell surface receptors. Additionally, various tumors display differential galectin expression profiles, which control various hallmarks of cancer, including invasion and metastasis, by acting as on–off switches.226 Gal-1, -3 and -9 are known to play pivotal roles in various aspects of cancer progression.159Gal-1 is expressed by various cancers in which Gal-1 is secreted by tumor cells, and is deposited into the ECM. In addition to this, Gal-1 is also expressed by various stromal components such as ECs, fibroblasts, neutrophils, MØs, DCs and T cells.227 Gal-1 has been associated with proliferation of ECs, migration, and adhesion, suggesting its specific function in angiogenesis. Gal-1 promotes EC migration by binding to neuropilin-1, activating vascular endothelial growth factor receptor (VEGFR-2), and modulating the c-Jun NH2-terminal kinase (JNK) signaling pathways in oral squamous cell carcinomas (OSCC).228 Gal-1 uptake by ECs promotes H-Ras signaling to the Raf/mitogen-activated protein kinase/ERK kinase (MEK) /extracellular-signal-regulated kinase (ERK) cascade and enhances EC proliferation and migration, which further leads to tumor angiogenesis and cancer progression.229 CAFs are a significant component of the stromal compartment of the TME, and their interaction with cancer cells is crucial for tumor growth and aggressiveness. CAFs overexpress Gal-1 and release microvesicles with elevated levels of Gal-1. Intracellular transport of Gal-1 to the tumor cells from CAFs via microvesicles results in upregulation of steady-state Gal-1 in cancer cells, which leads to an increased migration ability of tumor cells.230 Additionally, Gal-1 encourages T cell apoptosis, suppresses Th1-dependent immune responses, and promotes a shift towards the Th2 cytokine profile, favoring expansion of CD4+CD25highFoxp3+ Tregs.231 Gal-1 also polarizes MØs toward an M2 phenotype through the Janus kinase (JAK)-signal transducer and activator of transcription (STAT) signaling pathway, all of which inhibit immune surveillance and favor tumor growth.232
Gal-3 plays a significant role in cancer as it influences many biological processes such as tumor cell fitness, cell migration, EMT, as well as stemness by interacting with various glycosylated tumor-associated receptors such as epithelial growth factor (EGF), transforming growth factor (TGF)-β receptors, and cell surface integrins. It has been shown that the interaction of Gal-3 with MUC1 results in an activation of the mitogen-activated protein kinase and PI3K/Akt signaling pathways, which results in enhanced cell proliferation and motility.233 In another study, it was shown that interactions between Gal-3 and MUC1 enhance the dimerization and activation of the EGF receptor in epithelial cancer cells.234 Tumor-derived Gal-3 can facilitate invasion by disrupting interactions between the N-glycosylated proteins in the ECM, such as fibronectin and laminin, and adhesion molecules expressed on the surface of cancer cells.159 Additionally, Gal-3 has also been shown to promote cancer cell proliferation and anchorage-independent cell growth, and prevent cancer cell apoptosis through K-Ras-mediated activation of Raf-MEK-ERK signaling.235 Gal-3 plays a significant role in tumor progression and metastasis and acts as a tumor-promoting galectin. At pathological concentrations, Gal-3 has been shown to stimulate the secretion of various metastasis-promoting cytokines such as IL-6, soluble intercellular adhesion molecule-1 (sICAM-1), granulocyte colony stimulating factor (G-CSF), and granulocyte-macrophage colony stimulating factor (GM-CSF) from blood vascular endothelium in vitro and in vivo. These cytokines either interact in an autocrine or paracrine manner with the vascular endothelium to enhance the production of adhesion molecules on the EC surface. This increases cancer cell–endothelial adhesion, EC migration, and tubule formation, all of which are crucial events in the metastatic cascade.236 By interacting with the Nkp30 receptor, Gal-3 inhibits NK cell cytotoxicity, enabling tumor escape from NK cell immunity.237 Extracellular Gal-3 has also been shown to be responsible for T cell apoptosis, which plays a major role in immune escape.238
Gal-9 is expressed in a variety of cancers and is present on tumor cells, immune cells such as MØs, neutrophils and T cells, and stromal cells such as fibroblasts. Studies have shown that bone marrow mesenchymal stem cells (BMSCs) exhibit elevated Gal-9 expression, which can lead to the exhaustion of NK cells as seen in Fig. 5. This is achieved by downregulating the expression of EOMES and Notch1 through a TIM-3/Gal-9-mediated pathway. Additionally, certain subsets of CAFs demonstrate increased secretion of chemokines (CXCL9, CXCL10, and CXCL12), alongside an upregulation of MHC class I and enrichment of Gal-9. These factors collectively contribute to restricting CD8+ T cell infiltration and ultimately promote tumor progression by inhibiting the effector differentiation of the pre-effector CD8+ T cells, specifically those identified as TCF1+GZMK+CD8+ T cells.239–241
 | ||
| Fig. 5 Glycan–lectin circuits and their engagement in immunosuppression: aberrant glycosylation is a defining hallmark of cancer cells, as it is implicated in several cancer-related pathways. Essential cellular processes, including cell adhesion, signaling, and proliferation, are affected by the tumor-specific lectin–binding glycans. These pathways have been linked to various important cancer characteristics, such as triggering of invasion and metastasis, proliferative signaling, and resisting anti-tumor immunity. (a) CLRs are a vast family of pattern recognition receptors that are extensively expressed on myeloid cells. DC-SIGN recognizes Lewis antigens and boosts the secretion of IL-10, which is in charge of impairing DC maturation and the Th1 immune response. MGL interaction with Tn antigens is also evident in increasing IL-10 production, decreasing effector T cell proliferation, and promoting T cell apoptosis. The overexpression of MGL ligands also leads to enhanced infiltration of tumors with PD-L1+ TAMs; (b) Siglecs are lectins that identify sialic acids on tumor cells, such as STn and SLex or a. Siglecs expressed on NK cells interact with sialic acid-containing ligands to adversely affect NK cell activation and cytotoxic activity. Siglecs influence tolerogenic activities in DCs by interacting with tumor-associated sialoglycans, restricting the development of effector T cells. Siglecs are also implicated in producing TAM-like phenotypes, which produce pro-tumor cytokines like IL-10 and TGF-β, leading to suppression of CD8+ T cell responses, allowing tumor immune escape. Siglecs on neutrophils also contribute to the immune escape and prevent the neutrophil-mediated killing of tumor cells; (c) Galectins play a complex role in the development and dissemination of tumor cells. They are necessary for tumor cells to be able to evade the immune system. Galectins are involved in these processes by binding to LacNAc structures on immune cells and encouraging the development of tolerogenic DCs, Tregs, and MDSCs, decreasing antitumor immunity. Gal-1 induces the apoptosis of activated T cells, eliminates Th1 and Th17, and increases the expansion of MDSCs and Treg cells. Furthermore, Gal-1 favors the differentiation of IL-27- and IL-10-producing tDCs, which promotes Tr1 cell expansion. Gal-3 modifies antitumor immune responses by preventing plasmacytoid DC expansion and suppressing CD8+ T cells upon binding to LAG-3. Through TIM-3-dependent pathways, Gal-9 suppresses antitumor responses and promotes the development of MDSCs, apoptosis of T cells and inhibition of NK cell cytotoxicity (created in BioRender. Jain, M. (2025) https://BioRender.com/n23z034). | ||
Gal-2, -4, -7, and -8 are also studied for their expression in TMEs and role in tumor progression, immunosuppression, angiogenesis, and metastasis. Circulating Gal-2, -4, and-8 trigger the secretion of certain chemokines and cytokines from blood vascular ECs, such as G-CSF, IL-6, monocyte chemoattractant protein-1 (MCP-1), and growth-regulated oncogene alpha (GROα). Galectin-mediated secretion of these cytokines and chemokines results in enhanced expression of EC surface adhesion molecules, which leads to increased adhesion of cancer cells to ECs and endothelial tubule formation, which further leads to angiogenesis and metastasis.242 Gal-7 expression was observed in epithelial ovarian cancer histological subtypes, but the expression of Gal-7 was more prominent and frequent in high-grade and metastatic tumors. Gal-7 also elevates the invasiveness of ovarian cancer cells by inducing matrix metalloproteinases (MMP)-9 and enhancing cell motility.243
7.2. Galectin involvement in immunosuppressive TMEs
Galectins influence a range of immune cell activities and shape the immunosuppressive landscape via various intracellular or extracellular mechanisms. By coopting certain inhibitory receptors, disrupting various co-stimulatory pathways, and/or regulating immune cell activation, differentiation, and survival, galectins can orchestrate immunosuppressive circuits. Since they promote T cell exhaustion, restrict T cell survival, encourage growth of Treg cells, deactivate NK cells, and polarize myeloid cells toward an immunosuppressive phenotype, galectins have become recognized as new regulatory checkpoints that facilitate immune evasion.223Gal-1 is expressed by tumor cells and can inhibit the T cell effector activity by promoting cell growth arrest and activated T cell death. Furthermore, Gal-1 is also responsible for the aggressiveness of tumors and the acquisition of metastatic phenotypes. Functional Gal-1 secreted by melanoma cells extensively contributes to the immunosuppressive and proapoptotic activities of tumor cells. PBMCs, when cultured in Gal-1-containing serum-free conditioned media (SFCM) from melanoma cell lines, induced higher apoptosis of activated T cells compared with T cells cultured in the presence of anti-Gal-1 antibody-neutralized SFCM. A similar reduction in apoptosis of T cells was also observed when SFCM was incubated with thiodigalactoside (TDG), a galectin-specific inhibitor.244 In the case of CRC, both tumor- and stromal-derived Gal-1 influence antitumor immune responses and facilitate tumorigenesis by modulating the frequency and tumor-suppressive activity of CD8+CD122+PD-1+ Tregs.245
Gal-3 acts as an immune regulator that directly influences T cells’ activation and their function. Gal-3-mediated activation of T cells leads to T cell apoptosis. Additionally, the delivery of high doses of Gal-3 has been shown to inhibit tumor-reactive T cells and facilitate tumor growth.246 Gal-3 was shown to shift MØ polarization to a pro-tumor M2 MØ type. An IL-4-mediated Gal-3 feedback loop that causes sustained PI3K activation via activation of CD98 promotes a major mechanism involved in alternative M2 MØ activation.247–249
Several studies have revealed the immunosuppressive effects of Gal-9 in human cancers. Gal-9 has been found to cause immunosuppression in the TME, mediated by myeloid-lineage cells. In nasopharyngeal carcinoma, Gal-9 significantly enhances the expression of various pro-inflammatory cytokines, such as IL-1β and IL-6, which are crucial for the differentiation of MDSCs. This process is primarily driven by the accelerated degradation of the STING protein, which is due to the direct interaction of Gal-9 CRD1 with the C-terminus of the STING protein, leading to increased E3 ubiquitin ligase TRIM29-mediated K48-linked ubiquitination of STING. Additionally, Gal-9 strengthens the interaction between STING and TRIM29, which further accelerates the degradation of the STING protein. The mobilization of the Gal-9/TRIM29/STING pathway results in T cell suppression mediated by myeloid cells, ultimately supporting the survival of tumor cells.250 Activated Gal-9/TIM-3 negatively regulates CD4+ T cells and suppresses the effector function of Th1 T cells, and further promotes Treg cells, which are implicated in the immune escape of chronic lymphocytic leukemia.251 By crosslinking TIM-3 and promoting TIM-3 aggregation, Gal-9 has also been shown to trigger TIM-3-mediated T cell death.252
7.3. Galectin's role in tumor progression and metastasis
Metastasis, a multiphase process, is a major cause of cancer-mediated deaths. A crucial stage in tumor metastasis is increased cancer cell invasiveness, which necessitates several modifications. For instance, cancer cells need to become more motile, degrade surrounding tissues, and lose their adhesion to surrounding cells and ECM. Many genes, including MMP, integrins, and cadherins, can be altered by genetic or epigenetic modifications in tumor cells and their microenvironmental cells to enhance cancer growth and metastasis.253There are several lectins involved in metastasis, and Gal-1, being one of them, is engaged in each phase of the process. The degree of Gal-1 expression influences the invasiveness of cancer cells, and it increases as cancer cells develop a more malignant phenotype.254 Changes in the proteolytic degradation of the nearby tissue are necessary for tumor invasion. MMP-2 and MMP-9 are known to play significant roles in the degradation of gelatin and collagen ECM and are associated with LNM. It has been shown that Gal-1 expression is involved in the regulation of production and functions of MMP-2 and MMP-9 in OSCC cells, and that the increased expression of these MMPs induced by Gal-1 may be a key factor contributing to the high invasiveness and metastatic potential of oral cancer cells.253 Receptors for activated C kinase 1 (RACK1) in a Gal-1-dependent manner aid in tumor cell invasion and the formation of lymphatic tubes in vitro as well as encourage lymph angiogenesis and LNM in vivo. By lowering miR-1275 levels, RACK1 boosted the expression along with the secretion of Gal-1. Furthermore, in cervical cancer cells, RACK1 increased Gal-1-induced downstream MEK/ERK, focal adhesion kinase (FAK), and AKT signaling via integrin-β1.255 Expression of Gal-1 in low-metastatic lung cancer cell lines promoted cancer cell migration, invasion, and EMT. In highly invasive lung cancer cell lines such as CL1–5 and A549, suppression of Gal-1 inhibited migration and invasion and led to mesenchymal to epithelial transition. Additionally, Gal-1 enhances the expression and association of integrin β4 with α6 subunits, which results in increased phosphorylation of FAK and AKT. Gal-1 enhances Notch levels along with its ligand Jagged2 and also promotes AKT activation.256
Cancer cells escape from the primary tumor during the metastatic conversion process, infiltrate the lymphatic or blood vessels, and invade the tissues surrounding the primary tumor.253 Gal-3 overexpression subsequently increases tumor cell–ECM adherence and accelerates tumor cell escape from the primary location of the tumor.257 In ovarian, thyroid, colorectal, and melanoma cancer, elevated Gal-3 expression has been found to correlate with greater tumor invasiveness, and overexpression of Gal-3 showed enhanced tumor cell adhesion to ECM via ECM glycoproteins such as fibronectin, elastin, collagen IV, and laminin and promoted tumor escape from primary tumor sites.258 Furthermore, Gal-3 has been shown to influence angiogenesis and EMT-driven metastasis by activating the β-catenin signaling cascade by targeting insulin-like growth factor-binding protein 3 (IGFBP-3) and vimentin in the TME of HCC.259 It was shown that Gal-3 is a natural ligand of melanoma cell adhesion molecule (MCAM) in melanoma cells, and their interaction promotes MCAM clustering, which in turn activates AKT. This causes an increase in melanoma progression by cell proliferation, adhesion, migration, and invasion.260
A positive correlation has been shown between the expression of Gal-9 and an increase in the number of CD206 MØs in tumors. Melanoma survival was shown to be negatively linked with higher CD206 MØ concentrations. These findings demonstrate that Gal-9 interacts with CD206 on M2 MØs, promoting angiogenesis along with chemokine production, leading to tumor growth and a poor patient prognosis.261
8. Pentraxin, tetranexin and calnexin in the TME
Pentraxin 3 (PTX3), a prototypic long pentraxin, is a potential prognostic and diagnostic marker for various cancers such as lung carcinoma, pancreatic carcinoma and glioma malignancy. Autocrine synthesis of EGF-induced PTX3 improved binding of HNSCC tumor cells to ECs, boosting the capacity of tumor cells to penetrate blood vessels, and enhances the expression of fibronectin and MMP-9, resulting in promotion of metastasis.262 Tetranectin belongs to a distinct group of the CLR subfamily, identified as a biomarker for metastatic OSCC as its concentration is significantly reduced in the serum and saliva of these patients, which may contribute in activation of plasminogen to plasmin, which is involved in extracellular protein degradation and progression of cancer.263 Calnexin, an ER chaperone protein, is upregulated in OSCC and its enhanced expression on OSCC cells is associated with lower infiltration of T cells and poor survival for patients. Moreover, calnexin inhibits CD4+ and CD8+ T cell proliferation and production of IFN-γ, TNF-α, and IL-2.2649. Glycan–lectin dialogue: in failure of immunotherapies
In cancer, there is an unmet medical need to overcome both primary and acquired immunotherapy resistance. Immunotherapies may become more effective when targeted in combination with therapies suppressing the factors that promote resistance to immunotherapies, which can also lead to long-lasting therapeutic effects. Uncertainty surrounds the mechanisms behind both primary and acquired resistance; however, they might include any of the following: lack of sufficient antigen presentation, inadequate immune cell infiltration into the TME, exclusion of T cells, T cell anergy, reduced IFN-γ signaling, presence of immunosuppressive cells, expression of several inhibitory ICs, and diminished T cell functioning/T cell exhaustion.265The ability of lectins, particularly galectins, to recognize glycans is crucial for tumor-immune tolerance. It is interesting to note that galectins have been linked to several instances of immune regulation, playing significant roles in influencing T cell activity and encouraging tumor immune tolerance.266 Galectins have been identified as significant therapeutic factors of sensitivity or resistance to various anticancer treatments, including therapies like chemotherapy, radiotherapy, antiangiogenic therapies, targeted therapies and immunotherapies due to their ability to influence tumor development hallmarks.267 Many malignancies do not respond well to ICIs, owing to hostile TMEs restricting T cells, the most frequent effector for most immuno-activating therapies. Tumor samples examined from various cancer types demonstrate that T cell infiltration into tumors is a key indicator for anticipating ICI responses.268–270 Nambiar et al. found that Gal-1 levels had an inverse relationship with the treatment outcomes and survival in head and neck cancer patients receiving ICIs and demonstrated that Gal-1 aggressively altered the tumor endothelium to limit T cell invasion, resulting in exclusion of T cells from the TME. They further showed that the sustained release of Gal-1 from the tumor was enough to activate the STAT signaling pathway in ECs. These cells, in turn, increased the expression of immune checkpoint ligands, which reduced intratumoral T cell infiltration. Therapeutically targeting Gal-1 with Gal-1-specific antibody transformed T cell-depleted tumor into one abundant in T cells receptive to anti-PD-1 treatment and radiation therapy.271
Gal-3 was found to be essential for prostate tumor cells to acquire and maintain immune tolerance, which was accomplished by triggering dysregulation of CD8+ CTL responses. For anti-cancer vaccines to be successful, naïve T cells must be properly primed to develop effector functions that permit the elimination of tumor cells. Inhibition of Gal-3 in tumor cells has been shown to decrease the ratio between CD8+CD122+CD28 Tregs and total CD8+ T cells, allowing efficient activation and proliferation of CD8+ CTLs. These findings imply that Gal-3 has a significant impact on the ability of DC-based vaccines to prevent cancer.266
Remarkably, sialoglycans have been proposed to modulate several activities of DCs, which are essential immune system regulators. Ac53FaxNeu5Ac, a sialic acid mimetic, has been found to successfully inhibit sialic acid expression in moDCs, resulting in greater responsiveness to TLR activation as indicated by higher maturation and cytokine production. Consequently, Ac53FaxNeu5Ac-treated moDCs have been shown to be potent activators of allogeneic T cells. Furthermore, sialic acids on moDCs may serve as ligands for immunosuppressive siglecs, and Ac53FaxNeu5Ac treatment inhibits these interactions. Therefore, sialic acid blockage facilitates the development of potent DC-based vaccines that can elicit potent in vivo anticancer responses.272
10. Glyconanovaccine-mediated tumor suppression
In several physiological processes, including cell adhesion, signal transduction, molecular trafficking, and endocytosis, glycans play an essential role. Due to the widespread presence of glycans within the body, their immunogenicity is below par. When malignant cells with TAAs are encountered directly by CTLs, an insufficient cytotoxic immune response is elicited, whereas XPT results in a heightened, sustained, and comparably more effective immune response. In addition to antigens from apoptotic cells, DCs can also cross-present antigens encapsulated in nanocarriers that are targeted to specialized uptake receptors expressed by DCs. Glycans play a role in immune cell-to-cell communication carried out by CLRs. CLRs identify and bind to certain glycans, allowing them to take up the molecules and regulate immune responses.2,273,274 Therefore, developing glycan-based nanomedicines can offer future paths for fostering robust DC-based immune responses to eliminate cancer. Specialized cell surface receptors, called PRRs, are present on DCs and can recognize ligands which are antigenic subunits on pathogens and further trigger T cell immune responses. Several PRRs along with their associated ligands have been discovered. However, only CLRs and TLRs are known to engage in antigen recognition and are suitable targets that can be leveraged to develop vaccines designed for triggering DC-mediated immune responses.275 Such an immune response, however, might not be sufficient to target and eradicate the antigens effectively. Therefore, to revitalize DC-based nanomedicines against cancer, the glycan recognition ability of DCs can be harnessed.17,27610.1. Glycans, the complex carbohydrates for targeted immunotherapy
Vaccine delivery methods using polymeric particles have been widely used for antigen delivery to DCs. As these polymeric particles lack PAMPs or targeting molecules, their ability to interact with DCs is restricted.277,278 Initially, targeting of CLRs on APCs was achieved by mAbs as they provide receptor specificity as well as high affinity; however, their tissue penetration due to their size, Fc region-mediated non-specific uptake, Fc-R triggering or provoking of immune response are responsible for their elimination.279 Glycan-mediated targeting of CLRs has been popularized in recent times and is being used for cell-specific targeting and immunomodulatory activities. Immunogenicity of glycans is below par and glycans are present across the body.280 Glycan array data have confirmed the affinity of various glycans toward CLRs present on APCs. Additionally, the common affinity of glycans for multiple CLRs presents an unique opportunity to target several CLRs simultaneously.281–285 Therefore, glycans are considered as ideal in the preparation of glycan-modified synthetic materials.10.2. Glycomaterials with multivalent presentations of glycans and concept of glyconanovaccines
Recent advances in glycan-mediated targeting of CLRs on APCs have shown increasing interest in the design of glycan-modified synthetic materials, commonly known as glycomaterials.2,17,20,286,287 Although the interactions between monovalent carbohydrates and lectins have weak affinity, this effect is enhanced by the multivalent nature of densely packed carbohydrate molecules on the surface, commonly referred to as the “glycocluster effect”.288 Synthetic materials such as nanoparticles, liposomes, polymers, dendrimers, supramolecular assemblies, etc. possess a dense and highly repetitive molecular architecture and are ideal and better suited for the preparation of multivalent systems. Synthesis of multivalent glycomaterials is accomplished by conjugation of glycan to the central or core structure of synthetic materials which are already functionalized for glycan attachment.289–291 These synthetic glycomaterials, which feature multivalent presentations of glycans that mimic the immobilized carbohydrate clusters found in nature, possess an increased affinity and avidity for recognition events. Additionally, multivalent binding significantly enhances the affinities of monovalent interactions by many orders of magnitude (Fig. 1). In addition, glycomaterials have also shown superior binding specificity, flexibility, and spatial orientation.292 Therefore, carefully designed glycomaterials with these features are optimally required for efficient binding towards CLRs on APCs. Synthetic glycomaterials such as glycoliposomes, glycodendrimers, glycan-conjugated pH-responsive liposomes, glyconanoparticles, and so on are ideal for developing XPT-based immunotherapy. Nanosized glycomaterials with a multivalent presentation of glycans containing tumor antigen and appropriate immune stimulators can specifically target CLRs on APCs. Glycan-mediated targeting of CLRs on APCs exhibited efficient uptake of these glycomaterials harboring tumor antigen in a CLR-dependent pathway, resulting in APC maturation and improved XPT to tumor antigen-specific CD8+ and CD4+ T cells. Thus, combining a knowledge of XPT and nanosize synthetic glycomaterials with tumor antigens and appropriate stimulators (nanovaccines) can aid in the development of glyconanovaccines (GNVs) for targeting APCs and eliciting robust CD8+ T cell responses.293–29510.3. Understanding CLRs’ glycan specificity
The CLRs are transmembrane receptors involved in the uptake of glycosylated antigens. CLRs are expressed on specific DCs subsets. For instance, MR is primarily found on MØs, while langerins are present on Langerhans cells (LCs). CLRs exhibit a wide range of glycan specificity, and several CLRs, including DC-SIGN, MR, langerin, DEC-205, dendritic cell immunoreceptor (DCIR), and BDCA-2, have the ability to internalize glycosylated antigens effectively.183,296–301 The most studied CLR, DC-SIGN, has been shown to have a strong binding affinity for glycan structures comprising high-mannose and fucose, as well as Le antigens. Data from glycan arrays indicate that DC-SIGN preferentially binds to N-linked high-mannose oligosaccharides, with the highest binding observed for glycopeptides containing Man9GlcNAc2-Asn. This binding preference diminishes when dealing with glycan structures that contain fewer mannose residues.285 With DC-SIGN, binding of fucosylated glycan structures was also observed, and binding was in order of Leb > Ley > Lea.302 MR shows a strong affinity for glycoconjugates terminated with mannose, fucose, or GalNAc. It has also been reported that MR has an affinity for sulphated ligands, particularly those with galactose or sulphated GalNAc.284 Langerin CRD showed a binding preference towards mannose, fucose, and GlcNAc. This binding also requires the oligomeric structure formed via the neck region.282 Carbohydrate microarray analysis showed that langerin binds to sulphated Lex-glycan, while it exhibited weak binding to high-mannose glycans.303 MGL exhibits a strong preference for terminal α- or β-linked GalNAc residues found in glycoproteins and glycosphingolipids. Additionally, MGL shows affinity for the Tn antigen as well (α-GalNAc).281 DCIR exhibited glycan specificity for mannotriose, Lea, Leb, and sulfo-Lea.304 BDCA-2 demonstrated selective binding to galactose-terminated biantennary glycans containing Galβ1-3/4GlcNAcβ1-2Man. BDCA-2 has two distinct binding sites for mannose- and galactose-containing glycans.305 Dectin-1 shows a binding preference towards β-1,3 and/or β-1,6-linked glycans.306,307 As the understanding of CLR binding to various glycans has improved, there has been a focused effort in preparing glycomaterials that can specifically target antigens to CLRs.10.4. Glycomaterial-based strategies for targeting of CLRs
