
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMelatonin and the nervous system: nanomedicine perspectives
Fucen
Luo
a,
Yuru
Deng
b,
Borislav
Angelov
 c and
Angelina
Angelova
*a
c and
Angelina
Angelova
*a
aUniversité Paris-Saclay, CNRS, Institut Galien Paris-Saclay, F-91400 Orsay, France. E-mail: angelina.angelova@universite-paris-saclay.fr
bWenzhou Institute, University of Chinese Academy of Sciences, No.1, Jinlian Road, Longwan District, Wenzhou, Zhejiang 325001, China
cExtreme Light Infrastructure ERIC, Department of Structural Dynamics, CZ-25241 Dolni Brezany, Czech Republic
First published on 28th March 2025
Abstract
The mechanism of action of melatonin on the nervous system, sleep, cognitive deficits, and aging is not fully understood. Neurodegenerative diseases (ND) are one of the leading causes of disability and mortality worldwide. Sleeping and cognitive impairments also represent common and serious public health problems, particularly deteriorating with the aging process. Melatonin, as a neuromodulatory hormone, regulates circadian rhythms and the sleep–wake cycle, with functions extending to antioxidant, anti-inflammatory, neuroprotective, and anti-aging properties. However, melatonin is a hydrophobic compound with relatively low water solubility and a short half-life. While melatonin can cross the blood–brain barrier, exogenous melatonin administered orally or intravenously has poor bioavailability, undergoes rapid metabolism in the circulation, and shows limited brain accumulation, ultimately compromising its therapeutic efficacy. In recent years, the convergence of melatonin research with nanomedicine ensures safe therapeutic uses, limited drug degradation, and perspectives for targeted drug delivery to the central nervous system. Here we outline the promising neurotherapeutic properties of nanomaterials as carriers loaded with melatonin drug alone or in combinations with other active molecules.
1 Introduction
Neurodegenerative pathologies and cognitive damage represent common and serious public health problems. Over the past three decades, there has been a significant increase globally in the prevalence of neurological conditions such as stroke, Alzheimer's disease (AD), dementia, and meningitis attributed to factors like population aging and growth and heightened exposure to environmental, metabolic, and lifestyle risk factors. According to the latest analysis done by the 2021 Global Burden of Disease, Injuries, and Risk Factors Study, 37 neurological conditions (ranked as the leading cause of disability-adjusted life-years worldwide) affected 3.4 billion individuals and caused the death of 11.1 million people in 2021. Disorders affecting the nervous system are diverse and include neurodevelopmental disorders, late-life neurodegeneration, and newly emergent conditions, such as cognitive impairment following COVID-19.1 Aging is often associated with increasing impaired brain homeostasis and represents a major risk factor for most neurodegenerative disorders.An age-related decline of melatonin (MT) disrupting mitochondrial homeostasis and cytosolic DNA-mediated inflammatory reactions in neurons is the main contributory factor in the emergence of neurological abnormalities.2 In addition, the circadian clock exerts a notable influence on neurons during both developmental stages and throughout aging processes.3 The suprachiasmatic nucleus (SCN), located in the hypothalamus, is the principal circadian clock of the brain.4 Circadian rhythms (around 24 hours) significantly influence human lives, particularly through the sleep–wake cycle. Nearly all essential physiological functions and metabolic processes are regulated by circadian rhythms.5 Growing evidence reveals a bidirectional link between disturbances in circadian rhythms, sleep patterns, and neurodegenerative diseases (ND). Circadian disruptions and sleep disorders exacerbate neurodegeneration, and conversely, ND can disrupt circadian rhythms and sleep.6 Standard aging is associated with a reduced ability to initiate and maintain sleep. Sleep disruption beyond normal aging is especially prevalent in dementia as disrupted sleep is common in people with ND.7 The neuroprotective potential of MT to influence the connections between sleep disturbances, aging, and neurological conditions has not been fully exploited yet.
MT is a neuro-regulatory hormone that is essential for circadian rhythm. In humans, serum MT levels remain low during the daytime (10–20 pg mL−1). Around 10:00 PM, MT secretion increases significantly, rising continuously over 4 hours and reaching its peak concentration (80–120 pg mL−1) between 2:00 and 4:00 am. Thereafter, MT levels gradually decline, returning to the lower baseline concentrations observed during the daytime.8 The release of the endogenous hormone MT from the pineal gland progressively diminishes with aging and individuals with neurological disorders tend to have reduced levels of MT.9,10 MT is known for its preventive and therapeutic effects across a wide range of diseases including neurodegenerative disorders, aging, depression, and ocular, cardiac, immune, and orthopedic diseases.11–13 The susceptibility of people to these diseases increases with age, particularly neurological disorders. The brain is especially vulnerable to the detrimental effects of reactive oxygen species (ROS) due to its relatively high consumption of oxygen, elevated metabolic rate, excessive amount of iron, high content of polyunsaturated fatty acids, and comparatively limited ability for cellular regeneration when compared with other organs.14,15
The role of MT in various neurological disorders in in vitro or in vivo models is summarized in Table 1. In both short-term paradigms (24 to 72 hours) and long-term treatments (23 days to 9 months) in animal models, MT exhibits good therapeutic effects in some neurological disorders. Single-dose or short-term administration alleviates oxidative stress, inflammation, and cognitive deficits, as observed in 24-hour MCAO-mediated acute ischemic stroke16 and sleep deprivation models.17 In contrast, long-term MT treatment supports neuroprotection, mitigates neurodegeneration, and enhances hippocampal neurogenesis in aging-related and chronic disease models such as Alzheimer's18 and multiple sclerosis.19 MT exerts its therapeutic and neuroprotective effects through multiple mechanisms such as (i) downregulating inflammatory factors, (ii) inhibiting cell apoptosis, (iii) modulating signal transduction and gene expression, (iv) regulating enzyme and neurotrophic protein levels, (v) restoring the function of mitochondria and endoplasmic reticulum (ER), (vi) reducing oxidative stress, and (vii) maintaining circadian rhythms.
| Disease | In vitro/in vivo model | ROA/dosea | Effects | Year/ref. |
|---|---|---|---|---|
| a Route of administration (ROA); intraperitoneal injection (i.p.). | ||||
| Alzheimer's disease (AD) | In vitro: amyloid-β (Aβ)-induced inflammation in SH–SY5Y cells | 10 μM | MT played a protective role against Aβ–induced inflammation via an inflammasome–associated mechanism that is essential in inducing the active forms of cytokines and pyroptosis. | 202325 |
| Aging-related AD | In vivo: SAMP8 mice | 10 mg mL−1 kg−1 oral, 9 months | Chronic treatment with MT for 9 months decreased the neurodegenerative processes and the neurodegeneration-induced neurogenic response. MT induced recovery in the functionality of adult hippocampal neurogenesis in aged SAMP8 mice. | 202218 |
| Parkinson's disease (PD) | In vitro: (MPP+)-toxin-induced model in SH-SY5Y cells | 10 μg mL−1 in culture medium | MT showed promoting anti-oxidative and anti-apoptotic properties (increased SOD and GSH-Px activity, Bcl2 levels, and decreased ROS, MDA, Bax, and cleaved caspase-3 levels). MT hindered the toxic effects of MPP+ on dopaminergic neuronal cells via upregulation of the HSF1/HSP70 pathway, which could be a promising therapeutic strategy for PD. | 202226 |
| Japanese encephalitis virus (JEV) infection | In vitro: SH–SY5Y cells | 125–500 μM in culture medium | MT interfered with the replication cycle of JEV; abrogated the enzymatic function of the nonstructural proteins (NS3 and NS5); attenuated the JEV-induced upregulation of inflammatory factors (TNF-α, TLRs, NF-κB, and COX-2); and reduced the pro-apoptotic proteins levels and the caspase cascade activity. | 202327 |
| Cervical spondylotic myelopathy | In vitro: primary cultures of rat cortical neurons, SH-SY5Y cells. In vivo: a rat model of chronic cervical cord compression | 10 mg kg−1 (i.p.) | MT attenuated protein kinase R-like ER kinase–eukaryotic initiation factor 2α-C/EBP-homologous protein, inositol requiring enzyme 1, and transcription factor 6 signaling pathways to release ER stress and prevents Bax translocation to the mitochondrion. MT remodeled the ER morphology and restored homeostasis via ER-phagy (by activation of Sec. 62 receptor) in injured neurons. | 202328 |
| Huntington's disease (HD) | In vitro: N2a cells, Q7 and Q111 knockin mouse striatal cells. In vivo: AANAT knockout mice | 5 μM in culture medium | Insufficient MT levels impaired mitochondrial homeostasis resulting in mitochondrial DNA (mtDNA) release and activation of the cytosolic DNA-mediated inflammatory response in neurons. Exogenous MT inhibited the increased mtDNA release, cGAS activation, and inflammation in an HD mouse model. | 202029 |
| Ischemic stroke | In vivo: rats with MCAO model | 5 mg kg−1 (i.p.) a single dose, 24 h | MT attenuated MCAO-mediated stress associated with the MAPK p-P38/p-JNK pathways; restored the expression level of thioredoxin but did not affect the Nrf2 levels; alleviated NF-kB-induced inflammatory types (NOS-2 and COX-2). The elevated expression of p-NF-kB was accompanied by decreased thioredoxin expression. | 202016 |
| Multiple sclerosis (MS) | In vivo: male and female mice with experimental MS induced by cuprizone | 80 mg kg−1 day−1 (i.p.) 9 weeks | In the demyelination stage, MT showed neuroprotective effects in both male and female mice, improving motor ability and antioxidant levels (CAT, SOD, GPx, and GSH) and reducing MDA and inflammatory factors (IL-1β and TNF-α). In the remyelination stage, MT showed protective effects only in male mice. | 202019 |
| Neuro-degeneration | In vivo: PCBs mediated glutamate-induced neurodegeneration in rats | 5 mg kg−1 day−1 (i.p.) 30 days | MT protected the cerebral cortex from polychlorinated biphenyl (PCB)-impaired glutamate-BDNF signaling. It scavenged the ROS, decreased the NMDAR, and increased the level of CREB and BDNF leading to neuronal survival. | 201530 |
| Cognitive deficits | In vivo: chronic sleep deprivation-induced cognitive impairment in rat | 20 mg/kg day−1 (i.p.) 23 days | MT treatment attenuated chronic rapid eye movement sleep deprivation-induced cognitive impairment via regulating HDAC3-Bmal1/Clock interaction. | 202331 |
| Cognitive impairment | In vivo: sleep-deprived rats | 15 mg kg−1 day−1 (i.p.) 24/72 h | Short-term and large dose MT pre-treatment ameliorated cognitive impairment in 24 h and 72 h sleep-deprived rats. The possible mechanism may be associated with effects on oxidative stress, BDNF and CaMKII levels in the cerebral cortex (CC) and hippocampus. | 201317 |
MT is traditionally administered orally, but its bioavailability is limited due to its poor oral absorption, short biological half-life, and extensive first-pass metabolism.20 The oral transmucosal pathway resulted in high MT plasma concentrations, possibly due to avoiding the first-pass effect. Subcutaneous injection of free MT drug displayed a rapid absorption rate but showed no advantages compared with other administration routes. Transdermal delivery of MT was used in local applications requiring slow absorption and deposition in the skin.21 Limited MT absorption on mucosal and dermal surfaces hampered its efficient use as an antioxidant and neuroprotective compound. Overall, conventional approaches have failed to achieve appropriate pharmacokinetic and pharmacodynamic properties of MT at the target site, and thus produced low therapeutic efficacy and often elevated toxicity.21 The blood–brain barrier (BBB) is composed of brain capillary endothelium and effectively prevents over 98% of all small-molecule drugs and 100% of large-molecule neurotherapeutics from entering the brain.22 The presence of the BBB poses a unique challenge to the accessibility of drugs and antioxidants to the central nervous system (CNS), preventing them from effectively reaching therapeutic concentrations.23,24
Novel nanotechnologies exploiting the intranasal route are considered promising strategies for MT delivery to the brain.32,33 Nano-drug delivery systems effectively protect bioactive agents from degradation in physiological conditions while ensuring controlled release, prolonged efficacy, and further reduced side effects.34 MT-loaded nanocarriers have exhibited superior antioxidant, anti-inflammatory, and antitumor properties across different cell types and tissues compared with the free MT drug.35–37 A variety of nanocarriers, including solid lipid nanoparticles (SLNs), polymeric vesicles, silica nanoparticles (NPs), nanofibers, graphene-based nanocarriers, and metallic or non-metallic NPs, have been engineered to overcome MT's challenges such as poor solubility, stability, and bioavailability, enabling sustained delivery in some systems.38 Considering the vital functions of the nervous system, this review focuses on the nanocarrier-mediated delivery of MT to the brain and its application in treating and improving neurological disorders.