Glycomaterial-based strategies are popularized in recent times for targeted antigen delivery to CLRs. Various groups working world-wide in the targeted immunotherapy area developed materials-based strategies for targeted delivery with a focus on CLRs such as DC-SIGN, langerin, MR, MGL, and dectin-1. The detailed glycomaterial-based strategy for targeting of CLRs employed by various groups are as follows:Glycan-modified PEGylated liposomes and non-PEGylated liposomes containing OVA as a model antigen were prepared to study the formulation considerations in effective CLR targeting. Lex and Ley glycans were coupled to PEGylated and non-PEGylated liposomes via maleimide groups. Glycan-modified PEGylated liposomes were prepared from a mixture of egg phosphatidylcholine (EPC-35)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[maleimide (polyethylene glycol)-2000] (PEG-mal):PEG-DSPE:cholesterol (Chol) in a ratio of 1.85:0.075:0.075:1, whereas non-PEGylated liposomes were created from EPC-35:ethanolamine phosphoglyceride (EPG):1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-[4-(p-maleimidophenyl)butyramide] (sodium salt) (MPB-PE):Chol in a ratio of 1.5:0.4:0.075:1. Lipid film was formed by solvent evaporation and further hydration in buffer containing OVA molecules. The sizes of the glycan-coupled PEGylated and non-PEGylated liposomes were observed to be in the range of 200 and 220 nm, with a polydispersity index (PDI) under 0.20, showing a uniform size distribution. The average zeta potential for PEGylated and non-PEGylated liposomes was measured and found to be −25 mV and −55 mV, respectively. The presence of Lex and Leb glycans on PEGylated liposomes was detected by anti-Lex and anti-Leb antibodies using ELISA. To assess their proper orientation for DC-SIGN binding, a chimeric DC-SIGN molecule was employed; however, no binding was observed with the PEGylated liposomes. In contrast, glycan-modified non-PEGylated liposomes also showed detectable Lex and Leb by ELISA and effectively bound to the DC-SIGN-Fc chimeric molecule, confirming that PEG sterically hindered the binding of PEGylated liposomes. This is further confirmed by the binding of Lex and Leb-coupled non-PEGylated liposomes to DC-SIGN on bone marrow-derived DCs (BMDCs), while PEGylated liposomes did not bind to DC-SIGN on BMDCs. This highlights the importance of formulation considerations in specific CLR targeting.311
:1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[maleimide (polyethylene glycol)-2000] (PEG-mal):PEG-DSPE:cholesterol (Chol) in a ratio of 1.85:0.075:0.075:1, whereas non-PEGylated liposomes were created from EPC-35:ethanolamine phosphoglyceride (EPG):1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-[4-(p-maleimidophenyl)butyramide] (sodium salt) (MPB-PE):Chol in a ratio of 1.5:0.4:0.075:1. Lipid film was formed by solvent evaporation and further hydration in buffer containing OVA molecules. The sizes of the glycan-coupled PEGylated and non-PEGylated liposomes were observed to be in the range of 200 and 220 nm, with a polydispersity index (PDI) under 0.20, showing a uniform size distribution. The average zeta potential for PEGylated and non-PEGylated liposomes was measured and found to be −25 mV and −55 mV, respectively. The presence of Lex and Leb glycans on PEGylated liposomes was detected by anti-Lex and anti-Leb antibodies using ELISA. To assess their proper orientation for DC-SIGN binding, a chimeric DC-SIGN molecule was employed; however, no binding was observed with the PEGylated liposomes. In contrast, glycan-modified non-PEGylated liposomes also showed detectable Lex and Leb by ELISA and effectively bound to the DC-SIGN-Fc chimeric molecule, confirming that PEG sterically hindered the binding of PEGylated liposomes. This is further confirmed by the binding of Lex and Leb-coupled non-PEGylated liposomes to DC-SIGN on bone marrow-derived DCs (BMDCs), while PEGylated liposomes did not bind to DC-SIGN on BMDCs. This highlights the importance of formulation considerations in specific CLR targeting.311
Liposomes containing OVA/MART-1 peptides were prepared using a similar strategy. They were sized using a high-pressure extrusion device and sequentially extruded through stacked polycarbonate filters (800, 400, and 200 nm). Ultracentrifugation was used to eliminate the non-encapsulated antigen. Using thio-activated Lex and Leb (thio-Lex and Leb), glycan coupling to these liposomes was accomplished by a thiol–ene reaction with the maleimide groups of the MBP-PE lipid. The non-modified and Lex- and Leb-modified liposomes (extruded through 200 nm) showed a mean size of around 200 nm, with PDI < 0.2 for non-modified and Lex-modified liposomes, and <0.3 for Leb-modified liposomes. The zeta potential of these liposomes was also around −55 mV. The presence of Lex and Leb on the liposomes was verified using ELISA employing specific antibodies on the plate-bound liposomes. The correct orientation and affinity of Leb on the modified liposomes were validated with the DC-SIGN-Fc construct in ELISA assays. Notably, Leb-modified liposomes demonstrated significantly enhanced binding and higher uptake by human DC-SIGN-expressing BMDCs from hSIGN transgenic mice. Targeting OVA to DC-SIGN via glyco-liposomes enhances MHC class II presentation by approximately 100-fold. Additionally, incorporating LPS, a TLR4 ligand, further boosts the XPT of OVA to CD8+ T cells by around 100-fold. Similarly, Lex-coupled and melanoma antigen MART-1-encapsulated liposomes incubated with human moDC in the presence of lipopolysaccharide (LPS) significantly improved the antigen presentation to a MART-1-specific CD8+ T cell clone, as indicated by the increase in IFN-γ secretion. This demonstrates the effectiveness of DC-SIGN-targeted glyco-liposomes in CD4+ and CD8+ effector T cell activation.312
Another approach involved developing a single liposome formulation with glycan for CLR targeting, an adjuvant, and tumor antigen for DC maturation, and an antigen-specific immune response. Using the film extrusion method, glycan-modified liposomes containing TLR ligands were prepared from a mixture of Chol and phospholipids. In brief, EPC-35:EPG-Na:Chol in a 3.8:1:2.5 molar ratio were mixed with either MPLA (2 mol%), Pam3CysSK4 (1 mol%) or R848 (4 mol%) separately. 0.1 mol% of the lipophilic fluorescent tracer DiD (1′-dioctadecyl-3,3,3′,3′-tetramethyl indodicarbocyanine) was added during the first step of preparation. Hydrophilic TLR ligand Poly I:C and antigenic peptide gp100280–288 (YLEPGPVTA) were encapsulated into the liposomes during the hydration step. Liposomes were sized by sequential extrusion. Lex-glycolipid was inserted into the liposomes by the incubation of 1 ml of liposomes into 0.75 mg glycolipid (dissolved in 15 μl of methanol) followed by vigorous stirring, followed by ultracentrifugation and resuspension in buffer. The size, PDI and zeta potential for empty liposomes, Pam3CysSK4-encapsulated liposomes, Poly I:C-encapsulated liposomes, MPLA-encapsulated liposomes and R848-encapsulated liposomes were around the range of 212–216 nm, 0.04 to 0.12, and −45 to −52 mV, respectively. Similarly, the size, PDI, and zeta potential for empty Lex-glycolipid modified liposomes, Lex-glycolipid modified Poly I:C-encapsulated liposomes, Lex-glycolipid modified MPLA-encapsulated liposomes and Lex-glycolipid-modified R848 encapsulated-liposomes were in the range of 207–221 nm, 0.03–0.08, and −38.5 to −44.5 mV, respectively. The successful conjugation and orientation of Lex glycan on liposomes was validated through ELISA using a recombinant DC-SIGN-Fc construct. Lex-modified liposomes specifically bound to and were taken up by human moDCs via DC-SIGN. Incorporating MPLA as a TLR4 ligand into Lex-modified liposomes led to enhanced DC activation, marked by increased expression of CD83 and CD86. Additionally, targeting antigens and adjuvants to DC-SIGN with Lex-modified liposomes resulted in elevated IFN-γ production and a significantly improved XPT of gp100280–288 to CD8+ T cells.313 Human skin contains a large subset of dermal DCs (dDCs), with CD14+ dDCs specifically expressing DC-SIGN, which indicates their potential for antigen endocytosis and routing for MHC class I and class II presentation to CD8+ and CD4+ T cells. In a human skin explant model, intradermal delivery of Lex-modified liposomes carrying melanoma antigens (MART-1 or gp100) effectively targeted DC-SIGN+ CD14+ dDCs. Additionally, intradermal administration of GM-CSF and IL-4 enhanced the mobilization and surface expression of DC-SIGN on CD14+ DCs, leading to increased internalization of glycoliposomes, improved antigen presentation, and a heightened MART-1 and gp100 specific CD8+ T cell response.293
In another study, a combination of palmitoylated antigen and lipo-Ley, along with alpha-galactosylceramide (αGC), was successfully delivered to skin APCs using a single liposome. In order to prepare these liposomes, a mixture of EPC-35, EPG-Na, and Chol (in a molar ratio of 3.8:1:2.5) was dissolved in chloroform/methanol. DiD, a lipophilic fluorescent tracer (0.1% mol), palmitoyl-gp100/MART-1 (400 μg), lipo-Ley (1.5 mg), and αGC (30 μg) were then incorporated to this mixture. The resulting lipid film was hydrated in a HEPES buffer pH of 7.5, and the liposomes were formed by extrusion. Unencapsulated antigens and lipo-Ley were removed through ultracentrifugation. The mean size of the extruded liposomes was approximately 180 nm, with a zeta potential of around −53.9 mV. This liposomal vaccine resulted in significant activation of CD8+ and iNKT cells, as indicated by increased secretion of IFN-γ. Furthermore, the inclusion of lipo-Ley improved the uptake by moDCs, dDCs, and LCs, while also enhancing the activation of gp100-specific CD8+ T cells and iNKT cells in human skin-emigrated APCs. Loading moDCs with liposomes containing Ley resulted in the priming of MART-1126–35L-specific CD8+ T cells. Consequently, these liposomes represent a novel cancer vaccination approach.314
Another kind of multivalent platform for CLR targeting is PAMAM dendrimers. These dendrimers are symmetrical, highly branched, monodisperse polymers featuring a compact spherical shape, with diameters varying from 1.1 nm for the first generation (G0) to 9 nm for the seventh generation (G7). They are commercially available with their functional groups activated, which allows for the design and development of multivalent structures through simple chemical reactions. In their study, Garcia-Vallejo and colleagues modified poly(amide amine) PAMAM dendrimers using a peptide antigen that ends with Leb. Their findings indicated that third-generation dendrimers with 16–32 glycans had the optimal level of multivalency for enhanced binding to DC-SIGN, internalization by moDCs, and delivery to lysosomal compartments, leading to specific CD4+ and CD8+ T cell responses.315
:38:5 and also included 4.75% DSPE-PEG-GlcNTosyl and 0.25 mol% DSPE-PEG-AlexaFluor647 for langerin targeting and tracking of liposomes. Another set of liposomes incorporated 4.5 mol% DSPE-PEG-GlcNTosyl as a targeting ligand and 0.25 mol% pHrodo, along with A647, to monitor the uptake and routing of liposomes into langerin-expressing adherent HEK293 cells. The size of the liposomes was 140 ± 23 nm. Confocal imaging revealed their uptake into cells, evidenced by punctate fluorescence in endosomal compartments, while live cell imaging demonstrated accumulation in acidic late endosomal/lysosomal regions. This study highlighted that the influx of liposomes into late endosomal/lysosomal compartments was sustained by new liposomes entering early endosomal compartments. Notably, when protein antigen-loaded liposomes were incubated with langerin-expressing cells and primary human LCs, specific delivery mediated by langerin was observed, making this liposomal platform a promising tool for targeted vaccine delivery in the epidermis.318
Functional nanogels created through the self-assembly of associated polymers have gained significant attention as potential nanocarriers. Cholesteryl-group-bearing pullulan (CHP) forms physically cross-linked nanogels by self-assembling in water. These CHP nanogels effectively capture proteins mainly through hydrophobic interactions, functioning similarly to chaperones. This process allows proteins to be enclosed within the hydrated gel without aggregation, enabling their release in their native form. As a result, CHP nanogels exhibit remarkable capabilities for protein delivery, particularly in the development of cancer vaccines. Furthermore, a novel adjuvant intranasal vaccine delivery system has been developed using nanometer-sized hydrogels made from cationic CHP, which has demonstrated both systemic and mucosal antigen-specific immune responses.319 Prof. Kazunari Akiyoshi's group developed a carboxyl group-modified CHP (CHPCOOH) nanogel vaccine system for cancer immunotherapy. The CHPCOOH nanogel vaccine was created using the following method: CHP, CHPCOOH19/CHPCOOH40 were dissolved in phosphate-buffered saline (PBS) containing 6M urea, followed by addition of 1.6 mg ml−1 of ovalbumin (OVA), the mixture was incubated at 37 °C for 24 hours. The resulting OVA/CHPCOOH19 and OVA/CHPCOOH40 were dialyzed against PBS to remove urea, passed through a 0.22 μm filter, and stored at 4 °C. The diameters of OVA/CHPCOOH19 and OVA/CHPCOOH40 were 96 nm and 98 nm, respectively, while their zeta potentials were −15 mV and −21.8 mV, indicating a decrease in zeta potential depending upon the substitution degree of carboxyl group substitution. The interaction between CHPCOOH nanogels and DC2.4 cells increased in a time-dependent manner. In contrast, as RAW264.7 expressed the SR-A molecule, interactions with RAW264.7 were improved by increasing carboxyl group substitution. Incubation of RAW264.7 cells with fucoidan or poly I reduced their uptake, demonstrating SR-mediated interaction with the cells. Additionally, antigen XPT by mature DC2.4 cells occurred in the presence of CpG, a TLR9 agonist. The CHP nanogel with carboxyl groups induced significantly stronger CTL activation. The CHPCOOH nanogels exhibited interactions that were 2–4 times more significant with DCs and MØs. These nanogels also enhanced interactions with migratory langerin+ DCs, which migrate from the skin to lymph nodes. Therefore, CHPCOOH nanogel vaccines interacted with various APCs through mechanisms such as phagocytosis and via SRs, as well as through langerin. Furthermore, CHPCOOH nanogels improved interactions with CD103+ langerin+ DCs, a subset capable of XPT. These gels also presented antigens to DCs with langerin receptors in vivo, contributing to the production of killer T cells. In summary, CHPCOOH nanogels demonstrate potential as a novel therapeutic cancer vaccine320
A glycodendrimer-based approach was developed for dual targeting of CLRs (DC-SIGN and langerin). G0 and G3 PAMAM dendrimers were coupled with a melanoma-specific gp100 synthetic long peptide and modified with the glycan Ley, a common ligand for both receptors for creating multivalent glycodendrimers. The G3 glycodendrimers were effectively bound and internalised by moDCs and primary LCs in a DC-SIGN- and langerin-dependent manner, respectively. In a human skin explant model, langerin was expressed on LCs, and CD14+ DCs expressed DC-SIGN, both showing increased uptake of G3 glycodendrimers. CD1a+ dDC also efficiently took up glycodendrimers. Glycodendrimer-pulsed moDCs with the TLR4 stimulus MPLA exhibited enhanced degranulation and IFN-γ production. Similarly, G3 glycodendrimers targeting langerin on primary LCs increased CD8+ T cell activation. Thus, G3 glycodendrimers improved binding to moDCs and LCs in a DC-SIGN- and langerin-dependent manner, and antigens were efficiently routed to the XPT pathway in the presence of a TLR stimulation. Thus, glycodendrimer-based glycovaccine formulations can be employed to target numerous human skin DC subsets.294
These approaches of DC-SIGN and langerin are summarized in Fig. 6.
 | ||
| Fig. 6 Glycomaterial-based targeting of DC-SIGN and langerin for improved anticancer immune responses: (a) liposomes containing the glycan Lex which is highly specific for DC-SIGN expressed by DC. Lex-modified liposomes were taken up by DCs in a DC-SIGN-specific manner. The addition of LPS enhanced the presentation of encapsulated antigens to CD4+ and CD8+ T cells. Furthermore, incorporating MPLA promoted DC maturation and the production of pro-inflammatory cytokines, significantly improving XPT to CD8+ T cells. Ley-modified liposomes are endocytosed by DC-SIGN+ DCs and mediate efficient antigen presentation to CD4+ and CD8+ T cells. (b) Liposomal vaccine containing synthetic long peptides and αGC conjugated with Ley antigen promotes CD8+ T cell responses and induces iNKT cell activation, which enhances XPT. (c) The G3 glycodendrimers were effectively bound and internalised by moDCs and primary LCs in a DC-SIGN- and langerin-dependent manner, respectively and enhanced the CD8+ T cell activation and response. Additionally, moDCs pulsed with glycodendrimer displayed specific activation of gp100-specific CD4+ T cells, as indicated by IFNγ secretion, which increased both in the absence and presence of TLR4 stimulation (MPLA). (d) OVA/CHPCOOH is an anionic nanogel based vaccine designed to be delivered to draining lymph nodes, where it engages with APCs. This nanogel vaccine upon interacting with SR and langerin promotes the endocytosis and presentation of antigens via both MHC class I and class II pathways (created in BioRender. Jain, M. (2025) https://BioRender.com/edvy8q8). | ||
:1:2.5. Gangliosides (3 mol% GM1, GM3, GD3, GD1a and GT1b) and 0.1 mol% of lipophilic fluorescent tracer DiD were added to the mixture. TLR-ligand MPLA (2 mol%) or R848 (4 mol%) was also added. The pancreatic cancer-associated antigen Wilms’ Tumor 1 short peptide and gp100 long peptide, both at 3 mg ml−1, were encapsulated into liposomes. The size, PDI and zeta potential were 164–177 nm, 0.09–0.13, and −51.2 to −56.8 mV, respectively. Specifically, all ganglioside–liposomes bind to CD169 and the CD169-overexpressing THP-1 human monocytic cell line, with GD1a- and GT1b-liposomes showing the most effective binding. Monocyte-derived MØs (moMØs) highly expressing CD169 bound and internalized ganglioside–liposomes in a CD169-dependent manner. Moreover, human primary MØs also took up ganglioside–liposomes in a CD169-dependent manner, while moDCs demonstrated efficient binding and internalization. Furthermore, inclusion of TLR4 ligand MPLA and WT1 antigen in ganglioside–liposomes showed uptake of these liposomes by moDCs, stimulated IL-6 production indicating its activation, and furthermore these liposomes also stimulated IFNγ secretion by CD8+ T cells. GM3-containing liposomes encapsulating gp100 long peptide cocultured with moDCs induced similar levels of IFNγ by gp100-specific T cells, comparable to Ley liposomes. Additionally, ganglioside–liposomes were also internalized by Axl+ DCs in a CD169-dependent manner. GM3-liposomes with WT1 tumor antigen and TLR7 ligand R848 further promoted IFNγ secretion from WT1-specific CD8+ T cells. Axl+ DCs were also found in cancers of pancreatic ductal adenocarcinoma, gastrointestinal malignancies, hepatocellular carcinoma, melanoma and colorectal liver metastasis. High expression of CD169 in Axl+ DCs was observed and these DCs can also take up ganglioside liposmes.327 OVA-encapsulated and GM3- and αGC-containing liposomes were prepared to achieve CD169+ targeting and superior DC activation. Using a similar lipid mixture, EPC-35, EPG-Na and Chol in a molar ratio (3.8:1:2.5) were dissolved in chloroform/methanol (2:1 ratio) and combined with 3 mol% GM3 gangliosides and/or α-galactosylceramide (30 μg) and 0.1 mol% of the lipophilic fluorescent tracer DiD. The sizes, PDI, and zeta potential of GM3-OVA, αGC-OVA, and GM3-αGC-OVA liposomes ranged from 152 to 175 nm, 0.09 to 0.11, and −55 to 56 mV, respectively. GM3-modified liposomes were efficiently taken up by splenic CD169+ MØs. The inclusion of αGC in liposomes effectively activated NKT and NK cells, leading to IFNγ production. Immunization with GM3-αGC-OVA liposomes resulted in a high frequency of antigen-specific CD8+ T cells and higher CD8+ T cell response, and also stimulated CD4+ T cells and B cells. Following vaccination with GM3-αGC-OVA liposomes, CD169+ MØs are vital for activating antigen-specific CD8+ T cells, although their role in activating B cells, NKT cells, and NK cells is quite limited. Additionally, cDC1 cells play a crucial role in generating antigen-specific CD8+ T cells when immunized with GM3-αGC-OVA liposomes. Immunization using GM3-GC liposomes has been found to improve DC maturation. NKT cells can help mature DCs by increasing the expression of CD40L and secreting cytokines, as indicated by the presence of IFN-γ- and IL-4-producing cells and the upregulation of CD40L. A significant number of NK cells producing IFN-γ were also detected. Maturation markers such as CD40, CD80, and CD86 showed increased expression on both cDC1 and cDC2 cells. Moreover, GM3-αGC liposomes lead to elevated levels of IL-12, indicating their possible role in promoting a stronger immune response in mice.328
Extracellular vesicles (EVs) are nano- to micro-sized lipid bilayer assemblies released from nearly all cell types. EVs consist of DNA, RNA, and proteins derived from their donor cells, serving as mediators of intracellular communication. Recently, EVs have been shown to play significant roles in various processes, including immune responses, tumor growth and metastasis, and viral infections. Different types of EV have been identified based on their origin, including apoptotic EVs (nanometer-sized), endosome-derived vesicles known as exosomes (less than 200 nm), and microvesicles (MVs), which are formed by direct budding from the plasma membrane. Cell surface glycans play a crucial role in cell recognition, immunity, microbial infection, signal transduction, and cancer. Consequently, the glycans on the surface of EVs are also important in these processes. High-throughput analysis of EV surface glycans has been performed using lectin microarrays with evanescent field fluorescence (EEF) detection. The EEF-assisted lectin microarray binds to lectins without any washing steps, enabling accurate analysis of EVs even with small amounts less than 500 ng.329 Alterations in glycosylation are widely recognized as associated with cancer diagnosis, including N-linked branched glycans, fucosylation, sialylation, and truncated O-glycans. Sialyl-Tn from EVs can act as a marker for gastric cancer.330 EV surface glycans play a key role in their internalization. In a study, mesenchymal stem cell-derived EVs were injected subcutaneously into mice to assess their uptake by APCs in the lymph nodes. The results showed that EVs preferentially bound to CD11b-expressing cells, particularly those that were siglec-positive, highlighting the potential for cell-surface lectin-mediated entry. This indicates that the glycans on the surface of EVs can act as ligands for cell-specific targeting. Additionally, another study found that B-cell-derived EVs, which were enriched with α-2,3-linked sialic acids, successfully bound to siglec-1+ MØs in the spleen and lymph nodes. This suggests that glycoengineering of cells and EVs can be utilized to enhance cellular uptake and modulate biodistribution.331 Indeed, glycoengineering of cells and EVs has been applied for enhancing cellular uptake and modulation of biodistribution. The role of EV surface glycans in cellular uptake has been demonstrated. The potential role of EV glycan in uptake has been demonstrated using two murine hepatic cell lines, and cells treated with glycosidase PNGase F and neuraminidase showed enhanced affinity of EVs. Therefore, EV glycoengineering is another important strategy in enhancing their cellular uptake and for modified EVs as anti-cancer vaccines.332
EVs derived from glioblastoma may serve as an enriched, cell-free source of tumor-associated neoantigens to stimulate DCs for an anti-tumor immune response. EVs were isolated from two well-established glioblastoma cell lines, U87 and GBM8, using ultracentrifugation. The quality of the EV preparation was assessed using immune transmission electron microscopy (immune-TEM) with CD63 immunogold staining. The TEM images revealed the typical morphology of the EVs. Size distribution profiles of EVs were assessed using TEM images and nanoparticle tracking analysis. Surface analysis revealed the presence of α-2,3- and α-2,6-linked sialic acid-capped complex N-glycans, along with bi-antennary N-glycans. Screening for siglec ligands indicated preferential binding to siglec-9, which is predominantly expressed on DCs. Desialylation via pan-sialic acid hydrolase reduced sialic acid on EVs, and subsequent insertion of Ley, a high-affinity ligand for DC-SIGN, resulted in a four-fold increase in uptake by moDCs. This suggests that modified EVs could serve as a promising anti-cancer vaccine.333
Prof. Zhuang Liu's group has constructed a cancer vaccine by encapsulating the TLR7 agonist imiquimod (R837) in PLGA nanoparticles coated with cancer cell membrane whose surface proteins act as tumor-specific antigen and further modified them with mannose to prepare NP-R@M-M. In brief, PLGA NPs loaded with TLR7 agonist imiquimod (R837) were prepared. B16-OVA membrane in PBS was mixed with PLGA NPs at 4 °C overnight (NP-R@M). NPs were collected by centrifugation and further modified with mannose. The obtained membrane-coated NPs (NP-R@M) were mixed with mannose-conjugated 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-methoxy (polyethylene glycol) (DSPE-PEG-Man) to prepare mannose-modified and membrane-coated adjuvant NPs (NP-R@M-M). The solution was further stirred for 1 h and the obtained product (NP-R@M-M) was stored in PBS. The sizes of NP-R@M and NP-R@M-M were in the range of 140–160 nm. Membrane-coated NPs have a zeta potential of −23 mV. NP-R@M-M showed enhanced cellular uptake by BMDCs and were highly taken up by MØs. NP-R@M-M are also very effective in triggering DC maturation, secreting high amounts of TNF-α and IL-12 cytokines. NP-R@M-M showed high retention in lymph nodes due to high interaction with APC (MR positive), exhibited a very high antitumor effect and also triggered upregulation of IFN-γ generation.336
Prof. Wantong Song's group has developed TLR9 agonist CpG- and OVA-loaded mannan-decorated polylactic acid–polyethylenimine (PLA–PEI) assembled nanoparticles which have been shown to enhance the targeting of DCs, which can be the result of MR- and DC-SIGN-mediated endocytosis, stimulate DC maturation, promote XPT and enhance tumor-specific immune responses. Effective vaccine accumulation in lymph nodes is crucial for a strong T cell response. Particles sized 20–200 nm are optimal for this, and surface hydrophilic shielding can improve their lymph node drainage. Cationic polymers like branched PEI enhance this process through the proton sponge effect, facilitating endosomal rupture and antigen release into the cytosol, making PEI a valuable nanocarrier. The MPVax nanovaccine, developed for cancer immunotherapy, utilizes a PLA–PEI core loaded with CpG and antigens, shielded with oxidized mannan. In brief, conjugation of PLA to PEI was performed to create PLA–PEI, followed by the formation of a PLA–PEI inner core (PVax) through nanoprecipitation. The MPVax-CpG/OVA was prepared by mixing OVA and CpG with PVax (1.0 mg ml−1) in a weight ratio of 2:1:10, and then adding this mixture to mannan solution (5 mg ml−1) at a 1:5 volume ratio. The resultant MPVax-CpG/OVA had a size of 110 ± 20 nm, a zeta potential of 11 ± 2 mV, and an OVA loading efficiency of 96 ± 3%. The internalization behavior indicated stronger fluorescence intensity for MPVax-CpG/cy5-OVA, attributed to MR- and DC-SIGN-mediated endocytosis. This formulation exhibited the strongest stimulatory effects and improved DC activation due to the combined action of CpG and mannan. MPVax-CpG/OVA achieved the highest antigen XPT, with mannan enhancing the vaccine's ability to drain to lymph nodes. A greater population of OVA+ DCs was observed in the MPVax-CpG/OVA group, particularly an increase in OVA+CD8+ DCs in the deeper paracortex of lymph nodes, confirming superior antigen XPT. Treatment with MPVax-CpG/OVA resulted in a significantly increased IFN-γ+ CD4+ and CD8+ T cell population, as well as heightened CD4+ and CD8+ memory T cell levels, leading to enhanced T cell infiltration and effective tumor growth inhibition. In murine models, MPVax-CpG/MC38 demonstrated strong efficacy in preventing tumor relapse post-surgery, with elevated IFN-γ+ T cell populations and effective memory response induction. Similarly, in the EO771 tumor model, MPVax-CpG/EO771 nanovaccine improved survival rates. In both the murine tumor models a superior anti-tumor response was observed, with 50% of mice completely cured.337
In another study mannan-decorated stimulator of the interferon genes (STING)-activating vaccine carrier was developed for systematic, spatially coordinated stimulation of antigen-specific immune responses and induction of strong anti-tumor immunity. In brief, the azole molecule end-capped polyethyleneimine (PEI-4BImi), which was shown to have innate stimulating activity via activation of the STING pathway,338 was further improved. PLA–PEI-4BImi (referred to as PPB) was synthesized using amide condensation agents. The synthesis involved dissolving 4BImi (1 mmol, 20 equivalents), PLA–PEI (0.025 mmol, 1 equivalent), EDC–HCL (1.5 mmol, 30 equivalents), and NHS (2 mmol, 40 equivalents) in DMSO, which was dialyzed after 72 hours with water at a pH ranging from 3 to 7 and then lyophilized. To prepare PPB/OVA-M0.5/1, OVA solution (1.0 mg ml−1) was added dropwise to the PPB solution (3.0 mg ml−1) at a 1:1 volume ratio while vortexing, and then oxidized mannan (1.0 mg ml−1) was added at weight ratios of 1:0.5 and 1:1. This process conjugated hydrophobic PLA with branched PEI, enhancing nanoparticle stability. Additionally, the loading of OVA followed by mannan decoration resulted in an increase in the diameter of the nanoparticles and a decrease in their zeta potential. PPB/OVA-M0.5/1 remained stable for 48 hours due to branched PLA and effectively activated the STING pathway, as evidenced by IFN-β secretion, despite a slight reduction in IFN-β caused by mannan decoration. This formulation enhanced cellular uptake, DC maturation, and TLR4 interactions, promoting synergistic effects with the STING pathway as shown in Fig. 7. Mannan decoration further improved its accumulation and DC-targeting capacity in the lymph nodes. PPB/OVA-M1 resulted in a significant increase in OVA+CD8+ DC populations, along with enhanced accumulation in pDCs, crucial for IFN secretion and T cell differentiation. In vivo studies demonstrated that PPB/OVA-M1 had superior spatial synergy in activating antigen-specific immunity, indicated by better innate stimulation through STING pathway phosphorylation. In a B16-OVA melanoma model, PPB/OVA-M1 displayed a remarkable 93% tumor growth inhibition, increased DCs in the lymph nodes, and boosted CD8+ effector and central memory T cells, leading to a robust antigen-specific immune response. These findings highlight the superior efficacy of the mannan-decorated STING activating vaccine.339
 | ||
| Fig. 7 Glycomaterial-based targeting of siglec-1 and MR for improved anti-cancer immune response focusing on enhanced CD8+ T cell responses: (a) delivery of GM3/WT1/MPLA-liposomes to CD169+ moDCs and Axl+ DCs leads to XPT of antigen and activation of tumor antigen-specific CD8+ T cells. GM3/WT1/MPLA-liposomes were bound and taken up by human moDC in a CD169-dependent manner and stimulate IL-6 production by DCs, further promoting IFN-γ secretion by CD8+ T cells. Similarly, GM3/WT1/R848 liposomes are specifically bound and taken up by Axl+ DCs in a CD169-dependent manner, induce TNFα production and further drive IFN-γ production by CD8+ T cells; (b) Cancer cell membrane-coated adjuvant nanoparticles modified with mannose and containing the TLR7 agonist imiquimod (R837) were significantly taken up by MR-positive DCs. This approach was highly effective in triggering DC maturation and effective CTL responses, and when combined with PD-1 blockade, it exhibited a remarkable antitumor effect; (c) Mannan-decorated pathogen-like polymeric nanoparticles (MPVax), loaded with CpG and antigen, promote efficient accumulation in lymph nodes, enhance endocytosis, stimulate DC activation and promote antigen XPT, induce robust antigen-specific T cell responses, and effectively inhibit tumor growth; (d) Mannan-decorated STING-activating vaccines improve cellular uptake, DC maturation, and TLR4 interactions. This promotes synergistic effects with the STING pathway, resulting in nanovaccine accumulation in lymph nodes. Further mannan decoration improved DC targeting ability in lymph nodes and CD4+ and CD8+ T cells were greatly enhanced. Effector and central memory T cells were also significantly enhanced, ultimately leading to a strong antigen-specific immune response (created in BioRender. Jain, M. (2025) https://BioRender.com/78m3q01). | ||
Following a similar strategy, chondroitin sulfate (CS)-based pH-sensitive polymer-modified liposomes were prepared. CS derivatives were prepared by reaction of CS with 3-methylglutaric anhydride and 1,2-cyclohexanedicarboxylic anhydride to MGlu-CS and 2-carboxycyclohexane-1-carboxylated CS (CHex-CS). A dry, thin membrane made from a mixture of EYPC and polysaccharides at different lipid–polymer ratios was suspended in PBS with 4 mg mL−1 OVA through brief sonication. This was followed by hydration via freeze-thaw and extrusion through a 100 nm polycarbonate membrane. The liposome suspension was then purified by ultracentrifugation, resulting in liposomes with an average size of 130–200 nm and a negative zeta potential. CS derivative-modified liposomes containing high contents of MGlu or CHex units displayed elevated cellular association to DCs. Dextran sulfate, an inhibitor of scavenger receptor (SR), suppressed the cellular association, suggesting that liposomes were internalized by recognition of anionic molecules on liposomes by DC2.4 cell SR. Polymer-modified liposomes were taken up by the DC2.4 cells and they released FITC-OVA inside of endo/lysosomes in response to a weak acidic pH. Modified liposomes further destabilized the endo/lysosomal membranes over time, leading to the release of FITC-OVA into the cytosol. Introduction of CHex in CS increased their adjuvant properties. CHex77-CS-A8-modified liposomes showed higher cytokine production by DCs, confirming that CS derivatives showed an adjuvant effect upon modification which was responsible for tumor regression in tumor-bearing mice.341
Further study explored the combined antigen delivery and adjuvant capabilities of pH-sensitive liposomes incorporating the bioactive polysaccharides curdlan and mannan, which are recognized by the CLRs dectin-1 and dectin-2 on MØs and DCs. 3-Methyl glutarylated mannan (MGlu-Man) and 3-methyl glutarylated curdlan (MGlu-Curd) were prepared by reaction with 3-methylglutaric anhydride. Polymer-modified liposomes containing MGlu-Man and MGlu-Curd were prepared using OVA as a model antigen, similar to the dextran- and CS-based pH-sensitive liposomes. MPLA (4 g mol−1 lipids) was incorporated into the liposomal membrane to enhance the immune response. The modified liposomes exhibited a smaller diameter, ranging from 100 nm to 131 nm, compared with unmodified liposomes (157 nm). Additionally, the zeta potential of unmodified liposomes was −11.6 mV, while modified liposomes ranged from −20.0 mV to −50.5 mV. The decreased zeta potential after modification with polysaccharide derivatives suggests effective surface coverage with carboxylated compounds. For example, MGlu-Curd-C10 exhibited a strong negative charge of −50 mV, while MGlu-Man-C10 showed a lower zeta potential, indicating limited exposure of carboxylic acids due to steric hindrance from branched structures. Modified liposomes in the weakly acidic pH range (6.5–5.0, corresponding to early/late endosomes) displayed content release but became hydrophobic and destabilized at these pH levels. MGlu-Curd-C10-modified liposomes demonstrated significantly higher content release compared with MGlu-Man-C10 due to differences in hydrophobicity. MGlu-Curd-C10 exhibited 13 times greater cellular association than unmodified liposomes, while MGlu-Man-C10 had less cellular association due to its lower zeta potential, as SR on DCs preferentially recognize anionic molecules. The high affinity of SR for MGlu-Curd-C10 was confirmed by the inhibition of cellular association with dextran sulfate, and to a lesser extent by curdlan, indicating interaction with dectin-1. However, liposomes with high MGlu content were unaffected by curdlan, suggesting that higher MGlu levels inhibit the curdlan–dectin-1 interaction. Notably, the interaction of MGlu-Man-C10-modified liposomes was also inhibited by dextran sulfate, indicating SR-mediated interactions. Therefore, modification of mannan with MGlu completely abrogated its interaction with dectin-2. Furthermore, the antigen delivery performance of modified liposomes showed much lower colocalization values than unmodified liposomes, confirmed that polysaccharide derivatives became hydrophobic at the weakly acidic pH of early/late endosomes and destabilized efficiently their and the endosomal membranes and released the FITC-OVA in the cytosol of DCs. Moreover, curdlan- and mannan-modified liposomes induced higher levels of TNF-α, IL-10, IL-12, and enhanced IL-1β production, activating the inflammasome. In vivo, MGlu-Curd modified liposomes triggered a significant increase in IFN-γ and exhibited greater cytotoxicity as well as antitumor effects. Notably, treatment with MGlu-Curd-C10 without MPLA resulted in improved tumor regression and prolonged survival, while the presence of MPLA further amplified the antitumor response, with MGlu-Man-C10 showing the strongest effects.342
pH-Responsive hyaluronic acid (HA) derivatives are gaining popularity as an intracellular drug delivery system. HA selectively binds to the CD44 molecule on cancer cells, enabling the targeted release of anti-cancer drugs in response to endosomal pH changes. Additionally, CD44 is expressed on antigen-presenting cells (APCs), where HA acts as an inflammatory mediator via recognition of CD44 and/or TLR2/4. Liposomes modified with pH-sensitive HA derivatives (MGlu-HA-A and CHex-HA-A) were developed, averaging 120–140 nm in size with a negative zeta potential. These HA-modified liposomes exhibited enhanced cellular uptake by APCs, delivered antigens to DC cytosols, and demonstrated adjuvant properties, evidenced by increased production of Th1 cytokines (TNF-α and IL-12). Notably, CHex-HA derivatives significantly boosted TNF-α production with higher CHex units up to 100 resulting in increased cytokine production. Higher CHex units introduced in liposomes also showed high IL-12 from DC2.4 cells. Moreover, the modified liposomes enhanced IFN-γ production and showed promising therapeutic effects in tumor-bearing mice, leading to reduced tumor volume and promoting antigen-specific cellular immune responses.343
Incorporating mannose residues onto antigen delivery carriers enhances recognition by APCs. MR-mediated targeting facilitates antigen uptake by DCs via the weakly acidic compartments, also known as the “vacuolar pathway”, making mannose modification a key strategy for effective antigen delivery. The feasibility of mannose modification on pH-responsive curdlan was assessed. Curdlan derivatives containing MGlu as pH-sensitive units (MGlu-Curd) and decylamidated units for liposomal membranes (MGlu-Curd-A) were further modified with mannose to create MGlu-Curd-A-Man derivatives for antigen-loaded liposomes. These liposomes were prepared by hydrating a mixed thin film of EYPC and the curdlan derivatives, using OVA as a model antigen. The liposomes were extruded through a 200 nm polycarbonate membrane and purified using a Sepharose 4B column. Prepared liposomes were in a size range of 150–180 nm with a negative zeta potential showing modification of carboxylated curdlan derivatives. These modified liposomes demonstrated content release at weakly acidic pH levels (5–6), characteristic of late endosomes and lysosomes, facilitating intracellular release. Man-3-modified liposomes achieved over 90% content release at late endosome pH, showing 3–4 times higher cellular association in MR-expressing RAW264.7 cells. Cellular associations of primary cells from the spleen were also evaluated. Mannose-modified curdlan derivative-modified liposomes were highly internalized by F4/80+ MØs and CD11c+ DCs, thus showing the possibility of uptake of these liposomes by APCs in vivo also. Modified liposomes were internalized via endocytosis and localized in endo/lysosomes. When delivering the FITC-OVA model antigen, mannose-modified liposomes exhibited enhanced cellular uptake and effective XPT, leading to strong antitumor effects in tumor-bearing mice. However, no significant difference was noted between curdlan-modified pH-sensitive liposomes and mannose-modified variants in vivo, suggesting that further improvements are needed.344
To further improve the specific cell uptake, XPT efficiency, adjuvant effect, and antigen-specific immune response, a pH-sensitive polysaccharide derivative and Man9GlcNAc2-glycopeptide were introduced in antigen-loaded liposomes. Model antigen OVA-loaded liposomes modified with pH-responsive polysaccharide derivatives and soybean agglutinin (SBA)-derived glycopeptides were prepared by hydration of a mixed thin film composed of lipids and polysaccharide derivatives and further extruded with a polycarbonate filter with a 100 nm pore size to adjust the liposome size. Furthermore, maleimide-introduced SBA glycopeptide was reacted with the liposomal surface via a thiol–maleimide reaction. The size of the liposomes was in the range of 90–110 nm, corresponding to the pore size of the polycarbonate membrane. The PDI value of 0.10 showed a narrow size distribution. Modification of pH-responsive polysaccharide derivatives results in a decrease of zeta potential of liposomes and confirmed the modification of carboxylated polymers on the surface. The prepared liposomes showed content release below 6.5 pH. At pH 5.5 belonging to the endosomal pH, 100% of the contents were released. Glycopeptide- and pH-responsive polysaccharide-modified liposomes enhanced the cellular association of these liposomes and in the presence of excess glycopeptide the observed association was decreased, confirming the interaction of these liposomes with the lectins present on DC2.4 cells. Glycopeptide- and pH-responsive polysaccharide-modified liposomes demonstrated significantly enhanced XPT due to their high cellular association. These liposomes also boosted TNF-α and IL-6 production, and increased pro-inflammatory cytokines, and displayed pH-responsive content release, along with strong adjuvant properties. These liposomes elevated the populations of DCs, M1 MØs, and effector T cells in the spleen, while also promoting IFN-γ secretion from splenocytes. Notably, treatment with these liposomes reduced tumor volume in tumor-bearing mice. This study highlights the effectiveness of combining pH-responsive liposomes and glycopeptides in antigen-loaded formulations for improved XPT and cancer immunotherapy.295 The summary of these pH-sensitive liposomes is illustrated in Fig. 8.
 | ||
| Fig. 8 Overview of glycan-coated pH-sensitive liposomes in modulating immune responses: (a) dextran derivative-based pH-sensitive liposomes are endocytosed by DCs into the endosome and get destabilized upon exposure to weak acidic environments, triggering the release of antigen into the endosomes and their transfer to the cytosol. The antigens in the cytosol are presented via MHC class I to CD8+ T cells, enhancing the CTL response. Antigens in the endosome are presented through MHC class II to CD4+ T cell inducing antigen-specific Th1 and Th2 cells; (b) Chondroitin sulfate derivative-modified liposomes specifically target APCs through scavenger receptors, stimulating cytokine production and enabling the escape of antigenic proteins from endosomes via pH-responsive membrane destabilization leading to CD8+ T cell responses; (c) liposomes modified with curdlan and mannan derivative are recognized by the receptors dectin-1 and dectin-2. These liposomes are endocytosed by DCs, leading to the release of antigens into the cytosol due to the acidic environment within the endosomes, which promotes proteasomal degradation for subsequent presentation to CD8+ T cells; (d) HA-based pH-sensitive liposomes are recognized by CD44 found on APCs. These liposomes effectively deliver model antigenic proteins into the cytosol of DCs, resulting in the release of degraded antigenic peptides that are then loaded onto MHC class I molecules, eliciting recognition by CD8+ T cells; (e) The MR recognizes mannose-modified curdlan-coated liposomes and transports them to the endosome for degradation. The acidic environment further promotes release of the antigen into the cytosol, where it is degraded by proteasomes and then presented to CD8+ T cells. They are also recognized by β-glucan receptors, which leads to enhanced DC maturation; (f) soybean agglutinin-derived glycopeptide-modified pH-sensitive liposomes enhanced the liposome lectin-mediated uptake by DCs and cytosolic release of antigen via pH-responsive endosomal membrane disruption. These liposomes promote the XPT and induction of antigen-specific CD8+ and CD4+ T cell responses (created in BioRender. Jain, M. (2025) https://BioRender.com/wszgfjn). | ||
Indeed, XPT-based GNVs for cancer immunotherapy have shown their performance in antigen-specific CD8+ T cell response generation. However, achieving their full potential requires overcoming the immunosuppressive effects of immunomodulatory molecules such as siglecs and galectins.
Various nanocarriers can also be potentially functionalized to deliver galectin and siglec inhibitors to sites of tumor progression. The overall efficacy of the GNVs can be enhanced by either integrating galectin and siglec inhibitors into the GNVs or providing nanocarrier-encapsulated galectin and siglec inhibitors in combination with GNVs for obstructing the glycan–lectin interactions which can help to suppress tumor growth by inducing antitumor immunity and inhibiting aberrant angiogenesis.345Table 3 displays various nanoparticle (NP)-based therapies for inhibition of glycan–lectin interactions to decrease cancer progression.
| Target | Structures | Carrier | Cancer model | Combination | Mode of administration | Outcome | Ref. |
|---|---|---|---|---|---|---|---|
| Gal-1 |

|
Chitosan lipid nanocapsules | Glioblastoma | Anti-EGFR anti-Gal-1 siRNA, temozolomide | Intracranial injection by convection enhancement delivery | Reduced expression of EGFR and galectin-1 and enhanced survival | 346 |
| Gal-3 |

|
STn-targeted PLA–didodecyldimethylammonium bromide (PLA-DDAB) NPs | Gastric cancer | ST6GalNac-I- and Gal-3-targeting dsRNA | Intravenous injection | Downregulation of ST6GalNAc-I and Gal-3 RNA expression levels | 347 |
| Gal-1 |

|
Antibody-like polymeric NPs composed of albumin–polymer hybrid NPs and acid-responsive PEG shell | Melanoma | Anti-PD-1 antibody | Intravenous injection | Remove Gal-1 from TME and encourage T cell infiltration and T cell-mediated antitumor immune activation | 348 |
| Gal-3 |

|
Dialdehyde oligomer of citrus pectin CPDA-based core–shell NPs | TNBC | Doxorubicin and sulindac | Intravenous injection | Efficient Gal-3 targeting and binding, along with tumor responsiveness and synergistic delivery of doxorubicin and sulindac | 349 |
| Siglec-2 |

|
BPCNeuAc liposomes | B-cell lymphoma | Doxorubicin | Intravenous injection | Bind and kill malignant B cells from peripheral blood samples | 350 |
| Siglec-2 |

|
Polysialic acid-modified PLGA nanocarrier | B-cell lymphoma | Mitoxantrone | Intravenous injection | Promote immunogenic cell death and anti-tumor immune responses | 351 |
11. Ongoing approaches and upcoming ways to challenge the tumor glyco-code
One of the key tasks that must be addressed to enhance the effectiveness of immunotherapies, such as GNVs, is the challenge posed by the glyco-code, a complex system that influence how immune cells identify and respond to tumors. To address this challenge, a range of inhibitors that target the lectins responsible for recognizing these glycans is being developed. Although drug development in overcoming the role of glycan–lectin networks in immune suppression is on the rise, most of them are still in the preclinical and clinical trial stages, and only a small number have received FDA approval for the treatment of cancer patients.The FDA has authorized the use of two siglec inhibitors against siglec-2 and -3 for the treatment of cancer patients. More than 90% of B-cell acute lymphoblastic leukemia (B-ALL) cells and mature B lymphocytes express siglec-2, making it a potential target for immunotherapy. Inotuzumab ozogamicin (Besponsa) is a humanized mAb conjugated to calicheamicin targeted towards siglec-2, and after binding with Siglec-2, it gets internalized into lysosomal vesicles. Calicheamicin, being a highly potent cytotoxic antitumor antibiotic, binds to the DNA minor groove and cleaves double-strand DNA, further leading to cell apoptosis.352,353
A phase II inotuzumab ozogamicin single agent (ITCC-059) clinical trial for the activity of inotuzumab ozogamicin in pediatric patients having relapsed/refractory (R/R) B-cell precursor acute lymphoblastic leukemia (R/R BCP-ALL) showed that an overall response rate (ORR) of 81.5% was achieved after the first cycle and minimal residual disease (MRD) negativity as the best response was achieved in 81.8% of the responding subjects. One-year event-free survival (EFS) was found to be 36.7%, while overall survival (OS) was observed to be 55.1%. These results show that inotuzumab ozogamicin was mostly well tolerated with a low prevalence of infections occurring during the treatment; however, sinusoidal obstructive syndrome (SOS), occurring in 25% of patients, still remains the most serious adverse event (AE).354 The INO-Ped-ALL-1 phase I trial evaluated inotuzumab ozogamicin in pediatric patients with R/R CD22-positive ALL in Japan. Out of 6 enrolled patients (median age: 7.5 years), 5 patients achieved complete remission, among whom 3 were found to be MRD-negative. No dose-limiting toxicities were observed, but all patients experienced AEs, including an increase in alanine aminotransferase along with aspartate aminotransferase in 4 individuals. 3 individuals experienced serious AEs, which included hepatic veno-occlusive disease (VOD), ALL, and fever. Furthermore, no anti-drug antibodies were detected against inotuzumab ozogamicin. While inotuzumab ozogamicin was found to be well tolerated in pediatric R/R ALL, the risk of VOD requires close monitoring in future studies.355 The INITIAL-1 (NCT03460522), a phase II trial, evaluated inotuzumab ozogamicin along with dexamethasone as induction therapy in elderly individuals (≥55 years) having CD22+ Philadelphia chromosome-negative BCP-ALL. Among 43 patients (median age: 64 years), all achieved complete remission, with 23 (53%) and 30 (71%) patients being MRD-negative following the second and third rounds of treatment, respectively. The EFS after a median follow-up of 2.7 years at 1 and 3 years was 88% and 55%, while OS was 91% and 73%, respectively. There were no deaths during the 6 months after the start of the induction. The most common AEs were leukocytopenia, neutropenia, thrombocytopenia, anemia, and elevated liver enzymes having a common toxicity criteria grade ≥3 and only one case of nonfatal VOD after second induction. These results support inotuzumab ozogamicin as a viable and well tolerated first-line option for older B-ALL patients, resulting in high rates of remission and OS.356 A phase II (NCT03441061) trial demonstrated that inotuzumab ozogamicin is effective in eradicating MRD in CD22-positive ALL. Among 26 patients (median age: 46 years), 18 (69%) achieved MRD negativity, with 16 (89%) responding after the first cycle. At 2 years, relapse-free survival (RFS) was 54%, and OS was 60%. The treatment was generally well tolerated, though SOS occurred in 8% of patients. This trial shows that in patients with B-cell ALL who have persistent MRD or experience MRD recurrence, inotuzumab ozogamicin is a therapeutic approach that is both safe as well as effective in abolishing MRD.357
Gemtuzumab ozogamicin (Mylotarg) is another humanized anti-siglec-3 mAb antibody covalently linked to N-acetyl gamma calicheamicin. Most AML cells and immature cells of myelomonocytic lineage exhibit siglec-3, to which gemtuzumab ozogamicin binds and upon binding it gets internalized, with further release of calicheamicin, which intercalates DNA, and further shows double-strand DNA breaks ultimately leading to cell death.358 The AAML0531 (NCT01407757) phase III trial evaluated gemtuzumab ozogamicin in pediatric KMT2A-rearranged AML. EFS was found to be significantly superior after treatment with gemtuzumab ozogamicin. While EFS was 29% in the absence of gemtuzumab ozogamicin, it increased to 48% with gemtuzumab ozogamicin treatment. For patients treated with gemtuzumab ozogamicin and who had complete remission, gemtuzumab ozogamicin was found to be linked with better 5-year disease-free survival and lower relapse risk. Furthermore, a lower risk of relapse was linked to prior gemtuzumab ozogamicin exposure in patients who underwent hematopoietic stem cell transplant (HSCT). The results of this trial showed that gemtuzumab ozogamicin is independently associated with decreased relapse risk, improved EFS, and disease-free survival (DFS). Based on these findings, gemtuzumab ozogamicin may be helpful in combination with chemotherapy, particularly in patients who undergo HSCT consolidation.359 Another phase IV trial, NCT03727750, investigated the QT interval, pharmacokinetics, and safety following fractionated gemtuzumab ozogamicin dosing in R/R CD33+ AML patients. In 50 individuals (median age: 67 years), a gemtuzumab ozogamicin fractioned dosage regimen (3 mg per m2 per dose) was not found to pose any significant safety risk to QT interval prolongation. The most frequent grade 3–4 treatment-emergent AEs were thrombocytopenia and febrile neutropenia, with no instances of VOD. Additionally, the best overall response based on complete response (CR) with incomplete platelet recovery (CRi) was obtained in 9.8% of patients, with a median OS of 2.8 months.360
While these drugs are currently available to target specific siglecs within the TME, they are not completely potent. This implies that further studies and the development of major inhibitors of immunosuppressive lectins are necessary to facilitate an efficient anti-cancer immune response.