2 Neuroprotective and neuro-repairing effects of melatonin
Although every neurodegenerative disease exhibits unique molecular mechanisms and clinical characteristics, there are commonly recognized pathways across the various pathological processes. These pathways include protein misfolding and aggregation, oxidative stress, mitochondrial dysfunction, impaired phosphorylation, and dysregulation of metal homeostasis that often occur concurrently.39 MT shows a regulatory effect on the pathological processes through receptor-dependent and receptor-independent pathways (Fig. 1). By the receptor-independent pathway, MT can upregulate antioxidant defense mechanisms, directly remove ROS, inhibit inflammation, and prevent apoptosis. At physiological concentrations, MT exerts receptor-mediated actions, whereas receptor-independent actions usually require supra-physiological concentrations.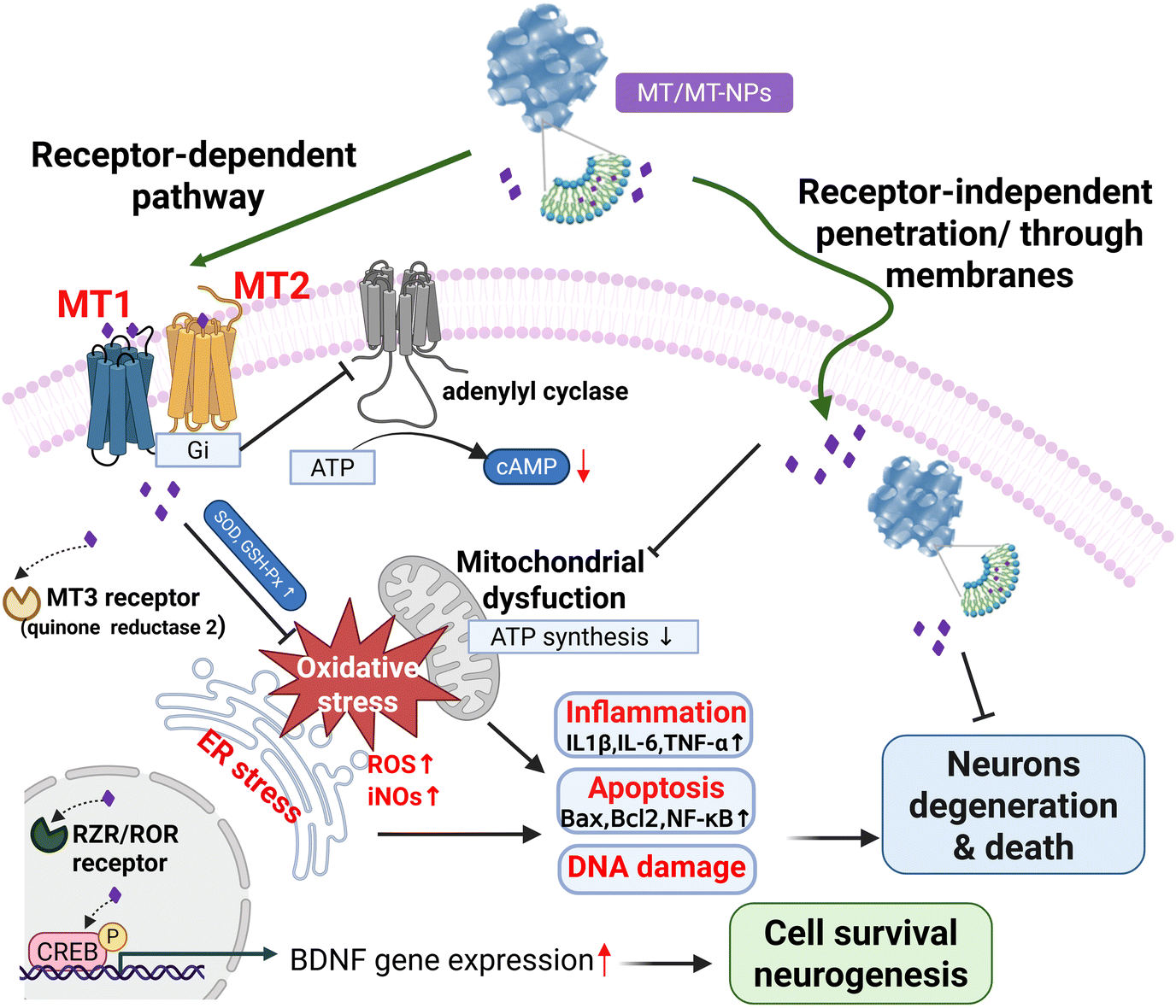 | ||
| Fig. 1 Melatonin (MT) and melatonin-loaded-nanoparticles(MT and MT-NPs) can exert neuroprotective and neuro-regenerative effects through receptor-dependent and receptor-independent pathways involving the alleviation of oxidative stress, mitochondrial dysfunction, endoplasmic reticulum (ER) stress-induced inflammation, apoptosis, DNA damage, and the regulation of signal transduction and gene expression to modulate the levels of enzymes and neurotrophins such as brain-derived neurotrophic factor (BDNF) (created with Biorender). | ||
There are four different MT receptor subtypes. Two of them are membrane-associated receptors, while the other two are nuclear receptors. Melatonin receptor 1 (MT1) and melatonin receptor 2 (MT2) subtypes are present in humans and other mammals. Melatonin receptor 3 (MT3) is a cytosolic enzyme known as quinone reductase 2 (QR2) rather than a membrane receptor. MT is also a ligand for retinoid orphan nuclear hormone receptors, referred to as ROR and RZR, at concentrations in the low nanomolar range. Both receptors are present in the central and peripheral nervous system and have been associated with cell differentiation and immune response regulation.40
2.1 MT1/MT2 receptor-dependent pathways of melatonin
Among the various molecular mechanisms of MT action, its most potent mechanism involves the activation of G protein-coupled receptors. These receptors, namely ML1 with high affinity and ML2 with low affinity, are now known as MT1 and MT2 receptors, respectively.41 They are members of the G protein–coupled receptor superfamily, preferentially coupling to Gi/o proteins. The MT1 and MT2 receptors have been localized to specific regions of the rodent and human nervous system, including the hippocampus, cerebellum, suprachiasmatic nucleus (SCN), and thalamus, as well as peripheral tissues. These localizations were determined by using receptor autoradiography with 2-[125I] iodomelatonin, in situ mRNA hybridization techniques, and immunohistochemistry. The functions of the MT1 and MT2 receptors have been studied through genetic deletion in mice lacking either the MT1 or MT2 receptor or through pharmacological methods using competitive MT receptor antagonists like luzindole and 4-phenyl-2-propionamidotetralin (4P-PDOT).42MT receptor binding regulates several second messengers, e.g. cyclic adenosine monophosphate (cAMP), cyclic guanosine monophosphate (cGMP), diacylglycerol, inositol trisphosphate, arachidonic acid, and intracellular Ca2+ concentration ([Ca2+]). In many cases, its effect is inhibitory and requires previous activation of the cell by a stimulatory agent.43 Activation of the MT1 and MT2 receptors primarily triggers Gi activation, which negatively regulates adenylyl cyclase (AC), leading to lower cAMP levels and reduced activity of cAMP-dependent kinase A (PKA). Additionally, both receptors can recruit β-arrestins. MT1 activation can also initiate the Gi/PI3K/Akt, Gi/PKC/ERK1/2, and Gq/PLCβ/IP3/Ca2+ pathways. Activation of MT2 receptors inhibits intracellular cGMP levels, subsequently reducing the activity of cGMP-dependent kinases or protein kinase G (PKG). The signaling of MT receptors within dimeric complexes involves distinct pathways depending on the receptor pairings.44
MT enhances PTEN-induced putative kinase 1-dependent mitophagy via the MT2/Akt/NF-κB pathway, and such mitophagy is critical for high glucose-induced mitochondrial impairment and apoptosis in neuronal cells.45 MT1 knockout led to increased loss of dopaminergic neurons and more severe motor dysfunction. It also suppressed the Sirt1/Nrf2/Ho1/Gpx4 pathway, reducing resistance to ferroptosis, and inhibited ferritin Fth1 expression, causing greater release of ferrous ions. MT1 activation prevents α-Syn-induced ferroptosis in PD, highlighting MT1's neuroprotective role in PD.46 Copper is crucial for generating ROS induced by Aβ peptide aggregation, making copper homeostasis a potential therapeutic target for AD. Research has shown that copper chelators (tetrathiomolybdate) facilitate the non-amyloidogenic processing of Aβ protein precursor (AβPP) via MT1/2/CREB-dependent signaling pathways.47 The inducible ADAM10 production caused by copper chelators can be blocked by a melatonin receptor (MT1/2) antagonist (luzindole) and an MT2 inhibitor (4P-PDOT), suggesting that the expression of ADAM10 depends on the activation of MT1/2 signaling pathways. Furthermore, MT1 and MT2 play opposite roles in brain cancer progression. Using an MT2-selective antagonist, DH97, a study demonstrated that MT1 impairs while MT2 promotes the proliferation of glioma and medulloblastoma cell lines. It provides the first evidence of the different roles of MT1 and MT2 in brain tumor progression, highlighting their relevance as druggable targets.48
2.2 Role of melatonin in neurological disorders
MT is considered a multi-pathway antioxidant and effective neuroprotective agent,50 which demonstrates remarkable efficacy in reducing oxidative stress under various conditions through multiple mechanisms summarized as follows:
2.2.1.1 Directly neutralizing free radical scavenging activity. Unlike the classic antioxidants, MT does not display pro-oxidative behavior and has a cascade-like antioxidant potential. Any intermediates formed during its interaction with reactive species also possess free radical scavenging properties. Consequently, a single MT molecule can potentially scavenge up to four or more reactive species, enhancing its efficacy as an antioxidant.51 For example, traditional antioxidants typically scavenge one or fewer ABTS˙+ radicals and react completely within a short period (<1 min), while every molecule of MT can scavenge up to four ABTS˙+ radicals with a sufficiently long reaction time (2 hours). This prolonged duration and enhanced capacity of scavenging has been attributed to the sequential generation of MT metabolites that also possess the ability to neutralize the ABTS+ radicals.52 The corresponding mechanism may involve multiple-electron donations via intermediates through a stepwise process. Intermediates including the melatoninyl cation radical, the melatoninyl neutral radical, 3-hydroxy-melatonin, and N1-AFMK appear to participate in these reactions.53
2.2.1.2 Indirectly regulating antioxidant enzyme activity and pro-oxidant enzyme activity. Several studies have reported that MT supplementation effectively reduces brain oxidative damage marked by decreased carbonyl, nitric oxide (NO), and malondialdehyde (MDA) content, and enhanced antioxidant enzyme activities, superoxide dismutase (SOD), catalase (CAT) and glutathione peroxidase (GPx) along with an overall antioxidant capacity.54,55 Interestingly, an earlier study reported opposite effects, with MT treatment decreasing antioxidant enzyme levels.56 Long-term MT administration to Alzheimer's transgenic mice reduced the expression of three antioxidant enzymes in the cerebral cortex (SOD, GPx, and CAT), suggesting decreased oxidative stress.56 This may occur because long-term use of MT is beneficial for the restoration of normal levels of antioxidant enzymes. Under conditions of short-term oxidative stress, MT promotes the activity of antioxidant enzymes, and this represents a dynamic regulatory process.
2.2.1.3 Metal-chelating activity. MT is reported to bind transition metals involved in harmful Fenton/Haber–Weiss reactions, thus reducing the production of toxic hydroxyl radicals and subsequently lowering oxidative stress.57 The Fenton reaction, involving divalent (Fe2+) and hydrogen peroxide (H2O2), generates ferric iron (Fe3+) and hydroxyl radicals (˙OH). These radicals then react with H2O2, producing superoxide and further ˙OH and anions in the Haber–Weiss reaction. Overall, metals like Fe, Cu, Zn, and Al catalyze the formation of highly reactive ˙OH radicals through these reactions. Fenton reactions can exacerbate ferroptosis (a form of cell death induced by iron accumulation and lipid peroxidation), which is involved in the pathogenesis of PD.46 The catalytic activities of transition metals, driven by Fenton reactions, play a role in the survival and pathological signaling pathways, neural plasticity, and neuroprotection.58 MT and its metabolites were demonstrated to completely inhibit oxidative stress caused by Cu(II)-ascorbate mixtures through Cu(II) chelation. Similarly, MT, N1-AFMK, and 3OHM prevented the initial step of the Haber–Weiss reaction, thereby stopping ˙OH production via the Fenton reaction. Moreover, it has been suggested that MT, alongside its known role in free-radical scavenging cascades, also participates in a concurrent ‘chelating cascade’, contributing to the reduction of oxidative stress.59
Strong evidence supports MT as a mitochondria-targeted antioxidant.61,62 MT accumulates in mitochondria at high concentrations against a gradient, probably through active transport by mitochondrial MT transporters. It protects mitochondria by scavenging ROS, inhibiting the mitochondrial permeability transition pore, and activating uncoupling proteins.63 MT reduces the mitochondrial membrane potential, thereby inhibiting the production of superoxide anions and hydrogen peroxide. Simultaneously, it maintains the efficiency of oxidative phosphorylation and ATP synthesis, while boosting the activity of respiratory complexes, mainly complexes I, III, and IV.64 Notably, a recent study has identified a mitochondrial GPCR mechanism contributing to the neuroprotective action of MT.65 MT synthesized exclusively in the mitochondrial matrix activates the mitochondrial MT1 signaling pathway, which inhibits stress-induced cytochrome c release and caspase activation. Consequently, this retards the neurodegenerative process.65
Multiple pieces of evidence suggests that MT may cure neurological disorders by alleviating mitochondrial dysfunction.66 MT and related indole metabolites can reverse mitochondrial dysfunction caused by Aβ peptide aggregation. This effect is partially mediated by MT binding to plasma membrane receptors, which then activate signaling pathways to the mitochondria.67 MT can modulate the SIRT1/Drp1 pathway, thereby ameliorating mitochondrial dysfunction, attenuating inflammation and apoptosis, and enhancing neural function after spinal cord injuries.68
In a study with a PD mouse model, MT displayed a protective effect against paraquat-induced motor deficits by alleviating the kinesin family member 5A-mediated axonal mitochondrial transport dysfunction in the midbrain.69 Moreover, the application of exogenous MT boosted mitochondrial ATP production and synergy, reducing abnormal phase separation and related mitochondrial dysfunction.70 It has been hypothesized that long-term daily MT supplementation may enhance survival, improve neurological function, and offer an alternative preventive measure against mitochondrial dysfunction after resuscitation.71
Other studies have shown that MT is crucial in protecting neurons, improving cognitive function, and treatment of traumatic brain injury and neurodegeneration against ER stress.74–76 C/EBP homologous protein (CHOP), also known as growth arrest- and DNA damage-inducible gene 153 (GADD153), is one of the main components of the ER stress-mediated apoptosis pathway.77 The primary target molecule of ER stress for MT is CHOP, and PERK and GRP78/BiP are the secondary target molecules.76 In a rat model of chronic cervical cord compression, MT reduced the activation of the signaling pathways involving PERK, eukaryotic initiation factor 2α-CHOP, inositol-requiring enzyme 1 (IRE1), and transcription factor 6 (ATF6), thereby alleviating ER stress. It also prevented Bax from translocating to the mitochondria, which supports motor recovery and protects neurons in vivo.
Additionally, MT can counteract ER stress-induced glutamate toxicity in primary rat cortical neurons in vitro and restore ER morphology and homeostasis through ER-phagy in damaged neurons.28 SIRT1 is an NAD+-dependent histone deacetylase, and its expression is up-regulated by ER stress contributing to ER stress-induced cellular damage.78 MT can attenuate spatial learning and memory dysfunction in developing rats by suppressing isoflurane-induced ER stress via the SIRT1/Mfn2/PERK signaling pathway.79 Under oxidative stress conditions, MT treatment inhibited the activation of ER stress-related and autophagy-related proteins by promoting the upregulation of cellular prion protein expression.80
Both in vivo and in vitro experiments have demonstrated that MT treatment markedly decreases hippocampal microglial activation and the expression of the inflammatory factors IL-1β and TNF-α induced by dim blue light. The beneficial effect of MT is associated with its interaction with the MT2 receptor.83 Another mechanism involves MT binding to its specific receptor (MT1) on microglia, which activates the JAK2/STAT3 pathways and boosts the expression of telomerase in the nucleus. This leads to a reduction in the production of proinflammatory cytokines like IL-1β, TNF-α, and iNOS by M1 microglia, while enhancing the production of anti-inflammatory cytokines such as CD206 and TGFβ by M2 microglia.84
The Toll-like receptors (TLRs) are transmembrane signaling proteins that play a crucial role in neuroinflammation. Exogenous MT provides strong neuroprotective effects by inhibiting TLR4 activation. The activation of TLR4 triggers autophagy and apoptosis in neuronal cells, facilitates the formation of the NOD-like receptor family pyrin domain containing 3 (NLRP3) inflammasome, and elevates the secretion of downstream inflammatory cytokines.85 The NLRP3 inflammasome is recognized as a new target for MT. This inflammasome plays a role in increasing IL-1β levels, activating caspase-1, and promoting pyroptosis.86 Administration of a 5 mg kg−1 dose of MT 30 min prior to ischemia reduced brain infarction associated with sequentially rescued neuronal apoptosis. Furthermore, MT attenuated neuroinflammatory markers and ROS, induced by ischemic stroke, by halting the key players of the mitogen stress family (p38/JNK).16
MT therapy has been efficient in improving cognitive and mood function in a rat model. The MT effect was dose-dependent, with lower (10–20 mg kg−1) doses improving several cognitive tasks and mood function with the suppression of oxidative stress and NLRP3 inflammasomes.87 Furthermore, moderate MT doses (40–80 mg kg−1) mediated robust anti-inflammatory activity with the modulation of the NF-kB–NLRP3–caspase-1 pathway, whereas 80 mg kg−1 MT activated the BDNF–ERK–CREB pathway and improved a more complex cognitive function.87
High doses of MT are recommended for treating the neuro-invasiveness associated with COVID-19 outbreak, as this may help regulate the immune response and neuroinflammation caused by SARS-CoV-2 (Fig. 2). MT-mediated signaling may influence reduced SARS-CoV-2 entry. When SARS-CoV-2 infects the CNS cells, it triggers the release of pro-inflammatory cytokines, for instance, (i) TNF-α, which acts by binding to TNFR receptor recruiting TRADD. This protein binds to TRAF2 to phosphorylate and activate IKK. Then, the IKK complex phosphorylates IKBα, resulting in the translocation of NF-κB to the nucleus, where it targets many genes coding for mediators of inflammatory responses. (ii) IL-6 induces gene activation in response to cytokine receptor stimulation. STAT3 proteins dimerize and translocate to the nucleus. JAK2/STAT3 signaling acts as a pivotal mediator of neuroinflammation. (iii) The binding of SARS-CoV-2 protein to the TLR (TLR3/7/9) upregulates the pro-inflammatory transcription factor NF-κB and causes the release of pro-IL-1β which is cleaved by caspase-1, followed by NLRP3 inflammasome activation. Interestingly, MT may revert these pro-inflammatory effects by inhibiting JAK2/STAT3 signaling pathway and NF-κB translocation.88 MT is also considered to be a particularly well-suited drug to modulate acute and chronic inflammatory activity around neural electrode implants, translating into a more stable and reliable interface.89
 | ||
| Fig. 2 Hypothetical diagram of potential therapeutic targets where melatonin may act against SARS-CoV-2 infection in the central nervous system (CNS). The virus enters neuronal cells via ACE2 and TMPRSS2, triggering clathrin-mediated endocytosis, and replicates inside the cell. This replication involves the synthesis of viral RNA and proteins, which are assembled in the ER and Golgi, forming new virions that are released by exocytosis. SARS-CoV-2 infection may disrupt mitochondrial metabolism, increase ROS, and induce ER stress. MT, with its high diffusibility, enters neuronal cells, binds to CaM, and may regulate the Ca2+/CaMKII system, modulate ACE2 expression, link ER stress to the inflammatory response, and scavenge ROS. MT-mediated signaling may influence viral entry through either MT1/MT2 and α7nAChR receptors. The virus triggers the release of pro-inflammatory cytokines like TNF-α and IL-6, and activates TLRs, leading to inflammation through pathways such as NF-κB and JAK2/STAT3. MT may counteract these effects by inhibiting these pathways and reducing inflammation (including the inhibition of NLRP3 inflammasome activation). The diagram shows stimulation (in blue) or inhibition (in red) of therapeutic targets by MT or SARS-CoV-2 protein. Organelles and structures are not drawn to scale88 (reproduced from ref. 88 under PMC Open Access License. Copyright © 2020, Springer Nature). | ||
Oxidative stress mediates chemical damage to DNA, resulting in a wide variety of byproducts. MT and several of its metabolites can repair the sites of direct (chemical) oxidation in DNA. They can repair guanine-centered radical cations through electron transfer at extremely high, diffusion-limited rates, and also repair carbon-centered radicals in the sugar component of 2′-deoxyguanosine via hydrogen atom transfer. Furthermore, the MT metabolites 6-hydroxymelatonin and 4-hydroxymelatonin are also predicted to repair OH adducts in the imidazole ring.92
Cuprizone (a copper-chelating agent) treatment disrupts hippocampal neurogenesis in the dentate gyrus by reducing BDNF levels and decreasing CREB phosphorylation, effects that are alleviated by MT treatment.95 Preventive MT treatment restored the propofol-induced inactivation of PKA/CREB/BDNF signaling and reversed synaptic dysfunction.96 MT1/MT2 receptor activation by agonists increases the neuronal content of BDNF regardless of which of the two MT receptors is expressed by the neurons. On the other hand, the BDNF-stimulatory action of ramelteon (melatonin receptor agonist) appeared to involve translational rather than transcriptional mechanisms.97 MT and its immediate precursor N-acetylserotonin (NAS) mediated the protective effect of ketamine-induced downregulation of BDNF protein levels as well as decreased phosphorylation of ERK and AKT. MT appears to increase BDNF levels via MT receptor(s) activation and stimulate TrkB, while NAS seems to activate TrkB directly.98
Recent studies have demonstrated that MT plays a key role in cell survival signaling pathways. Prolonged drug administration improved neuronal survival through the AKT and MAPK signaling pathways after focal brain ischemia in mice. The elevated expression of CREB and Atf-1 highlighted the significant role of MT in promoting neuronal regeneration.99 MT treatment counteracts energy depletion and protects against brain damage through the regulation of p-AMPK/p-CREB signaling pathways in the mouse brain.100 MicroRNA-132 is essential for neuronal survival and significantly contributes to the pathological process of AD. Treatment with MT restores the expression of miR-132 and reduces the levels of PTEN and FOXO3a. MT also prevents the nuclear translocation of FOXO3a, thereby inhibiting its pro-apoptotic pathways.101 Thus, MT is also able to provide neuroprotection against Aβ-induced neurotoxicity through the miR-132/PTEN/AKT/FOXO3a pathway.
2.3 Clinical trials and limitations of melatonin
While preclinical studies strongly support the safety and efficacy of MT as a neuroprotective agent, clinical research remains limited. Most of the clinical trials have focused on the effects of MT on improving sleep and cognitive impairment, or on its use as an adjuvant therapeutic molecule. A meta-analysis has underscored the potential importance of MT in treating specific primary sleep disorders. MT needs to be prescribed early and used long-term at a dose of 2 to 5 or 10 mg.102 MT supplementation (2 mg day−1) improved subjective sleep quality and actigraphic sleep efficiency in traumatic brain injury patients.103 Clinical studies have concluded that MT is effective in treating the irregular sleep/wake cycles associated with AD. A daily dose of 3–12 mg of MT at bedtime has proved to be particularly effective for managing REM (rapid eye movement) sleep behavior disorder in PD.104 MT at doses of 10 mg day−1 or more for at least 12 weeks in immediate-release form has shown significant benefits in improving motor symptoms and sleep disturbances in Parkinson's disease (PD).105 Evidence indicates that MT treatment for over 12 weeks may improve cognitive functioning in mild AD.106 MT (3 mg) administration resulted in clinical improvement in patients with ischemic stroke.107 Combined delivery of MT (3 mg day−1) with valproate for adults with generalized epilepsy and generalized onset motor seizures has led to significantly improved clinical outcomes.108 Another study has reported that prolonged MT release at a 4 mg dose did not reduce REM sleep behavior disorder in PD.In the context of the COVID-19 pandemic, clinical research on MT for the symptomatic treatment or adjuvant treatment of SARS-CoV-2 viral infection has become a hot topic. Several clinical trials have tested the efficacy of MT in coronavirus inhibition, providing an understanding of the appropriate doses and effectiveness of MT against the virus.109,110 In a randomized, double-blind, placebo-controlled trial, patients received either 3 mg of MT three times daily (n = 42) or a placebo (n = 39) for 2 weeks. MT significantly increased oxygen saturation, reduced respiratory rate, and lowered inflammatory markers such as CRP, ESR, LDH, CPK, ferritin, and D-dimer, most of which are prognostic for COVID-19. Importantly, no adverse side effects were observed. These findings suggested that MT is an effective and safe adjunctive therapy for mild to moderate COVID-19 infection.111 Another clinical trial demonstrated that combining the oral administration of MT tablets (3 mg, three times daily) with standard treatment could significantly enhance sleep quality and blood oxygen saturation in hospitalized COVID-19 patients.112
The positive effects of MT on the nervous system have been demonstrated in cellular and animal models of various diseases (Table 1). Most of these studies used supraphysiological concentrations of MT and the required doses were significantly higher than those used for sleep regulation. In principle, the established role of MT in rhythmic function is not necessarily incompatible with the use of high doses to achieve ‘protective’ effects.113 However, one needs to carefully distinguish the effects of endogenous MT from the effects observed with high doses of exogenous MT.114 MT is available as a supplement in several Western countries (including the United States and Western Europe) in doses ranging from 1 to 10 mg. Limited clinical trials have demonstrated the potential application of MT in AD and PD, especially in the early stages of the disease. Calculations derived from animal studies suggest that cytoprotective MT doses are in the range of 40–100 mg day−1. Clinical trials have reported doses as high as 100 mg, which is regarded as a substantial dosage—the highest level documented so far.115 Evidently, there is an urgent need for clinical studies and safety assessments within this dosage range.104,116 Although MT is considered a non-toxic natural molecule, the safety of its long-term use, especially for the additional risks that may arise from the elderly and young children, requires more intensive clinical research117,118
It is worth mentioning that MT has low water solubility, short half-life, and poor bioavailability. In healthy volunteers, the elimination half-lives of oral administration or intravenous administration of 10 mg of MT were 54 minutes and 39 minutes, respectively. Although there were significant individual differences, the median absolute bioavailability of oral MT was as low as ∼2.5% (1.7–4.7%).119 An early study found that even with higher oral doses of MT (100 mg), the peak concentration in human plasma was only 0.435 μM after 60 min.120 However, MT was typically administered at dosages greater than 1 μM in preclinical investigations, and a dose of 1 mM served as the foundation for most trials. Thus, it is unlikely that either humans or animals would naturally attain such concentrations, and certainly not over an extended period of time.114
In recent strategies, the undesirable poor bioavailability of small molecule drugs such as MT in clinical applications could be overcome by introducing nanoencapsulation and delivery technologies. These strategies aim to construct nanocarrier systems with the potential for targeting and controlled release of drugs. Since the Food and Drug Administration approval of Doxil (a stealth liposome encapsulating doxorubicin) in 1995, more than 50 nanopharmaceuticals have entered clinical practice, with many more under investigation for various therapeutic indications.121,122 Diverse nanoformulations, including liposomes (LP), polymers, nanocrystals, inorganic nanoparticles, micelles, and protein-based carriers, have been explored primarily for cancer therapy, oncology, fungal infections, and pain management, with lipid-based nanoparticles leading the field.121 However, the safety and efficacy of nanopharmaceuticals, along with the lack of specific regulatory guidelines and cost–benefit considerations, remain key concerns.122,123
Several MT nanomedicines have been studied at the cellular and animal levels. However, a significant limitation remains the lack of pharmacokinetic and pharmacodynamics data on MT nanocarriers in animal models (in both blood and brain),124 particularly in neurodegenerative and neurological disorders. A recent study used a microdialysis system to measure real-time melatonin (MT) concentrations in the brains of freely moving animals.125 Each animal received an equivalent MT dose (10 mg kg−1) from three different formulations. MT-loaded LP and nanoemulsions (NE) demonstrated superior pharmacokinetic parameters—longer half-life (T1/2), higher maximum plasma concentration (Cmax), shorter time to peak concentration (Tmax), and greater overall drug exposure (AUC)—compared with MT dissolved in DMSO.125 These findings provide crucial insights for preclinical research and could facilitate the clinical translation of MT-based nanocarrier formulations.
3 Nanocarrier-mediated melatonin delivery
Many nanoparticles (NPs) can encapsulate either lipophilic or hydrophilic drugs, genes, proteins, and peptides, protecting them from degradation, improving drug stability, prolonging plasma half-life, reducing side effects, and controlling the release of active components at the desired site.126Liposomes (LP), solid lipid nanoparticles (SLNs), and nanostructured lipid carriers (NLCs) have been extensively studied as lipid-based nanocarriers for brain disease treatment over the past 30 years. The lipid composition provides superior biocompatibility and safety compared with polymeric and inorganic nanoparticles, allowing them to cross the BBB without requiring additional functionalization.128 Polymeric nanoparticles offer tunable size, shape, physicochemical properties, and surface modifications, enabling controlled and sustained drug delivery across the BBB. However, their degradation can generate acidic by-products, leading to potential toxicity that limits long-term brain applications.128 Inorganic nanoparticles, while highly stable and chemically versatile, also pose inherent toxicity concerns. Despite their advantages, lipid-based nanoparticles face challenges such as oxidation and drug leakage, which hinder their clinical translation. Complex hybrid systems (multi-lipid, lipid-polymer, etc.) offer a promising solution, balancing stability, safety, and efficacy for brain-targeted drug delivery. For example, ionizable lipids have been designed to form strong ionic interactions with encapsulated drugs, while cholesterol enhances the structural integrity to ensure tighter drug packing. Additionally, modifying the particle surface with polyethylene glycol prolongs circulation time.121
Fig. 3 summarizes a variety of nanocarriers for MT encapsulation and delivery including lipid-based nanocarriers, polymer-based nanocarriers, hybrid lipid–polymer nanocarriers, inorganic NPs, and mesoporous nonlamellar liquid crystalline lipid NPs (cubosomes). MT-loaded NPs have received attention due to beneficial features such as (i) improving drug bioavailability by increasing water-solubility and prolonging the duration of MT action; (ii) providing a biphasic release profile (an early burst release phase followed by a slow release phase), (iii) environmentally sensitive (pH- or temperature-) responsive release; or (iv) targeted delivery of MT facilitated by ligands or peptides anchored on the NP surface.
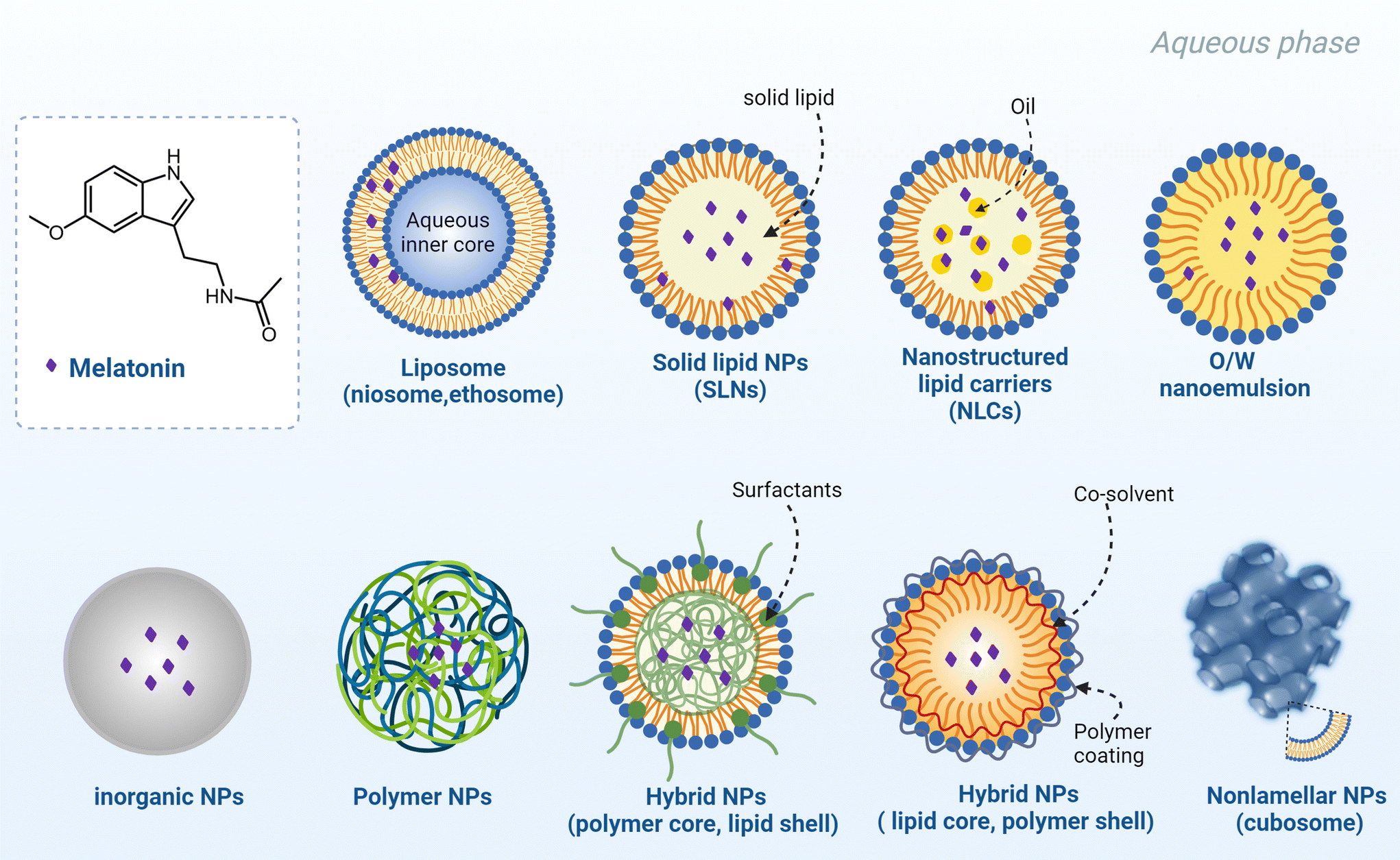 | ||
| Fig. 3 Chemical structure of MT (N-acetyl-5-methoxyindolamine) and a schematic presentation of different types of nanocarrier suitable for the encapsulation and protection of MT as a drug. MT was initially discovered in 1958 by Lerner who isolated it from the extract of bovine pineal tissue.127 MT is a derivative of tryptophan, an essential amino acid for mammals, which contains a indole heterocycle and two side chains, namely, a 5-methoxy group and 3-amide group.51 Promising nanocarriers of MT include lipid-based nanocarriers like liposomes, solid lipid nanoparticles (SLNs), nanostructured lipid carriers (NLCs), and nanoemulsions, inorganic NPs, polymer NPs, lipid–polymer hybrid nanoparticles, and non-lamellar liquid crystalline lipid NPs (e.g., cubosomes) (created with BioRender). | ||
3.1 Lipid-based nanocarrier-mediated melatonin delivery
The lipid-based nanocarrier systems for drug delivery mainly include vesicle-type systems like LP, ethosomes (ET), transfersomes (TF) and niosomes (NS), NE systems, and particulate systems such as SLNs and NLCs.129Table 2 summarizes the lipid-based nanocarrier systems investigated for efficient MT delivery. These carriers are typically made of naturally occurring lipids that are well-tolerated by the human body. Incorporating water-insoluble drugs into such formulations enhances their solubility and stability in aqueous environments, removing the need for harmful co-solvents or pH adjustments to dissolve hydrophobic drugs.130 These NPs confer safety for therapeutic use and limit the drug degradation phenomena.131| Type | Formulation | Studied model | ROA | EE% | LE% | Release profile | Outcome |
|---|---|---|---|---|---|---|---|
| Nanocarrier types: liposome (LP), solid lipid nanoparticle (SLN), nanostructured lipid carrier (NLC), transfersomes (TF), niosomes (NS), ethosomes (ET), ethanolic liposome (ET-LP), nanoemulsion (NE); phosphatidyl choline (PC); route of administration (ROA): intraperitoneal injection (i.p.), transdermal administration (TD), intravenous injection (i.v.); not available (N.A.). | |||||||
| LP, NE | LP: DSPC/cholesterol/PEG2000-DSPE | In vivo brain microdialysis: female Sprague–Dawley rats aged 12 weeks | 25 mg kg−1 (i.v.) | N.A. | N.A. | Prolonged release, T1/2: 81, 50, 26 min for MT-LP, MT-NE, and MT-EtOH | Lipidic NPs provide higher levels of MT in the brain, a longer time above critical pharmacological concentrations, and a similar MT concentration in the circulation compared with MT dissolved in DMSO.125 |
| NE: tricaproin, soybean oil, medium-chain triglycerides (MCT) | |||||||
| EL-LP | PC, cholesterol, sodium deoxycholate | Permeation: isolated dermis of mouse | TD | 73.9 | 9.92 | Cumulative penetration of EL 1.5 times higher than that of conventional LP | EL more easily penetrate into the body; MT-EL enhanced the skin hydration level and preserved the integrity of dermal collagen and elastic fibers.111 |
| In vivo: photoaging mouse model | |||||||
| ET-LP | Soya PC, ethanol | Skin irritancy: male albino rabbits | TD | 70.7 | N.A. | N.A. | MT-loaded ETs provided an enhanced transdermal flux, lower lag time, higher entrapment efficiency and low skin irritancy potential.133 |
| TF NS | Cholesterol | HFF-1 cells | TD | TF | N.A. | Higuchi model; delayed and sustained release (TF/NS 74.8/66.8% at 24 h) vs. MT solution rapid release (102.3% at 8 h) | MT-loaded TF showed greater permeability, MT deposition, higher NO inhibition and stimulation of collagen than NS and MT solution.134 |
| PC | RAW 264.7 cells | 74.9 | |||||
| Tween 80 | NS | ||||||
| PEG 400 | 66.8 | ||||||
| SLN | Compritol 888, Span80, Tween80 | In vivo: male Wistar rats with testicular trauma | 25 mg kg−1 (i.p.) | 20 | 5 | Burst release at the first 30 min followed by a sustained release pattern | Testicular trauma disturbed spermatogenesis, morphometric, and oxidative parameters. MT and especially MT-SLN improved traumatic damage.135 |
| SLN | N.A. | In vivo: male Wistar rats with cyclosporine A (CsA)-induced cardiac damage | 1 mg kg−1 (i.p.) | N.A. | N.A. | N.A. | (i) MT significantly reduces CsA cardiotoxicity acting also on apoptotic processes, and (ii) the reduction in CsA-induced cardiotoxicity is mediated mainly by its antioxidant effect.136 |
| NLC | Caprylic/capric, triglycerides (CCT), octyl, Compritol 888 ATO, glyceryldistearate, polaxamer 407 | Breast cancer | N.A. | 84.3 | 18.7 | N.A. | Co-treatment of the cells with MT-NLC and tamoxifen caused a 2-fold increase in the percentage of apoptosis; MT-NLCs on activation of apoptosis are associated with alterations in the cell cycle progression by triggering sub-G1 arrest; MT induces marked increase in Bid and Bax mRNA expression level.137 |
| MCF-7 cells | |||||||
LP are spherical lipid bilayer structures with an aqueous core, where the aqueous volume is enclosed by one or more lipid bilayers. The bilayer typically consists of phospholipids. Based on their structure, number of bilayers, and size, LP can be categorized as small unilamellar vesicles (25–50 nm), large unilamellar vesicles (100 nm–1 μm), multilamellar (0.1–15 μm), and multivesicular (1.6–10.5 μm) particles.132
NE is a thermodynamically unstable colloidal mixture of two immiscible liquids, where one liquid acts as the dispersed phase and the other as the dispersing medium. NE usually consists of fine oil droplets dispersed in water, with droplet sizes typically between 100 and 600 nm.138