Many of the glycan–lectin interaction inhibitors are currently undergoing clinical trials and hold great promise in the treatment of cancer patients. By disrupting the glyco-code, these inhibitors could help to enhance the immune response to cancer and improve the efficacy of the immunotherapies.
SGN-2FF is a first-in-class and first in-human, oral small-molecule inhibitor targeting glycoprotein fucosylation in individuals having advanced solid tumors. The purpose of this phase 1 study (NCT02952989), which included 46 patients, was to assess the safety, tolerability, maximum tolerated dose (MTD), pharmacokinetics (PK), pharmacodynamics (PD), and antitumor activity of SGN-2FF both as monotherapy and as combination therapy with pembrolizumab. The trial comprised 4 parts: SGN-2FF monotherapy dose-escalation (part A) and expansion (part B), and SGN-2FF + pembrolizumab dose-escalation (part C) and expansion (part D). SGN-2FF was administered to 32 individuals in part A at doses varying from 1–15 g once daily (QD) and 2–5 g twice daily (b.i.d.). 10 g per day was found to be the maximum tolerated dose (MTD). Fatigue, nausea, and diarrhea in grades 1–2 were common toxicities. However, the study was terminated because 16% of patients in part A and 14% of patients in part C experienced thromboembolic events (grades 2–5) during the SGN-2FF lead-in period. The study showed that there was evidence of pharmacodynamic target inhibition, as levels of IgG fucosylation decreased with higher doses of SGN-2FF and longer treatment periods. Preliminary antitumor activity was observed, including one complete response in a patient with advanced HNSCC and stable disease in 36% of evaluable patients. Notably, an individual with advanced triple-negative breast cancer experienced a 51% reduction in tumor burden.361
AMG 330 is a bispecific T cell engager (BiTE®) designed to target CD33 along with CD3 on T cells, facilitating T cell-mediated cytotoxicity towards CD33+ cells. This first-in-human, open-label, dose-escalation study evaluated the safety, pharmacokinetics, and preliminary efficacy of AMG 330 in 77 adult patients with R/R AML, receiving doses from 0.5 μg day−1 to 1.6 mg day−1 in 14-day or 28-day cycles. The maximum tolerated dose (MTD) was not established, and the most frequently observed adverse effect was cytokine release syndrome (CRS), which occurred in 78% of participants, with 10% experiencing Grade 3/4 CRS. This was effectively managed with stepwise AMG 330 dosing, dexamethasone, and early treatment of tocilizumab. Out of 60 patients evaluated, 8 achieved complete remission or were in a morphologic leukemia-free state (MLFS), while 37% of those who did not respond showed a reduction of 50% or more in AML blasts. Thus AMG 330 was considered as promising CD33-targeting therapeutic strategy for R/R AML.362 JNJ-67571244 is a CD33 and CD3 bispecific antibody which was designed to engage CD3 protein on T cells and target CD33-expressing AML cells to selectively eliminate cancerous cells. The first-in-human, phase I, dose-escalation/dose-expansion study aimed to identify the MTD, recommended phase II dose (RP2D), safety profile, and preliminary clinical activity of JNJ-67571244. The study enrolled 68 patients who received JNJ-67571244 either intravenously (IV) or subcutaneously (SC) using a step-up dosing schedule. However, the study encountered significant challenges with toxicity, particularly CRS, infusion-related reactions (IRR), and elevated liver function tests. These AEs limited dose escalation and prevented reaching the projected exposure level for efficacy. No patient achieved a significant response, though some experienced temporary disease burden reductions. The study was terminated after assessing ten dose-escalation cohorts and prior to initiating dose-expansion, without determining the MTD or RP2D.363
NC318 is a humanized IgG1 mAb that inhibits siglec-15-mediated immune suppression and prevents tumor growth by reestablishing anti-tumor immunity in the TME and normalizing T cell activity.364 In a Phase 1/2 dose escalation trial (NCT03665285), an earlier monotherapy study from NextCure demonstrated single-agent activity of NC318 for patients with advanced solid tumors. A randomized trial was conducted to study the efficacy and safety of NC318 alone or in combination with pembrolizumab. The combined portion of this study was to assess the efficacy in NSCLC patients who have seen disease progression on/after PD-1 axis inhibitor therapy (NCT04699123). The trial's key findings show that pembrolizumab and NC318 perform effectively together in advanced PD-1 axis inhibitor-refractory NSCLC. Durable clinical benefit was reported by 28% of patients (5/18), with three of these responses being confirmed. Despite six Grade 3 treatment-related adverse events (TRAEs), including transverse myelitis, infusion reactions, rash, and pneumonitis, and four Grade 2 TRAEs, including pericarditis, psoriasis, and infusion reactions, the monotherapy and combination arms were well tolerated. Seven patients (three Grade 3 and four Grade 2) had infusion reactions to NC318; six received the combination, and one received solely NC318. There were no further infusion reactions after NC318's infusion time was increased from 30 to 60 minutes. Gal-3 can be utilized as a prognostic or diagnostic biomarker to monitor the progression of a disease or the effectiveness of a treatment. The concentration of the circulating Gal-3 in the serum of cancer patients has been shown to be greater compared with healthy persons, and the amount of Gal-3, which can vary in various malignancies, has been demonstrated to correlate with tumor growth. GB1211, a small molecule Gal-3 inhibitor, was developed as part of a new class of galactopyranosides in which non-natural aromatic substituents are added to the 1- and 3-positions of α-D-galactopyranosides, and its affinity towards Gal-3 is then optimized by specific interactions such as fluorine–amide, sulphur–π, phenyl–arginine and halogen bonds. With a significant oral bioavailability in animals (68% in mice) and a high affinity for Gal-3 (0.025 ± 0.0017 M), GB1211 is now being explored for the treatment of cancer, particularly NSCLC (NCT05240131). Gal-3 inhibition with the structural counterpart of GB1211, GB1107, which increases the infiltration and activity of CD8+ CTLs within NSCLC tumors and decreases the number of M2-like TAMs, provides evidence in favor of this. This reduces tumor development and metastasis while increasing responsiveness to anti-PD-L1 treatment. GB1211 also reverses Gal-3-induced suppression of ICIs, such as pembrolizumab and atezolizumab, by binding to PD-1 and PD-L1. This lowers tumor resistance towards these drugs and restores responsiveness for ICIs.365–367 Calixarene derivative OTX008 binds to the amphipathic β-sheet structure of Gal-1.368 Nuclear magnetic resonance (NMR) studies revealed that OTX008 binds Gal-1 in a region remote from the lectin CRD and works as an allosteric inhibitor of glycan binding.369 At micromolar concentrations, OTX008 exhibited anti-proliferative and anti-invasive characteristics in colon, head and neck, breast, prostate, renal, ovarian, and lung cancer cell lines.368,370 The La Jolla Pharmaceutical Company developed GCS-100 as a potential inhibitor of Gal-3. GCS-100 exhibits antitumor action, including the induction of apoptosis in multiple myeloma cells. GCS-100 stimulates AML cell apoptosis either by itself or in combination with BCL-2 homology domain 3 (BH3) mimetics. Intravenous administration of GCS-100 to patients with chronic lymphocytic leukemia in a phase II clinical study (NCT00514696) demonstrated that it was well tolerated and that it led to a more than 50% decrease of lymph node lesions in 16% of patients and partial remission in 25% of patients. GR-MD-02, also known as belapectin, is a pectin-derived galactoarabino-rhamnogalacturonate with mainly β(1–4)-D-galactose and α(1–4)-L-arabinose side chains developed by Galectin Therapeutics and has been proposed as a Gal-3 inhibitor.159 A phase I clinical trial (NCT02575404) on patients with metastatic melanoma or HNSCC to test the effectiveness of belapectin in combination with pembrolizumab showed belapectin plus pembrolizumab treatment is safe, linked to improved clinical outcomes, enhanced T cell activation, and limited expansion of monocytic MDSCs.371 A chemically altered galactomannan from the plant Cyamopsis tetragonoloba (Guar Gum), GM-CT-01, Davanat, has been demonstrated to bind Gal-3 and increase the effectiveness of the chemotherapy drug 5-fluorouracil in breast and colon cancer models. These results further led to phase I (NCT00054977) and phase II (NCT00110721) clinical trials which showed no toxicities and a 46% improvement in survival for CRC patients who took this treatment combination.159 These strategies of overcoming the immunosuppressive glycan–lectin circuits are shown in Fig. 9.
 | ||
| Fig. 9 Strategies to inhibit immunosuppressive TMEs and enhance glyconanovaccine efficacy: GNVs are promising immunotherapeutic vaccines for the treatment of cancer. However, for the success of the GNVs, a highly enhanced CTL response is required but various immunomodulatory molecules present in the immunosuppressive TME can be detrimental to its efficacy. Galectins, siglecs, and altered glycosylation are a few targets that can be focused upon to increase the CTL response, and here, two approaches have been highlighted: (1) galectin inhibitors and (2) siglec inhibitors. Various galectins are implicated in suppressing antitumor immunity, and approaches like NPs, peptides, carbohydrates, nucleic acids, and monoclonal antibody-based inhibitors can inhibit galectins. (i) The NP-based inhibitors like LacNAc expressing self-assembled nanofibers and CTL-PEG Au NPs can target Gal-1; (ii) peptide-based inhibitors directly inhibit Gal-1 and -3 by binding to them; (iii) various carbohydrate-based inhibitors, which bind and inhibit various galectins, are under clinical trials; (iv) nucleic acid-based strategies, such as siRNA-loaded liposomes, inhibit the synthesis of galectins and DNA aptamers, which inhibit galectins by directly binding to them; (v) anti-Gal antibodies such as F8.G7 and LYT-200 also inhibit galectins by directly binding to them. In the end, these approaches conclude in increasing the T cell activity by preventing the immunosuppressive roles of galectins and killing the tumor. In the case of siglec inhibitors, there are different ways, such as anti-siglec-antibody, sialic acid mimetic-based inhibition, and CAR-T cell therapy, and there are two FDA-approved drug-conjugates through which interaction between the sialic acids and siglecs can be inhibited. (vi) There are different antibodies like NC318, which targets siglec-15; AMG330/BiTE is a bispecific antibody that targets CD-3 present in T cells and siglec-3; anti-siglec antibody targets siglec-7 and -9 present on myeloid cells; (vii) in sialic acid mimetic (SAM)-based inhibition, the liposome is decorated with SAMs that bind to siglec-1, or -7 on the target cells, increasing T-cell activation; (viii) in CAR-T-cell therapy, anti-siglec-2 CAR-T cell binds to the siglec-2 of the malignant B-cell, further leading to its apoptosis; (ix) Besponsa and Mylotarg are the two FDA-approved drug-conjugated antibodies targeting siglec-2 and -3, respectively, and upon their binding, they get internalized into the lysosomes, where they are cleaved, and the drug is released, which leads to DNA damage leading to cell death. Apart from these targets, various ICs are also immunosuppressive in nature, which also needs to be blocked by various ICIs to increase the efficacy of the GNVs. Using these therapies in combination can be an effective approach to take full advantage of these therapies to enhance the efficacy of GNVs and CTL responses (created in BioRender. Jain, M. (2025) https://BioRender.com/e46y897). | ||
For maximum effectiveness, these inhibitors of the immunomodulatory glycan–lectin checkpoints can be used in conjunction with DC-based cancer vaccines such as GNVs. Due to the immunosuppressive factors offered by glycan–lectin interactions, only a small percentage of patients are benefiting from immunotherapies, despite their widespread success. A personalized patient-tailored regimen of immunotherapy and glycan–lectin checkpoint inhibition may ensure enhanced efficacy of DC-based vaccination strategies and improved survival rates.
12. Future prospects
The emergence of glycan–lectin interactions in tumor microenvironments as novel immune checkpoints represents a paradigm shift in the field of cancer biology and cancer immunotherapeutics. Aberrant glycosylation and immunomodulatory lectins are increasingly being acknowledged as major factors in altering the anti-cancer immune response, allowing the tumor to flourish by evading immune surveillance. By interrupting these immunosuppressive pathways, inhibitors of these glycan–lectin interactions have the potential to reinvigorate anti-tumor immunity and boost the effectiveness of existing and developing cancer immunotherapies.18Even though a lot of research regarding the aberrant glycosylation of tumors is being done, there is still scope for deeper exploration of tumor glycosylation patterns and their corresponding receptors expressed in the TME. At present, clinically approved cancer biomarkers are most effective for patients with advanced or widespread cancer. Unfortunately, some biomarkers do not reliably detect cancer in its earlier stages. There is still a need for individual biomarkers that demonstrate sufficient sensitivity and specificity for the most common types of cancer.372 According to a study, the degree of glycosylation of serum prostate-specific antigen (PSA) was measured using an advanced dual-functional aptamer assay. This was linked to cancer-associated breakage of PSA complexes with serum-circulating proteins, and it demonstrated its ability to classify primary and metastatic prostate cancer. The “Glycan Score” of PSA provided a 100% correct assessment of prostate cancer status in a group of 30 patients. Thus, the liquid biopsy glycan score biomarker has great promise for precise prostate cancer diagnosis and staging.373 Furthermore, it is necessary to create cell-based high-throughput screening assays that can accurately identify the glycosylation pattern and specific biomarkers and drug targets. This is essential for evaluating the comprehensive impact of glycosylation and therapy on the cells. For example, high-throughput mass spectrometry analysis of the SW480/SW620 shFUT8 CRC cell model led to new insights into the molecular characteristics of CRC. This analysis revealed significant alterations in the N-glycome and proteome of the SW480/SW620 cells following shRNA-induced knockdown of the FUT8 gene. However, existing research has not fully addressed the glycomic profile of each cancer stage, leaving many questions unanswered.374 Further detailed research is needed to gain a comprehensive understanding of the molecular evolution of different cancer stages. Specifically, thorough examination of the extensive microheterogeneity of N-glycomes is necessary to gain clearer insight into how aberrant glycosylation affects tumor progression. The precise identification of glycosylation signatures and comprehensive profiling of evolving glycosylation profiles of tumors that are specific to the type and stage of cancer can lead to the development of GNVs tailored to particular cancer types and stages. This approach can improve effectiveness and stimulate a specific immune response that aligns with the current immune landscape by targeting the stage-specific aberrations.
Aberrant glycosylation has emerged as a hallmark of cancer. This alteration in glycosylation patterns of cell surface and secreted glycoproteins occurs during malignant transformation and tumor progression. Growing evidence highlights glycosylation's crucial role in all stages of cancer development. Therefore, understanding this complex glycosylation machinery could provide valuable insights for the diagnosis, prognosis, and treatment of cancer.375 Artificial intelligence (AI) offers a robust analysis framework which can be used for glycosylation analysis, which has been challenging due to the intricate nature of glycosylation. Several glycosylation-related resources such as GlyTouCan, SugarBind, GlyCosmos, GlycoEpitope, Carbohydrate-Active Enzymes Database (CAZy) and GlycoGene DataBase can be used in computational models. Thus, AI models can be used in glycomics data analysis in five fundamental ways: (i) to comprehend glycosylation phenotypes to predict glycan structure, location site and site occupancy; (ii) to understand the aberrant glycosylation machinery for deciphering the pathophysiology of glycan-linked diseases, (iii) to gain insight into the intricate glycosylation mechanism by elucidating the mechanisms of glycosyltransferases and glycosidases; (iv) to develop glycan-targeted therapies to counteract the effects of abnormal glycosylation; and (v) to enhance the performance of the current predictive model of glycosylation and aid in glycomics data analysis.376 Artificial neural networks (ANNs) have been applied for predicting high-accuracy protein glycosylation. The ANN model has been proved to accurately predict site-specific distributions of glycoforms for as many as eighteen glycan species with an average absolute error of 1.1%. It accurately replicates the impact of metabolic disruption in a hybrid, kinetic/ANN, glycosylation model (HyGlycoM) and also the impact of manganese supplementation and glycosyltransferase knockout experiments as a stand-alone machine learning (ML) method.377 ML and bioinformatics have been applied to study the function of glycosylation-related genes in the development and prognosis of CRC. The integration of these strategies allowed identifying the most relevant glycosylation-related genes. Moreover, a strong prognostic risk model was established that successfully divided patients into high-risk and low-risk categories and this division also indicated significant survival rate differences and correlations with immune cell infiltration in the TME.378 Moreover, for the elucidation of functional differences in glycosylation patterns in lung adenocarcinoma (LUAD) and to identify the key genes expressed in relation to glycosylation, a predictive model based on ANNs was developed.379 Cellular dormancy is associated with a high recurrence rate of LUAD, which encourages resistance to chemotherapy and evasion of immune cell destruction. Single-cell RNA sequencing (scRNA-seq) data from LUAD patients were utilized to categorise dormant and active cells. The density weighted gene co-expression network analysis and pseudo-time cell trajectory were used to identify aberrant expression of genes related to protein O-glycosylation in dormant cells. A risk score model was created employing the hub genes and gene set variation analysis and ML which displayed excellent accuracy in predicting LUAD prognosis.380 LectinOracle is a deep learning model designed to predict lectin–glycan interactions by combining transformer-based protein representations with graph convolutional networks for glycans. It demonstrated high accuracy in cross-validation against experimental data, effectively predicting known lectin binding specificities. The model generalizes well to new lectins and glycans with qualitative and quantitative agreement with experimental data, and it can be used in applications such as lectin classification, lectin-directed evolution, prediction of epidemiological outcomes, and host–microbe interactions and was further helpful in lectin study and roles in glycobiology.381 These studies have demonstrated the potential of AI and ML, especially through hybrid approaches, for creating highly effective models of protein glycosylation. The integration of ML with bioinformatics tools and other techniques has enabled a deeper understanding of glycosylation-related genes and their patterns and holds great promise for identifying prognostic and therapeutic targets in various types of cancer.
Additionally, the development of precise and selective inhibitors that target siglecs or galectins has potential for attaining therapeutic effectiveness while maintaining normal immune function. One of the primary challenges with inhibitory compounds is avoiding off-target effects, which prevents them from targeting other biological molecules or even other members of the same family. Understanding the complex distinctions in the target molecule's structures and ligand binding specificity can help build high-specificity inhibitors. The clinical use of galectin inhibitors faces several challenges. Key issues include the need for specificity to avoid off-target effects, the development of degradation-resistant inhibitors with selective biodistribution, and the design of inhibitors that can withstand the evolving immunosuppressive tumor glycome. Additionally, creating inhibitors that precisely target relevant pathological roles without negatively impacting normal functions is a significant challenge that must be addressed for the effective translation of these inhibitors into reliable cancer therapies.382 For instance, the lack of selectivity and structural diversity of MCP still limits its usage as an anticancer drug. Despite the fact that MCPs have a shown affinity for Gal-3, there is little to no knowledge about their selectivity against other lectins.159 More research is needed to evaluate the affinity of MCPs and other inhibitors in cancer-related processes to fully comprehend the molecular mechanisms of action of these compounds. Addressing this challenge, and incorporating highly specific inhibitors into the GNVs, can enhance their targeting to APCs and can reprogram the microenvironment from pro-tumor to anti-tumor, ultimately leading to enhanced T cell cytotoxicity, enhancing the overall efficacy of the GNVs.
A further unanswered question is if there is a particular immune response that would benefit most from targeting immunomodulatory receptors like siglecs and galectins. This may influence the choice of combination therapy which can offer a solid foundation for the treatment of cancer, going beyond the use of a single modality. The interaction between sialic acid and siglec has been demonstrated to influence the effector functions of DCs by affecting their maturation, XPT, and ability to prime T cells.212 It has been shown that coexpression of PD-1 and TIM-3 on T cells results in their exhausted state. PD-1 interacts with the TIM-3 ligand Gal-9, which decreases TIM-3/Gal-9-induced T cell death, thereby enabling the exhausted T cells to survive.383 Therefore, it can be advantageous to combine siglec or galectin inhibitors with T cell-, DC-, or XPT-based therapies to convert a cold tumor to hot tumor. This way the combination strategy has the power to fully harness the therapeutic potential of GNVs to increase their efficacy and improve outcomes for a wider spectrum of patients.
Finally, it remains a question of what is the most efficient approach to target immunomodulatory receptors. Will blocking the binding receptors be more beneficial, or will blocking their ligands be more successful? Will completely inhibiting their synthesis have any effect on the cancer hallmarks? For instance, a phase 2 trial is currently testing the efficacy of a siglec-15-blocking antibody (NCT04699123), while siglec-7 and -9 blocking antibodies are in preclinical development.384 A study has shown that complete tumor desialylation unexpectedly led to a significant increase in in vivo colorectal cancer tumor growth.385 Additionally, different types of galectin inhibitor, such as carbohydrate, peptide, and nanoparticle-based inhibitors, are being created as anti-cancer drugs.159 Furthermore, nucleic acid-based galectin inhibitors that inhibit galectin synthesis are also in the pre-clinical stage.386 This suggests that further research is needed to fully comprehend the overall effects of different inhibitory strategies in order to determine the most suitable approach in specific contexts. It is important to understand the effects of combining various therapeutic strategies before implementing them from bench to bed. By understanding this, personalized GNVs co-delivering various ligands, inhibitors and adjuvants can be developed personalized to the specific tumor microenvironments to ensure the desired immune response avoiding any adverse side effects.