Medium-chain triglyceride NE (MCT-NE) has gained popularity as a carrier in recent years because of its unique properties, such as its capacity for enhanced solubilization of insoluble drugs, slow release rates, and increased transdermal absorption.139 LP and NE are excellent solubilizers for lipophilic substances like MT. It has been demonstrated that intravenous administration of MT-nanoformulations (LP or MCT-NE) improves the drug bioavailability in the brain compared with the administration of MT dissolved in organic solvents like DMSO. Interestingly, MT pharmacokinetics in the brain is influenced by the type of nanocarrier. Fig. 4 shows that MCT-NE yielded a higher Cmax of MT, while LP provided higher MT levels in the brain and maintained pharmacological concentrations for a longer duration.125 Conventional LP typically do not cross the BBB in vivo unless disrupted or ligand-modified. A potential mechanism underlying this enhanced brain accumulation may involve NP fusion with the endothelial luminal membrane, leading to intracellular release and facilitated brain uptake.125
 | ||
| Fig. 4 Example of pharmacokinetic profiles and in vivo brain characterization data. (a) Melatonin concentration in the brain (mean ± SD) after a bolus injection of 10 mg kg−1 MT in DMSO, liposomes (LP), or medium-chain triglycerides nanoemulsion (MCT-NE). (b) Estimated pharmacokinetic parameters of MT in DMSO, LP or MCT-NE (mean ± SD); (b) maximal plasma concentration of drug (Cmax)125 (reproduced with permission from ref. 125. Copyright © 2020, Elsevier). | ||
Considering the low drug entrapment efficiency and challenging large-scale manufacturing of vesicle systems, like LP, SLNs have been investigated as advanced CNS drug delivery systems.140 SLNs are lipid-based biocompatible nanocarriers consisting of a condensed-phase lipid matrix (triglycerides, fatty acids, or waxes) that can be organized into a crystalline nanostructure. SLNs, prepared by the probe ultrasonication method, range in size from 10 to 1000 nm in diameter.141 MT-loaded SLNs showed burst release behavior of MT at the initial stage (the first 30 min) followed by a sustained release pattern. The burst release could be attributed to the diffusion of the drug located on the surface of the SLN. Thereafter, the drug is released from the core of the SLNs, where MT is suggested to be homogeneously distributed within the NPs.135
NLCs have been developed as a modified version of SLNs. NLCs feature a combination of solid-phase and liquid-phase lipids, resulting in a different internal core structure compared with SLNs, which have only a solid lipid core. This structural difference introduces imperfections in the core, leading to a more performant formulation.142 For instance, NLCs showed much higher MT entrapment efficiency (EE) (84.3 versus 20%) and loading efficiency (LE) (18.7 versus 5%) than the SLNs.135,137
3.2 Polymer-based nanocarrier-mediated MT delivery
Polymeric NPs, providing numerous functions such as targeting, pH response, and control release, have been extensively investigated as drug delivery systems. They range in size from 1 to 1000 nm and can encapsulate active compounds either within the polymeric core or adsorbed on the surface.143 By utilizing polymer nanomaterials and drug-loading nanoparticle technology, MT has been either encapsulated or attached to the NPs, thereby altering the dissolution rate and enhancing drug absorption in vivo. The designed MT formulations improved its therapeutic efficacy against multiple diseases (Table 3).| Nanocarriers type | Formulation | Studied model | ROA | EE% | DE% | Release profile | Outcome |
|---|---|---|---|---|---|---|---|
| COLBP: Type II collagen-binding peptide; CTS: chitosan; HPMC: hydroxypropyl methylcellulose; TPP: tripolyphosphate; route of administration (ROA): intravenous injection (i.v.); intraperitoneal injection (i.p.); not available (N.A.). | |||||||
| MT@PLGA-COLBP NPs | PLGA, DSPC, DSPE-PEG2000, type II collagen-binding peptide WYRGRL (COLBP) | Osteoarthritis model; cells: chondrocytes; in vivo: female C57BL/6 mice | MT 10 μL, 50 mM; NPs10 μL, 2 mg mL−1 injection into the knee joint | N.A. | N.A. | Continuous release during 14 days | MT protects chondrocytes by inhibiting TLR2/4-MyD88-NFκB signal pathway; NPs targeting cartilage and sustained release of MT in the articular cavity.149 |
| Fucoidan/chitosan layered PLGA NPs (MNPs@C@F) | PLGA, polyvinyl alcohol (PVA), chitosan, fucoidan | Breast cancer mouse 4T1 cells; in vivo: BALB/c mice were orthotopically inoculated with 4T1 cells | Oral | 15.1 | 4.1 | Overcome the barriers of the GI tract and reach an effective cumulative dose at the tumor site (pH5) | MNPs@C@F promoted intestinal microfold cell transcytosis for the delivery of MT and fucoidan into tumors; MT and fucoidan in the tumors could regulate the tumor microenvironment by decreasing P-gp, Twist, HIF-1α, and anti-inflammatory immune cell expression while increasing cytotoxic T cell populations following doxorubicin treatment.151 |
| Polydopamine NPs (MPDANPs) | Dopamine, cyclohexane, Tween20, ammonium hydroxide | In vitro: high glucose (HG)-induced DR model in ARPE-19 cells; in vivo: male SD rats with DR | MT 10 mg kg−1; MPDANPs 20 mg kg−1 (i.p.) | 94.6 | 50 | Higher release at pH 6.7; controlled and extended drug release until 160 h | MPDANPs have demonstrated a reduction in VEGF- and PKCδ- levels, and ROS-mediated DR pathogenesis.152 |
| Chitosan NPs | Sodium tripolyphosphate (TPP), chitosan | Cells: RAW 264.7; in vivo: female Balb/C mice with bowel disease | (i.v.) | N.A. | 20.4 | Initial burst release followed by prolonged release; cumulative release at 24 h: 76.35% at pH7.4 and 96.22% at pH4.5 | Significant anti-inflammatory activity of MT-CS NPs is attributed to nitric oxide (NO) reduction, inhibited nuclear translocation of NF-kB p65 and reduced IL-1β and IL-6 expression. 2021153 |
| Chitosan-TPP NPs | Chitosan (CTS), TPP | Etoposide-induced genotoxicity; cells: HepG2 | N.A. | 75 | N.A. | A biphasic release: an early burst release phase and a slow-release phase | MT reduced the effects of etoposide significantly through the reduction of the level of DNA damage. MT decreased the intracellular ROS generation but increased the intracellular GSH levels in HepG2 cells. MT-loaded NPs were more effective than the free MT drug. |
| Chitosan/HPMC NPs | Chitosan, TPP, hydroxypropyl methylcellulose (HPMC) | Breast cancer; cells: MDA-MB-231 | N.A. | N.A. | N.A. | Cumulative release of MT at pH 5.5 was 61%, while it was 18% at pH 7.5. | The toxicity of MT encapsulated in CTS/HPMC NPs was found to be higher than that of the free MLT, indicating that MT encapsulation facilitated its uptake in the cancer cells.146 |
| Gelatin-PLA-CTS NPs | PLA, gelatin, chitosan, Span-80, Tween-80 | In vivo: pinealectomized male Wistar rats | MT, 10 mg kg−1; NPs, 60 mg kg−1 (s.c.) | 33.82 | 15.77 | Controlled-release; pH-sensitivity | Drug release from MT-loaded NPs was more sensitive in simulated intestinal fluid (pH 7.4) and blood (pH 6.8) and displayed better antidepressant actions compared with the free MT.148 |
| Sericin-based NPs (MR-SNC) | Sericin, resveratrol | Breast cancer; cells: MCF-7 | N.A. | 98 | 27 | An apparent burst release (67%) of MT occurred in pH 6 at 55 h. | The efficient cellular uptake of MR-SNC, DNA fragmentation and chromatin condensation was found at pH 6. Proficient release of the entrapped drugs occurred from MR-SNC in an acidic environment leading to cell apoptosis.145 |
Several natural (chitosan, hyaluronic acid, or sericin) and synthetic (PLGA, PLA, or PCL) polymers have been used as delivery systems for MT to treat a variety of pathophysiological processes.144,145 The pH dependence of MT release was linked to the pH sensitivity of the NPs’ supramolecular structure as determined by their chemical nature, for instance, the amine groups of the chitosan chains, which have a pKa value of 6.5. In an acidic medium, these amine groups become protonated, increasing the electrostatic repulsive forces between the cationic moieties. The repulsive forces then promote the release and diffusion of MT from the nanocarriers to the acidic environment.146
Cyclodextrin (CD)-based molecularly imprinted nanosponges (MIP-NSs) have been used to overcome the limitations associated with MT release.147 Gelatin, polylactic acid (PLA), and chitosan were utilized to formulate MT-loaded nanoparticles (MTNPs), which demonstrated varied controlled release profiles in different pH environments.148 The type II collagen-targeting peptide was attached to the surface of PLGA NPs to create a nano-delivery system for MT. In vivo, this system remained stable for at least 21 days and persisted in the joint cavities of mice, releasing MT for at least 14 days. As a result, the injection frequency of this nano-delivery system was reduced by 75% compared with injections of MT alone.149
3.3 Hybrid nanocarrier-mediated melatonin delivery
Lipid and polymeric NPs are the most studied NPs and have been shown to be the most efficient drug delivery systems. Depending on the composition, some of them display instability, rapid drug release, limited drug loading capacity, low biocompatibility, and problems for large-scale production. To overcome these limitations, lipid–polymer hybrid nanoparticles (LPHNPs) have been developed to converge the advantages of lipid- and polymer-based nanocarriers in terms of biocompatibility and stability, improved drug loading and control release, providing an increased drug half-life and therapeutic effects.150Hybrid NPs (LPHNPs) have been classified into two main groups based on the different arrangements of lipids and polymers, e.g. a polymer core with a lipid shell or a lipid core with a polymer shell. LPHNPs comprising a polymer core and a lipid shell are the simplest forms of LPHNPs. These carriers consist of three different ingredients, as shown in Fig. 3: (i) the inner polymer core that encapsulates the active therapeutic compounds; (ii) the lipid monolayer that encapsulates the polymer core; and (iii) the external lipid–PEG layer, whose role is to stabilize and prolong the systemic circulation (i.e., to ensure that nanoparticles stay in the body for a long time).154 The natural or synthetic biomimetic lipids and the biodegradable polymer core together constitute an advanced and beneficial delivery system that can be used to treat a variety of diseases through systemic or local drug delivery.155
Typically, lipid-core/polymer-shell systems include a lipid core and one or more polymer surface layers. The core encapsulates the active ingredient, while the surface coating improves the NP stability and interaction with biological barriers. Several studies have functionalized lipid nanocarriers (SLNs, NLCs, or LP) with polymer shells (chitosan, PEG) to fabricate chitosan-coated lipid NPs, PEGylated lipid NPs, or multilamellar objects.156 Chitosan-coated LP were used as carriers of MT, with an encapsulation efficiency between 34.4% and 60.8%. The presence of chitosan on the LP surface led to a decrease in the thickness of the lipid bilayer, suggesting that the biopolymer modifies the lipid nanostructure or its hydration.157
Table 4 summarizes the hybrid types of LPHNP used as MT carriers with controlled and sustained release characteristics.158,159 Previous research with chosen experimental models has focused on evaluating the antioxidant effects160 and the anti-apoptotic properties161 of MT-encapsulated NPs.162 As an example, the conjugated yne-ene chain of the polymeric backbone of polydiacetylene has enabled the formation of stable nanoaggregates from which only 50% of the encapsulated MT has been released after 72 h under physiological conditions.159 A 2-hydroxypropyl-β-cyclodextrin (HPβCD) grafted solid lipid nanoparticle (SLN)-based bioconjugate has been synthesized and used for administering a combination of MT and amphotericin B (AmB) orally for effective visceral Leishmaniasis treatment. The formulations (HPCDMT-AmB SLN) showed a high loading capacity and a high entrapment efficiency of AmB (%DL = 9.0 ± 0.6 and %EE = 87.9 ± 0.6) and MT (%DL = 7.5 ± 0.5 and % EE = 63 ± 6.2). The cumulative percent release of AmB and MT was 66.1% and 73.1%, respectively, up to 72 h.158
| Nanocarriers | Formulation | Studied model | ROA | EE% | DE% | Release profile | Outcome |
|---|---|---|---|---|---|---|---|
| Not available (N.A.). | |||||||
| HPCD-MT-AmB SLN | 2-Hydroxy 3-propyl beta-cyclodextrin, glyceryl monostearate, PVA, AmB | Visceral Leishmaniasis cells: L. donovani-infected J774A.1 macrophages, in vivo: BALB/c mice and Swiss albino mice | Oral 10–20 mg kg−1 | AmB ∼87.9; MT ∼63 | AmB ∼9.0; MT ∼7.5 | Burst release and then slow sustained diffusion until 72 h, AmB 66.1%; MT 73.1% | Efficiency of MT in combination with amphotericin B, delivered by HPβCD-modified SLN, to inhibit the infection.158 |
| Polymer-coated lipid-core nanocapsules (LNCs) | CCT, sorbitan monostearate, PCL, Tween 80 | In vivo: paraquat (PQ) intoxicated Caenorhabditis elegans | Oral 0.96 mg mL−1 | 39 | N.A. | N.A. | Pretreatment with MT-LNC increased the survival rate, reduced the ROS, and maintained the development in C. elegans exposed to PQ; demonstrated uptake and distribution of MT-LNC in a nematode; while LNC was not toxic, MT-LNC prevented the effects of PQ poisoning.169 |
| Ethylcellulose NPs (NCECMEs) | Ethylcellulose, Span 60, MCT | Retinal degeneration; in vivo: white female rabbits | Topical in eyes 50 μL per /2 h; 8 h per day for 9 days | 67–73 | N.A. | Drug release fitted by a Korsmeyer–Peppas model | Greater permeation capacity of MT observed in NCECMEs providing higher neuroprotective and antiapoptotic effects on RGCs.161 |
| Polymeric (NC) and lipid-core nanocapsules (LNC) | CCT, PCL, Tween 80, Span 60 | In vitro cultured bovine embryos | N.A. | N.A. | N.A. | N.A. | MT-LNC increased embryo cell number at the concentration (10−9 M), decreased cell apoptosis and ROS levels, down-regulated mRNA levels of BAX, CASP3, and SHC1 genes, and upregulated mRNA levels of CAT and SOD2 genes.160 |
| Polymer/lipid hybrid nanovesicles (Lip-MT) | PCDA; DMPC | Bone formation; in vivo: zebrafish; cells: C3H10T1/2, mouse mesenchymal stem cells | N.A. | 47.26%. | N.A. | A steady increase over time up to 108 h | The Lip-MT elevated the expression of key transcription factors (Runx2, type 1 col mRNAs) and the secretion of extracellular matrix proteins that are related to osteoblast differentiation. The bone formation in a zebrafish model was enhanced after exposure to Lip-MT compared with MT.159 |
A nanoscale system, consisting of ethylcellulose, medium chain triglyceride (MCT), and surfactants (Span60 and Tween80), has been developed for the controlled topical administration of MT in the retina. It enabled an enhanced neuroprotective effect of MT on the retinal ganglion.161 MT-loaded lipid-core nanocapsules (MT-LNCs) upregulated mRNA levels of catalase (CAT) and superoxide dismutase 2 (SOD2) genes (Fig. 5), while down-regulating the transcription levels of the pro-apoptotic BCL2-associated X protein (BAX), cysteine peptidase 3 (CASP3), and SHC-transforming protein 1 (SHC1) genes.160 Such effects have not been substantial enough for polymer carriers (MT-NC) alone.
 | ||
| Fig. 5 Effects of free and nanoencapsulated melatonin (10−9 M) on the relative mRNA abundance of oxidative stress-related genes.160 (A) catalase (CAT) gene; (B) glutathione peroxidase (GPX) gene; (C) peroxiredoxin (PRDX5) gene, and (D) superoxide dismutase 2 (SOD2) gene. The small symbols a and b above the histograms indicate significant differences between groups (P < 0.05). Mel = non-encapsulated MT, Mel-NC = melatonin-loaded polymeric nanocapsules, Mel-LNC = melatonin-loaded lipid-core nanocapsules (reproduced from ref. 160 UNDER open science license. Copyright © 2016, PLOS Open Access Publisher). | ||
3.4 Other types of nanocarrier for melatonin delivery
Other types of nanocarrier, including silica NPs,163 nanofibers,164 metallic or non-metallic NPs,165 protein nanodots,166 graphene-dendrimeric systems,167 and cell-derived exosomes,168 have been engineered to overcome MT's challenges such as poor solubility, stability, and bioavailability, and enabling sustained delivery in certain systems. Magnetite–silica–carbon quantum dot nanocomposites were synthesized for MT controlled-release as an anticancer drug.163 Yadav et al. evaluated the biological efficacy of MT-loaded protein nanodots for live-cell imaging and enhanced nano-drug delivery efficacy in a breast cancer cell line.166 MT-selenium NPs showed hepatic protective activity associated with antioxidant properties such as inhibiting lipid peroxidation and NO production as well as increasing antioxidant enzyme action.165 A functionalized graphene-dendrimeric system was designed by using Fe3O4 NP as a magnetic nanocarrier for co-delivery of doxorubicin and MT.167 Engineered M2 macrophage-derived exosomes (M2-exos) were used for MT loading and effective treatment of periodontitis thanks to the fact that M2-exos can target inflammatory sites and modulate immune microenvironments. MT-loaded M2-exos have positively affected inflammatory bone loss in periodontitis by mediating ER stress and immune reprogramming.168 Moreover, studies have shown increased cytotoxicity and apoptosis when MT was used in combination with some metal NPs such as ZnO170 and Pd NPs.171 This effect helped achieving anti-cancer cell proliferation outcome.4 Perspectives for development of melatonin-based nanomedicines for the nervous system
Despite the fact that MT is thought to cross the BBB, nanomedicines aiming at the improvement of MT therapeutic efficacy face big challenges. Nanomaterial-based BBB-crossing strategies have been broadly used for the brain delivery of theranostic agents, including intranasal delivery, temporary disruption of the BBB, local delivery, cell-penetrating peptide (CPP)-mediated BBB crossing, receptor-mediated BBB-crossing, shuttle peptide mediated BBB-crossing, and cell-mediated BBB crossing.1724.1 Nose-to-brain delivery of nanomedicines
Nose-to-brain delivery has been explored as a non-invasive method that bypasses the BBB, allowing direct access to the brain via the olfactory and trigeminal nerves (Fig. 6A).173,174 The five major pathways through which drugs are transported across the BBB are summarized in Fig. 6B. Particularly, lipid-based and polymer-based carriers are beneficial for CNS delivery due to their safety, high drug-loading capacity, and controlled-release features.174 The intranasal route of nanocarrier administration, e.g. NLCs, SLNs, or NE, appeared to be advantageous for targeted drug delivery to the brain. Such NPs were employed for MT loading and demonstrated the ability to cross the BBB.175 MT-loaded vehicles for intranasal delivery have been studied in vitro and in animal models representing conditions such as ischemic stroke,176 cerebral ischemia177 and glioblastoma178 (Table 5). The intranasal drug delivery systems have proved to be more effective for CNS delivery as compared with other routes or suspensions of non-encapsulated drugs.179 It has been reported that MT-loaded lipidic nanocapsules (MT-LNCs) have a 10.35 times higher permeation of MT than the drug solution across sheep nasal mucosa. This helped promoting neuronal survival by lowering oxidative stress and anti-inflammation. | ||
| Fig. 6 (A) (1) Direct transport of intranasal drugs from the nasal mucosa to the olfactory bulb may occur by axonal transport along olfactory neurons or para- or transcellular transport across the nasal epithelium. (2) In the respiratory region of the nasal cavity, drugs can be endocytosed by the trigeminal nerve and travel along the axon to reach the CNS. They may also cross the epithelial cell layer to reach the blood. (3) Once in the blood, drugs administered intranasally need to cross the BBB to reach the CNS; (B) transport across the BBB primarily occurs by paracellular transport (1), passive diffusion (2), receptor-mediated transcytosis (3) or carrier-mediated transport (4). Nanoparticles (NPs) can also cross the BBB for CNS drug delivery under certain conditions (5); biomimetic NPs synthesized using physiological proteins, cell membranes or viruses take advantage of the natural uptake of these biomaterials. Additionally, synthetic nanoparticles can be coated by targeting ligands such as transferrin, P-glycoprotein, and angiopep-2 that bind to receptors located on the BBB cells to facilitate permeation of the BBB and drug release within the brain parenchyma174 (reproduced under open access Creative Commons Attribution (CC BY) license. Copyright © 2023, MDPI). | ||
| Nanocarrier | Disease/state | Studied model | Administration route | Outcome | Year/ref. |
|---|---|---|---|---|---|
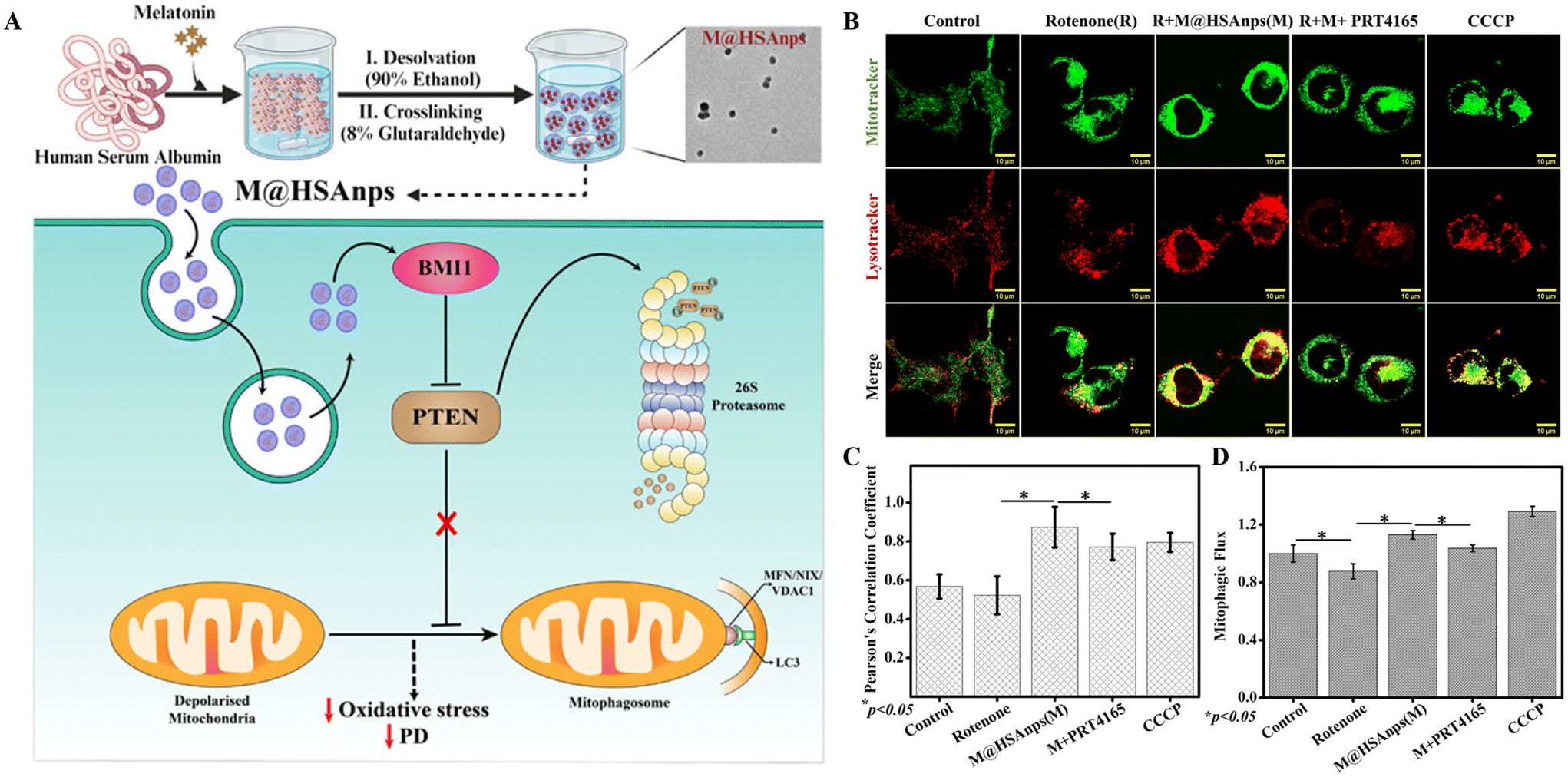
| MT-loaded human serum albumin nanoparticles (MT@HSAnps) | PD | In vitro release: dialysis bag (12 kDa); cell line: SH-SY5Y cells; in vivo: BalB/C mice | i.p. | MT@HSAnps showed a controlled drug release profile (initial burst release and sustained release later). The neurotherapeutic efficacy of MT-loaded HSAnps in preventing rotenone-induced PD was attributed to enhancing BMI1-mediated PTEN degradation and mitophagy induction. | 2024180 |
| Lipidic nanocapsules (LNCs) | Ischemic stroke | Ex vivo permeation: sheep nasal mucosa; in vivo (male rats): cerebral ischemia/reperfusion (I/R) | Intranasal | LNCs exhibited 10.35-fold higher permeation of MT than free drug solution; MT-LNCs favored reduced oxidative stress with lower MDA levels, higher GSH and SOD levels, decreased inflammatory markers (TNF-α, NO, MPO), and significant inhibition of caspase-3 activity. WB analysis showed recovery of Nrf-2 and HO-1 protein expression, downregulation of key inflammatory markers (NF-κB, p65, iNOS, Bax, Cyt C), and upregulation of Bcl-2, promoting neuronal survival. | 2022176 |
| Polymeric nanocapsules (PNCs) | Brain ischemia | Ex vivo permeation: sheep nasal mucosa; in vivo (male rats): global cerebral ischemia/reperfusion | Intranasal | MT-PNCs displayed 8-fold higher permeation than the free drug solution; MT revived the I/R-mediated disruption of Nrf-2/HO-1 as a protective pathway; the neuroprotective/anti-apoptotic effects of MT and MT-PNCs counteracted neuronal death, decreased apoptotic markers, and increased the pro-surviving protein Bcl-2; MT-PNCs displayed better therapeutic performance as compared with MT solution alone. | 2022177 |
| Poly-caprolactone nanoparticles (PCL) | Glioblastoma | In vitro drug release: dialysis tubes (12–14 kDa); cell lines: U87MG and MRC-5 cells; in vivo (male rats): brain fluorescence tomography, MT quantification in brain and plasma | Intranasal and oral | MT-NP increased the drug apparent water solubility ∼35 fold; MT-NPs demonstrated strong activity against U87MG cells, resulting in an IC50 ∼2500 fold lower than that of the free drug; FMT images revealed rapid translocation and accumulation of NPs from nasal cavity to the brain. Intranasal administration of MT-NPs resulted in higher AUC brain and drug targeting index compared with the free drug by either intranasal or oral routes. | 2019178 |
| Melatonin niosomes (MN) | Preclinical evaluation | Cell lines: IMR-32 (neuroblastoma) and RPMI 2650 (human nasal septum carcinoma) cells; in vivo (male rates): evaluated acute and subchronic toxicity | Intranasal and i.v. | Intranasal MN was bioequivalent to i.v. MT providing therapeutic level doses. Acute and subchronic toxicity screening showed no abnormal signs, symptoms or hematological effects in any animals. The intranasal MN could deliver MT to the brain to induce sleep and provide delayed systemic circulation (relative to i.v.) as well as distribution to peripheral tissues. | 201732 |
Fig. 7 shows the effects of free MT and MT-LNC treatments on hippocampal neurons in an ischemic stroke model.176 The sham-operated rat sections revealed intact neurons with a robust architecture in the hippocampal region (Fig. 7(A,a)). In contrast, following ischemia–reperfusion, there was evident selective and extensive damage in the hippocampal CA1 area 5 days post-ischemic insult (Fig. 7(B,b)), characterized by neurons showing shrunken cytoplasm with pyknotic nuclei (indicated by black arrows), accompanied by a decrease in the number of viable neurons. Post-ischemia administration of MT-LNCs (Fig. 7(D,d)) significantly restored the pathological changes in the hippocampal neurons. The treatment with MT-LNC was more effective as compared with MT solution (free drug) (Fig. 7(C,c)).176
 | ||
| Fig. 7 Photomicrographs (×400) of hematoxylin and eosin (H&E) staining revealing the neuroprotective effect of melatonin (MT) on the hippocampal CA1 region. (A,a) Sham, (B,b) ischemia/reperfusion (I/R), (C,c) MT solution-treated group, and (D,d) MT-lipid nanocapsule (LNC)-treated group176 (reproduced with permission from ref. 176 under PMC Open Access License. Copyright © 2022, Taylor & Francis Publisher). | ||
4.2 Innovative strategies with melatonin-based nanomedicines for neurological diseases
Table 5 summarizes recent advances in the development of MT nanomedicines for treating neurological diseases. Targeted delivery, multi-drug loading, and multifunctionality are several potential and attractive strategies, which enable MT-based nanomedicines to better exert their neuroprotective and neuroregenerative effects, promoting their role in the prevention and treatment of neurological disorders.Targeted formulations play a crucial role in enhancing efficacy by precisely directing the delivered drug to specific tissues. Many approaches can create targeted nanoformulations, including custom biomolecules such as antibodies or aptamers. Recently, much attention has been paid to biomimetic nanoparticle platforms that exploit targeting specificities, like small molecules (folate and riboflavin), carbohydrates, cell-penetrating peptides, proteins (transferrin or lipoproteins) and even mammalian cell membranes naturally found in living systems. These strategies have evolved over time to maximize functionality.181 The biomimetic targeting nanosystems have received widespread interest in the therapeutic field of neurological diseases. The use of cell membrane-coated NPs for targeted drug delivery to the brain and treatment of neurological disorders has been recently summarized.182
Intracellular targeting is an effective method to improve drugs’ therapeutic index by delivering the active therapeutic substance directly to its intracellular site of action. This involves targeting not just tissues or cells, but specifically the subcellular organelles such as lysosomes, mitochondria, nuclei, and Golgi/ER. It is considered as the third level of drug targeting.183 Mitochondria-targeted biomimetic NPs have been reported to control mitochondrial dysfunction in neurons and improve PD treatment. These Cu2-xSe-based NPs, functionalized with curcumin and enveloped in a DSPE-PEG2000-TPP-modified macrophage membrane, were able to efficiently target the mitochondria of damaged neurons in an inflammatory environment. It has been concluded that targeting mitochondrial biogenesis to alleviate mitochondrial dysfunction holds significant potential for treating PD and other mitochondria-related diseases.184
Melatonin-loaded human serum albumin nanoparticles (MT@HSAnps) have been reported to enhance both mitophagy (removing depolarized mitochondria) and mitochondrial biogenesis. Hence, their potential as neurotherapeutic agents has been investigated.180 The data in Fig. 8A indicate that MT@HSAnps prevent rotenone-induced PD by enhancing BMI1-mediated PTEN degradation and inducing mitochondrial autophagy. Mitophagy was assessed through colocalization analysis of mitochondria and lysosomes using confocal scanning microscopy (Fig. 8B). The rotenone-treated group exhibited reduced mitophagy, which was reversed in MT@HSAnp-cotreated cells. However, additional PRT4165 (a chemical inhibitor of BMI1) cotreatment with MT@HSAnps hindered the effect of nanoformulation and inhibited mitophagy (Fig. 8B and C). Furthermore, the PRT4165 treatment group showed relatively lower mitophagy compared with the untreated control group. Considering that the downregulation of BMI1 inhibited mitophagy, it has been suggested as a novel crucial regulator of mitophagy (Fig. 8A). Mitophagic flux, an important parameter for assessing both the formation of mitophagosomes and their degradation in the lysosomes, was quantified by flow cytometry using MitoTracker Green FM and hydroxychloroquine (HCQ). It was established that MT@HSAnp cotreatment enhanced the flux, which was diminished by rotenone treatment (Fig. 8D).