13. Conclusion
GNVs hold a revolutionary potential in the cancer immunotherapy landscape. Though there are several new strategies being developed for cancer treatment, GNVs can be viewed as the next-generation cancer vaccine in the coming era of immunotherapeutics. One of the key reasons is that GNVs substantially improve CTL-mediated immunity, most notably by facilitating XPT of the TAAs by APCs boosting the anti-cancer immune response. With the goal of significantly amplifying the efficacy of GNVs, blocking different immunosuppressive elements and signals in the TME has great promise. Galectins and siglecs, which are often expressed in immunosuppressive TMEs, along with aberrant glycosylation profiles of the tumors, as discussed in this review, are the major targets that should be looked upon to harness the complete efficacy of the GNVs. Blocking various ICs, such as CTLA-4, PD-1, PD-L1, etc., can also significantly help in enhanced efficacy of the GNVs. By focusing on these targets, a robust CTL response can be achieved, which is quintessential for the success of the GNVs.Taking these together, a combination therapy approach with GNVs, galectin and siglec inhibitors, and ICIs represents a promising frontier in cancer immunotherapy and warrants further investigation to address the clinical outcome of these combinations in inducing robust anti-cancer immune responses. Such cancer immunotherapy has the potential to transform cancer treatment by offering highly personalized and tailored treatment to each patient.
Author contributions
Conceptualization, M. J., E. Y., and R. K. G.; writing—original draft preparation, M. J., I. M. J., S. V. D., S. R. S., and R. K. G.; writing—review and editing, E. Y., G. S., and R. K. G.Data availability
No primary research results, software or code has been included and no new data were generated or analysed as part of this review.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors wish to express their gratitude to the India-Japan collaborative project on DST and JSPS grant number: DST/INT/JSPS/P-307/2020 and JPJSBP120207716. Rajesh Kumar Gupta would like to dedicate this review paper to his mother, Mrs. Gyanmati, who passed away recently.References
- F. Bray, M. Laversanne, H. Sung, J. Ferlay, R. L. Siegel, I. Soerjomataram and A. Jemal, CA Cancer J. Clin., 2024, 74, 229–263 CrossRef PubMed.
- A. I. Makandar, M. Jain, E. Yuba, G. Sethi and R. K. Gupta, Vaccines, 2022, 10, 2049 CrossRef CAS PubMed.
- T. L. Whiteside, Oncogene, 2008, 27, 5904–5912 CrossRef CAS PubMed.
- T. Gutting, E. Burgermeister, N. Hartel and M. P. Ebert, Semin. Cancer Biol., 2019, 55, 78–89 CrossRef CAS PubMed.
- E. Khatoon, D. Parama, A. Kumar, M. S. Alqahtani, M. Abbas, S. Girisa, G. Sethi and A. B. Kunnumakkara, Life Sci., 2022, 306, 120827 CrossRef CAS PubMed.
- S. Bagchi, R. Yuan and E. G. Engleman, Annu. Rev. Pathol., 2021, 16, 223–249 CrossRef CAS PubMed.
- D. S. Chen and I. Mellman, Immunity, 2013, 39, 1–10 CrossRef CAS PubMed.
- M. A. Stanczak, N. Rodrigues Mantuano, N. Kirchhammer, D. E. Sanin, F. Jacob, R. Coelho, A. V. Everest-Dass, J. Wang, M. P. Trefny, G. Monaco, A. Barenwaldt, M. A. Gray, A. Petrone, A. S. Kashyap, K. Glatz, B. Kasenda, K. Normington, J. Broderick, L. Peng, O. M. T. Pearce, E. L. Pearce, C. R. Bertozzi, A. Zippelius and H. Laubli, Sci. Transl. Med., 2022, 14, eabj1270 CrossRef CAS PubMed.
- A. Nicolini and P. Ferrari, Front. Immunol., 2024, 15, 1353787 CrossRef CAS PubMed.
- L. Qiong, X. Shuyao, X. Shan, F. Qian, T. Jiaying, X. Yao and L. Hui, Curr. Mol. Pharmacol., 2024, 17, e18761429308361 CrossRef CAS PubMed.
- A. Glaviano, H. S. Lau, L. M. Carter, E. H. C. Lee, H. Y. Lam, E. Okina, D. J. J. Tan, W. Tan, H. L. Ang, D. Carbone, M. Y. Yee, M. K. Shanmugam, X. Z. Huang, G. Sethi, T. Z. Tan, L. H. K. Lim, R. Y. Huang, H. Ungefroren, E. Giovannetti, D. G. Tang, T. C. Bruno, P. Luo, M. H. Andersen, B. Z. Qian, J. Ishihara, D. C. Radisky, S. Elias, S. Yadav, M. Kim, C. Robert, P. Diana, K. A. Schalper, T. Shi, T. Merghoub, S. Krebs, A. P. Kusumbe, M. S. Davids, J. R. Brown and A. P. Kumar, J. Hematol. Oncol., 2025, 18, 6 CrossRef PubMed.
- C. L. Yin and Y. J. Ma, Curr. Mol. Pharmacol., 2024, 17, e18761429266116 CrossRef CAS PubMed.
- C. M. Bailey, Y. Liu, M. Liu, X. Du, M. Devenport, P. Zheng, Y. Liu and Y. Wang, J. Clin. Invest., 2022, 132, e150846 CrossRef CAS PubMed.
- S. M. Manohar, Curr. Mol. Pharmacol., 2024, 17, e060923220758 Search PubMed.
- L. Liu, Y. Xie, H. Yang, A. Lin, M. Dong, H. Wang, C. Zhang, Z. Liu, Q. Cheng, J. Zhang, S. Yuan and P. Luo, iMeta, 2023, 2, e130 CrossRef CAS PubMed.
- C. Liu, J. Lu, H. Tian, W. Du, L. Zhao, J. Feng, D. Yuan and Z. Li, Mol. Med. Rep., 2017, 15, 1063–1070 CrossRef CAS PubMed.
- V. U. Warrier, A. I. Makandar, M. Garg, G. Sethi, R. Kant, J. K. Pal, E. Yuba and R. K. Gupta, Biosci. Rep., 2019, 39, BSR20193220 CrossRef CAS PubMed.
- E. RodrÍguez, S. T. T. Schetters and Y. van Kooyk, Nat. Rev. Immunol., 2018, 18, 204–211 CrossRef PubMed.
- D. Thomas, A. K. Rathinavel and P. Radhakrishnan, Biochim. Biophys. Acta, Rev. Cancer, 2021, 1875, 188464 CrossRef CAS PubMed.
- B. Gupta, D. Sadaria, V. U. Warrier, A. Kirtonia, R. Kant, A. Awasthi, P. Baligar, J. K. Pal, E. Yuba, G. Sethi, M. Garg and R. K. Gupta, Semin. Cancer Biol., 2022, 80, 87–106 CrossRef CAS PubMed.
- X. Cai, H. Zhan, Y. Ye, J. Yang, M. Zhang, J. Li and Y. Zhuang, Front. Genet., 2021, 12, 785153 CrossRef CAS PubMed.
- S. Qin, L. Xu, M. Yi, S. Yu, K. Wu and S. Luo, Mol. Cancer, 2019, 18, 155 CrossRef PubMed.
- G. Lizée, W. W. Overwijk, L. Radvanyi, J. Gao, P. Sharma and P. Hwu, Annu. Rev. Med., 2013, 64, 71–90 CrossRef PubMed.
- X. B. Wang, Z. Z. Fan, D. Anton, A. V. Vollenhoven, Z. H. Ni, X. F. Chen and A. K. Lefvert, BMC Immunol., 2011, 12, 21 CrossRef CAS PubMed.
- S. Liu, F. Wang, W. Tan, L. Zhang, F. Dai, Y. Wang, Y. Fan, M. Yuan, D. Yang, Y. Zheng, Z. Deng, Y. Liu and Y. Cheng, Cancer Cell Int., 2020, 20, 519 CrossRef CAS PubMed.
- K. Hudson, N. Cross, N. Jordan-Mahy and R. Leyland, Front. Immunol., 2020, 11, 568931 CrossRef CAS PubMed.
- J. Wu, C. Liu, S. Qian and H. Hou, DNA Cell Biol., 2013, 32, 648–653 CrossRef CAS PubMed.
- L. Tu, R. Guan, H. Yang, Y. Zhou, W. Hong, L. Ma, G. Zhao and M. Yu, Int. J. Cancer, 2020, 147, 423–439 CrossRef CAS PubMed.
- B. Huard, P. Prigent, M. Tournier, D. Bruniquel and F. Triebel, Eur. J. Immunol., 1995, 25, 2718–2721 CrossRef CAS PubMed.
- S. R. Woo, M. E. Turnis, M. V. Goldberg, J. Bankoti, M. Selby, C. J. Nirschl, M. L. Bettini, D. M. Gravano, P. Vogel, C. L. Liu, S. Tangsombatvisit, J. F. Grosso, G. Netto, M. P. Smeltzer, A. Chaux, P. J. Utz, C. J. Workman, D. M. Pardoll, A. J. Korman, C. G. Drake and D. A. Vignali, Cancer Res., 2012, 72, 917–927 CrossRef CAS PubMed.
- H. Wu, Y. Chen, H. Liu, L. L. Xu, P. Teuscher, S. Wang, S. Lu and A. L. Dent, Eur. J. Immunol., 2016, 46, 1152–1161 CrossRef CAS PubMed.
- X. Yu, K. Harden, L. C. Gonzalez, M. Francesco, E. Chiang, B. Irving, I. Tom, S. Ivelja, C. J. Refino, H. Clark, D. Eaton and J. L. Grogan, Nat. Immunol., 2009, 10, 48–57 CrossRef CAS PubMed.
- W. He, H. Zhang, F. Han, X. Chen, R. Lin, W. Wang, H. Qiu, Z. Zhuang, Q. Liao, W. Zhang, Q. Cai, Y. Cui, W. Jiang, H. Wang and Z. Ke, Cancer Res., 2017, 77, 6375–6388 CrossRef CAS PubMed.
- X. Li, Z. Xu, G. Cui, L. Yu and X. Zhang, OncoTargets Ther., 2020, 13, 215–224 CrossRef CAS PubMed.
- X. Lan, S. Li, H. Gao, A. Nanding, L. Quan, C. Yang, S. Ding and Y. Xue, OncoTargets Ther., 2017, 10, 919–926 CrossRef CAS PubMed.
- J. R. Sedy, M. Gavrieli, K. G. Potter, M. A. Hurchla, R. C. Lindsley, K. Hildner, S. Scheu, K. Pfeffer, C. F. Ware, T. L. Murphy and K. M. Murphy, Nat. Immunol., 2005, 6, 90–98 CrossRef CAS PubMed.
- J. R. Šedý, R. L. Bjordahl, V. Bekiaris, M. G. Macauley, B. C. Ware, P. S. Norris, N. S. Lurain, C. A. Benedict and C. F. Ware, J. Immunol., 2013, 191, 828–836 CrossRef PubMed.
- L. Dyck and K. H. G. Mills, Eur. J. Immunol., 2017, 47, 765–779 CrossRef CAS PubMed.
- X. Shi, C. W. Li, L. C. Tan, S. S. Wen, T. Liao, Y. Zhang, T. Z. Chen, B. Ma, P. C. Yu, Z. W. Lu, N. Qu, Y. Wang, R. L. Shi, Y. L. Wang, Q. H. Ji and W. J. Wei, J. Clin. Endocrinol. Metab., 2021, 106, 120–132 CrossRef PubMed.
- A. Kassardjian and N. A. Moatamed, Int. J. Clin. Exp. Pathol., 2021, 14, 1038–1047 CAS.
- S. Yoshida, H. Nagatsuka, K. Nakano, Y. Kogashiwa, Y. Ebihara, M. Yano and M. Yasuda, Int. J. Med. Sci., 2018, 15, 1723–1730 CrossRef CAS PubMed.
- E. A. Mittendorf, A. V. Philips, F. Meric-Bernstam, N. Qiao, Y. Wu, S. Harrington, X. Su, Y. Wang, A. M. Gonzalez-Angulo, A. Akcakanat, A. Chawla, M. Curran, P. Hwu, P. Sharma, J. K. Litton, J. J. Molldrem and G. Alatrash, Cancer Immunol. Res., 2014, 2, 361–370 CrossRef CAS PubMed.
- R. H. Thompson, H. Dong and E. D. Kwon, Clin. Cancer Res., 2007, 13, 709s–715s CrossRef CAS PubMed.
- Y. Cao, X. Zhou, X. Huang, Q. Li, L. Gao, L. Jiang, M. Huang and J. Zhou, PLoS One, 2013, 8, e53834 CrossRef PubMed.
- Q. Guo, S. Shen, G. Guan, C. Zhu, C. Zou, J. Cao, W. Cheng, X. Xu, J. Yu, Z. Lin, G. Wang, L. Chen, P. Cheng and A. Wu, iScience, 2022, 25, 105329 CrossRef CAS PubMed.
- X. Gao, Y. Zhu, G. Li, H. Huang, G. Zhang, F. Wang, J. Sun, Q. Yang, X. Zhang and B. Lu, PLoS One, 2012, 7, e30676 CrossRef CAS PubMed.
- Y. He, H. Yu, L. Rozeboom, C. J. Rivard, K. Ellison, R. Dziadziuszko, K. Suda, S. Ren, C. Wu, L. Hou, C. Zhou and F. R. Hirsch, J. Thorac. Oncol., 2017, 12, 814–823 CrossRef PubMed.
- J. Tian, Y. Liu, T. L. Zhang, Y. N. Xiao, C. Y. Guo, Y. H. Xie and Z. J. An, Indian J. Pathol. Microbiol., 2022, 65, 581–588 CrossRef PubMed.
- W. A. Freed-Pastor, L. J. Lambert, Z. A. Ely, N. B. Pattada, A. Bhutkar, G. Eng, K. L. Mercer, A. P. Garcia, L. Lin, W. M. Rideout 3rd, W. L. Hwang, J. M. Schenkel, A. M. Jaeger, R. T. Bronson, P. M. K. Westcott, T. D. Hether, P. Divakar, J. W. Reeves, V. Deshpande, T. Delorey, D. Phillips, O. H. Yilmaz, A. Regev and T. Jacks, Cancer Cell, 2021, 39, 1342–1360 CrossRef CAS PubMed.
- S. Laurent, P. Carrega, D. Saverino, P. Piccioli, M. Camoriano, A. Morabito, B. Dozin, V. Fontana, R. Simone, L. Mortara, M. C. Mingari, G. Ferlazzo and M. P. Pistillo, Hum. Immunol., 2010, 71, 934–941 CrossRef CAS PubMed.
- M. Tekguc, J. B. Wing, M. Osaki, J. Long and S. Sakaguchi, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2023739118 CrossRef CAS PubMed.
- X. Chen, Q. Shao, S. Hao, Z. Zhao, Y. Wang, X. Guo, Y. He, W. Gao and H. Mao, Oncotarget, 2017, 8, 13703–13715 CrossRef PubMed.
- M. Ahmadzadeh, L. A. Johnson, B. Heemskerk, J. R. Wunderlich, M. E. Dudley, D. E. White and S. A. Rosenberg, Blood, 2009, 114, 1537–1544 CrossRef CAS PubMed.
- V. R. Juneja, K. A. McGuire, R. T. Manguso, M. W. LaFleur, N. Collins, W. N. Haining, G. J. Freeman and A. H. Sharpe, J. Exp. Med., 2017, 214, 895–904 CrossRef CAS PubMed.
- O. Pagliano, R. M. Morrison, J. M. Chauvin, H. Banerjee, D. Davar, Q. Ding, T. Tanegashima, W. Gao, S. R. Chakka, R. DeBlasio, A. Lowin, K. Kara, M. Ka, B. Zidi, R. Amin, I. Raphael, S. Zhang, S. C. Watkins, C. Sander, J. M. Kirkwood, M. Bosenberg, A. C. Anderson, V. K. Kuchroo, L. P. Kane, A. J. Korman, A. Rajpal, S. M. West, M. Han, C. Bee, X. Deng, X. M. Schebye, P. Strop and H. M. Zarour, J. Clin. Invest., 2022, 132, e152864 CrossRef CAS PubMed.
- Y. Zhang, C. J. Ma, J. M. Wang, X. J. Ji, X. Y. Wu, J. P. Moorman and Z. Q. Yao, J. Leukoc. Biol., 2012, 91, 189–196 CrossRef CAS PubMed.
- A. C. Anderson, D. E. Anderson, L. Bregoli, W. D. Hastings, N. Kassam, C. Lei, R. Chandwaskar, J. Karman, E. W. Su, M. Hirashima, J. N. Bruce, L. P. Kane, V. K. Kuchroo and D. A. Hafler, Science, 2007, 318, 1141–1143 CrossRef CAS PubMed.
- N. Joller and V. K. Kuchroo, Curr. Top. Microbiol. Immunol., 2017, 410, 127–156 CAS.
- Y. Zhang and Z. Zhang, Cell. Mol. Immunol., 2020, 17, 807–821 CrossRef CAS PubMed.
- F. Petitprez, M. Meylan, A. de Reyniès, C. Sautès-Fridman and W. H. Fridman, Front. Immunol., 2020, 11, 784 CrossRef CAS PubMed.
- U. A. Ramagopal, W. Liu, S. C. Garrett-Thomson, J. B. Bonanno, Q. Yan, M. Srinivasan, S. C. Wong, A. Bell, S. Mankikar, V. S. Rangan, S. Deshpande, A. J. Korman and S. C. Almo, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E4223–E4232 CrossRef CAS PubMed.
- D. Schadendorf, F. S. Hodi, C. Robert, J. S. Weber, K. Margolin, O. Hamid, D. Patt, T. T. Chen, D. M. Berman and J. D. Wolchok, J. Clin. Oncol., 2015, 33, 1889–1894 CrossRef CAS PubMed.
- S. J. Keam, Drugs, 2023, 83, 93–102 CrossRef CAS PubMed.
- L. A. Raedler, Am. Health Drug Benefits, 2015, 8, 96–100 Search PubMed.
- J. Larkin, D. Minor, S. D'Angelo, B. Neyns, M. Smylie, W. H. Miller Jr., R. Gutzmer, G. Linette, B. Chmielowski, C. D. Lao, P. Lorigan, K. Grossmann, J. C. Hassel, M. Sznol, A. Daud, J. Sosman, N. Khushalani, D. Schadendorf, C. Hoeller, D. Walker, G. Kong, C. Horak and J. Weber, J. Clin. Oncol., 2018, 36, 383–390 CrossRef CAS PubMed.
- L. A. Raedler, Am. Health Drug Benefits, 2015, 8, 180–183 Search PubMed.
- S. R. Ahmed, E. Petersen, R. Patel and M. R. Migden, Expert Rev. Clin. Pharmacol., 2019, 12, 947–951 CrossRef CAS PubMed.
- T. Damsin, E. Lebas, N. Marchal, A. Rorive and A. F. Nikkels, Expert Rev. Anticancer Ther., 2022, 22, 243–248 CrossRef CAS PubMed.
- Y. M. Ning, D. Suzman, V. E. Maher, L. Zhang, S. Tang, T. Ricks, T. Palmby, W. Fu, Q. Liu, K. B. Goldberg, G. Kim and R. Pazdur, Oncologist, 2017, 22, 743–749 CrossRef CAS PubMed.
- M. Reck, G. Shankar, A. Lee, S. Coleman, M. McCleland, V. A. Papadimitrakopoulou, M. A. Socinski and A. Sandler, Expert Rev. Respir. Med., 2020, 14, 125–136 CrossRef CAS PubMed.
- S. J. Casak, M. Donoghue, L. Fashoyin-Aje, X. Jiang, L. Rodriguez, Y. L. Shen, Y. Xu, X. Jiang, J. Liu, H. Zhao, W. F. Pierce, S. Mehta, K. B. Goldberg, M. R. Theoret, P. G. Kluetz, R. Pazdur and S. J. Lemery, Clin. Cancer Res., 2021, 27, 1836–1841 CrossRef CAS PubMed.
- S. V. Liu, M. Reck, A. S. Mansfield, T. Mok, A. Scherpereel, N. Reinmuth, M. C. Garassino, J. De Castro Carpeno, R. Califano, M. Nishio, F. Orlandi, J. Alatorre-Alexander, T. Leal, Y. Cheng, J. S. Lee, S. Lam, M. McCleland, Y. Deng, S. Phan and L. Horn, J. Clin. Oncol., 2021, 39, 619–630 CrossRef CAS PubMed.
- R. Gutzmer, D. Stroyakovskiy, H. Gogas, C. Robert, K. Lewis, S. Protsenko, R. P. Pereira, T. Eigentler, P. Rutkowski, L. Demidov, G. M. Manikhas, Y. Yan, K. C. Huang, A. Uyei, V. McNally, G. A. McArthur and P. A. Ascierto, Lancet, 2020, 395, 1835–1844 CrossRef CAS PubMed.
- H. Sui, N. Ma, Y. Wang, H. Li, X. Liu, Y. Su and J. Yang, J. Immunol. Res., 2018, 2018, 6984948 Search PubMed.
- L. Paz-Ares, Y. Chen, N. Reinmuth, K. Hotta, D. Trukhin, G. Statsenko, M. J. Hochmair, M. Özgüroğlu, J. H. Ji, M. C. Garassino, O. Voitko, A. Poltoratskiy, E. Musso, L. Havel, I. Bondarenko, G. Losonczy, N. Conev, H. Mann, T. B. Dalvi, H. Jiang and J. W. Goldman, ESMO Open, 2022, 7, 100408 CrossRef CAS PubMed.
- M. R. Gaiser, M. Bongiorno and I. Brownell, Expert Rev. Clin. Pharmacol., 2018, 11, 345–359 CrossRef CAS PubMed.
- J. Paik, Drugs, 2022, 82, 925–931 CrossRef CAS PubMed.
- Y. J. Park, D. S. Kuen and Y. Chung, Exp. Mol. Med., 2018, 50, 1–13 Search PubMed.
- H. N. Bell and W. Zou, Annu. Rev. Immunol., 2024, 42, 521–550 CrossRef CAS PubMed.
- A. Bessede, F. Peyraud, B. Besse, S. Cousin, M. Cabart, F. Chomy, C. Rey, O. Lara, O. Odin, I. Nafia, L. Vanhersecke, F. Barlesi, J. P. Guégan and A. Italiano, Clin. Cancer Res., 2024, 30, 779–785 CrossRef CAS PubMed.
- S. Y. Lim, E. Shklovskaya, J. H. Lee, B. Pedersen, A. Stewart, Z. Ming, M. Irvine, B. Shivalingam, R. P. M. Saw, A. M. Menzies, M. S. Carlino, R. A. Scolyer, G. V. Long and H. Rizos, Nat. Commun., 2023, 14, 1516 CrossRef CAS PubMed.
- M. Lauss, B. Phung, T. H. Borch, K. Harbst, K. Kaminska, A. Ebbesson, I. Hedenfalk, J. Yuan, K. Nielsen, C. Ingvar, A. Carneiro, K. Isaksson, K. Pietras, I. M. Svane, M. Donia and G. Jönsson, Nat. Commun., 2024, 15, 3075 CrossRef CAS PubMed.
- D. Memon, A. J. Schoenfeld, D. Ye, G. Fromm, H. Rizvi, X. Zhang, M. R. Keddar, D. Mathew, K. J. Yoo, J. Qiu, J. Lihm, J. Miriyala, J. L. Sauter, J. Luo, A. Chow, U. K. Bhanot, C. McCarthy, C. M. Vanderbilt, C. Liu, M. Abu-Akeel, A. J. Plodkowski, N. McGranahan, M. Łuksza, B. D. Greenbaum, T. Merghoub, I. Achour, J. C. Barrett, R. Stewart, P. Beltrao, T. H. Schreiber, A. J. Minn, M. L. Miller and M. D. Hellmann, Cancer Cell, 2024, 42, 209–224 CrossRef CAS PubMed.
- B. H. Herzog, J. M. Baer, N. Borcherding, N. L. Kingston, J. I. Belle, B. L. Knolhoff, G. D. Hogg, F. Ahmad, L. I. Kang, J. Petrone, C. Y. Lin, R. Govindan and D. G. DeNardo, Sci. Transl. Med., 2023, 15, eadh8005 CrossRef CAS PubMed.
- Y. Li, Y. Zheng, J. Huang, R. C. Nie, Q. N. Wu, Z. Zuo, S. Yuan, K. Yu, C. C. Liang, Y. Q. Pan, B. W. Zhao, Y. Xu, Q. Zhang, Y. Zheng, J. Chen, Z. L. Zeng, W. Wei, Z. X. Liu, R. H. Xu and H. Y. Luo, Gut, 2025, 74, 350–363 CrossRef CAS PubMed.
- J. Qiu, B. Xu, D. Ye, D. Ren, S. Wang, J. L. Benci, Y. Xu, H. Ishwaran, J. C. Beltra, E. J. Wherry, J. Shi and A. J. Minn, Nat. Cancer, 2023, 4, 43–61 CAS.
- S. S. Pinho and C. A. Reis, Nat. Rev. Cancer, 2015, 15, 540–555 CrossRef CAS PubMed.
- R. Kannagi, J. Yin, K. Miyazaki and M. Izawa, Biochim. Biophys. Acta, 2008, 1780, 525–531 CrossRef CAS PubMed.
- S. Hakomori and R. Kannagi, J. Natl. Cancer Inst., 1983, 71, 231–251 CAS.
- T. Freire, S. Bay, S. von Mensdorff-Pouilly and E. Osinaga, Cancer Res., 2005, 65, 7880–7887 CrossRef CAS PubMed.