 | ||
| Fig. 8 (A) Schematic diagram of the preparation method of melatonin-loaded human serum albumin nanoparticles (M@HSAnps) and their mechanism of action exploiting enhanced mitophagy to alleviate PD (B–D). Evaluation of mitophagy: (B) confocal microscopy to analyze mitophagy, where MitoTracker and LysoTracker were used to stain the mitochondria and lysosomes, respectively. (C) Analysis of colocalization of mitochondria and lysosomes represented through the Pearson's correlation coefficients. (D) Estimation of the fold change in the mitophagic flux using a flow cytometer180 (reproduced with permission from ref. 180. Copyright © 2024, American Chemical Society). | ||
The development of MT-based nanomedicines accounts also for the fact that the neurological disorders are multifactorial. Nanomedicines that can act on multiple molecular targets comprise a promising strategy to halt the progression of these diseases. Combination therapies can be envisioned by synthesizing new compounds that exert diverse activities. For example, a series of MT–alkylbenzylamine hybrids have been designed and synthesized as multitarget agents for the treatment of AD.185 However, the development of novel drugs is a long and costly path. Hence, straightforward combining of existing drugs, by encapsulating them in the same nanocontainer, presents an intriguing alternatove.186
Recently, smart nanocomplexes have been fabricated by combining targeted and multi-drug properties. Fig. 9 presents nanospheres for the targeted delivery of acetylcholine and MT via a C5a-targeted aptamer which effectively reduces the reperfusion injury in ischemic stroke. The SiO2@PAA-MT/ACh-aC5a nanospheres, guided by anti-C5a aptamers, were administered via intravenous injection and were found to cross the damaged BBB to target ischemic regions. Additionally, the specific binding of anti-C5a aptamers to C5a selectively reduced C5a-mediated inflammatory cytokines from microglia. Importantly, the low pH in ischemic regions triggered the release of acetylcholine (ACh) and MT molecules, which polarize microglia from the M1 to M2 phenotype, thereby suppressing the inflammatory response, and eliminating reactive oxygen species (ROS) to alleviate oxidative stress. This approach may enable the SiO2@PAA-MT/ACh-aC5a nanospheres to ultimately protect neurons from inflammatory damage and oxidative stress in cerebral ischemia–reperfusion injury.187
 | ||
| Fig. 9 Schematic illustration of cerebral ischemia/reperfusion injury treatment with SiO2@PAA-MT/ACh-aC5a biocompatible nanospheres to attenuate the inflammatory response and oxidative stress on neurons in vivo187 (reproduced with permission from ref. 187. Copyright © 2023, Wiley). | ||
4.3 Lipid liquid crystalline nanoparticles as promising CNS drug delivery vehicles
Lyotropic lipid liquid crystalline nanoparticles (LCNPs) comprise an advanced class of nanomaterials for drug delivery. They are the next generation of bilayer membrane structures forming more complex 2D- and 3-dimensional nonlamelllar architectures that may involve inverted cubic and hexagonal mesophase inner organizations.188 Compared with liposomal drug delivery nanocarriers, lipid-based cubosomes, hexosomes and spongosomes involve multiple internal compartments, which represent a structural advantage enabling an enhanced encapsulation efficacy. The entrapment of biomolecules of various sizes and hydrophilicities can be achieved as well as sustained drug release.189 Selachyl alcohol-based cubosome and hexosome LCNPs have demonstrated high encapsulation efficiencies for the model anti-seizure drug phenytoin, exceeding 97% in both types of particle. For this type of drug, cubosomes proved to be more effective than hexosomes for delivering the therapeutic molecules to the brain, highlighting the significant potential of cubosomes as LCNPs for drug delivery.190Nonlamellar liquid crystalline organizations and topologies have been proved as favorable for the encapsulation of various types of therapeutic compound (anti-viral, anti-oxidant, antibiotic, anti-cancer, etc.) of interest for nanomedicine development.191,192 Lipid nanocarriers have attracted considerable attention in the field of neuroprotection and neuro-regeneration as delivery vehicles through the BBB.193,194 LCNPs enhanced the nose-to-brain delivery of lipophilic drugs such as tranilast,195 plasmalogens,196 duloxetine hydrochloride,197 and hydrophilic drugs such as almotriptan malate.198 Curcumin, fish oil, and BDNF have been successfully co-encapsulated in monoolein-based cubosome and spongosome LCNPs and have shown in vitro neuroprotective potential to alleviate endoplasmic reticulum stress.199 Vesicular and nonlamellar-type LCNPs, and lipid–peptide nanocarriers encapsulating a PUFA-plasmalogen and a neurotrophic peptide, have been shown to promote neuronal cell regeneration after oxidative stress through a survival mechanism involving CREB phosphorylation.200
5 Conclusions
MT exerts neuroprotective, neuromodulatory, antioxidant, anti-inflammatory, and anti-aging properties, but has poor pharmacological characteristics. MT-loaded nanocarriers, compared with a free MT drug, have demonstrated superior antioxidant, anti-inflammatory, and neuroprotective activities across various cell types and biological tissues. Among the reported nanocarrier systems, lipid nanocarriers have received significant interest due to their solubilizing properties, drug protective potential, biocompatibility, tolerability, and ability to bypass physiological barriers. Strategies such as multi-drug loading, targeted delivery, and multifunctionality can enhance the potential of MT-based nanomedicines to exert neuroprotective and neuro-regenerative effects. While various nanosystems have been utilized for MT encapsulation and delivery, LCNPs stand out as promising CNS drug delivery vehicles, offering advantages like enhanced stability, controlled release, improved bioavailability, and reduced undesirable side effects. These characteristics make LCNPs particularly beneficial for brain-targeted MT delivery in view of the treatment of neurological diseases. Therefore, MT-loaded LCNPs may provide a novel avenue in preventing and treating neurological disorders, thus necessitating further research to substantiate this potential.Abbreviations
| AC | Adenylyl cyclase |
| ACE2 | Angiotensin-converting enzyme 2 |
| ACh | Acetylcholine |
| AD | Alzheimer's disease |
| Akt/PKB | Protein kinase B |
| AmB | Amphotericin B |
| ANA12 | An antagonist of the TrkB receptor |
| ATF6 | Transcription factor 6 |
| ATP | Adenosine triphosphate |
| Aβ | Amyloid-β |
| Bax | Bcl2-associated X protein |
| BBB | Blood–brain barrier |
| Bcl2 | B-cell lymphoma 2 |
| BDNF | Brain-derived neurotrophic factor |
| BH3 | Bcl-2 homology 3 domain |
| Bmal1 | Brain and muscle aryl hydrocarbon receptor nuclear translocator-like 1 |
| C/EBP | CCAAT/enhancer binding protein |
| Ca2+ | Calcium ions |
| CaMKII | Calcium/calmodulin-dependent protein kinase II |
| cAMP | Cyclic adenosine monophosphate |
| CAT | Catalase |
| CC | Cerebral cortex |
| CCT | Capric/caprylic triglyceride |
| CD206 | Cluster of differentiation 206 |
| CDK5 | Cyclin-dependent kinase 5 |
| cGAS | Cyclic GMP-AMP synthase |
| cGMP | Cyclic guanosine monophosphate |
| CHOP | C/EBP homologous protein |
| CNS | Central nervous system |
| COX-2 | Cyclooxygenase-2 |
| CPK | Creatine phosphokinase 4 |
| CREB | cAMP response element-binding protein |
| CRP | C-reactive protein 1 |
| CsA | Cyclosporine A |
| DMPC | Dimyristoyl phosphatidyl choline |
| Drp1 | Dynamin-related protein 1 |
| ER | Endoplasmic reticulum |
| ERK1/2 | Extracellular signal-regulated kinases 1 and 2 |
| ESR | Erythrocyte sedimentation rate |
| ET | Ethosome |
| ET-LP | Ethanolic liposomes |
| Fe3+ | Ferric iron |
| FOXO3a | Forkhead box protein O3 |
| Fth1 | Ferritin heavy chain 1 |
| GADD153 | Growth arrest- and DNA damage-inducible gene 153 |
| Gi/o proteins | G-protein alpha inhibitory/other subunits |
| Gpx4 | Glutathione peroxidase 4 |
| GRP78/BiP | Glucose-regulated protein 78/binding immunoglobulin protein |
| GSH-Px | Glutathione peroxidase |
| GSK3 | Glycogen synthase kinase 3 |
| H2O2 | Hydrogen peroxide |
| HD | Huntington's disease |
| HDAC3 | Histone deacetylase 3 |
| Ho1 | Heme oxygenase 1 |
| HPMC | Hydroxypropyl methylcellulose |
| HSF1 | Heat shock factor-1 |
| HSP70 | Heat shock protein 70 |
| i.p. | Intraperitoneal injection |
| i.v. | Intravenous injection |
| IL-1β | Intereukin-1β |
| IL-6 | Interleukin-6 |
| iNOS | Inducible nitric oxide synthase |
| IP3 | Inositol trisphosphate |
| IRE1 | Inositol-requiring enzyme 1 |
| JAK2 | Janus kinase 2 |
| JEV | Japanese encephalitis virus |
| KIF5A | Kinesin family member 5A |
| LC3-II | Microtubule-associated protein 1 light chain 3-II |
| LCNPs | Lipid lyotropic liquid crystalline nanoparticles |
| LDH | Lactate dehydrogenase3 |
| LNC | Lipid nanocasules |
| LP | Liposomes |
| LPHNPs | Lipid–polymer hybrid nanoparticles |
| MAPK | Mitogen-activated protein kinase |
| MCAO | Middle cerebral artery occlusion |
| MCT | Medium chain triglycerides |
| MDA | Malondialdehyde |
| Mfn2 | Mitofusin 2 |
| MPP+ | 1-Methyl-4-phenylpyridinium |
| MS | Multiple sclerosis |
| MT | Melatonin |
| MT1 | Melatonin receptor 1 |
| MT2 | Melatonin receptor 2 |
| mtDNA | Mitochondrial DNA |
| mTOR | Mammalian target of rapamycin |
| N.A. | Not applicable |
| N1-AFMK | N1-acetyl-N2-formyl-5-methoxykynuramine |
| NAD+ | Nicotinamide adenine dinucleotide |
| NAS | N-Acetylserotonin |
| NC | Nanocapsules |
| NE | Nanoemulsions |
| NF-κB | Nuclear factor kappa B |
| NLCs | Nanostructured lipid carriers |
| NLRP3 | NOD-like receptor family pyrin domain containing 3 |
| NMDAR | N-Methyl-D-aspartate receptor |
| NOD | Domain nucleotide-binding oligomerization |
| NOS-2 | Nitric oxide synthase 2 |
| NPs | Nanoparticles |
| Nrf2 | Nuclear factor erythroid 2-related factor 2 |
| NS | Niosomes |
| NS3 | Nonstructural protein 3 |
| NS5 | Nonstructural protein 5 |
| p53 | Tumor protein p53 |
| p62 | Sequestosome 1 |
| p-AMPK | Phospho-5′AMP-activated protein kinase |
| PC | Phosphatidyl choline |
| PCBs | Polychlorinated biphenyls |
| PCDA | 10,12-Pentacosadiynoic acid |
| PCL | Polycaprolactone |
| p-CREB | Phospho-CAMP-response element-binding |
| PD | Parkinson's disease |
| PERK | Protein kinase RNA-like endoplasmic reticulum kinase |
| p-JNK | Phosphorylated c-Jun N-terminal kinase |
| PKA | cAMP-dependent kinase A/protein kinase C |
| PKG | cGMP-dependent kinases/protein kinase G |
| PLA | Polylactic acid |
| PLCβ | Phospholipase C beta |
| PLGA | Poly(lactic-co-glycolic acid) |
| p-P38 | Phosphorylated P38 MAPK |
| PrPC | Cellular prion protein |
| PTEN | Phosphatase and tensin homolog |
| PVA | Polyvinyl alcohol |
| REM | Rapid eye movement (sleep disorder) |
| RNS | Reactive nitrogen species |
| ROA | Route of administration |
| ROR | Retinoic acid-related orphan receptor |
| ROS | Reactive oxygen species |
| RZR | Retinoid Z receptor |
| s.c. | Subcutaneously |
| SCN | Suprachiasmatic nucleus |
| Sec. 62 | Sec. 62 receptor (related to ER-phagy) |
| Sirt1 | Sirtuin 1 |
| SLNs | Solid lipid nanoparticles |
| SOD | Superoxide dismutase |
| STAT3 | Signal transducer and activator of transcription 3 |
| TD | Transdermal administration |
| TF | Transfersomes |
| TGFβ | Transforming growth factor-beta |
| THC | Δ9-Tetrahydrocannabinol |
| TLRs | Toll-like receptors |
| TMPRSS2 | Transmembrane protease, serine 2 |
| TNF-α | Tumor necrosis factor-α |
| TPP | Tripolyphosphate |
| VEGF | Vascular endothelial growth factor |
| ˙OH | Hydroxyl radicals |
| 4P-PDOT | 4-Phenyl-2-propionamidotetralin |
Author contributions
Fucen Luo: investigation, visualization, writing – original draft, writing – review & editing; Yuru Deng: writing – review & editing; Borislav Angelov: funding acquisition, writing – review & editing; Angelina Angelova: conceptualization, supervision, project administration, writing – review & editing.Data availability
No primary research results, software, or code have been included and no new data were generated or analyzed as part of this review.All cited data were included in the article.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
F. L. acknowledges a PhD fellowship from the China Scholarship Council (CSC). A. A. acknowledges membership of the CNRS GDR2088 BIOMIM research network. B. A. was funded by the project “Structural dynamics of biomolecular systems (ELIBIO)” (No. CZ.02.1.01/0.0/0.0/15_003/0000447) from the European Regional Development Fund, by the Czech Science Foundation (GACR project no. 24-10671S), and the Johannes Amos Comenius Operational Program OPJAK (project no. SENDISO CZ.02.01.01/00/22_008/0004596). Fig. 1 and 3 were generated with BioRender.com.References
- J. D. Steinmetz, K. M. Seeher, N. Schiess, E. Nichols, B. Cao, C. Servili, V. Cavallera, E. Cousin, H. Hagins and M. E. Moberg, Lancet Neurol., 2024, 23, 344–381 CrossRef PubMed
.
- A. K. Verma, S. Singh and S. I. Rizvi, Biogerontology, 2023, 24, 183–206 CrossRef CAS PubMed
- R. Van Drunen and K. Eckel-Mahan, Front. Neural Circuits, 2022, 16, 1059229 CrossRef CAS PubMed
- M. H. Hastings, E. S. Maywood and M. Brancaccio, Nat. Rev. Neurosci., 2018, 19, 453–469 CrossRef CAS PubMed
- G. D. Potter, D. J. Skene, J. Arendt, J. E. Cade, P. J. Grant and L. J. Hardie, Endocr. Rev., 2016, 37, 584–608 CrossRef CAS PubMed
- Y. Shen, Q. K. Lv, W. Y. Xie, S. Y. Gong, S. Zhuang, J. Y. Liu, C. J. Mao and C. F. Liu, Transl. Neurodegener., 2023, 12, 8 CrossRef PubMed
- B. A. Mander, J. R. Winer and M. P. Walker, Neuron, 2017, 94, 19–36 CrossRef CAS PubMed
- M. Karasek and K. Winczyk, J. Physiol. Pharmacol., 2006, 57, 19 Search PubMed
- J. Tchekalarova and R. Tzoneva, Int. J. Mol. Sci., 2023, 24, 3022 CrossRef CAS PubMed
- M. Gunata, H. Parlakpinar and H. A. Acet, Rev. Neurol., 2020, 176, 148–165 CrossRef CAS PubMed
- M. Shukla, P. Govitrapong, P. Boontem, R. J. Reiter and J. Satayavivad, Curr. Neuropharmacol., 2017, 15, 1010–1031 CAS
- S. B. Ahmad, A. Ali, M. Bilal, S. M. Rashid, A. B. Wani, R. R. Bhat and M. U. Rehman, Cell. Mol. Neurobiol., 2023, 43, 2437–2458 CrossRef CAS PubMed
- M. Hou, Y. Zhang, Y. Liu, X. Ge, X. Hu, Z. Zhao, X. Tian, T. Liu, H. Yang, X. Chen, F. He and X. Zhu, J. Mater. Sci. Technol., 2023, 146, 102–112 CrossRef CAS
- J. K. Andersen, Nat. Med., 2004, 10(Suppl), S18–S25 CrossRef PubMed
- R. Dhapola, S. K. Beura, P. Sharma, S. K. Singh and D. HariKrishnaReddy, Mol. Biol. Rep., 2024, 51, 48 CrossRef CAS PubMed
- L. Ling, A. Alattar, Z. Tan, F. A. Shah, T. Ali, R. Alshaman, P. O. Koh and S. Li, Front. Pharmacol., 2020, 11, 1220 CrossRef CAS PubMed
- L. Zhang, H. Q. Zhang, X. Y. Liang, H. F. Zhang, T. Zhang and F. E. Liu, Behav. Brain Res., 2013, 256, 72–81 CrossRef CAS PubMed
- C. Cachan-Vega, I. Vega-Naredo, Y. Potes, J. C. Bermejo-Millo, A. Rubio-Gonzalez, C. Garcia-Gonzalez, E. Antuna, M. Bermudez, J. Gutierrez-Rodriguez, J. A. Boga, A. Coto-Montes and B. Caballero, Molecules, 2022, 27, 5543 CrossRef CAS PubMed
- H. A. Abo Taleb and B. S. Alghamdi, J. Mol. Neurosci., 2020, 70, 386–402 CrossRef CAS PubMed
- L. Kikwai, N. Kanikkannan, R. J. Babu and M. Singh, J. Controlled Release, 2002, 83, 307–311 CrossRef CAS PubMed
- D. Zetner, L. Andersen and J. Rosenberg, Drug Res., 2015, 66, 169–173 CrossRef PubMed
- W. M. Pardridge, NeuroRx, 2005, 2, 3–14 CrossRef PubMed
- S. Fakhri, S. Abdian, S. N. Zarneshan, S. Z. Moradi, M. H. Farzaei and M. Abdollahi, Int. J. Nanomed., 2022, 17, 299–331 CrossRef CAS PubMed
- D. S. Hersh, A. S. Wadajkar, N. B. Roberts, J. G. Perez, N. P. Connolly, V. Frenkel, J. A. Winkles, G. F. Woodworth and A. J. Kim, Curr. Pharm. Des., 2016, 22, 1177–1193 CrossRef CAS PubMed
- C. Nopparat, A. Boontor, S. Kutpruek and P. Govitrapong, Sci. Rep., 2023, 13, 17841 CrossRef CAS PubMed
- Y. J. Jung, H. Choi and E. Oh, Biochem. Biophys. Res. Commun., 2022, 621, 59–66 CrossRef CAS PubMed
- K. Kitidee, A. Samutpong, N. Pakpian, T. Wisitponchai, P. Govitrapong, R. J. Reiter and P. Wongchitrat, Sci. Rep., 2023, 13, 6063 CrossRef CAS PubMed
- M. Yao, P. M. Pu, Z. Y. Li, K. Zhu, L. Y. Zhou, Y. L. Sun, Y. X. Dai, X. J. Cui and Y. J. Wang, J. Pineal Res., 2023, 74, e12859 CrossRef CAS PubMed
- A. Jauhari, S. V. Baranov, Y. Suofu, J. Kim, T. Singh, S. Yablonska, F. Li, X. Wang, P. Oberly, M. B. Minnigh, S. M. Poloyac, D. L. Carlisle and R. M. Friedlander, J. Clin. Invest., 2020, 130, 3124–3136 CrossRef CAS PubMed
- S. Bavithra, E. Sugantha Priya, K. Selvakumar, G. Krishnamoorthy and J. Arunakaran, Neurochem. Res., 2015, 40, 1858–1869 CrossRef CAS PubMed
- Y. Hu, Y. Lv, X. Long, G. Yang and J. Zhou, CNS Neurosci. Ther., 2023, 30, e14474 CrossRef PubMed
- A. Priprem, J. R. Johns, S. Limsitthichaikoon, W. Limphirat, P. Mahakunakorn and N. P. Johns, Ther. Delivery, 2017, 8, 373–390 CrossRef CAS PubMed
- R. P. Patil, D. D. Pawara, C. S. Gudewar and A. R. Tekade, J. Liposome Res., 2019, 29, 264–273 CrossRef CAS PubMed
- Z. Edis, J. Wang, M. K. Waqas, M. Ijaz and M. Ijaz, Int. J. Nanomed., 2021, 16, 1313–1330 CrossRef PubMed
- Q. Zhang, C. Ou, S. Ye, X. Song and S. Luo, J. Microencapsulation, 2017, 34, 687–698 CrossRef CAS PubMed
- D. C. Altindal and M. Gumusderelioglu, J. Microencapsulation, 2016, 33, 53–63 CrossRef CAS PubMed
- S. K. Pramanik, U. Pal, P. Choudhary, H. Singh, R. J. Reiter, A. Ethirajan, S. Swarnakar and A. Das, ACS Appl. Bio Mater., 2019, 2, 5218–5226 CrossRef CAS PubMed
- L. G. D. A. Chuffa, F. R. F. Seiva, A. A. Novais, V. A. Simão, V. M. Martín Giménez, W. Manucha, D. A. P. D. C. Zuccari and R. J. Reiter, Molecules, 2021, 26, 3562 CrossRef PubMed
- A. Cavalli, M. L. Bolognesi, A. Minarini, M. Rosini, V. Tumiatti, M. Recanatini and C. Melchiorre, J. Med. Chem., 2008, 51, 347–372 CrossRef CAS PubMed
- E. Esposito and S. Cuzzocrea, Curr. Neuropharmacol., 2010, 8, 228–242 CrossRef CAS PubMed
- M. L. Dubocovich and M. Markowska, Endocrine, 2005, 27, 101–110 CrossRef CAS PubMed
- J. Liu, S. J. Clough, A. J. Hutchinson, E. B. Adamah-Biassi, M. Popovska-Gorevski and M. L. Dubocovich, Annu. Rev. Pharmacol. Toxicol., 2016, 56, 361–383 CrossRef CAS PubMed
- J. Vanecek, Physiol. Rev., 1998, 78, 687–721 CrossRef CAS PubMed
- H. H. Okamoto, E. Cecon, O. Nureki, S. Rivara and R. Jockers, J. Pineal Res., 2024, 76, e12952 CrossRef CAS PubMed
- X. Onphachanh, H. J. Lee, J. R. Lim, Y. H. Jung, J. S. Kim, C. W. Chae, S. J. Lee, A. A. Gabr and H. J. Han, J. Pineal Res., 2017, 63, e12427 CrossRef PubMed
- Q. K. Lv, K. X. Tao, X. Y. Yao, M. Z. Pang, B. E. Cao, C. F. Liu and F. Wang, J. Pineal Res., 2024, 76, e12948 CrossRef CAS PubMed
- Z. Wang, Y. H. Zhang, W. Zhang, H. L. Gao, M. L. Zhong, T. T. Huang, R. F. Guo, N. N. Liu, D. D. Li, Y. Li, Z. Y. Wang and P. Zhao, J. Pineal Res., 2018, 65, e12502 CrossRef PubMed
- G. S. Kinker, L. H. Ostrowski, P. A. C. Ribeiro, R. Chanoch, S. M. Muxel, I. Tirosh, G. Spadoni, S. Rivara, V. R. Martins, T. G. Santos, R. P. Markus and P. Fernandes, J. Mol. Med., 2021, 99, 289–301 CrossRef CAS PubMed
- A. Galano, E. G. Guzman-Lopez and R. J. Reiter, Int. J. Mol. Sci., 2021, 22, 11584 CrossRef CAS PubMed
- D. Bebbington, N. J. T. Monck, S. Gaur, A. M. Palmer, K. Benwell, V. Harvey, C. S. Malcolm and R. H. P. Porter, J. Med. Chem., 2000, 43, 2779–2782 CrossRef CAS PubMed
- D. X. Tan, R. J. Reiter, L. C. Manchester, M. T. Yan, M. El-Sawi, R. M. Sainz, J. C. Mayo, R. Kohen, M. Allegra and R. Hardeland, Curr. Top. Med. Chem., 2002, 2, 181–197 CrossRef CAS PubMed
- L. C. Manchester, A. Coto-Montes, J. A. Boga, L. P. Andersen, Z. Zhou, A. Galano, J. Vriend, D. X. Tan and R. J. Reiter, J. Pineal Res., 2015, 59, 403–419 CrossRef CAS PubMed
- D. X. Tan, R. Hardeland, L. C. Manchester, B. Poeggeler, S. Lopez-Burillo, J. C. Mayo, R. M. Sainz and R. J. Reiter, J. Pineal Res., 2003, 34, 249–259 CrossRef CAS PubMed
- E. Motallebzadeh, A. A. Tameh, S. A. T. Zavareh, B. Farhood, A. Aliasgharzedeh and M. Mohseni, J. Cell Physiol., 2020, 235, 8791–8798 CrossRef CAS PubMed
- Y. Liu, L. Zhang, H. Zhang, B. Liu, Z. Wu, W. Zhao and Z. Wang, J. Pineal Res., 2012, 52, 47–56 CrossRef PubMed
- J. M. Olcese, C. Cao, T. Mori, M. B. Mamcarz, A. Maxwell, M. J. Runfeldt, L. Wang, C. Zhang, X. Lin, G. Zhang and G. W. Arendash, J. Pineal Res., 2009, 47, 82–96 CrossRef CAS PubMed
- J. Limson, T. Nyokong and S. Daya, J. Pineal Res., 1998, 24, 15–21 CrossRef CAS PubMed
- T. Kanti Das, M. R. Wati and K. Fatima-Shad, Arch. Neurosci., 2014, 2, e60038 Search PubMed
- A. Galano, M. E. Medina, D. X. Tan and R. J. Reiter, J. Pineal Res., 2015, 58, 107–116 CrossRef CAS PubMed
- R. J. Reiter, R. N. Sharma, L. G. A. Chuffa, D. G. H. da Silva and S. Rosales-Corral, Histol. Histopathol., 2024, 40, 271–282 Search PubMed
- R. J. Reiter, J. C. Mayo, D. X. Tan, R. M. Sainz, M. Alatorre-Jimenez and L. Qin, J. Pineal Res., 2016, 61, 253–278 CrossRef CAS PubMed
- R. J. Reiter, S. Rosales-Corral, D. X. Tan, M. J. Jou, A. Galano and B. Xu, Cell. Mol. Life Sci., 2017, 74, 3863–3881 CrossRef CAS PubMed
- D. X. Tan, L. C. Manchester, L. Qin and R. J. Reiter, Int. J. Mol. Sci., 2016, 17, 2124 CrossRef PubMed
- A. Lopez, J. A. Garcia, G. Escames, C. Venegas, F. Ortiz, L. C. Lopez and D. Acuna-Castroviejo, J. Pineal Res., 2009, 46, 188–198 CrossRef CAS PubMed
- Y. Suofu, W. Li, F. G. Jean-Alphonse, J. Jia, N. K. Khattar, J. Li, S. V. Baranov, D. Leronni, A. C. Mihalik, Y. He, E. Cecon, V. L. Wehbi, J. Kim, B. E. Heath, O. V. Baranova, X. Wang, M. J. Gable, E. S. Kretz, G. Di Benedetto, T. R. Lezon, L. M. Ferrando, T. M. Larkin, M. Sullivan, S. Yablonska, J. Wang, M. B. Minnigh, G. Guillaumet, F. Suzenet, R. M. Richardson, S. M. Poloyac, D. B. Stolz, R. Jockers, P. A. Witt-Enderby, D. L. Carlisle, J. P. Vilardaga and R. M. Friedlander, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E7997–E8006 CrossRef CAS PubMed
- D. P. Cardinali, E. S. Pagano, P. A. Scacchi Bernasconi, R. Reynoso and P. Scacchi, Horm. Behav., 2013, 63, 322–330 CrossRef CAS PubMed
- N. Dragicevic, N. Copes, G. O'Neal-Moffitt, J. Jin, R. Buzzeo, M. Mamcarz, J. Tan, C. Cao, J. M. Olcese, G. W. Arendash and P. C. Bradshaw, J. Pineal Res., 2011, 51, 75–86 CrossRef CAS PubMed
- G. Zhong, Y. Yang, D. Feng, K. Wei, J. Chen, J. Chen and C. Deng, Neuroscience, 2023, 534, 54–65 CrossRef CAS PubMed
- H. Hong, J. Li, T. Tong, T. Yang, H. Wang, Y. Xu, X. Lin, J. Lin, S. Liu, K. Luo, Z. Yu, W. Yuan, H. Pi and Z. Zhou, Sci. Total Environ., 2024, 934, 173119 CrossRef CAS PubMed
- D. Loh and R. J. Reiter, Commun., 2024, 2, 67–84 Search PubMed
- Y. Hu, X. Zhao, G. Jiang, M. Jin, W. Jiang and F. Han, Sci. Rep., 2023, 13, 20100 CrossRef CAS PubMed
- R. Singh, N. Kaur, V. Choubey, N. Dhingra and T. Kaur, Brain Res., 2024, 1826, 148742 CrossRef CAS PubMed
- N. Abolhasanpour, S. Alihosseini, S. Golipourkhalili, R. Badalzadeh, J. Mahmoudi and L. Hosseini, Arch. Med. Res., 2021, 52, 673–682 CrossRef CAS PubMed
- C. Wu, M. Du, R. Yu, Y. Cheng, B. Wu, J. Fu, W. Tan, Q. Zhou, E. Balawi and Z. B. Liao, Free Radicals Biol. Med., 2022, 178, 271–294 CrossRef CAS PubMed
- P. Thangwong, P. Jearjaroen, P. Govitrapong, C. Tocharus and J. Tocharus, Biochem. Pharmacol., 2022, 198, 114980 CrossRef CAS PubMed
- Y. M. Yoo and S. S. Joo, Int. J. Mol. Sci., 2023, 24, 2381 CrossRef CAS PubMed
- S. Oyadomari and M. Mori, Cell Death Differ., 2004, 11, 381–389 CrossRef CAS PubMed
- T. Koga, M. A. Suico, S. Shimasaki, E. Watanabe, Y. Kai, K. Koyama, K. Omachi, S. Morino-Koga, T. Sato, T. Shuto, K. Mori, S. Hino, M. Nakao and H. Kai, J. Biol. Chem., 2015, 290, 30366–30374 CrossRef CAS PubMed
- X. Fang, Q. Han, S. Li and A. Luo, Heliyon, 2022, 8, e10326 CrossRef CAS PubMed
- J. H. Lee, Y. M. Yoon, Y. S. Han, S. K. Jung and S. H. Lee, Cell Proliferation, 2019, 52, e12545 CrossRef PubMed
- D. J. DiSabato, N. Quan and J. P. Godbout, J. Neurochem., 2016, 139(Suppl 2), 136–153 CrossRef CAS PubMed
- S. M. Nabavi, S. F. Nabavi, A. Sureda, J. Xiao, A. R. Dehpour, S. Shirooie, A. S. Silva, A. Baldi, H. Khan and M. Daglia, Crit. Rev. Food Sci. Nutr., 2019, 59, S4–S16 CrossRef CAS PubMed
- C. Song, Z. Suo, Z. Wang, J. Cao, Y. Dong and Y. Chen, Front. Pharmacol., 2024, 15, 1416350 CrossRef CAS PubMed
- Q. Zhou, L. Lin, H. Li, H. Wang, S. Jiang, P. Huang, Q. Lin, X. Chen and Y. Deng, Mol. Neurobiol., 2021, 58, 6552–6576 CrossRef CAS PubMed
- Y. Huang, C. Jiang, X. Liu, W. Tang, H. Gui, T. Sun, D. Xu, M. He, M. Han, H. Qiu, M. Chen and S. Huang, J. Cell. Mol. Med., 2024, 28, e18338 CrossRef CAS PubMed
- M. Ashrafizadeh, M. Najafi, N. Kavyiani, R. Mohammadinejad, T. Farkhondeh and S. Samarghandian, Inflammation, 2021, 44, 1207–1222 CrossRef CAS PubMed
- L. N. Madhu, M. Kodali, S. Attaluri, B. Shuai, L. Melissari, X. Rao and A. K. Shetty, Redox Biol., 2021, 43, 101973 CrossRef CAS PubMed
- A. Romero, E. Ramos, F. Lopez-Munoz, E. Gil-Martin, G. Escames and R. J. Reiter, Cell. Mol. Neurobiol., 2022, 42, 489–500 CrossRef CAS PubMed
- D. D. Krahe, K. M. Woeppel, Q. Yang, N. Kushwah and X. T. Cui, Antioxidants, 2022, 11, 1628 CrossRef CAS PubMed
- R. Gupta, R. K. Ambasta and K. Pravir, Cell. Mol. Life Sci., 2021, 78, 8001–8047 CrossRef CAS PubMed
- F. Luo, A. F. Sandhu, W. Rungratanawanich, G. E. Williams, M. Akbar, S. Zhou, B. J. Song and X. Wang, Int. J. Mol. Sci., 2020, 21, 7174 CrossRef CAS PubMed
- A. Perez-Gonzalez, R. Castaneda-Arriaga, J. R. Alvarez-Idaboy, R. J. Reiter and A. Galano, J. Pineal Res., 2019, 66, e12539 CrossRef PubMed
- A. Angelova and B. Angelov, Neural Regener. Res., 2017, 12, 886–889 CrossRef CAS PubMed
- R. Huang, Y. Xu, X. Lu, X. Tang, J. Lin, K. Cui, S. Yu, Y. Shi, D. Ye, Y. Liu and X. Liang, J. Pineal Res., 2021, 71, e12716 CrossRef CAS PubMed
- W. Kim, K. R. Hahn, H. Y. Jung, H. J. Kwon, S. M. Nam, J. W. Kim, J. H. Park, D. Y. Yoo, D. W. Kim, M. H. Won, Y. S. Yoon and I. K. Hwang, Brain Behav., 2019, 9, e01388 CrossRef PubMed
- J. Li, G. Wu, W. Song, Y. Liu, Z. Han, Z. Shen and Y. Li, Neurotoxic. Res., 2021, 39, 227–239 CrossRef CAS PubMed
- M. Imbesi, T. Uz, S. Dzitoyeva and H. Manev, Neurosci. Lett., 2008, 439, 34–36 CrossRef CAS PubMed
- A. Choudhury, S. Singh, G. Palit, S. Shukla and S. Ganguly, Behav. Brain Res., 2016, 297, 204–212 CrossRef CAS PubMed
- U. Kilic, B. Elibol, A. B. Caglayan, M. C. Beker, M. Beker, B. Altug-Tasa, O. Uysal, B. Yilmaz and E. Kilic, J. Mol. Neurosci., 2022, 72, 994–1007 CrossRef CAS PubMed
- S. U. Rehman, M. Ikram, N. Ullah, S. I. Alam, H. Y. Park, H. Badshah, K. Choe and M. O. Kim, Cells, 2019, 8, 760 CrossRef PubMed
- Y. Zhao, R. Zhao, J. Wu, Q. Wang, K. Pang, Q. Shi, Q. Gao, Y. Hu, X. Dong, J. Zhang and J. Sun, BioFactors, 2018, 44, 609–618 CrossRef CAS PubMed
- M. F. Vecchierini, U. Kilic-Huck, M. A. Quera-Salva and M. E. L. C. G. O. T. S. Members of the, Rev. Neurol., 2021, 177, 245–259 CrossRef CAS PubMed
- N. A. Grima, S. M. W. Rajaratnam, D. Mansfield, T. L. Sletten, G. Spitz and J. L. Ponsford, BMC Med., 2018, 16, 8 CrossRef PubMed
- D. P. Cardinali, Front. Endocrinol., 2019, 10, 480 CrossRef PubMed
- S. Iftikhar, H. M. Sameer and Zainab, Front. Neurol., 2023, 14, 1265789 CrossRef PubMed
- D. M. Sumsuzzman, J. Choi, Y. Jin and Y. Hong, Neurosci.
Biobehav. Rev., 2021, 127, 459–473 CrossRef CAS PubMed
- F. Faraji, A. T. Zanjani, A. R. Ashtiani, G. Motamedi and S. Nourigheimasi, Iran Red Crescent Med. J., 2023, 25, e1471 Search PubMed
- N. Verma, R. Maiti, B. R. Mishra, M. Jha, M. Jena and A. Mishra, J. Neurosci. Res., 2021, 99, 1618–1631 CrossRef CAS PubMed
- D. Acuna-Castroviejo, G. Escames, J. C. Figueira, P. de la Oliva, A. M. Borobia and C. Acuna-Fernandez, J. Pineal Res., 2020, 69, e12683 CrossRef CAS PubMed
- G. Farnoosh, M. Akbariqomi, T. Badri, M. Bagheri, M. Izadi, A. Saeedi-Boroujeni, E. Rezaie, H. E. G. Ghaleh, H. Aghamollaei, M. Fasihi-Ramandi, K. Hassanpour and G. Alishiri, Arch. Med. Res., 2022, 53, 79–85 CrossRef CAS PubMed
- X. Hou, X. Qiu, Y. Wang, S. Song, Y. Cong and J. Hao, Oxid. Med. Cell. Longevity, 2022, 2022, 7135125 Search PubMed
- S. A. Mousavi, K. Heydari, H. Mehravaran, M. Saeedi, R. Alizadeh-Navaei, A. Hedayatizadeh-Omran and A. Shamshirian, J. Med. Virol., 2022, 94, 263–271 CrossRef CAS PubMed
- J. Arendt, Front. Endocrinol., 2019, 10, 391 CrossRef PubMed
- J. A. Boutin, D. J. Kennaway and R. Jockers, Biomolecules, 2023, 13, 943 CrossRef CAS PubMed
- N. G. Harpsoe, L. P. Andersen, I. Gogenur and J. Rosenberg, Eur. J. Clin. Pharmacol., 2015, 71, 901–909 CrossRef PubMed
- D. P. Cardinali, D. E. Vigo, N. Olivar, M. F. Vidal and L. I. Brusco, Antioxidants, 2014, 3, 245–277 CrossRef PubMed
- C. Tuft, E. Matar, Z. Menczel Schrire, R. R. Grunstein, B. J. Yee and C. M. Hoyos, Clin. Interventions Aging, 2023, 18, 49–59 CrossRef CAS PubMed
- J. Paprocka, M. Kijonka, B. Rzepka and M. Sokol, Int. J. Endocrinol., 2019, 2019, 9626715 Search PubMed
- L. P. Andersen, M. U. Werner, M. M. Rosenkilde, N. G. Harpsoe, H. Fuglsang, J. Rosenberg and I. Gogenur, BMC Pharmacol. Toxicol., 2016, 17, 8 CrossRef PubMed
- O. Vakkuri, J. Leppäluoto and A. Kauppila, Life Sci., 1985, 37, 489–495 CrossRef CAS PubMed
- T. T. H. Thi, E. J. A. Suys, J. S. Lee, D. H. Nguyen, K. D. Park and N. P. Truong, Vaccines, 2021, 9, 359 CrossRef CAS PubMed
- C. L. Ventola, Pharm. Ther., 2017, 42, 742 Search PubMed
- R. Foulkes, E. Man, J. Thind, S. Yeung, A. Joy and C. Hoskins, Biomater. Sci., 2020, 8, 4653–4664 RSC
- E. A. Bseiso, S. A. AbdEl-Aal, M. Nasr, O. A. Sammour and N. A. A. El Gawad, Drug Delivery, 2022, 29, 2469–2480 CrossRef CAS PubMed
- A. Molska, A. K. G. Nyman, A. M. Sofias, K. A. Kristiansen, S. Hak and M. Wideroe, Eur. J. Pharm. Biopharm., 2020, 152, 248–256 CrossRef CAS PubMed
- M. H. Akhter, M. Rizwanullah, J. Ahmad, S. Amin, M. Z. Ahmad, M. A. Minhaj, M. A. Mujtaba and J. Ali, Drug Res., 2021, 71, 122–137 CrossRef CAS PubMed
- A. B. Lerner, J. D. Case, Y. Takahashi, T. H. Lee and W. Mori, J. Am. Chem. Soc., 1958, 80, 2587–2587 CrossRef CAS
- C. Tapeinos, M. Battaglini and G. Ciofani, J. Controlled Release, 2017, 264, 306–332 CrossRef CAS PubMed
- D. K. Mishra, R. Shandilya and P. K. Mishra, Nanomedicine, 2018, 14, 2023–2050 CrossRef CAS PubMed
- S. B. Lim, A. Banerjee and H. Önyüksel, J. Controlled Release, 2012, 163, 34–45 CrossRef CAS PubMed
- M. C. Teixeira, C. Carbone and E. B. Souto, Prog. Lipid Res., 2017, 68, 1–11 CrossRef CAS PubMed
- H. Nsairat, A. A. Ibrahim, A. M. Jaber, S. Abdelghany, R. Atwan, N. Shalan, H. Abdelnabi, F. Odeh, M. El-Tanani and W. Alshaer, J. Liposome Res., 2024, 34, 178–202 CrossRef CAS PubMed
- V. Dubey, D. Mishra and N. K. Jain, Eur. J. Pharm. Biopharm., 2007, 67, 398–405 CrossRef CAS PubMed
- P. Phanphothong, N. Kanpipit and S. Thapphasaraphong, Int. J. Pharm.:X, 2023, 6, 100217 CAS
- M. Mirhoseini, Z. Rezanejad Gatabi, M. Saeedi, K. Morteza-Semnani, F. Talebpour Amiri, H. R. Kelidari and A. A. Karimpour Malekshah, Res. Pharm. Sci., 2019, 14, 201–208 CrossRef PubMed
- R. Rezzani, L. F. Rodella, F. Fraschini, M. R. Gasco, G. Demartini, C. Musicanti and R. J. Reiter, J. Pineal Res., 2009, 46, 255–261 CrossRef CAS PubMed
- M. Sabzichi, N. Samadi, J. Mohammadian, H. Hamishehkar, M. Akbarzadeh and O. Molavi, Colloids Surf., B, 2016, 145, 64–71 CrossRef CAS PubMed
- M. Kumar, N. Chauhan, D. Kumar, S. Mahmood, S. Chopra and A. Bhatia, J. Dispersion Sci. Technol., 2024, 6, 1–26 Search PubMed
- N. Wu, Z. Ye, K. Zhou, F. Wang, C. Lian and Y. Shang, Langmuir, 2024, 40, 7723–7732 CrossRef CAS PubMed
- Y. Duan, A. Dhar, C. Patel, M. Khimani, S. Neogi, P. Sharma, N. S. Kumar and R. L. Vekariya, RSC Adv., 2020, 10, 26777–26791 RSC
- M. K. Satapathy, T. L. Yen, J. S. Jan, R. D. Tang, J. Y. Wang, R. Taliyan and C. H. Yang, Pharmaceutics, 2021, 13, 1183 CrossRef CAS PubMed
- R. Paliwal, S. R. Paliwal, R. Kenwat, B. D. Kurmi and M. K. Sahu, Expert Opin. Ther. Pat., 2020, 30, 179–194 CrossRef CAS PubMed
- A. Zielinska, F. Carreiro, A. M. Oliveira, A. Neves, B. Pires, D. N. Venkatesh, A. Durazzo, M. Lucarini, P. Eder, A. M. Silva, A. Santini and E. B. Souto, Molecules, 2020, 25, 3731 CrossRef CAS PubMed
- P. I. R. Franco, J. R. do Carmo Neto, V. L. Rocha, J. R. Machado, A. C. Amaral and M. P. Miguel, Curr. Med. Chem., 2023, 30, 3315–3334 CrossRef CAS PubMed
- F. Aghaz, Z. Asadi, S. Sajadimajd, K. Kashfi, E. Arkan and Z. Rahimi, Sci. Rep., 2023, 13, 11090 CrossRef CAS PubMed
- H. Jafari, M. Hassanpour, A. Akbari, J. Rezaie, G. Gohari, G. R. Mahdavinia and E. Jabbari, Mater. Lett., 2021, 282, 128818 CrossRef CAS
- G. Hoti, R. Ferrero, F. Caldera, F. Trotta, M. Corno, S. Pantaleone, M. M. H. Desoky and V. Brunella, Polymers, 2023, 15, 1543 CrossRef CAS PubMed
- M. Si, Q. Sun, H. Ding, C. Cao, M. Huang, Q. Wang, H. Yang and Y. Yao, Adv. Polym. Technol., 2020, 2020, 1–9 CrossRef
- H. Liang, Y. Yan, W. Sun, X. Ma, Z. Su, Z. Liu, Y. Chen and B. Yu, Int. J. Mol. Sci., 2023, 24, 8740 CrossRef CAS PubMed
- F. Persano, G. Gigli and S. Leporatti, Nano Express, 2021, 2, 012006 CrossRef CAS
- Y. W. Yen, Y. L. Lee, L. Y. Yu, C. E. Li, P. W. Shueng, H. C. Chiu and C. L. Lo, Int. J. Biol. Macromol., 2023, 250, 126211 CrossRef CAS PubMed
- M. N. Sardoiwala, S. Nagpal, B. Bhatt, S. Roy Choudhury and S. Karmakar, Mol. Pharm., 2023, 20, 2899–2910 CrossRef CAS PubMed
- J. M. Soni, M. N. Sardoiwala, S. R. Choudhury, S. S. Sharma and S. Karmakar, Mater. Sci. Eng., C, 2021, 124, 112038 CrossRef CAS PubMed
- D. C. F. Soares, S. C. Domingues, D. B. Viana and M. L. Tebaldi, Biomed. Pharmacother., 2020, 131, 110695 CrossRef PubMed
- V. Dave, K. Tak, A. Sohgaura, A. Gupta, V. Sadhu and K. R. Reddy, J. Microbiol. Methods, 2019, 160, 130–142 CrossRef CAS PubMed
- S. Rao and C. A. Prestidge, Expert Opin. Drug Delivery, 2016, 13, 691–707 CrossRef CAS PubMed
- M. C. Goncalves, O. Mertins, A. R. Pohlmann, N. P. Silveira and S. S. Guterres, J. Biomed. Nanotechnol., 2012, 8, 240–250 CrossRef CAS PubMed
- S. Parvez, G. Yadagiri, K. Arora, A. Javaid, A. K. Kushwaha, O. P. Singh, S. Sundar and S. L. Mudavath, ACS Biomater. Sci. Eng., 2023, 9, 2902–2910 CrossRef CAS PubMed
- W. Ishaniya, C. Sumithaa, S. Raghunandhakumar, S. Vimalraj and M. Ganeshpandian, J. Drug Delivery Sci. Technol., 2022, 72, 103415 CrossRef CAS
- E. R. Komninou, M. H. Remiao, C. G. Lucas, W. B. Domingues, A. C. Basso, D. S. Jornada, J. C. Deschamps, R. C. Beck, A. R. Pohlmann, V. Bordignon, F. K. Seixas, V. F. Campos, S. S. Guterres and T. Collares, PLoS One, 2016, 11, e0157561 CrossRef PubMed
- C. D. V. Bessone, S. M. Martinez, J. D. Luna, M. A. Marquez, M. L. Ramirez, D. A. Allemandi, A. R. Carpentieri and D. A. Quinteros, Exp. Eye Res., 2020, 200, 108222 CrossRef CAS PubMed
- L. G. A. Chuffa, F. R. F. Seiva, A. A. Novais, V. A. Simao, V. M. Martin Giménez, W. Manucha, D. Zuccari and R. J. Reiter, Molecules, 2021, 26, 3562 CrossRef PubMed
- A. Faeghinia, H. Nuranian and M. Eslami, Synth. Sintering, 2023, 3, 79–87 CrossRef
- Z. Wang, T. Chen, Z. Wu, X. Jiang, Q. Hou, S. Miao, R. Xia and L. Wang, J. Mater. Chem. B, 2022, 10, 4020–4030 RSC
- H. Wang, W. Wei, S. Y. Zhang, Y. X. Shen, L. Yue, N. P. Wang and S. Y. Xu, J. Pineal Res., 2005, 39, 156–163 CrossRef CAS PubMed
- K. Yadav, M. Das, N. Hassan, A. Mishra, J. Lahiri, A. K. Dubey, S. K. Yadav and A. S. Parmar, RSC Adv., 2021, 11, 9076–9085 RSC
- G. Niu, B. Yousefi, D. Qujeq, A. Marjani, J. Asadi, Z. Wang and S. M. Mir, Mater. Sci. Eng., C, 2021, 119, 111554 CrossRef CAS PubMed
- Y. Cui, S. Hong, Y. Xia, X. Li, X. He, X. Hu, Y. Li, X. Wang, K. Lin and L. Mao, Adv. Sci., 2023, 10, e2302029 CrossRef PubMed
- M. F. Charao, C. Souto, N. Brucker, A. Barth, D. S. Jornada, D. Fagundez, D. S. Avila, V. L. Eifler-Lima, S. S. Guterres, A. R. Pohlmann and S. C. Garcia, Int. J. Nanomed., 2015, 10, 5093–5106 CrossRef CAS PubMed
- S. Sruthi, N. Millot and P. V. Mohanan, Int. J. Biol. Macromol., 2017, 103, 808–818 CrossRef CAS PubMed
- S. Gurunathan, M. Jeyaraj, M. H. Kang and J. H. Kim, Antioxidants, 2020, 9, 357 CrossRef CAS PubMed
- J. Xie, Z. Shen, Y. Anraku, K. Kataoka and X. Chen, Biomaterials, 2019, 224, 119491 CrossRef CAS PubMed
- S. Ganger and K. Schindowski, Pharmaceutics, 2018, 10, 116 CrossRef PubMed
- R. Maher, A. Moreno-Borrallo, D. Jindal, B. T. Mai, E. Ruiz-Hernandez and A. Harkin, Pharmaceutics, 2023, 15, 746 CrossRef CAS PubMed
- A. C. Correia, A. R. Monteiro, R. Silva, J. N. Moreira, J. M. Sousa Lobo and A. C. Silva, Adv. Drug Delivery Rev., 2022, 189, 114485 CrossRef CAS PubMed
- E. A. Bseiso, S. A. AbdEl-Aal, M. Nasr, O. A. Sammour and N. A. A. El Gawad, Drug Delivery, 2022, 29, 2469–2480 CrossRef CAS PubMed
- E. A. Bseiso, S. A. Abd El-Aal, M. Nasr, O. A. Sammour and N. A. Abd El Gawad, Life Sci., 2022, 306, 120797 CrossRef CAS PubMed
- E. R. de Oliveira Junior, T. L. Nascimento, M. A. Salomao, A. C. G. da Silva, M. C. Valadares and E. M. Lima, Pharm. Res., 2019, 36, 131 CrossRef PubMed
- C. P. Costa, J. N. Moreira, J. M. Sousa Lobo and A. C. Silva, Acta Pharm. Sin. B, 2021, 11, 925–940 CrossRef CAS PubMed
- L. Biswal, M. N. Sardoiwala, A. C. Kushwaha, S. Mukherjee and S. Karmakar, ACS Appl. Mater. Interfaces, 2024, 16, 8417–8429 CrossRef CAS PubMed
- D. Dehaini, R. H. Fang and L. Zhang, Bioeng. Transl. Med., 2016, 1, 30–46 CrossRef PubMed
- J. Li, Y. Wei, C. Zhang, R. Bi, Y. Qiu, Y. Li and B. Hu, Pharmaceutics, 2023, 15, 621 CrossRef CAS PubMed
- N. M. Sakhrani and H. Padh, Drug Des., Dev. Ther., 2013, 7, 585–599 CAS
- Q. Zheng, H. Liu, H. Zhang, Y. Han, J. Yuan, T. Wang, Y. Gao and Z. Li, Adv Sci (Weinh), 2023, 10, e2300758 CrossRef PubMed
- P. Liu, M. Cheng, J. Guo, D. Cao, J. Luo, Y. Wan, Y. Fang, Y. Jin, S. S. Xie and J. Liu, Bioorg. Med. Chem., 2023, 78, 117146 CrossRef CAS PubMed
- L. Vasileva, G. Gaynanova, F. Valeeva, G. Belyaev, I. Zueva, K. Bushmeleva, G. Sibgatullina, D. Samigullin, A. Vyshtakalyuk, K. Petrov, L. Zakharova and O. Sinyashin, Int. J. Mol. Sci., 2023, 24, 10494 CrossRef CAS PubMed
- X. Xiao, Y. Gao, S. Liu, M. Wang, M. Zhong, J. Wang, N. Jiang and Q. Peng, Adv. Funct. Mater., 2023, 33, 2213633 CrossRef CAS
- J. Zhai, C. Fong, N. Tran and C. J. Drummond, ACS Nano, 2019, 13, 6178–6206 CrossRef CAS PubMed
- M. Rakotoarisoa, B. Angelov, S. Espinoza, K. Khakurel, T. Bizien and A. Angelova, Molecules, 2019, 24, 3058 CrossRef CAS PubMed
- Y. Mohammad, R. N. Prentice, B. J. Boyd and S. B. Rizwan, J. Colloid Interface Sci., 2022, 605, 146–154 CrossRef CAS PubMed
- S. Urandur, D. Marwaha, S. Gautam, V. T. Banala, M. Sharma and P. R. Mishra, Ther. Delivery, 2018, 9, 667–689 CrossRef CAS PubMed
- S. Murgia, S. Biffi and R. Mezzenga, Curr. Opin. Colloid Interface Sci., 2020, 48, 28–39 CrossRef CAS
- J. S. D'Arrigo, Biomimetics, 2018, 3, 4 CrossRef PubMed
- H. Azhari, M. Strauss, S. Hook, B. J. Boyd and S. B. Rizwan, Eur. J. Pharm. Biopharm., 2016, 104, 148–155 CrossRef CAS PubMed
- G. L. See, F. Arce Jr., S. Dahlizar, A. Okada, M. Fadli, I. Hijikuro, S. Itakura, M. Katakura, H. Todo and K. Sugibayashi, J. Controlled Release, 2020, 325, 1–9 CrossRef CAS PubMed
- Y. Wu, J. Wang, Y. Deng, B. Angelov, T. Fujino, M. S. Hossain and A. Angelova, Adv. Healthc. Mater., 2024, 13, e2304588 CrossRef PubMed
- F. M. Elsenosy, G. A. Abdelbary, A. H. Elshafeey, I. Elsayed and A. R. Fares, Int. J. Nanomed., 2020, 15, 9517–9537 CrossRef CAS PubMed
- G. N. Desai, P. M. Dandagi and T. M. Kazi, J. Pharm. Innovation, 2022, 18, 934–951 CrossRef
- M. Rakotoarisoa, B. Angelov, M. Drechsler, V. Nicolas, T. Bizien, Y. E. Gorshkova, Y. Deng and A. Angelova, Smart Mater. Med., 2022, 3, 274–288 Search PubMed
- Y. Wu, B. Angelov, Y. Deng, T. Fujino, M. S. Hossain, M. Drechsler and A. Angelova, Commun. Chem., 2023, 6, 241 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2025 |