- P. Buckhaults, L. Chen, N. Fregien and M. Pierce, J. Biol. Chem., 1997, 272, 19575–19581 CrossRef CAS PubMed.
- A. Schietinger, M. Philip, B. A. Yoshida, P. Azadi, H. Liu, S. C. Meredith and H. Schreiber, Science, 2006, 314, 304–308 CrossRef CAS PubMed.
- J. Ashkani and K. J. Naidoo, Sci. Rep., 2016, 6, 26451 CrossRef CAS PubMed.
- R. Saldova, V. D. Haakensen, E. Rodland, I. Walsh, H. Stockmann, O. Engebraaten, A. L. Borresen-Dale and P. M. Rudd, Mol. Oncol., 2017, 11, 1361–1379 CrossRef CAS PubMed.
- D. A. Scott and R. R. Drake, Expert Rev. Proteomics, 2019, 16, 665–680 CrossRef CAS PubMed.
- S. Mirzaei, S. Saghari, F. Bassiri, R. Raesi, A. Zarrabi, K. Hushmandi, G. Sethi and V. Tergaonkar, J. Cell Physiol., 2022, 237, 2770–2795 CrossRef CAS PubMed.
- J. G. Rodrigues, M. Balmana, J. A. Macedo, J. Pocas, A. Fernandes, J. C. M. de-Freitas-Junior, S. S. Pinho, J. Gomes, A. Magalhaes, C. Gomes, S. Mereiter and C. A. Reis, Cell. Immunol., 2018, 333, 46–57 CrossRef CAS PubMed.
- S. Carvalho, T. Oliveira, M. F. Bartels, E. Miyoshi, M. Pierce, N. Taniguchi, F. Carneiro, R. Seruca, C. A. Reis, S. Strahl and S. S. Pinho, Oncotarget, 2016, 7, 65231–65246 CrossRef PubMed.
- A. Liwosz, T. Lei and M. A. Kukuruzinska, J. Biol. Chem., 2006, 281, 23138–23149 CrossRef CAS PubMed.
- M. Nita-Lazar, V. Noonan, I. Rebustini, J. Walker, A. S. Menko and M. A. Kukuruzinska, Cancer Res., 2009, 69, 5673–5680 CrossRef CAS PubMed.
- S. S. Pinho, J. Figueiredo, J. Cabral, S. Carvalho, J. Dourado, A. Magalhaes, F. Gartner, A. M. Mendonfa, T. Isaji, J. Gu, F. Carneiro, R. Seruca, N. Taniguchi and C. A. Reis, Biochim. Biophys. Acta, 2013, 1830, 2690–2700 CrossRef CAS PubMed.
- S. B. Harosh-Davidovich and I. Khalaila, Exp. Cell Res., 2018, 364, 42–49 CrossRef CAS PubMed.
- F. Z. Jin, C. Yu, D. Z. Zhao, M. J. Wu and Z. Yang, Exp. Cell Res., 2013, 319, 1482–1490 CrossRef CAS PubMed.
- C. Fu, H. Zhao, Y. Wang, H. Cai, Y. Xiao, Y. Zeng and H. Chen, HLA, 2016, 88, 275–286 CrossRef CAS PubMed.
- J. Zhang, H. Wang, J. Wu, C. Yuan, S. Chen, S. Liu, M. Huo, C. Zhang and Y. He, Int. J. Biol. Sci., 2022, 18, 6068–6083 CrossRef CAS PubMed.
- M. C. Lin, M. J. Huang, C. H. Liu, T. L. Yang and M. C. Huang, Oral Oncol., 2014, 50, 478–484 CrossRef CAS PubMed.
- K. Taniuchi, R. L. Cerny, A. Tanouchi, K. Kohno, N. Kotani, K. Honke, T. Saibara and M. A. Hollingsworth, Oncogene, 2011, 30, 4843–4854 CrossRef CAS PubMed.
- X. Yan, J. Lu, X. Zou, S. Zhang, Y. Cui, L. Zhou, F. Liu, A. Shan, J. Lu, M. Zheng, B. Feng and Y. Zhang, FEBS J., 2018, 285, 3041–3055 CrossRef CAS PubMed.
- J. Song, W. Liu, J. Wang, J. Hao, Y. Wang, X. You, X. Du, Y. Zhou, J. Ben, X. Zhang, M. Ye and Q. Wang, Cell Death Dis., 2020, 11, 352 CrossRef CAS PubMed.
- E. Scott, K. Hodgson, B. Calle, H. Turner, K. Cheung, A. Bermudez, F. J. G. Marques, H. Pye, E. C. Yo, K. Islam, H. Z. Oo, U. L. McClurg, L. Wilson, H. Thomas, F. M. Frame, M. Orozco-Moreno, K. Bastian, H. M. Arredondo, C. Roustan, M. A. Gray, L. Kelly, A. Tolson, E. Mellor, G. Hysenaj, E. A. Goode, R. Garnham, A. Duxfield, S. Heavey, U. Stopka-Farooqui, A. Haider, A. Freeman, S. Singh, E. W. Johnston, S. Punwani, B. Knight, P. McCullagh, J. McGrath, M. Crundwell, L. Harries, D. Bogdan, D. Westaby, G. Fowler, P. Flohr, W. Yuan, A. Sharp, J. de Bono, N. J. Maitland, S. Wisnovsky, C. R. Bertozzi, R. Heer, R. H. Guerrero, M. Daugaard, J. Leivo, H. Whitaker, S. Pitteri, N. Wang, D. J. Elliott, B. Schumann and J. Munkley, Oncogene, 2023, 42, 926–937 CrossRef CAS PubMed.
- G. Xu, Y. L. Wu, N. Li, R. Xu, J. B. Zhang, H. Ming and Y. Zhang, Eur. Rev. Med. Pharmacol. Sci., 2020, 24, 11610–11619 Search PubMed.
- Y. Zheng, M. Liang, B. Wang, L. Kang, Y. Yuan, Y. Mao and S. Wang, Biochem. Biophys. Res. Commun., 2022, 610, 99–106 CrossRef CAS PubMed.
- T. Huanna, Z. Tao, W. Xiangfei, A. Longfei, X. Yuanyuan, W. Jianhua, Z. Cuifang, J. Manjing, C. Wenjing, Q. Shaochuan, X. Feifei, L. Naikang, Z. Jinchao and W. Chen, Mol. Carcinog., 2015, 54, 1159–1171 CrossRef PubMed.
- J. S. Hung, J. Huang, Y. C. Lin, M. J. Huang, P. H. Lee, H. S. Lai, J. T. Liang and M. C. Huang, Oncotarget, 2014, 5, 2096–2106 CrossRef PubMed.
- I. Lakshmanan, S. Chaudhary, R. Vengoji, P. Seshacharyulu, S. Rachagani, J. Carmicheal, R. Jahan, P. Atri, R. Chirravuri-Venkata, R. Gupta, S. Marimuthu, N. Perumal, S. Rauth, S. Kaur, K. Mallya, L. M. Smith, S. M. Lele, M. P. Ponnusamy, M. W. Nasser, R. Salgia, S. K. Batra and A. K. Ganti, Mol. Oncol., 2021, 15, 1866–1881 CrossRef CAS PubMed.
- X. Wu, J. Zhao, Y. Ruan, L. Sun, C. Xu and H. Jiang, Cell Death Dis., 2018, 9, 1102 CrossRef PubMed.
- N. Bhalerao, A. Chakraborty, M. P. Marciel, J. Hwang, C. M. Britain, A. D. Silva, I. E. Eltoum, R. B. Jones, K. L. Alexander, L. E. Smythies, P. D. Smith, D. K. Crossman, M. R. Crowley, B. Shin, L. E. Harrington, Z. Yan, M. M. Bethea, C. S. Hunter, C. A. Klug, D. J. Buchsbaum and S. L. Bellis, JCI Insight, 2023, 8, e161563 CrossRef PubMed.
- T. Okamoto, M. S. Yoneyama, S. Hatakeyama, K. Mori, H. Yamamoto, T. Koie, H. Saitoh, K. Yamaya, T. Funyu, M. Fukuda, C. Ohyama and S. Tsuboi, Mol. Med. Rep., 2013, 7, 359–364 Search PubMed.
- X. Yang, Z. Zhang, S. Jia, Y. Liu, X. Wang and Q. Yan, Int. J. Biochem. Cell Biol., 2007, 39, 1722–1730 Search PubMed.
- Q. Guo, B. Guo, Y. Wang, J. Wu, W. Jiang, S. Zhao, S. Qiao and Y. Wu, Biochem. Biophys. Res. Commun., 2012, 417, 311–317 CrossRef CAS PubMed.
- M. Liu, Q. Zheng, S. Chen, J. Liu and S. Li, J. Inflamm. Res., 2021, 14, 1069–1084 CrossRef PubMed.
- K. Tada, M. Ohta, S. Hidano, K. Watanabe, T. Hirashita, Y. Oshima, A. Fujnaga, H. Nakanuma, T. Masuda, Y. Endo, Y. Takeuchi, Y. Iwashita, T. Kobayashi and M. Inomata, Surg. Today, 2020, 50, 767–777 CrossRef CAS PubMed.
- Z. Liu, J. Liu, X. Dong, X. Hu, Y. Jiang, L. Li, T. Du, L. Yang, T. Wen, G. An and G. Feng, J. Cell. Mol. Med., 2019, 23, 2083–2092 CrossRef CAS PubMed.
- D. Bapu, J. Runions, M. Kadhim and S. A. Brooks, Cancer Lett., 2016, 375, 367–374 CrossRef CAS PubMed.
- V. da Costa, S. J. van Vliet, P. Carasi, S. Frigerio, P. A. Garcia, D. O. Croci, M. F. Festari, M. Costa, M. Landeira, S. A. Rodriguez-Zraquia, A. J. Cagnoni, A. M. Cutine, G. A. Rabinovich, E. Osinaga, K. V. Marino and T. Freire, Cancer Lett., 2021, 518, 72–81 CrossRef CAS PubMed.
- M. F. Festari, V. da Costa, S. A. Rodríguez-Zraquia, M. Costa, M. Landeira, P. Lores, P. Solari-Saquieres, M. G. Kramer and T. Freire, Glycobiology, 2021, 32, 366–379 CrossRef PubMed.
- S. Julien, E. Adriaenssens, K. Ottenberg, A. Furlan, G. Courtand, A. S. Vercoutter-Edouart, F. G. Hanisch, P. Delannoy and X. Le Bourhis, Glycobiology, 2006, 16, 54–64 Search PubMed.
- H. Ozaki, H. Matsuzaki, H. Ando, H. Kaji, H. Nakanishi, Y. Ikehara and H. Narimatsu, Clin. Exp. Metastasis, 2012, 29, 229–238 Search PubMed.
- B. Davidson, A. Berner, J. M. Nesland, B. Risberg, G. B. Kristensen, C. G. Trope and M. Bryne, Hum. Pathol., 2000, 31, 1081–1087 Search PubMed.
- D. Thomas, S. Sagar, T. Caffrey, P. M. Grandgenett and P. Radhakrishnan, J. Cell. Mol. Med., 2019, 23, 6885–6896 Search PubMed.
- S. Pinho, N. T. Marcos, B. Ferreira, A. S. Carvalho, M. J. Oliveira, F. Santos-Silva, A. Harduin-Lepers and C. A. Reis, Cancer Lett., 2007, 249, 157–170 Search PubMed.
- A. Blanas, N. M. Sahasrabudhe, E. Rodríguez, Y. van Kooyk and S. J. van Vliet, Front. Oncol., 2018, 8, 39 CrossRef PubMed.
- A. Kadota, M. Masutani, M. Takei and T. Horie, Int. J. Oncol., 1999, 15, 1081–1089 CAS.
- K. Fukuoka, N. Narita and N. Saijo, Lung Cancer, 1998, 20, 109–116 CrossRef CAS PubMed.
- F. Tanaka, R. Miyahara, Y. Ohtake, K. Yanagihara, T. Fukuse, S. Hitomi and H. Wada, Ann. Thorac. Surg., 1998, 66, 1745–1750 CrossRef CAS PubMed.
- Y. W. Koh, H. J. Lee, J. H. Ahn, J. W. Lee and G. Gong, Am. J. Clin. Pathol., 2013, 139, 746–753 CrossRef CAS PubMed.
- K. Steplewska-Mazur, A. Gabriel, W. Zajecki, M. Wylezol and M. Gluck, Hybridoma, 2000, 19, 129–133 CrossRef CAS PubMed.
- Z. Madjd, T. Parsons, N. F. Watson, I. Spendlove, I. Ellis and L. G. Durrant, Breast Cancer Res., 2005, 7, R780–R787 CrossRef CAS PubMed.
- T. Ben-David, O. Sagi-Assif, T. Meshel, V. Lifshitz, I. Yron and I. P. Witz, Immunol. Lett., 2008, 116, 218–224 CrossRef CAS PubMed.
- S. Nakamori, M. Kameyama, S. Imaoka, H. Furukawa, O. Ishikawa, Y. Sasaki, Y. Izumi and T. Irimura, Dis. Colon Rectum, 1997, 40, 420–431 CrossRef CAS PubMed.
- H. J. Crespo, J. T. Lau and P. A. Videira, Front. Immunol., 2013, 4, 491 Search PubMed.
- M. Silva, Z. Silva, G. Marques, T. Ferro, M. Goncalves, M. Monteiro, S. J. van Vliet, E. Mohr, A. C. Lino, A. R. Fernandes, F. A. Lima, Y. van Kooyk, T. Matos, C. E. Tadokoro and P. A. Videira, Oncotarget, 2016, 7, 41053–41066 CrossRef PubMed.
- M. Bax, J. J. García-Vallejo, J. Jang-Lee, S. J. North, T. J. Gilmartin, G. Hernández, P. R. Crocker, H. Leffler, S. R. Head, S. M. Haslam, A. Dell and Y. van Kooyk, J. Immunol., 2007, 179, 8216–8224 CrossRef CAS PubMed.
- N. Balneger, L. A. M. Cornelissen, M. Wassink, S. J. Moons, T. J. Boltje, Y. E. Bar-Ephraim, K. K. Das, J. N. Sondergaard, C. Bull and G. J. Adema, Cell. Mol. Life Sci., 2022, 79, 98 CrossRef CAS PubMed.
- H. J. Crespo, M. G. Cabral, A. V. Teixeira, J. T. Lau, H. Trindade and P. A. Videira, Immunology, 2009, 128, e621–e631 CrossRef PubMed.
- K. Lynch, O. Treacy, J. Q. Gerlach, H. Annuk, P. Lohan, J. Cabral, L. Joshi, A. E. Ryan and T. Ritter, Front. Immunol., 2017, 8, 1427 Search PubMed.
- L. J. Edgar, A. J. Thompson, V. F. Vartabedian, C. Kikuchi, J. L. Woehl, J. R. Teijaro and J. C. Paulson, ACS Cent. Sci., 2021, 7, 1508–1515 CrossRef CAS PubMed.
- S. Mereiter, M. Balmaña, D. Campos, J. Gomes and C. A. Reis, Cancer Cell, 2019, 36, 6–16 CrossRef CAS PubMed.
- R. Sun, A. M. J. Kim and S. O. Lim, Cells, 2021, 10, 1100 CrossRef CAS PubMed.
- Y. Huang, H. L. Zhang, Z. L. Li, T. Du, Y. H. Chen, Y. Wang, H. H. Ni, K. M. Zhang, J. Mai, B. X. Hu, J. H. Huang, L. H. Zhou, D. Yang, X. D. Peng, G. K. Feng, J. Tang, X. F. Zhu and R. Deng, Nat. Commun., 2021, 12, 2672 CrossRef CAS PubMed.
- C. B. Madsen, C. Petersen, K. Lavrsen, M. Harndahl, S. Buus, H. Clausen, A. E. Pedersen and H. H. Wandall, PLoS One, 2012, 7, e50139 CrossRef CAS PubMed.
- S. Tsuboi, M. Sutoh, S. Hatakeyama, N. Hiraoka, T. Habuchi, Y. Horikawa, Y. Hashimoto, T. Yoneyama, K. Mori, T. Koie, T. Nakamura, H. Saitoh, K. Yamaya, T. Funyu, M. Fukuda and C. Ohyama, EMBO J., 2011, 30, 3173–3185 CrossRef CAS PubMed.
- S. H. Itzkowitz, M. Yuan, C. K. Montgomery, T. Kjeldsen, H. K. Takahashi, W. L. Bigbee and Y. S. Kim, Cancer Res., 1989, 49, 197–204 CAS.
- T. Freire, R. Lo-Man, S. Bay and C. Leclerc, J. Biol. Chem., 2011, 286, 7797–7811 CrossRef CAS PubMed.
- S. A. Dusoswa, J. Verhoeff, E. Abels, S. P. Mendez-Huergo, D. O. Croci, L. H. Kuijper, E. de Miguel, V. Wouters, M. G. Best, E. Rodriguez, L. A. M. Cornelissen, S. J. van Vliet, P. Wesseling, X. O. Breakefield, D. P. Noske, T. Wurdinger, M. L. D. Broekman, G. A. Rabinovich, Y. van Kooyk and J. J. Garcia-Vallejo, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 3693–3703 CrossRef CAS PubMed.
- N. Lopes, V. G. Correia, A. S. Palma and C. Brito, Int. J. Mol. Sci., 2021, 22, 1972 CrossRef CAS PubMed.
- R. K. Gupta, A. H. Pande, K. C. Gulla, H. J. Gabius and K. Hajela, FEBS Lett., 2006, 580, 1691–1695 CrossRef CAS PubMed.
- Y. van Kooyk and G. A. Rabinovich, Nat. Immunol., 2008, 9, 593–601 Search PubMed.
- K. V. Mariño, A. J. Cagnoni, D. O. Croci and G. A. Rabinovich, Nat. Rev. Drug Discovery, 2023, 22, 295–316 CrossRef PubMed.
- S. Nandi, S. Ghosh, A. Ranjan, R. S. Sood, J. K. Pal, K. Hajela and R. K. Gupta, in Lectins: Innate immune defense and Therapeutics, ed. P. Elumalai and S. Lakshmi, Springer Singapore, Singapore, 2021, pp. 215–271. DOI:10.1007/978-981-16-7462-4_11.
- F. T. Liu and G. A. Rabinovich, Nat. Rev. Cancer, 2005, 5, 29–41 CrossRef CAS PubMed.
- S. P. Mendez-Huergo, A. G. Blidner and G. A. Rabinovich, Curr. Opin. Immunol., 2017, 45, 8–15 Search PubMed.
- S. J. van Vliet, S. I. Gringhuis, T. B. Geijtenbeek and Y. van Kooyk, Nat. Immunol., 2006, 7, 1200–1208 CrossRef CAS PubMed.
- G. D. Brown, J. A. Willment and L. Whitehead, Nat. Rev. Immunol., 2018, 18, 374–389 CrossRef CAS PubMed.
- I. González-Chavarría, R. P. Cerro, N. P. Parra, F. A. Sandoval, F. A. Zuñiga, V. A. Omazábal, L. I. Lamperti, S. P. Jiménez, E. A. Fernandez, N. A. Gutiérrez, F. S. Rodriguez, S. A. Onate, O. Sánchez, J. C. Vera and J. R. Toledo, PLoS One, 2014, 9, e106219 CrossRef PubMed.
- Y. Pan, C. Wang, S. Wang, X. Wu, L. Sheng and Z. Qi, J. Gastrointest. Oncol., 2023, 14, 1331–1345 Search PubMed.
- B. A. H. Smith and C. R. Bertozzi, Nat. Rev. Drug Discovery, 2021, 20, 217–243 Search PubMed.
- L. Borsig, Glycobiology, 2018, 28, 648–655 CrossRef CAS PubMed.
- P. R. Crocker, J. C. Paulson and A. Varki, Nat. Rev. Immunol., 2007, 7, 255–266 CrossRef CAS PubMed.
- S. Benmerzoug, M. F. Chevalier, L. Villier, S. Nguyen, V. Cesson, A. K. Schneider, F. Dartiguenave, S. C. Rodrigues-Dias, I. Lucca, P. Jichlinski, B. Roth, D. Nardelli-Haefliger and L. Derre, Eur. Urol. Open Sci., 2021, 34, 79–82 CrossRef PubMed.
- S. van de Wall, K. C. M. Santegoets, E. J. H. van Houtum, C. Büll and G. J. Adema, Trends Immunol., 2020, 41, 274–285 CrossRef CAS PubMed.
- S. Nakahara and A. Raz, Anticancer Agents Med. Chem., 2008, 8, 22–36 CrossRef CAS PubMed.
- J. Ren, Z. Hu, G. Niu, J. Xia, X. Wang, R. Hong, J. Gu, D. Wang and C. Ke, FASEB J., 2023, 37, e22790 CrossRef CAS PubMed.
- Y. Wang, K. Chen, Y. Cai, Y. Cai, X. Yuan, L. Wang, Z. Wu and Y. Wu, J. Exp. Clin. Cancer Res., 2017, 36, 111 CrossRef PubMed.
- Q. Z. Pan, K. Pan, D. S. Weng, J. J. Zhao, X. F. Zhang, D. D. Wang, L. Lv, S. S. Jiang, H. X. Zheng and J. C. Xia, Mol. Carcinog., 2015, 54, 598–607 CrossRef CAS PubMed.
- K. P. van Gisbergen, C. A. Aarnoudse, G. A. Meijer, T. B. Geijtenbeek and Y. van Kooyk, Cancer Res., 2005, 65, 5935–5944 CrossRef CAS PubMed.
- L. A. M. Cornelissen, A. Blanas, A. Zaal, J. C. van der Horst, L. J. W. Kruijssen, T. O'Toole, Y. van Kooyk and S. J. van Vliet, Front. Oncol., 2020, 10, 1622 CrossRef PubMed.
- M. Perdicchio, L. A. Cornelissen, I. Streng-Ouwehand, S. Engels, M. I. Verstege, L. Boon, D. Geerts, Y. van Kooyk and W. W. Unger, Oncotarget, 2016, 7, 8771–8782 CrossRef PubMed.
- C. Jandus, K. F. Boligan, O. Chijioke, H. Liu, M. Dahlhaus, T. Demoulins, C. Schneider, M. Wehrli, R. E. Hunger, G. M. Baerlocher, H. U. Simon, P. Romero, C. Munz and S. von Gunten, J. Clin. Invest., 2014, 124, 1810–1820 CrossRef CAS PubMed.
- J. Wang, J. Sun, L. N. Liu, D. B. Flies, X. Nie, M. Toki, J. Zhang, C. Song, M. Zarr, X. Zhou, X. Han, K. A. Archer, T. O'Neill, R. S. Herbst, A. N. Boto, M. F. Sanmamed, S. Langermann, D. L. Rimm and L. Chen, Nat. Med., 2019, 25, 656–666 CrossRef CAS PubMed.
- T. Dalotto-Moreno, D. O. Croci, J. P. Cerliani, V. C. Martinez-Allo, S. Dergan-Dylon, S. P. Mendez-Huergo, J. C. Stupirski, D. Mazal, E. Osinaga, M. A. Toscano, V. Sundblad, G. A. Rabinovich and M. Salatino, Cancer Res., 2013, 73, 1107–1117 CrossRef CAS PubMed.
- M. T. Elola, F. Ferragut, S. P. Méndez-Huergo, D. O. Croci, C. Bracalente and G. A. Rabinovich, Cell. Immunol., 2018, 333, 34–45 CrossRef CAS PubMed.
- S. J. van Vliet, E. Saeland and Y. van Kooyk, Trends Immunol., 2008, 29, 83–90 CrossRef CAS PubMed.
- Y. van Kooyk, J. M. Ilarregui and S. J. van Vliet, Immunobiology, 2015, 220, 185–192 CrossRef CAS PubMed.
- A. Dominguez-Soto, E. Sierra-Filardi, A. Puig-Kroger, B. Perez-Maceda, F. Gomez-Aguado, M. T. Corcuera, P. Sanchez-Mateos and A. L. Corbi, J. Immunol., 2011, 186, 2192–2200 CrossRef CAS PubMed.
- E. Rodriguez, K. Boelaars, K. Brown, K. Madunic, T. van Ee, F. Dijk, J. Verheij, R. J. E. Li, S. T. T. Schetters, L. L. Meijer, T. Y. S. Le Large, E. Driehuis, H. Clevers, S. C. M. Bruijns, T. O'Toole, S. J. van Vliet, M. F. Bijlsma, M. Wuhrer, G. Kazemier, E. Giovannetti, J. J. Garcia-Vallejo and Y. van Kooyk, Commun. Biol., 2022, 5, 41 CrossRef CAS PubMed.
- H. Laubli and A. Varki, Cell. Mol. Life Sci., 2020, 77, 593–605 CrossRef CAS PubMed.
- S. Wisnovsky, L. Mockl, S. A. Malaker, K. Pedram, G. T. Hess, N. M. Riley, M. A. Gray, B. A. H. Smith, M. C. Bassik, W. E. Moerner and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2015024118 CrossRef CAS PubMed.
- H. Läubli, K. Kawanishi, C. George Vazhappilly, R. Matar, M. Merheb and S. Sarwar Siddiqui, FEBS J., 2021, 288, 6206–6225 CrossRef PubMed.
- E. J. H. van Houtum, C. Bull, L. A. M. Cornelissen and G. J. Adema, Front. Immunol., 2021, 12, 790317 CrossRef CAS PubMed.
- M. Perdicchio, J. M. Ilarregui, M. I. Verstege, L. A. Cornelissen, S. T. Schetters, S. Engels, M. Ambrosini, H. Kalay, H. Veninga, J. M. den Haan, L. A. van Berkel, J. N. Samsom, P. R. Crocker, T. Sparwasser, L. Berod, J. J. Garcia-Vallejo, Y. van Kooyk and W. W. Unger, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 3329–3334 CrossRef CAS PubMed.
- C. Traving and R. Schauer, Cell. Mol. Life Sci., 1998, 54, 1330–1349 CrossRef CAS PubMed.
- C. Bull, M. H. den Brok and G. J. Adema, Biochim. Biophys. Acta, 2014, 1846, 238–246 Search PubMed.
- S. van de Wall, K. C. M. Santegoets, E. J. H. van Houtum, C. Bull and G. J. Adema, Trends Immunol., 2020, 41, 274–285 CrossRef CAS PubMed.
- M. C. Schmid and J. A. Varner, J. Oncol., 2010, 2010, 201026 Search PubMed.
- R. Beatson, V. Tajadura-Ortega, D. Achkova, G. Picco, T. D. Tsourouktsoglou, S. Klausing, M. Hillier, J. Maher, T. Noll, P. R. Crocker, J. Taylor-Papadimitriou and J. M. Burchell, Nat. Immunol., 2016, 17, 1273–1281 CrossRef CAS PubMed.
- E. Rodriguez, K. Boelaars, K. Brown, R. J. Eveline Li, L. Kruijssen, S. C. M. Bruijns, T. van Ee, S. T. T. Schetters, M. H. W. Crommentuijn, J. C. van der Horst, N. C. T. van Grieken, S. J. van Vliet, G. Kazemier, E. Giovannetti, J. J. Garcia-Vallejo and Y. van Kooyk, Nat. Commun., 2021, 12, 1270 CrossRef CAS PubMed.
- J. Wang, M. Manni, A. Barenwaldt, R. Wieboldt, N. Kirchhammer, R. Ivanek, M. Stanczak, A. Zippelius, D. Konig, N. Rodrigues Manutano and H. Laubli, Front. Cell Dev. Biol., 2022, 10, 828916 CrossRef PubMed.
- W. Jing, X. Guo, G. Wang, Y. Bi, L. Han, Q. Zhu, C. Qiu, M. Tanaka and Y. Zhao, Int. Immunopharmacol., 2020, 78, 106012 CrossRef CAS PubMed.
- C. Li, X. Luo, Y. Lin, X. Tang, L. Ling, L. Wang and Y. Jiang, PLoS One, 2015, 10, e0141817 CrossRef PubMed.
- R. Singh and B. K. Choi, eLife, 2019, 8, e48916 CrossRef CAS PubMed.
- S. J. Meyer, A. T. Linder, C. Brandl and L. Nitschke, Front. Immunol., 2018, 9, 2820 CrossRef PubMed.
- T. Hernández-Caselles, R. C. Miguel, A. J. Ruiz-Alcaraz and P. García-Peñarrubia, J. Immunol. Res., 2019, 2019, 6032141 CrossRef PubMed.
- B. Biedermann, D. Gil, D. T. Bowen and P. R. Crocker, Leuk. Res., 2007, 31, 211–220 CrossRef CAS PubMed.
- S. Benmerzoug, M. F. Chevalier, M. Verardo, S. Nguyen, V. Cesson, A. K. Schneider, F. Dartiguenave, S. C. Rodrigues-Dias, I. Lucca, P. Jichlinski, B. Roth, D. Nardelli-Haefliger and L. Derré, Eur. Urol. Focus, 2022, 8, 748–751 CrossRef PubMed.
- H. Jetani, A. Navarro-Bailón, M. Maucher, S. Frenz, C. Verbruggen, A. Yeguas, M. B. Vidriales, M. González, J. Rial Saborido, S. Kraus, K. Mestermann, S. Thomas, H. Bonig, M. Luu, R. Monjezi, D. Mougiakakos, M. Sauer, H. Einsele and M. Hudecek, Blood, 2021, 138, 1830–1842 CrossRef CAS PubMed.
- E. C. Brinkman-Van der Linden and A. Varki, J. Biol. Chem., 2000, 275, 8625–8632 CrossRef CAS PubMed.
- S. Benmerzoug, M. F. Chevalier, L. Villier, S. Nguyen, V. Cesson, A. K. Schneider, F. Dartiguenave, S. C. Rodrigues-Dias, I. Lucca, P. Jichlinski, B. Roth, D. Nardelli-Haefliger and L. Derré, Eur. Urol. Open Sci., 2021, 34, 79–82 CrossRef PubMed.
- D. Lamprinaki, P. Garcia-Vello, R. Marchetti, C. Hellmich, K. A. McCord, K. M. Bowles, M. S. Macauley, A. Silipo, C. De Castro, P. R. Crocker and N. Juge, Front. Immunol., 2021, 12, 744184 CrossRef CAS PubMed.
- L. Yang, Y. Feng, S. Wang, S. Jiang, L. Tao, J. Li and X. Wang, Int. Immunopharmacol., 2021, 99, 107965 CrossRef CAS PubMed.
- D. Mele, G. Pessino, G. Trisolini, A. Luchena, M. Benazzo, P. Morbini, S. Mantovani, B. Oliviero, M. U. Mondelli and S. Varchetta, Front. Immunol., 2022, 13, 997806 CrossRef CAS PubMed.
- K. Boelaars and Y. van Kooyk, Trends Cancer, 2024, 10, 230–241 CrossRef CAS PubMed.
- A. Trebo, N. Ditsch, T. Degenhardt, C. Kuhn, M. Rahmeh, E. Schmoeckel, D. Mayr, B. Czogalla, T. Kolben, S. Meister, S. Mahner, U. Jeschke and A. Hester, Int. J. Mol. Sci., 2021, 22, 2000 CrossRef CAS PubMed.
- B. S. Bochner, R. A. Alvarez, P. Mehta, N. V. Bovin, O. Blixt, J. R. White and R. L. Schnaar, J. Biol. Chem., 2005, 280, 4307–4312 CrossRef CAS PubMed.
- C. Ou, L. Liu, J. Wang, S. Dai, Y. Qu, Y. Xiong, W. Xi, J. Xu and J. Guo, Urol. Oncol., 2017, 35, 607 Search PubMed.
- Q. Haas, K. F. Boligan, C. Jandus, C. Schneider, C. Simillion, M. A. Stanczak, M. Haubitz, S. M. Seyed Jafari, A. Zippelius, G. M. Baerlocher, H. Läubli, R. E. Hunger, P. Romero, H. U. Simon and S. von Gunten, Cancer Immunol. Res., 2019, 7, 707–718 CrossRef CAS PubMed.
- M. A. Stanczak, S. S. Siddiqui, M. P. Trefny, D. S. Thommen, K. F. Boligan, S. von Gunten, A. Tzankov, L. Tietze, D. Lardinois, V. Heinzelmann-Schwarz, M. von Bergwelt-Baildon, W. Zhang, H. J. Lenz, Y. Han, C. I. Amos, M. Syedbasha, A. Egli, F. Stenner, D. E. Speiser, A. Varki, A. Zippelius and H. Läubli, J. Clin. Invest., 2018, 128, 4912–4923 CrossRef PubMed.
- V. Barriga, N. Kuol, K. Nurgali and V. Apostolopoulos, Cancers, 2019, 11, 1205 CrossRef CAS PubMed.
- A. A. Barkal, R. E. Brewer, M. Markovic, M. Kowarsky, S. A. Barkal, B. W. Zaro, V. Krishnan, J. Hatakeyama, O. Dorigo, L. J. Barkal and I. L. Weissman, Nature, 2019, 572, 392–396 CrossRef CAS PubMed.
- J. Freile, N. Ustyanovska Avtenyuk, M. G. Corrales, H. J. Lourens, G. Huls, T. van Meerten, E. Cendrowicz and E. Bremer, Biomedicines, 2022, 10, 1175 CrossRef CAS PubMed.
- S. S. Yin and F. H. Gao, Front. Immunol., 2020, 11, 1324 CrossRef CAS PubMed.
- S. Shafi, T. N. Aung, V. Xirou, N. Gavrielatou, I. A. Vathiotis, A. Fernandez, M. Moutafi, V. Yaghoobi, R. S. Herbst, L. N. Liu, S. Langermann and D. L. Rimm, Lab. Invest., 2022, 102, 1143–1149 CrossRef CAS PubMed.
- S. P. Méndez-Huergo, A. G. Blidner and G. A. Rabinovich, Curr. Opin. Immunol., 2017, 45, 8–15 CrossRef PubMed.
- M. Salatino, D. O. Croci, G. A. Bianco, J. M. Ilarregui, M. A. Toscano and G. A. Rabinovich, Expert Opin Biol. Ther., 2008, 8, 45–57 CrossRef CAS PubMed.
- D. Hanahan and R. A. Weinberg, Cell, 2011, 144, 646–674 CrossRef CAS PubMed.
- R. M. Perrotta, C. A. Bach, M. Salatino and G. A. Rabinovich, Biochem. J., 2021, 478, 597–617 CrossRef CAS PubMed.
- N. Martínez-Bosch and P. Navarro, Adv. Exp. Med. Biol., 2020, 1259, 17–38 CrossRef PubMed.
- S. H. Hsieh, N. W. Ying, M. H. Wu, W. F. Chiang, C. L. Hsu, T. Y. Wong, Y. T. Jin, T. M. Hong and Y. L. Chen, Oncogene, 2008, 27, 3746–3753 CrossRef CAS PubMed.
- V. L. Thijssen, B. Barkan, H. Shoji, I. M. Aries, V. Mathieu, L. Deltour, T. M. Hackeng, R. Kiss, Y. Kloog, F. Poirier and A. W. Griffioen, Cancer Res., 2010, 70, 6216–6224 CrossRef CAS PubMed.
- A. Toti, A. Santi, E. Pardella, I. Nesi, R. Tomasini, T. Mello, P. Paoli, A. Caselli and P. Cirri, J. Cell Commun. Signal, 2021, 15, 405–419 CrossRef CAS PubMed.
- P. Juszczynski, J. Ouyang, S. Monti, S. J. Rodig, K. Takeyama, J. Abramson, W. Chen, J. L. Kutok, G. A. Rabinovich and M. A. Shipp, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 13134–13139 CrossRef CAS PubMed.
- J. Li, Y. Pan, J. Yang, J. Wang, Q. Jiang, H. Dou and Y. Hou, Front. Immunol., 2022, 13, 945234 CrossRef CAS PubMed.
- Y. Mori, K. Akita, M. Yashiro, T. Sawada, K. Hirakawa, T. Murata and H. Nakada, J. Biol. Chem., 2015, 290, 26125–26140 CrossRef CAS PubMed.
- T. Piyush, A. R. Chacko, P. Sindrewicz, J. Hilkens, J. M. Rhodes and L. G. Yu, Cell Death Differ., 2017, 24, 1937–1947 CrossRef CAS PubMed.
- K. L. Wu, E. Y. Huang, E. W. Jhu, Y. H. Huang, W. H. Su, P. C. Chuang and K. D. Yang, J. Gastroenterol., 2013, 48, 350–359 CrossRef CAS PubMed.
- C. Chen, C. A. Duckworth, Q. Zhao, D. M. Pritchard, J. M. Rhodes and L. G. Yu, Clin. Cancer Res., 2013, 19, 1693–1704 Search PubMed.
- W. Wang, H. Guo, J. Geng, X. Zheng, H. Wei, R. Sun and Z. Tian, J. Biol. Chem., 2014, 289, 33311–33319 CrossRef CAS PubMed.
- B. N. Stillman, D. K. Hsu, M. Pang, C. F. Brewer, P. Johnson, F. T. Liu and L. G. Baum, J. Immunol., 2006, 176, 778–789 Search PubMed.
- C. Li, H. Guo, P. Zhai, M. Yan, C. Liu, X. Wang, C. Shi, J. Li, T. Tong, Z. Zhang, H. Ma and J. Zhang, Cancer Res., 2024, 84, 258–275 Search PubMed.
- S. Punt, V. L. Thijssen, J. Vrolijk, C. D. de Kroon, A. Gorter and E. S. Jordanova, PLoS One, 2015, 10, e0129119 CrossRef PubMed.
- Z. Y. Liu, N. H. Meng, P. P. Cao, Y. Jia, H. Wang, Y. H. Zhang, H. Liu and R. Fu, Clin. Transl. Med., 2023, 13, e1224 CrossRef CAS PubMed.
- C. Chen, C. A. Duckworth, B. Fu, D. M. Pritchard, J. M. Rhodes and L. G. Yu, Br. J. Cancer, 2014, 110, 741–752 CrossRef CAS PubMed.
- M. Labrie, M. C. Vladoiu, A. A. Grosset, L. Gaboury and Y. St-Pierre, Oncotarget, 2014, 5, 7705–7721 Search PubMed.
- N. Rubinstein, M. Alvarez, N. W. Zwirner, M. A. Toscano, J. M. Ilarregui, A. Bravo, J. Mordoh, L. Fainboim, O. L. Podhajcer and G. A. Rabinovich, Cancer Cell, 2004, 5, 241–251 Search PubMed.
- A. J. Cagnoni, M. L. Giribaldi, A. G. Blidner, A. M. Cutine, S. G. Gatto, R. M. Morales, M. Salatino, M. C. Abba, D. O. Croci, K. V. Mariño and G. A. Rabinovich, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2102950118 CrossRef CAS PubMed.
- W. Peng, H. Y. Wang, Y. Miyahara, G. Peng and R. F. Wang, Cancer Res., 2008, 68, 7228–7236 CrossRef CAS PubMed.
- T. Wang, Z. Chu, H. Lin, J. Jiang, X. Zhou and X. Liang, Mol. Biol. Rep., 2014, 41, 4069–4076 CrossRef CAS PubMed.
- A. C. MacKinnon, S. L. Farnworth, P. S. Hodkinson, N. C. Henderson, K. M. Atkinson, H. Leffler, U. J. Nilsson, C. Haslett, S. J. Forbes and T. Sethi, J. Immunol., 2008, 180, 2650–2658 CrossRef CAS PubMed.
- L. Díaz-Alvarez and E. Ortega, Mediators Inflammation, 2017, 2017, 9247574 Search PubMed.
- C. X. Zhang, D. J. Huang, V. Baloche, L. Zhang, J. X. Xu, B. W. Li, X. R. Zhao, J. He, H. Q. Mai, Q. Y. Chen, X. S. Zhang, P. Busson, J. Cui and J. Li, Oncogenesis, 2020, 9, 65 CrossRef CAS PubMed.
- N. Pang, X. Alimu, R. Chen, M. Muhashi, J. Ma, G. Chen, F. Zhao, L. Wang, J. Qu and J. Ding, FASEB J., 2021, 35, e21556 CAS.
- R. Yang, L. Sun, C. F. Li, Y. H. Wang, J. Yao, H. Li, M. Yan, W. C. Chang, J. M. Hsu, J. H. Cha, J. L. Hsu, C. W. Chou, X. Sun, Y. Deng, C. K. Chou, D. Yu and M. C. Hung, Nat. Commun., 2021, 12, 832 CrossRef CAS PubMed.
- M. H. Wu, T. M. Hong, H. W. Cheng, S. H. Pan, Y. R. Liang, H. C. Hong, W. F. Chiang, T. Y. Wong, D. B. Shieh, A. L. Shiau, Y. T. Jin and Y. L. Chen, Mol. Cancer Res., 2009, 7, 311–318 CrossRef CAS PubMed.
- V. Salunkhe, A. Mahajan, N. Prakash, G. L. Pradeep, R. Patil and S. K. Gajdhar, J. Oral Maxillofac. Pathol., 2020, 24, 186 CrossRef PubMed.
- H. Wu, S. Song, A. Yan, X. Guo, L. Chang, L. Xu, L. Hu, M. Kuang, B. Liu, D. He, R. Zhao, L. Wang, X. Wu, J. Gu and Y. Ruan, Cancer Lett., 2020, 469, 287–300 CrossRef CAS PubMed.
- Y. L. Hsu, C. Y. Wu, J. Y. Hung, Y. S. Lin, M. S. Huang and P. L. Kuo, Carcinogenesis, 2013, 34, 1370–1381 CrossRef CAS PubMed.
- M. Xin, X. W. Dong and X. L. Guo, Biomed. Pharmacother., 2015, 69, 179–185 CrossRef CAS PubMed.
- S. Pokhare, U. C. Sharma, K. Attwood and S. Mansoor, J. Cancer Sci. Clin. Ther., 2022, 6, 322–327 Search PubMed.
- M. Song, Q. Pan, J. Yang, J. He, J. Zeng, S. Cheng, Y. Huang, Z. Q. Zhou, Q. Zhu, C. Yang, Y. Han, Y. Tang, H. Chen, D. S. Weng and J. C. Xia, Br. J. Cancer, 2020, 123, 1521–1534 CrossRef CAS PubMed.
- Y. Pang, E. Maxwell, P. Sindrewicz-Goral, A. Shapanis, S. Li, M. Morgan and L. G. Yu, Biomolecules, 2022, 12, 1451 CrossRef CAS PubMed.
- E. A. L. Enninga, K. Chatzopoulos, J. T. Butterfield, S. L. Sutor, A. A. Leontovich, W. K. Nevala, T. J. Flotte and S. N. Markovic, J. Pathol., 2018, 245, 468–477 CrossRef CAS PubMed.
- W. C. Chang, S. L. Wu, W. C. Huang, J. Y. Hsu, S. H. Chan, J. M. Wang, J. P. Tsai and B. K. Chen, Oncotarget, 2015, 6, 7741–7757 CrossRef PubMed.
- M. E. Arellano-Garcia, R. Li, X. Liu, Y. Xie, X. Yan, J. A. Loo and S. Hu, Int. J. Mol. Sci., 2010, 11, 3106–3121 CrossRef CAS PubMed.
- Y. Chen, D. Ma, X. Wang, J. Fang, X. Liu, J. Song, X. Li, X. Ren, Q. Li, Q. Li, S. Wen, L. Luo, J. Xia, J. Cui, G. Zeng, L. Chen, B. Cheng and Z. Wang, Cancer Immunol. Res., 2019, 7, 123–135 CrossRef CAS PubMed.
- J. Yuan, A. Khilnani, J. Brody, R. H. I. Andtbacka, S. Hu-Lieskovan, J. J. Luke, A. Diab, A. Marabelle, A. Snyder, Z. A. Cao and F. S. Hodi, Eur. J. Cancer, 2021, 157, 493–510 CrossRef CAS PubMed.
- C. Tiraboschi, L. Gentilini, C. Velazquez, E. Corapi, F. M. Jaworski, J. D. Garcia Garcia, Y. Rondon, A. Chauchereau, D. J. Laderach and D. Compagno, J. Immunother. Cancer, 2020, 8, e001535 CrossRef PubMed.
- P. Navarro, N. Martinez-Bosch, A. G. Blidner and G. A. Rabinovich, Clin. Cancer Res., 2020, 26, 6086–6101 CrossRef CAS PubMed.
- R. S. Herbst, J.-C. Soria, M. Kowanetz, G. D. Fine, O. Hamid, M. S. Gordon, J. A. Sosman, D. F. McDermott, J. D. Powderly, S. N. Gettinger, H. E. K. Kohrt, L. Horn, D. P. Lawrence, S. Rost, M. Leabman, Y. Xiao, A. Mokatrin, H. Koeppen, P. S. Hegde, I. Mellman, D. S. Chen and F. S. Hodi, Nature, 2014, 515, 563–567 CrossRef CAS PubMed.
- J. Galon, A. Costes, F. Sanchez-Cabo, A. Kirilovsky, B. Mlecnik, C. Lagorce-Pagès, M. Tosolini, M. Camus, A. Berger, P. Wind, F. Zinzindohoué, P. Bruneval, P.-H. Cugnenc, Z. Trajanoski, W.-H. Fridman and F. Pagès, Science, 2006, 313, 1960–1964 CrossRef CAS PubMed.
- W.-T. Hwang, S. F. Adams, E. Tahirovic, I. S. Hagemann and G. Coukos, Gynecol. Oncol., 2012, 124, 192–198 CrossRef PubMed.
- D. K. Nambiar, T. Aguilera, H. Cao, S. Kwok, C. Kong, J. Bloomstein, Z. Wang, V. S. Rangan, D. Jiang, R. von Eyben, R. Liang, S. Agarwal, A. D. Colevas, A. Korman, C. T. Allen, R. Uppaluri, A. C. Koong, A. Giaccia and Q. T. Le, J. Clin. Invest., 2019, 129, 5553–5567 CrossRef CAS PubMed.
- C. Büll, E. Collado-Camps, E. D. Kers-Rebel, T. Heise, J. N. Søndergaard, M. H. den Brok, B. M. Schulte, T. J. Boltje and G. J. Adema, Immunol. Cell Biol., 2017, 95, 408–415 CrossRef PubMed.
- K. Ohtsubo and J. D. Marth, Cell, 2006, 126, 855–867 CrossRef CAS PubMed.
- A. Arina, I. Tirapu, C. Alfaro, M. Rodriguez-Calvillo, G. Mazzolini, S. Inoges, A. Lopez, E. Feijoo, M. Bendandi and I. Melero, Exp. Hematol., 2002, 30, 1355–1364 CrossRef CAS PubMed.
- P. Chen, X. Liu, Y. Sun, P. Zhou, Y. Wang and Y. Zhang, Hum. Vaccines Immunother., 2016, 12, 612–622 CrossRef PubMed.
- P. Stoitzner, N. Romani, C. Rademacher, H. C. Probst and K. Mahnke, Eur. J. Immunol., 2022, 52, 1909–1924 CrossRef CAS PubMed.
- J. E. Babensee, Semin. Immunol., 2008, 20, 101–108 CrossRef CAS PubMed.
- P. M. Kou and J. E. Babensee, J. Biomed. Mater. Res., Part A, 2011, 96, 239–260 CrossRef PubMed.
- C. H. Lehmann, L. Heger, G. F. Heidkamp, A. Baranska, J. J. Luhr, A. Hoffmann and D. Dudziak, Vaccines, 2016, 4, 8 CrossRef PubMed.
- W. W. Unger, C. T. Mayer, S. Engels, C. Hesse, M. Perdicchio, F. Puttur, I. Streng-Ouwehand, M. Litjens, H. Kalay, L. Berod, T. Sparwasser and Y. van Kooyk, OncoImmunology, 2015, 4, e970462 CrossRef PubMed.
- S. J. van Vliet, E. van Liempt, E. Saeland, C. A. Aarnoudse, B. Appelmelk, T. Irimura, T. B. Geijtenbeek, O. Blixt, R. Alvarez, I. van Die and Y. van Kooyk, Int. Immunol., 2005, 17, 661–669 CrossRef CAS PubMed.
- N. S. Stambach and M. E. Taylor, Glycobiology, 2003, 13, 401–410 CrossRef CAS PubMed.
- K. Bloem, I. M. Vuist, M. van den Berk, E. J. Klaver, I. van Die, L. M. Knippels, J. Garssen, J. J. Garcia-Vallejo, S. J. van Vliet and Y. van Kooyk, Immunol. Lett., 2014, 158, 33–41 CrossRef CAS PubMed.
- L. Martinez-Pomares, J. Leukoc. Biol., 2012, 92, 1177–1186 CrossRef CAS PubMed.
- Y. Guo, H. Feinberg, E. Conroy, D. A. Mitchell, R. Alvarez, O. Blixt, M. E. Taylor, W. I. Weis and K. Drickamer, Nat. Struct. Mol. Biol., 2004, 11, 591–598 CrossRef CAS PubMed.
- W. W. Unger and Y. van Kooyk, Curr. Opin. Immunol., 2011, 23, 131–137 CrossRef CAS PubMed.
- E. Yuba, Mol. Immunol., 2018, 98, 8–12 CrossRef CAS PubMed.
- J. J. Lundquist and E. J. Toone, Chem. Rev., 2002, 102, 555–578 CrossRef CAS PubMed.
- A. Restuccia, M. M. Fettis and G. A. Hudalla, J. Mater. Chem. B, 2016, 4, 1569–1585 RSC.
- K. G. Leslie, S. S. Berry, G. J. Miller and C. S. Mahon, J. Am. Chem. Soc., 2024, 146, 27215–27232 CrossRef CAS PubMed.
- S. J. Richards and M. I. Gibson, JACS Au, 2021, 1, 2089–2099 CrossRef CAS PubMed.
- C. Fasting, C. A. Schalley, M. Weber, O. Seitz, S. Hecht, B. Koksch, J. Dernedde, C. Graf, E. W. Knapp and R. Haag, Angew. Chem., Int. Ed., 2012, 51, 10472–10498 CrossRef CAS PubMed.
- C. M. Fehres, A. J. van Beelen, S. C. M. Bruijns, M. Ambrosini, H. Kalay, L. V. Bloois, W. W. J. Unger, J. J. Garcia-Vallejo, G. Storm, T. D. de Gruijl and Y. V. Kooyk, J. Invest. Dermatol., 2015, 135, 2228–2236 CrossRef CAS PubMed.
- S. Duinkerken, S. K. Horrevorts, H. Kalay, M. Ambrosini, L. Rutte, T. D. de Gruijl, J. J. Garcia-Vallejo and Y. van Kooyk, Theranostics, 2019, 9, 5797–5809 CrossRef CAS PubMed.
- E. Yuba and R. K. Gupta, Biomater. Sci., 2024, 12, 1490–1501 RSC.
- A. Engering, T. B. Geijtenbeek, S. J. van Vliet, M. Wijers, E. van Liempt, N. Demaurex, A. Lanzavecchia, J. Fransen, C. G. Figdor, V. Piguet and Y. van Kooyk, J. Immunol., 2002, 168, 2118–2126 CrossRef CAS PubMed.
- A. J. Engering, M. Cella, D. M. Fluitsma, E. C. Hoefsmit, A. Lanzavecchia and J. Pieters, Adv. Exp. Med. Biol., 1997, 417, 183–187 CrossRef CAS PubMed.
- K. Mahnke, M. Guo, S. Lee, H. Sepulveda, S. L. Swain, M. Nussenzweig and R. M. Steinman, J. Cell Biol., 2000, 151, 673–684 CrossRef CAS PubMed.
- E. P. McGreal, J. L. Miller and S. Gordon, Curr. Opin. Immunol., 2005, 17, 18–24 CrossRef CAS PubMed.
- M. M. Weck, S. Appel, D. Werth, C. Sinzger, A. Bringmann, F. Grunebach and P. Brossart, Blood, 2008, 111, 4264–4272 CrossRef CAS PubMed.
- C. A. Wells, J. A. Salvage-Jones, X. Li, K. Hitchens, S. Butcher, R. Z. Murray, A. G. Beckhouse, Y. L. Lo, S. Manzanero, C. Cobbold, K. Schroder, B. Ma, S. Orr, L. Stewart, D. Lebus, P. Sobieszczuk, D. A. Hume, J. Stow, H. Blanchard and R. B. Ashman, J. Immunol., 2008, 180, 7404–7413 CrossRef CAS PubMed.
- E. van Liempt, C. M. Bank, P. Mehta, J. J. Garcia-Vallejo, Z. S. Kawar, R. Geyer, R. A. Alvarez, R. D. Cummings, Y. Kooyk and I. van Die, FEBS Lett., 2006, 580, 6123–6131 CrossRef CAS PubMed.
- C. Galustian, C. G. Park, W. Chai, M. Kiso, S. A. Bruening, Y. S. Kang, R. M. Steinman and T. Feizi, Int. Immunol., 2004, 16, 853–866 CrossRef CAS PubMed.
- K. Bloem, I. M. Vuist, M. van den Berk, E. J. Klaver, I. van Die, L. M. Knippels, J. Garssen, J. J. García-Vallejo, S. J. van Vliet and Y. van Kooyk, Immunol. Lett., 2014, 158, 33–41 CrossRef CAS PubMed.
- S. A. Jégouzo, H. Feinberg, T. Dungarwalla, K. Drickamer, W. I. Weis and M. E. Taylor, J. Biol. Chem., 2015, 290, 16759–16771 CrossRef PubMed.
- G. D. Brown and S. Gordon, Nature, 2001, 413, 36–37 CrossRef CAS PubMed.
- M. Ujita, H. Nagayama, S. Kanie, S. Koike, Y. Ikeyama, T. Ozaki and H. Okumura, Biosci., Biotechnol., Biochem., 2009, 73, 237–240 CrossRef CAS PubMed.
- M. D. Joshi, W. J. Unger, G. Storm, Y. van Kooyk and E. Mastrobattista, J. Controlled Release, 2012, 161, 25–37 CrossRef CAS PubMed.
- M. Maji, S. Mazumder, S. Bhattacharya, S. T. Choudhury, A. Sabur, M. Shadab, P. Bhattacharya and N. Ali, Sci. Rep., 2016, 6, 27206 CrossRef CAS PubMed.
- E. Yuba, J. Mater. Chem. B, 2020, 8, 1093–1107 RSC.
- M. D. Joshi, W. W. Unger, A. J. van Beelen, S. C. Bruijns, M. Litjens, L. van Bloois, H. Kalay, Y. van Kooyk and G. Storm, Int. J. Pharm., 2011, 416, 426–432 CrossRef CAS PubMed.
- W. W. Unger, A. J. van Beelen, S. C. Bruijns, M. Joshi, C. M. Fehres, L. van Bloois, M. I. Verstege, M. Ambrosini, H. Kalay, K. Nazmi, J. G. Bolscher, E. Hooijberg, T. D. de Gruijl, G. Storm and Y. van Kooyk, J. Controlled Release, 2012, 160, 88–95 CrossRef CAS PubMed.
- M. A. Boks, M. Ambrosini, S. C. Bruijns, H. Kalay, L. van Bloois, G. Storm, J. J. Garcia-Vallejo and Y. van Kooyk, J. Controlled Release, 2015, 216, 37–46 CrossRef CAS PubMed.
- D. A. Stolk, A. de Haas, J. Vree, S. Duinkerken, J. Lubbers, R. van de Ven, M. Ambrosini, H. Kalay, S. Bruijns, H. J. van der Vliet, T. D. de Gruijl and Y. van Kooyk, Front. Immunol., 2020, 11, 990 CrossRef CAS PubMed.
- J. J. García-Vallejo, M. Ambrosini, A. Overbeek, W. E. van Riel, K. Bloem, W. W. Unger, F. Chiodo, J. G. Bolscher, K. Nazmi, H. Kalay and Y. van Kooyk, Mol. Immunol., 2013, 53, 387–397 CrossRef PubMed.
- P. Stoitzner, C. H. Tripp, A. Eberhart, K. M. Price, J. Y. Jung, L. Bursch, F. Ronchese and N. Romani, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 7783–7788 CrossRef CAS PubMed.
- L. Bellmann, H. Strandt, C. Zelle-Rieser, D. Ortner, C. H. Tripp, S. Schmid, J. Rühl, G. Cappellano, S. Schaffenrath, A. Prokopi, S. Spoeck, A. Seretis, B. Del Frari, S. Sigl, J. Krapf, C. Heufler, T. Keler, C. Münz, N. Romani and P. Stoitzner, Eur. J. Immunol., 2022, 52, 1829–1841 CrossRef CAS PubMed.
- J. Schulze, M. Rentzsch, D. Kim, L. Bellmann, P. Stoitzner and C. Rademacher, Biochemistry, 2019, 58, 2576–2580 CrossRef CAS PubMed.
- T. Nochi, Y. Yuki, H. Takahashi, S. Sawada, M. Mejima, T. Kohda, N. Harada, I. G. Kong, A. Sato, N. Kataoka, D. Tokuhara, S. Kurokawa, Y. Takahashi, H. Tsukada, S. Kozaki, K. Akiyoshi and H. Kiyono, Nat. Mater., 2010, 9, 572–578 CrossRef CAS PubMed.
- R. Miura, S. I. Sawada, S. A. Mukai, Y. Sasaki and K. Akiyoshi, Biomacromolecules, 2020, 21, 621–629 CrossRef CAS PubMed.
- M. Andreini, D. Doknic, I. Sutkeviciute, J. J. Reina, J. Duan, E. Chabrol, M. Thepaut, E. Moroni, F. Doro, L. Belvisi, J. Weiser, J. Rojo, F. Fieschi and A. Bernardi, Org. Biomol. Chem., 2011, 9, 5778–5786 RSC.
- H. Feinberg, A. S. Powlesland, M. E. Taylor and W. I. Weis, J. Biol. Chem., 2010, 285, 13285–13293 CrossRef CAS PubMed.
- C. G. Figdor, Y. van Kooyk and G. J. Adema, Nat. Rev. Immunol., 2002, 2, 77–84 CrossRef CAS PubMed.
- C. M. Fehres, H. Kalay, S. C. Bruijns, S. A. Musaafir, M. Ambrosini, L. van Bloois, S. J. van Vliet, G. Storm, J. J. Garcia-Vallejo and Y. van Kooyk, J. Controlled Release, 2015, 203, 67–76 CrossRef CAS PubMed.
- A. Cambi, F. de Lange, N. M. van Maarseveen, M. Nijhuis, B. Joosten, E. M. van Dijk, B. I. de Bakker, J. A. Fransen, P. H. Bovee-Geurts, F. N. van Leeuwen, N. F. Van Hulst and C. G. Figdor, J. Cell Biol., 2004, 164, 145–155 CrossRef CAS PubMed.
- J. J. Garcia-Vallejo and Y. van Kooyk, Trends Immunol., 2013, 34, 482–486 CrossRef CAS PubMed.
- A. J. Affandi, J. Grabowska, K. Olesek, M. Lopez Venegas, A. Barbaria, E. Rodríguez, P. P. G. Mulder, H. J. Pijffers, M. Ambrosini, H. Kalay, T. O'Toole, E. S. Zwart, G. Kazemier, K. Nazmi, F. J. Bikker, J. Stöckl, A. J. M. van den Eertwegh, T. D. de Gruijl, G. Storm, Y. van Kooyk and J. M. M. den Haan, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 27528–27539 CrossRef CAS PubMed.
- J. Grabowska, D. A. Stolk, M. K. Nijen Twilhaar, M. Ambrosini, G. Storm, H. J. van der Vliet, T. D. de Gruijl, Y. van Kooyk and J. M. M. den Haan, Vaccines, 2021, 9, 56 CrossRef CAS PubMed.
- A. Shimoda, Y. Tahara, S. I. Sawada, Y. Sasaki and K. Akiyoshi, Biochem. Biophys. Res. Commun., 2017, 491, 701–707 CrossRef CAS PubMed.
- A. Shimoda, S. I. Sawada, Y. Sasaki and K. Akiyoshi, Sci. Rep., 2019, 9, 11497 CrossRef PubMed.
- S. C. Saunderson, A. C. Dunn, P. R. Crocker and A. D. McLellan, Blood, 2014, 123, 208–216 CrossRef CAS PubMed.
- A. Shimoda and K. Akiyoshi, Pharm. Res., 2023, 40, 795–800 CrossRef CAS PubMed.
- S. A. Dusoswa, S. K. Horrevorts, M. Ambrosini, H. Kalay, N. J. Paauw, R. Nieuwland, M. D. Pegtel, T. Würdinger, Y. Van Kooyk and J. J. Garcia-Vallejo, J. Extracell. Vesicles, 2019, 8, 1648995 CrossRef CAS PubMed.
- S. Vassilaros, A. Tsibanis, A. Tsikkinis, G. A. Pietersz, I. F. McKenzie and V. Apostolopoulos, Immunotherapy, 2013, 5, 1177–1182 CrossRef CAS PubMed.
- V. Apostolopoulos, N. Barnes, G. A. Pietersz and I. F. McKenzie, Vaccine, 2000, 18, 3174–3184 CrossRef CAS PubMed.
- R. Yang, J. Xu, L. Xu, X. Sun, Q. Chen, Y. Zhao, R. Peng and Z. Liu, ACS Nano, 2018, 12, 5121–5129 CrossRef CAS PubMed.
- Y. Xu, S. Ma, J. Zhao, H. Chen, X. Si, Z. Huang, Z. Yu, W. Song, Z. Tang and X. Chen, Biomaterials, 2022, 284, 121489 CrossRef CAS PubMed.
- J. Zhao, Y. Xu, S. Ma, Y. Wang, Z. Huang, H. Qu, H. Yao, Y. Zhang, G. Wu, L. Huang, W. Song, Z. Tang and X. Chen, Adv. Mater., 2022, 34, e2109254 CrossRef PubMed.
- L. Liu, J. Zhao, Z. Huang, Y. Xu, H. Chen, R. Qiao, W. Song, Z. Tang, T. P. Davis and X. Chen, Fundam. Res., 2025, 5, 183–191 CrossRef CAS PubMed.
- E. Yuba, N. Tajima, Y. Yoshizaki, A. Harada, H. Hayashi and K. Kono, Biomaterials, 2014, 35, 3091–3101 CrossRef CAS PubMed.
- M. Okubo, M. Miyazaki, E. Yuba and A. Harada, Bioconjugate Chem., 2019, 30, 1518–1529 CrossRef CAS PubMed.
- E. Yuba, A. Yamaguchi, Y. Yoshizaki, A. Harada and K. Kono, Biomaterials, 2017, 120, 32–45 CrossRef CAS PubMed.
- M. Miyazaki, E. Yuba, H. Hayashi, A. Harada and K. Kono, ACS Biomater. Sci. Eng., 2019, 5, 5790–5797 CrossRef CAS PubMed.
- E. Yuba, Y. Fukaya, S. Yanagihara, N. Kasho and A. Harada, Pharmaceutics, 2020, 12, 754 CrossRef CAS PubMed.
- P. F. Hockl, A. Wolosiuk, J. M. Pérez-Sáez, A. V. Bordoni, D. O. Croci, Y. Toum-Terrones, G. J. Soler-Illia and G. A. Rabinovich, Pharmacol. Res., 2016, 109, 45–54 CrossRef CAS PubMed.
- F. Danhier, K. Messaoudi, L. Lemaire, J. P. Benoit and F. Lagarce, Int. J. Pharm., 2015, 481, 154–161 CrossRef CAS PubMed.
- S. N. dos Santos, D. S. G. Junior, J. P. M. Pereira, N. M. Iadocicco, A. H. Silva, T. do Nascimento, L. A. P. Dias, F. R. de Oliveira Silva, E. Ricci-Junior, R. Santos-Oliveira and E. S. Bernardes, Cancer Nanotechnol., 2023, 14, 26 CrossRef CAS.
- Y. Gu, Y. Zhao, Z. Zhang, J. Hao, Y. Zheng, Q. Liu, Y. Liu and L. Shi, ACS Appl. Mater. Interfaces, 2021, 13, 22159–22168 CrossRef CAS PubMed.
- B. Balakrishnan, S. Subramanian, M. B. Mallia, K. Repaka, S. Kaur, R. Chandan, P. Bhardwaj, A. Dash and R. Banerjee, Biomacromolecules, 2020, 21, 2645–2660 CrossRef CAS PubMed.
- W. C. Chen, G. C. Completo, D. S. Sigal, P. R. Crocker, A. Saven and J. C. Paulson, Blood, 2010, 115, 4778–4786 CrossRef CAS PubMed.
- Q. Zhang, S. Li, K. Guo, X. Yin and R. Tong, Acta Biomater., 2022, 149, 321–333 CrossRef CAS PubMed.
- H. Inaba and C. H. Pui, Cancer Metastasis Rev., 2019, 38, 595–610 CrossRef PubMed.
- H. Läubli, S. C. Nalle and D. Maslyar, Cancer Immunol. Res., 2022, 10, 1423–1432 CrossRef PubMed.
- E. Pennesi, N. Michels, E. Brivio, V. H. J. van der Velden, Y. Jiang, A. Thano, A. J. C. Ammerlaan, J. M. Boer, H. B. Beverloo, B. Sleight, Y. Chen, B. Vormoor-Bürger, S. Rives, B. Bielorai, C. Rössig, A. Petit, C. Rizzari, G. Engstler, J. Starý, F. J. Bautista Sirvent, C. Chen-Santel, B. Bruno, Y. Bertrand, F. Rialland, G. Plat, D. Reinhardt, L. Vinti, A. Von Stackelberg, F. Locatelli and C. M. Zwaan, Leukemia, 2022, 36, 1516–1524 CrossRef CAS PubMed.
- H. Nakayama, C. Ogawa, M. Sekimizu, H. Fujisaki, Y. Kosaka, H. Hashimoto, A. M. Saito and K. Horibe, Int. J. Hematol., 2022, 116, 612–621 CrossRef CAS PubMed.
- M. Stelljes, S. Raffel, N. Alakel, R. Wäsch, M. Kondakci, S. Scholl, A. Rank, M. Hänel, B. Spriewald, M. Hanoun, S. Martin, K. Schwab, H. Serve, L. Reiser, J. Knaden, H. Pfeifer, J. Marx, T. Sauer, W. E. Berdel, G. Lenz, M. Brüggemann, N. Gökbuget and K. Wethmar, J. Clin. Oncol., 2024, 42, 273–282 CrossRef CAS PubMed.
- E. Jabbour, F. G. Haddad, N. J. Short, J. Senapati, N. Jain, K. Sasaki, J. Jorgensen, S. A. Wang, Y. Alvarado, X. Wang, C. DiNardo, L. Masarova, T. Kadia, R. S. Garris, F. Ravandi and H. Kantarjian, Blood, 2024, 143, 417–421 CrossRef CAS PubMed.
- K. J. Norsworthy, C. W. Ko, J. E. Lee, J. Liu, C. S. John, D. Przepiorka, A. T. Farrell and R. Pazdur, Oncologist, 2018, 23, 1103–1108 CrossRef CAS PubMed.
- J. A. Pollard, E. Guest, T. A. Alonzo, R. B. Gerbing, M. R. Loken, L. E. Brodersen, E. A. Kolb, R. Aplenc, S. Meshinchi, S. C. Raimondi, B. Hirsch and A. S. Gamis, J. Clin. Oncol., 2021, 39, 3149–3160 CrossRef CAS PubMed.
- P. Montesinos, V. Kota, J. Brandwein, P. Bousset, R. J. Benner, E. Vandendries, Y. Chen and M. F. McMullin, Cancer Chemother. Pharmacol., 2023, 91, 441–446 CrossRef CAS PubMed.
- K. T. Do, L. Q. M. Chow, K. Reckamp, R. E. Sanborn, H. Burris, F. Robert, D. R. Camidge, C. E. Steuer, J. H. Strickler, A. Weise, J. M. Specht, M. Gutierrez, P. Haughney, S. Hengel, C. L. Derleth and T. A. Yap, Oncologist, 2021, 26, 925–e1918 CrossRef CAS PubMed.
- F. Ravandi, M. Subklewe, R. B. Walter, P. Vachhani, G. Ossenkoppele, V. Buecklein, H. Döhner, M. Jongen-Lavrencic, C. D. Baldus, L. Fransecky, T. S. Pardee, H. Kantarjian, P. K. Yen, L. Mukundan, B. Panwar, M. R. Yago, S. Agarwal, S. K. Khaldoyanidi and A. Stein, Leuk. Lymphoma, 2024, 65, 1281–1291 CrossRef CAS PubMed.
- R. Narayan, A. A. Piérola, W. B. Donnellan, A. M. Yordi, M. Abdul-Hay, U. Platzbecker, M. Subklewe, T. M. Kadia, J. M. Alonso-Domínguez, J. McCloskey, K. Bradford, M. Curtis, N. Daskalakis, C. Guttke, K. Safer, B. Hiebert, J. Murphy, X. Li, K. Duchin and D. Esteban, Clin. Transl. Sci., 2024, 17, e13742 CrossRef CAS PubMed.
- M. Gutierrez, O. Hamid, E. Shum, D. R. Wise, A. V. Balar, J. S. Weber, P. LoRusso, S. Shafi, D. L. Rimm, A. W. Tolcher, D. Basudhar, M. E. Dujka and K. N. Heller, J. Clin. Oncol., 2020, 38, TPS3166–TPS3166 Search PubMed.
- V. Aslanis, R. J. Slack, A. C. MacKinnon, C. McClinton, S. Tantawi, L. Gravelle, U. J. Nilsson, H. Leffler, A. Brooks, S. K. Khindri, R. P. Marshall, A. Pedersen, H. Schambye and F. Zetterberg, Cancer Chemother. Pharmacol., 2023, 91, 267–280 CrossRef CAS PubMed.
- L. Vuong, E. Kouverianou, C. M. Rooney, B. J. McHugh, S. E. M. Howie, C. D. Gregory, S. J. Forbes, N. C. Henderson, F. R. Zetterberg, U. J. Nilsson, H. Leffler, P. Ford, A. Pedersen, L. Gravelle, S. Tantawi, H. Schambye, T. Sethi and A. C. MacKinnon, Cancer Res., 2019, 79, 1480–1492 CrossRef CAS PubMed.
- J. Mabbitt, I. D. Holyer, J. A. Roper, U. J. Nilsson, F. R. Zetterberg, L. Vuong, A. C. Mackinnon, A. Pedersen and R. J. Slack, Front. Immunol., 2023, 14, 1250559 CrossRef CAS PubMed.
- L. Astorgues-Xerri, M. E. Riveiro, A. Tijeras-Raballand, M. Serova, G. A. Rabinovich, I. Bieche, M. Vidaud, A. de Gramont, M. Martinet, E. Cvitkovic, S. Faivre and E. Raymond, Eur. J. Cancer, 2014, 50, 2463–2477 CrossRef CAS PubMed.
- R. P. Dings, M. C. Miller, I. Nesmelova, L. Astorgues-Xerri, N. Kumar, M. Serova, X. Chen, E. Raymond, T. R. Hoye and K. H. Mayo, J. Med. Chem., 2012, 55, 5121–5129 CrossRef CAS PubMed.
- L. Gheysen, L. Soumoy, A. Trelcat, L. Verset, F. Journe and S. Saussez, Cells, 2021, 10, 1112 CrossRef CAS PubMed.
- B. D. Curti, Y. Koguchi, R. S. Leidner, A. S. Rolig, E. R. Sturgill, Z. Sun, Y. Wu, V. Rajamanickam, B. Bernard, I. Hilgart-Martiszus, C. B. Fountain, G. Morris, N. Iwamoto, T. Shimada, S. Chang, P. G. Traber, E. Zomer, J. R. Horton, H. Shlevin and W. L. Redmond, J. Immunother. Cancer, 2021, 9, e002371 CrossRef PubMed.
- A. Kirwan, M. Utratna, M. E. O'Dwyer, L. Joshi and M. Kilcoyne, BioMed Res. Int., 2015, 2015, 490531 Search PubMed.
- A. Díaz-Fernández, M. Ryø Jochumsen, N. L. Christensen, K. Dalsgaard Sørensen, K. Bouchelouche, M. Borre, M. Holm Vendelbo and E. E. Ferapontova, Anal. Chem., 2024, 96, 18815–18823 CrossRef PubMed.
- R. López-Cortés, L. Muinelo-Romay, A. Fernández-Briera and E. Gil Martín, J. Proteome Res., 2024, 23, 1379–1398 CrossRef PubMed.
- J. Munkley and D. J. Elliott, Oncotarget, 2016, 7, 35478–35489 CrossRef PubMed.
- H. Li, A. W. T. Chiang and N. E. Lewis, Biotechnol. Adv., 2022, 60, 108008 CrossRef CAS PubMed.
- P. Kotidis and C. Kontoravdi, Metab. Eng. Commun., 2020, 10, e00131 CrossRef PubMed.
- P. K. Chuang, K. F. Chang, C. H. Chang, T. Y. Chen, Y. J. Wu, H. R. Lin, C. J. Wu, C. C. Wu, Y. C. Ho, C. C. Lin, C. H. Yuan, C. Y. Wang, Y. K. Lee and T. Y. Chen, Int. J. Mol. Sci., 2025, 26, 1648 CrossRef CAS PubMed.
- P. Zhang, L. Wang, H. Liu, S. Lin and D. Guo, BMC Cancer, 2025, 25, 249 CrossRef CAS PubMed.
- C. Dong, Y. Liu, S. Chong, J. Zeng, Z. Bian, X. Chen and S. Fan, Int. J. Mol. Sci., 2024, 25, 9502 CrossRef CAS PubMed.
- J. Lundstrøm, E. Korhonen, F. Lisacek and D. Bojar, Adv. Sci., 2022, 9, e2103807 CrossRef PubMed.
- D. J. Laderach and D. Compagno, Front. Immunol., 2022, 13, 1104625 CrossRef CAS PubMed.
- R. Yang, L. Sun, C.-F. Li, Y.-H. Wang, J. Yao, H. Li, M. Yan, W.-C. Chang, J.-M. Hsu, J.-H. Cha, J. L. Hsu, C.-W. Chou, X. Sun, Y. Deng, C.-K. Chou, D. Yu and M.-C. Hung, Nat. Commun., 2021, 12, 832 CrossRef CAS PubMed.
- I. Ibarlucea-Benitez, P. Weitzenfeld, P. Smith and J. V. Ravetch, Proc. Natl. Acad. Sci. U. S. A., 2021, 118 CrossRef CAS PubMed.
- L. A. M. Cornelissen, A. Blanas, J. C. van der Horst, L. Kruijssen, A. Zaal, T. O'Toole, L. Wiercx, Y. van Kooyk and S. J. van Vliet, Int. J. Cancer, 2019, 144, 2290–2302 CrossRef CAS PubMed.
- Y. T. Tsai, C. H. Liang, J. H. Yu, K. C. Huang, C. H. Tung, J. E. Wu, Y. Y. Wu, C. H. Chang, T. M. Hong and Y. L. Chen, Mol. Ther. Nucleic Acids, 2019, 18, 991–998 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |