
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Multilamellar hyaluronic acid-b-poly(lactic acid) polymersomes for pathology-responsive MRI enhancement†
Dorian
Foster
*a,
Naisha
Shah
b,
Alaura
Cakley
a,
Ronald
Beyers
c and
Jessica
Larsen
 ad
ad
aDepartment of Chemical and Biomolecular Engineering, Clemson University, Clemson, SC 29634, USA. E-mail: larsenj@clemson.edu
bSouth Carolina Governor's School for Science and Mathematics, Hartsville, SC 29550, USA
cAuburn University MRI Research Center, Department of Electrical and Computer Engineering, Auburn University, Auburn, AL 36849, USA
dDepartment of Bioengineering, Clemson University, Clemson, SC 29634, USA
First published on 17th April 2025
Abstract
This study introduces a biocompatible, stimuli-responsive imaging and therapeutic delivery system using ultrasmall iron oxide nanoparticles (USPIONs) encapsulated within the hyaluronic acid-b-poly(lactic acid) (HA–PLA) polymersome membrane, with a model protein bovine serum albumin in the core. These multilamellar vesicles exhibit enhanced T2-weighted MRI contrast, achieving a relaxivity 3-fold higher than existing agents. The polymersomes demonstrate acid- and enzyme-triggered degradation, enabling controlled release and measurable contrast changes in pathological environments. Preliminary in vivo and postmortem studies confirm their strong imaging performance, high biocompatibility, and targeted response to enzymatic, acidic microenvironments, paving the way for theranostic applications in disease diagnosis and treatment monitoring.
Introduction
Imaging plays an integral role in diagnostics, disease and treatment monitoring. Magnetic resonance imaging (MRI), which generates two- and three-dimensional images using the natural magnetic properties of water content throughout the body, is a leading choice for soft tissue imaging.1 It is non-invasive and does not require non-ionizing radiation. Although it has high temporal and spatial resolution, MRI has low sensitivity. Contrast agents (CAs) are often necessary to compensate for this low sensitivity. Although other mechanisms exist, MRI CAs typically enhance contrast by altering the relaxation time of nearby water protons.1 Stimuli-responsive CAs can further improve the sensitivity and specificity of CA-enhanced MRI, as they experience a measurable change in relaxivity in response to a change in environment, usually one that is characteristic of pathology or organelle-specific physiology.Some of the most common stimuli around which responsive systems are designed are pH and enzyme activity. Polymer functionalization can integrate stimuli-responsivity, improving CA performance upon pathologic exposure. Solubility-switching polymers have been applied to directly enhance CA-water interactions2 or cause CA aggregation.3 Polymeric encapsulation has also been used to shield CAs from aqueous exposure temporarily,4 muting contrast in neutral conditions. Here, we have developed an acid and enzyme-responsive iron oxide (IO) CA through polymersome (PS) encapsulation. Although IO CAs are not predisposed to stimuli-responsive activity, polymer functionalization has been able to induce tuneable, stimuli-triggered relaxivity changes.1 PSs, nanoparticle vesicles formed from the self-assembly of amphiphilic block co-polymers, can co-encapsulate both hydrophilic and hydrophobic drugs,5 which could enable the future addition of therapeutics alongside IO CAs.
Previously, our lab has developed highly biocompatible, stimuli-responsive PSs made from hyaluronic acid (HA) and polylactic acid (PLA).6,7 HA–PLA PSs degrade under acidic conditions and upon exposure to enzymes like hyaluronidase or hexosaminidase A, with quantitative release profiles changing in response to changing HA molecular weight and hyaluronidase concentration.7 Building on this, we co-encapsulated ultrasmall IO nanoparticles (USPIONs) into HA(5 kDa)–PLA(15 kDa) PSs with bovine serum albumin (BSA) as a model protein to enable nanoparticle tracking via MRI. Our work lays the foundation for theranostic PSs for simultaneous protein delivery and disease monitoring.
Materials & methods
Materials
Amine functionalized iron oxide (Fe3O4) nanoparticles (3 nm) were purchased from US Research Nanomaterials, Inc (Houston, TX) due to their contrast enhancement properties when used with MRI (Fig. S1†). Tween20 (Sigma Aldrich, MO, USA) was used as a surfactant to prevent USPION aggregation for size characterization. N-Hydroxysuccinimide (NHS) activated poly(lactic acid) (PLA-NHS, MW 15 kDa) and hyaluronic acid (HA, MW 5 kDa) were purchased from Creative PEGworks (Durham, NC. USA). Float-a-lyzer dialysis devices (encapsulation studies) and slide-a-lyzers (release studies), both of MWCO 100 kDa, came from Thermo Fisher Scientific (Waltham, MA, USA). Phosphotungstic acid (PTA, Polysciences, Inc, Warrington, PA, USA) was provided by the Clemson Electron Microscopy Facility for polymersome staining. Ferrozine (Cayman Chemical Company, MI, USA) was used in iron quantification assays. Fluorescein isothiocyanate (FITC) tagged BSA and hyaluronidase (HYAL) were purchased from Sigma Aldrich (St Louis, MO, USA). Agarose (VWR International, PA, USA) was used to suspend iron nanoparticles for MRI phantom imaging. HYAL was purchased from Sigma Aldrich (MO, USA).SHSY-5Y cells were purchased from American Type Culture Collection (VA, USA) for in vitro studies. Media was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of Eagle's Minimum Essential Medium and F12 Medium (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% Fetal Bovine Serum (FBS) (Sigma Aldrich, MO, USA) and 1× Penicillin–Streptomycin (Thermo Fisher Scientific, Waltham, MA, USA). Passages were performed using 0.05% Trypsin (Corning Inc., Corning, NY, USA). Cytotoxicity was evaluated using an MTS Cell Proliferation Assay kit from BioVision (San Francisco, CA, USA). βgal−/− heterozygous mutant mice (Strain #:037063) were obtained from The Jackson Laboratory (Bar Harbor, ME, USA) for in vivo studies (AUP-2023-0215). Age-matched (five-month-old) mutant βgal−/− mice and wild-type (WT) mice from our breeding colony were used for live imaging studies; three WT mice (6 months of age, 1 female, 2 male) were used in the postmortem imaging study (AUP-2023-0402). All chemicals and reagents were of analytical grade.
:1 mixture of Eagle's Minimum Essential Medium and F12 Medium (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% Fetal Bovine Serum (FBS) (Sigma Aldrich, MO, USA) and 1× Penicillin–Streptomycin (Thermo Fisher Scientific, Waltham, MA, USA). Passages were performed using 0.05% Trypsin (Corning Inc., Corning, NY, USA). Cytotoxicity was evaluated using an MTS Cell Proliferation Assay kit from BioVision (San Francisco, CA, USA). βgal−/− heterozygous mutant mice (Strain #:037063) were obtained from The Jackson Laboratory (Bar Harbor, ME, USA) for in vivo studies (AUP-2023-0215). Age-matched (five-month-old) mutant βgal−/− mice and wild-type (WT) mice from our breeding colony were used for live imaging studies; three WT mice (6 months of age, 1 female, 2 male) were used in the postmortem imaging study (AUP-2023-0402). All chemicals and reagents were of analytical grade.
Methods
PSs were co-loaded with FITC-BSA, serving as a model hydrophilic drug/protein. 10 μL of 2 mg mL−1 FITC-BSA solution was vortexed with 10 mg of lyophilized PSs; 990 μL of Type 1 deionized water was then added and mixed to obtain a total volume of 1 mL of the loaded PS solution. The loaded PSs were then added into a float-a-lyzer (MWCO 100 kDa) for dialysis against deionized water. Samples of the dialysate were taken hourly to identify FITC-BSA release over time. Each time a sample was taken, the buffer was changed to maintain a concentration gradient and encourage complete diffusion of the released protein into the dialysate. The total FITC-BSA released into the buffer (Cr) was quantitatively analyzed by using UV-vis spectroscopy (BioteK Synergy H1); any FITC-BSA not released through dialysis was assumed to be encapsulated in the PSs. EE was calculated using the following equation:
 | (1) |
Results
USPIONs were coated with PLA to moderate hydrophobicity and enable loading into the PLA membrane. The PLA coating was confirmed by an increase in HD from the reported 3 nm to 6.85 ± 0.35 nm. PLA-coated USPIONs (PLA-USPIONs) were then loaded into PSs by dissolution into the organic phase during PS synthesis.6–8 The physical parameters of each PS formulation are given in Table 1. Upon USPION loading, the PS HD increased slightly from that of unloaded HA–PLA PSs. Furthermore, the surface charge became less negative in all cases versus the unloaded control.| PLA-USPION [μg] | HD [nm] | PDI | ZP [mV] |
|---|---|---|---|
| *p < 0.05, **p < 0.005 when compared to reference (unloaded PSs). | |||
| 0 | 82.0 ± 10.2 | 0.395 ± 0.05 | −24.9 ± 2.8 |
| 1 | 115.8 ± 17.3 | 0.499 ± 0.16 | −9.9 ± 1.2* |
| 5 | 112.0 ± 10.0* | 0.329 ± 0.11 | −7.3 ± 2.7** |
| 10 | 109.7 ± 19.7 | 0.367 ± 0.10 | −12.6 ± 2.1* |
| 15 | 100.6 ± 13.9 | 0.340 ± 0.10 | −13.3 ± 4.0 |
| 20 | 113.6 ± 21.1 | 0.369 ± 0.09 | −12.0 ± 5.7 |
USPION content in each PS sample was determined via ferrozine assay. The mass of USPION loaded in PSs increased with increasing PLA-USPION mass added. However, a plateau was observed following the addition of 10 μg PLA-USPION (Fig. 1), after which the deviation increased with increasing PLA-USPION content. Encapsulation efficiency, as calculated from mass content values, decreases nearly linearly with increasing USPION mass supplied. This data suggests that the PSs become saturated with USPIONs with a maximum loaded content of 0.52 ± 0.14 μg USPION/mL PS.
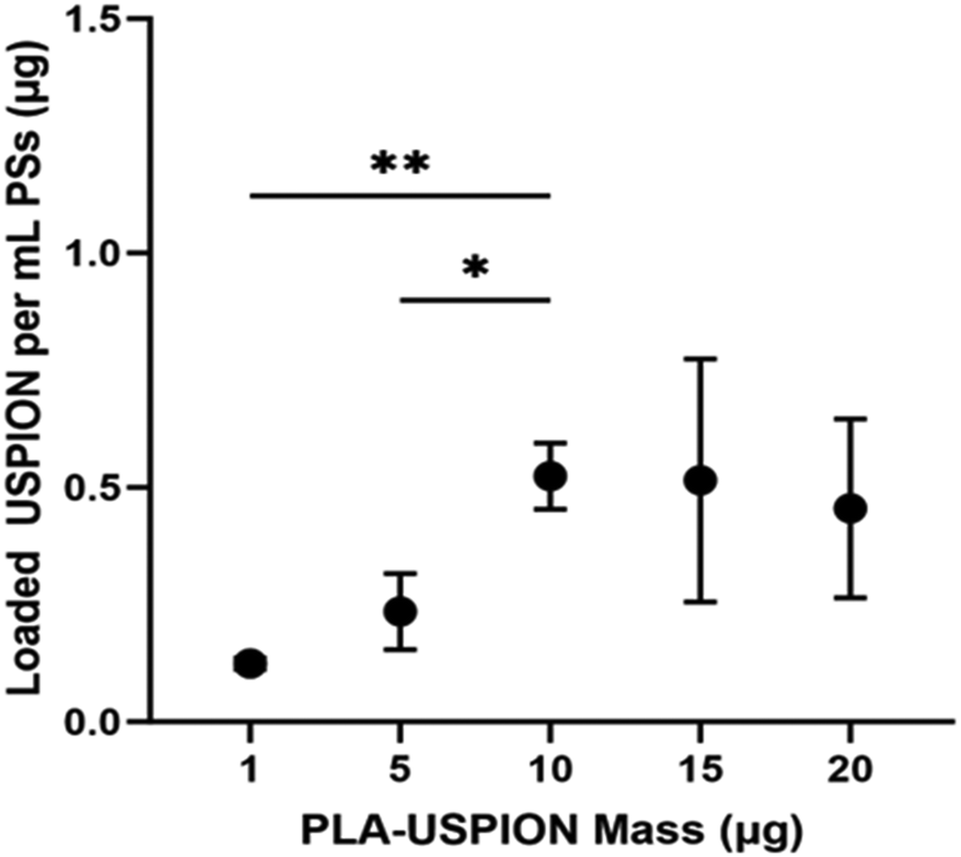 | ||
| Fig. 1 USPION mass in 1 mL of PSs (μg) plotted versus PLA-USPION mass (μg) used in synthesis. *p < 0.05; **p < 0.005. | ||
Transmission electron microscopy (TEM) images were taken to further confirm PS formation as well as verify membrane-localized loading (Fig. 2A). The images confirmed PS formation, with the presentation of detectable membranes, in good agreement with size and PDI measurements from DLS. USPION localization to the membrane, which was desired to leave the aqueous core available for hydrophilic drug or protein loading, was evidenced by the dark ring surrounding an otherwise light structure. TEM creates images based on the electron density of a sample, so this dark ring is assumed to be the electron-dense IO particles that have been trapped in the membrane. That dark ring is not observed in PSs without USPIONs.7 The most interesting observation from these images, however, was the multilamellar structure, which has not been previously observed in HA–PLA PSs to our knowledge.
 | ||
| Fig. 2 (A) PS-USPIONs and (B) No-PLA-PS-USPIONs. Samples were prepared with PTA before imaging, allowing visualization of the external soft PEG brush, while the internal dark rings are likely from USPIONs. | ||
All formulations examined here exhibited the multilamellar structure, although the PSs synthesized using greater PLA-USPION concentrations structurally deviated even more from the non-USPION-loaded, unilamellar PSs. The PSs synthesized with 15 and 20 μg of USPIONs exhibited a high incidence of multilamellar and unilamellar PSs as well as structures resembling an inverse micelle but with a separate, all-encapsulating outer membrane. Based on all PLA-USPION masses added to PSs and the observed overall nano-structures, we present a proposed phase diagram (Fig. 3) demonstrating the role that increasing PLA content has on the overall PS presentation, which follows expected behavior that occurs with increasing hydrophobicity.5
 | ||
| Fig. 3 Proposed phase diagram of PSs loaded with PLA-USPIONs with corresponding TEM images. We hypothesize that the increased PLA content introduced with increasing PLA-USPIONs changes the critical packing parameter and, thus, the surface chemistry of PS self-assembly, leading to the formation of alternative structures. However, multilamellar PSs were observed at all PLA-USPION masses loaded. | ||
Previous studies attribute the formation of these structures to temperature or pressure differences12,13 but these variables were held constant in our studies. We posit that at these higher PLA-USPION levels, there is a high level of supersaturation of hydrophobic entities that pushes the critical packing parameter above 1 such that an inverted micelle is formed,14 which is ultimately encapsulated within a unilamellar PS to render it stable in aqueous solution. The micelle-like vesicles pack PLA-USPIONs at a high concentration within the extra hydrophobic loading space; a trade-off to this structure is a notable loss in hydrophilic loading capacity. The respective combinations of vesicular structures, each exhibiting such different loading capacities, explain the high variability seen in loaded USPION mass for these two formulations (Fig. 1).
With clinical translatability in mind, PS-USPION loaded with 10 μg of PLA-USPIONs were chosen for continued development based on uniformity in structure and reproducibility. This formulation is referred to only as PS-USPION going forward. Even so, in considering all five formulations and the consistent incidence of multilamellarity, it figures that some consistent aspect must be driving the formation of multiple membranes and we hypothesize that this aspect is the PLA coating on the USPIONs.
To investigate PLA as the driving factor for multilamellarity, we loaded the PSs with unaltered, NH2-activated USPIONs under the same loading protocol as before and with the same USPION concentration as the PS-USPIONs (10 μg). TEM images of the No-PLA-PSs supported membrane internalization based on the dark ring outline on a slight inset from the PS exterior, but most notably, only unilamellar morphologies were observed (Fig. 2B). The same physical characteristics were calculated with the No-PLA-PS. Additionally, co-loading capacities were evaluated with FITC-BSA as a model hydrophilic drug – an important performance parameter for this system to apply in the theranostic space. Data is presented in Table 2. Physical parameters suggested that the PSs without additional PLA assembled more consistently at a larger size, further confirming our hypothesis that increased hydrophobicity led to increased cohesion. Loading capacity was not statistically different for either USPION content or co-loading with FITC-BSA. With both formulations being largely similar, the original formulation, PS-USPIONs, was chosen for further characterization development.
| With PLA | Without PLA | |
|---|---|---|
| *p < 0.05 when compared to with PLA, indicating statistical differences in properties. | ||
| HD [nm] | 109.72 ± 19.69 | 132.1 ± 22.23* |
| PDI | 0.367 ± 0.095 | 0.088 ± 0.023* |
| ZP [mV] | −12.6 ± 2.1 | −23.8 ± 2.43* |
| Loaded USPIONs [μg mg−1 PS] | 0.0198 ± 0.0051 | 0.0289 ± 0.0067 |
| FITC-BSA EE [%] | 36.65 ± 10.97 | 42.01 ± 3.84 |
Phantom images were obtained at 3.0T of the USPIONs and PLA-USPIONs suspended in gel (Fig. 4A), as well as of PS-USPIONs before and after incubation in a pathological environment (1 mg mL−1 HYAL, pH 4.8 at 37 °C for 72 hours) to characterize MRI performance (Fig. 4B). Rapid settling of the USPIONs and PLA-USPIONs, likely due to their very small size and hydrophobicity, necessitated the use of a gelling agent. Images clearly show that our CAs were T2-oriented. It is important to note that due to the increased viscosity of the 2% agarose solutions, the relaxivity will be different than what would be expected in situ.15 The use of the gelling agent has, therefore, prevented the quantitative characterization of relaxivity. However, these images serve as a qualitative verification of contrast-altering effects. There is a slightly more pronounced darkening effect with the USPIONs prior to PLA coating, suggesting some degree of hydration blocking by the PLA, which would reduce the interaction between the CA and surrounding water protons, effectively reducing influence. However, it is difficult to compare the USPION performance before and after PLA coating without quantification, especially accounting for gel interference and potential suspension inhomogeneity, so our analysis focuses on the PS-USPIONs.
 | ||
| Fig. 4 T 2-Weighted MRI contrast according to concentration. (A) USPIONs and PLA-USPIONs suspended in gel at increasing concentrations. A slight decrease in contrast effect is noticeable with the addition of PLA coating. (B) PS-USPIONs suspended at multiple dilutions in salt-based buffers – PBS or Citrate buffer with HYAL for acidic. Significant loss of contrast enhancing effect observed upon incubation in disease-model environment. Appropriate control buffer references (concentration = 0 μg mL−1) are included with each formulation/condition set. Image still from TE = 150 ms. | ||
The hydrophilic exterior of the PSs allowed for direct suspension and imaging of PS-USPIONs in PBS; however, PS concentration was significantly restricted by solubility limitations. Despite this, iron concentration-dependent darkening is obvious in the PS-USPION (pH 7.4) formulation (Fig. 4B); the transverse relaxivity (r2) was calculated to be 211.14 mM−1 s−1 (Fig. S2†). Regarding stimuli-responsivity, there is a loss of darkening following the incubation of PS-USPIONs in enzymatic and acidic conditions, which matches what was observed when optimizing HA–PLA polymersomes for their release behavior in pathologic conditions.7 Responsivity was investigated further to determine the impact of the kinetics of HA–PLA PS degradation on corresponding T2 weighted contrast, with images taken daily for five days.
PS-USPIONs at the highest soluble concentration, corresponding to ∼3.58 μg Fe per mL, were incubated in pathologic release conditions. At all time points in the MRI time release study, an obvious difference was observable between samples suspended in neutral (pH 7.4) and acidic (pH 4.8) + enzymatic (1 mg mL−1 HYAL) conditions. The PBS-suspended samples maintained a constant near-black contrasting effect, similar to what was observed when calculating relaxivity (Fig. 4), while the HYAL/citrate samples were notably less dark as early as 6 hours after incubation. By hour 6, relaxivity decreased from the maximum value of 211.14 mM−1 s−1 to 26.936 mM−1 s−1, a decrease of around 87%. This lower relaxivity was maintained over the course of the study (Fig. S3†). Even so, a gradual loss of negative contrast could be observed with day-to-day direct comparisons at matched concentrations (Fig. 5).
 | ||
| Fig. 5 T 2-weighted MRI daily response study. PS-USPIONs in neutral pH yields a consistent black contrast versus incubation in hyaluronidase enriched, acidic pH showed a gradual decrease in negative effect, approaching the contrast levels of a saline control. Image stills from TE of 550 ms. | ||
To ensure clinical translatability, we examined the biocompatibility of PS-USPIONs via MTS assay. Cell survival following treatment was examined in SH-SY5Y (neuroblastoma) cells to reflect our eventual goal of functionalizing a CA for central nervous system use. Dosages up to 100 μg Fe per mL were examined. The USPIONs and PLA-USPIONs showed high biocompatibility at all treatment levels and after both 4 and 24 hours of incubation (Fig. 6), with no statistical difference observed between any dose. The PS-USPIONs showed slight but obvious dose-dependent toxicity after 4 hours, with a higher dose showing more signs of cytotoxicity, although not statistically significant. Note that high contrast is observed at iron doses as low as 3.55 μg mL−1 (Fig. 4), and toxicity is not observed until iron doses that are 10-fold higher. With the high strength of the PS-based CA, dosing requirements may be so low that acute inflammation conditions are never approached.
 | ||
| Fig. 6 Viability analysis via MTS Assay over 4 and 24 hours of (A) USPION and PLA-USPION and (B) PS-USPIONs incubated. | ||
Postmortem MRI following PS-USPION injection was then performed to evaluate the general, untargeted contrast effects in tissue. The mouse was injected with PS-USPIONs (red arrow) on the right side such that the left could be injected with PBS at the same volume (blue arrow) as an internal control. In the resulting image (Fig. 7A), T2 hyperintensity can be observed in the right leg, an obvious difference from the control leg. As will be discussed in more detail below, mice were imaged in a 0.25T MRI, which changes contrast presentation. This hyperintensity was quantified, with observable increases in integrated density in the PS-USPION injected leg compared to internal PBS controls (Fig. 7B). A fold increase in integrated density of 1.63 ± 0.24 was calculated, indicating the consistent T2 hyperintensity occurring across animal replicates.
 | ||
| Fig. 7 (A) Transverse view (left) and coronal view (right) of C57BL/6 mouse injected in the posterior leg muscles with either PS-USPIONs (red) or PBS (blue) taken under 0.25T MRI. The low magnet strength of 0.25T makes T2 weight contrast agents appear light, with a dramatic lightening apparent in the posterior muscle injected with PS-USPIONs. (B) Quantified integrated density in each posterior leg muscle taken under 0.25T MRI. The integrated density in both the left and right hind leg muscles were quantified using ImageJ. Measurements indicate a clear observable contrast in the PS-USPION injected leg. When normalized to the internal PBS injection, PS-USPIONs led to a fold increase in integrated density of 1.63 ± 0.24 indicating a consistent increase in contrast. (C) Representative timed IVIS images of mice following tail vein injection of PS-USPIONs. IVIS imaging indicates that PS-USPIONs are more responsive in a diseased animal, with increased release occurring in the βgal−/− mouse at all observed time points. Scale bar of radiant efficiency: min = 1.76 × 107, max = 3.79 × 108. | ||
Finally, pharmacokinetic behavior of PS-USPIONs was evaluated for 24 hours post injection through fluorescent monitoring via IVIS imaging at selected time points. WT (healthy) and βgal−/− (diseased) mice were both injected with PS-USPIONs simultaneously encapsulating AF647. βgal−/− mice mimic a lysosomal storage disorder called GM1 gangliosidosis16 which is marked by high acidity and upregulated hexosaminidase A17 throughout the entire body, including the brain,18 causing an acidic and enzymatic environment that should lyse HA–PLA PSs and therefore PS-USPIONs. IVIS images (Fig. 7C) demonstrate greater fluorescent intensity detectible in the βgal−/− model mouse versus the WT mouse at all timepoints. Spikes in fluorescent signals were taken to indicate the release of dye from the PS, so it is clear to see that some aspect of the knock-out model mouse's physiology is driving preferential release over that of a normal mouse.
Discussion
HA–PLA PSs are clearly capable of encapsulating and delivering USPIONs, with maximum loaded content of 0.52 ± 0.14 μg mL−1 of PLA-USPIONs determined (Fig. 1). This maximum loaded content is likely observed due to the membranous loading of the PLA-USPIONs by design, confirmed via TEM (Fig. 2A). The membrane volume of a polymersome is typically greater than its lipid analog the liposome,19 with thickness controlled by hydrophobic polymer molecular weight.5 Despite this, the volume of the polymersome membrane is still only a fraction of the overall polymersome system and significantly smaller than the interior core,20 leading to a maximum payload of PLA-USPIONs in this finite space. Despite the slight increase in size from unloaded controls (Table 1), the small size of these PSs with USPIONs makes them attractive for transport and diffusion in hard-to-reach structures, which could include the brain21 and cancerous tumors22 – structures for which there is a great demand for improved imaging options. Multilamellar vesicles, in general, have been shown to have higher stability than unilamellar ones, which could be beneficial for cellular delivery.23–25 Multilamellar structures have also been shown to not only maintain the capacity for dual hydrophobic and hydrophilic loading but do so at higher encapsulation efficiencies than unilamellar PSs.26–29 Additionally, release tends to be more sustained29,30 due to the requirement of successive membrane degradation to free all encapsulated drug. In fact, theranostic multilamellar PSs citing all of those benefits have been developed with poly(ethylene glycol)-bl-poly(propylene sulfide).31The consistent incidence of multilamellarity suggests that the formation of multiple membranes is not PLA-USPION mass-dependent (Fig. 2A). Krack et al. explained the formation of multilamellar PSs upon nanoparticle loading as thermodynamically driven by a disruption to the hydrophobic/hydrophilic interface such that enclosure by multiple membranes yielded lower energy.32 While we agree that the hydrophobic space may be experiencing an incomplete closure from the surrounding hydrophilic cavities, especially due to the integration of USPIONs and disordered PLA chains, we also posit that the additional PLA coating abnormally alters the effective hydrophilic ratio which could be enough to alter PS morphology given that this is known to be such a crucial factor,5 leading to our proposed a theoretical phase diagram for this system (Fig. 3). The formation of additional membranes and aqueous core cavities could explain the size decrease observed with PLA-USPION loading when compared to uncoated USPION loading. If PLA-coated USPIONs are introduced within the large cavity of a unilamellar PS, the resulting increase in hydrophobicity may adjust the hydrogen-bonding patterns of encapsulated water making the unilamellar structure less entropically favorable by decreasing the possible hydrogen bonds that can be formed.33 Therefore, to minimize this loss, PS membranes may pinch inwards to form a multilamellar structure and re-maximize hydrogen bonding, which can be more easily maintained near a smaller hydrophobic region.
Additional support was provided for this hypothesis by examining the resulting loaded PS with USPIONs not coated by PLA. TEM images served as the primary source of confirmation; images portrayed generally circular vesicles with sizes in agreement with DLS, with interior dark rings representing electron density in the membrane due to IO packing. The rings were singular in these PSs, however (Fig. 2B). A thermodynamic explanation having to do with the hydrophilic fraction is likely involved, but it is most obvious to us that the phenomenon is in line with the hypothesis from Krack – the PLA-USPIONs, much bulkier than uncoated USPIONs, made it more difficult for the hydrophobic layer to become completely sealed off from hydrophilic spaces, motivating the formation of additional layers.
Physical properties, as determined by DLS, further support unilamellarity and the aforementioned hypothesis. The PSs with uncoated USPIONs were larger because they lacked the internal forces pinching the layers together to minimize unfavourable interactions. Additionally, the lower PDI indicates a more uniform self-assembly which is compatible with a simpler interaction between amphiphilic parts as allowed by the absence of additional packing of PLA strands. From this, we can tell that PLA coating was not necessary to ensure membrane localization of the USPIONs. In a comparison of the respective octanol–water partition coefficient (logP) of each component, the amine-activated surface is much closer to the hydrophobicity of PLA than HA, although, of course, not as close as a perfect PLA-to-PLA match with same MW. Therefore, membrane internalization is not entirely surprising but does imply that there may be a wider range of logP that are suited to membrane internalization for a given block copolymer, a concept that could also be worth exploring. Ultimately, the ability to co-load a therapeutic protein alongside the USPIONs, both PS-USPIONs and No-PLA-PSs were statistically indifferentiable (Table 2), making them both promising candidates for theranostic applications. With the benefits of multilamellarity in mind, we progressed to MRI performance evaluation on the PS-USPIONs.
The high relaxivity of the PS-USPION formulation suggests that encapsulation into the PS strengthened the original USPION. We hypothesize that this is because by forcing the structured aggregation of USPIONs into a particle, the PS-USPION acts as a singular particle with a much larger effective diameter. In this case, the decrease in tumbling rate would account for transverse contrast differences. It is known that increasing hydrodynamic diameter will increase r2,34 even just by adding dense polymer coatings, like polyethylene glycol.35 Because PSs are made up completely of amphiphilic block copolymer, the surface is a highly dense polymer coating of HA.
Currently, of the several IO CAs that initially received FDA approval, only Resovist remains on the market, although its use is limited to use in certain countries.33 It has a transverse relaxivity of 151 mM−1 s−1, but the magnetic strength under which this was measured is unclear, making direct comparison challenging. As an alternative, ferumoxytol is FDA-approved for the treatment of iron deficiency but is being explored for use as a T2 CA. Ferumoxytol has been reported to have an r2 value of 62.3 ± 3 mM−1 s−1 under similar conditions to those explored in this paper (3.0 T, saline).36 Therefore, compared to ferumoxytol, we have achieved a system with T2 contrast enhancement 3 times as strong as the leading alternative (Fig. 4). This high-strength T2 CA could offer the benefit of decreased dosage requirements and easier imaging discernibility.
The responsivity following PS release further strengthens this system. Within only 6 hours of incubation in the model acidic/enzymatic environment, the contrast enhancement decreased to 26.936 mM−1 s−1, less than 15% of its original value (Fig. 5). Considering our lab's characterization of HA–PLA PS release profiles,7 it is most likely that the contrast enhancement has decreased in the PS-USPION system following incubation in the model disease environment which leads to PS degradation. On a magnetic basis, we believe that while encapsulated into the PS, the USPIONs act collectively as one large IO nanoparticle. Upon release, the USPIONs returned to their natural strength, and because the iron content in the PS-USPIONs is lower than the 12.5 μg mL−1 used to image USPIONs and PLA-USPIONs (Fig. 4), and heuristically used as a minimum in many CA publications, the lower concentrations of USPIONs released from PSs are not detectible, leading to the lack of contrast. Additionally, given prior observations that unencapsulated USPIONs aggregate, it is likely that some degree of USPION settling occurred at the tube bottom, rendering them less measurable in the upper region of the tube. Either way, this change in relaxivity could then be monitored in situ with applicability in real-time drug tracking. If USPIONs are settling, this further substantiates our polymer degradation hypothesis and suggests facile USPION clearance following PS release in vivo. Until complete clearance, however, the lasting contrast enhancement could be applied to treatment monitoring or diagnostic imaging following therapeutic delivery.
Although most stimuli-responsive CAs become brighter in response to stimulus exposure, this reversal could be beneficial in a T2 CA; the CA will be very dark and easy to identify as it is moving throughout the body. Relaxivity reduction or a decrease in contrast enhancement would provide evidence of PS degradation and USPION release. With the addition of a hydrophilic drug, it would also provide evidence of successful drug delivery. Liu et al. developed an IO-containing PS37 that has similar sizing, contrast effects, and intended applications (targeting and dual drug loading) to those discussed here. These similarities lend credence to the translation of our system, especially with the success of their in vivo studies. While we have not achieved a CA quite as strong as theirs (611.6 mM−1 s−1), the comparative strength of our system lies in its pathologically-specific stimuli-responsivity. Our PS-USPIONs lead to changing contrast in direct response to relevant biomarkers, the enzyme- or acid-catalysed degradation of HA, as opposed to general biodegradability observed in their system. Furthermore, in their study, stimuli-responsive behavior was not confirmed via MRI. The multilamellar structure we observed is also unique and demonstrates a greater propensity for sustained release. It is also worth emphasizing, as Liu did, that r2 is a function of both the copolymer properties and the overall diameter and composition of USPIONs, which can vary.
The in vitro trends presented no cytotoxicity concerns in the USPION or PLA-USPION forms, with slight decreases in cell viability only being observed in the PS-USPION system. These results are in line with our prior observations when incubating HA–PLA nanostructures with SHSY-5Y cells.7 Because low MW HA causes inflammation,38,39 we expect a greater amount of HA–PLA PSs, which degrade into more low MW HA fragments, could result in minor cytotoxicity. Even so, the trend disappears by 24-hours, suggesting that any cytotoxicity is due to acute inflammation and is expected to be temporary. However, in vivo clearance studies as well as chronic dosing exposure would need to be performed to confirm elimination and biocompatibility, focusing on cytokines and anti-PEG antibodies produced over time and dose.
Postmortem imaging exposed a clear contrast effect observable in PS-USPION injected vs. PBS injected muscles, supporting the translatability of PS-USPIONs in tissue systems. The elicited effect of the CA under 0.25T MRI was to brighten versus darken, which was observed under 3T MRI, as is clearly shown on all phantom images. The reversal of influence is due to the extremely low strength of the MRI at only 0.25 T. It has been shown that IO nanoparticles can become positive T2 agents when imaged under low-field MRI,40 as is the case here. Therefore, these preliminary images confirm that our system generates a readable difference in tissue even on a weaker MRI, supporting its general strength in altering relaxivity. This increase in contrast was also measurable, with a 1.63 ± 0.24-fold increase in measurable integrated density when comparing PS-USPION-injected legs to PBS-injected internal controls on the same animal. It is promising that this increase in contrast enhancement is highly repeatable in vivo, demonstrated by a low animal-to-animal deviation. Additional animal imaging at higher magnetic strength (3.0 T or 7.0 T) will be required to gauge clinical effects more realistically.
Pharmacokinetics were examined by synchronized IVIS imaging in both healthy (Wild-Type (WT)) and GM1-affected (βgal−/−) mice to assess release of AF-647 from PS-USPIONs after dose-matched tail vein injections (Fig. 7C). Previously, fluorescein (FITC), encapsulation by PS has been shown to quench fluorescent signals;41 accordingly, an increase in detectable fluorescent signal could then be reliably used to quantify payload release from PSs. Based on our previous studies,42 we expect to see the same quenching effect when our loaded dye, AF-647, is encapsulated in PS-USPIONS. Therefore, we monitored mice post-injection to identify increases in fluorescence over time to indicate in vivo release profiles. We expected to see increased fluorescent release near the injection site in βgal−/− mice, which model GM1 gangliosidosis, leading to an acidic and enzymatic environment43–45 that should lyse HA–PLA PSs and, therefore, PS-USPIONs. The greater radiant efficiency in the GM1-affected mouse tail compared to WT at each time point after tail vein injection suggests preferential PS degradation with disease as the fluorescent signal from free AF-647 becomes detectible. Assuming differences in release are due to differences in the circulatory microenvironment, which is reasonable given the nonspecific administration route and lack of targeting ligands, the greater payload release is likely a result of elevated Hexosaminidase A levels in blood serum in GM1-affected mice.6 This observance lends strong in vivo support for the PS-USPIONs’ performance as a responsive release system, which mimics what was observed in our benchtop release studies performed when optimizing HA–PLA PSs for protein delivery.7 It is encouraging that the in vivo performance of our system matches release profiles expected in pathologic acidic and enzymatic conditions. To increase applicability as a theranostic tool, we could identify time points that tend to represent the greatest PS degradation/payload release to establish an optimum window for real-time MRI monitoring in clinical applications. For this, it would be helpful to establish the time of absolute clearance of the PSs. Our previous work indicates that PSs are cleared at different rates depending on injection routes and targeting ligands.42
Conclusions
We have created a biocompatible, responsive CA-PS system based on USPIONs and HA–PLA PSs. Polymer-matched USPION coating based on the hydrophilic block of the PS copolymer, PLA, was successfully added to ensure the membranous encapsulation of these USPIONs. Using logP matching, have not only left the PS open for co-loading of a model protein, but we have also created a multilamellar vesicle system that will allow for sustained release for continued MRI monitoring, possibly facilitating decreased administration frequency. Through the tight packing into the PS structure, effectively forcing the nanoparticles to act as a single large aggregate, the contrast enhancement observed using PS-USPION is 3-fold greater than the most promising clinical candidate, ferumoxytol. Preliminary in vivo and ex vivo studies in GM1-affected mice and environments confirmed contrast strength and preferential release towards enzymatic, acidic microenvironments. The ultimate result is a targetable, theranostic system that can be tracked until the time of delivery to pathological sites, after which carrier degradation triggers a measurable relaxivity change.
Author contributions
Dorian Foster – conceptualization, data curation, formal analysis, investigation, methodology, validation, visualization, writing – original draft, writing – review & editing; Naisha Shah – investigation, methodology, validation; Alaura Cakley – investigation, methodology, validation; Ronald Beyers – methodology, validation, visualization; Jessica Larsen – conceptualization, data curation, formal analysis, funding acquisition, project administration, resources, supervision, validation, visualization, writing – review & editing.Data availability
The data supporting this article have been included as part of the ESI.† Any additional data requests should be made by contacting the corresponding author.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported in part by the National Science Foundation CAREER program under NSF Award #2047697 and #2308851, and the National Institutes of Health under NIH awards 1R21NS133477, 5P20GM146584, and P20GM139769. Support was also received from Clemson University's Creative Inquiry program and the South Carolina Governor's School for Science and Mathematics Summer Program for Research Interns (SPRI), a program managed by the Clemson University Honors College.We would like to thank Dr Jerryl Jones and Dr Cerano Harrison for their help in designing the postmortem MRI study and analyzing the resulting images.
References
- D. Foster and J. Larsen, Polymeric Metal Contrast Agents for T1-Weighted Magnetic Resonance Imaging of the Brain, in ACS Biomaterials Science and Engineering, American Chemical Society, 2023, vol. 9, pp. 1224–1242 Search PubMed.
- L. Zhu, Y. Yang, K. Farquhar, J. Wang, C. Tian and J. Ranville, et al., Surface Modification of Gd Nanoparticles with pH-Responsive Block Copolymers for Use As Smart MRI Contrast Agents, ACS Appl. Mater. Interfaces, 2016, 8(7), 5040–5050 CrossRef CAS PubMed.
- Y. He, Z. Mao, Z. Lu, J. Yan, Y. Zhang and A. Bianco, et al., Extremely Small Iron Oxide Nanoparticles with pH-Dependent Solubility Transition as T1/T2Switchable Contrast Agents for MRI, ACS Appl. Nano Mater., 2022, 5(10), 15826–15836 CrossRef CAS.
- M. L. Viger, J. Sankaranarayanan, C. De Gracia Lux, M. Chan and A. Almutairi, Collective activation of MRI agents via encapsulation and disease-triggered release, J. Am. Chem. Soc., 2013, 135(21), 7847–7850 CrossRef CAS PubMed.
- D. E. Discher and F. Ahmed, Polymersomes, Annu. Rev. Biomed. Eng., 2006, 8, 323–341 CrossRef CAS PubMed . Available from: https://www.ncbi.nlm.nih.gov/pubmed/16834559.
- B. C. Paruchuri, S. Smith and J. Larsen, Enzyme-responsive polymersomes ameliorate autophagic failure in a cellular model of GM1 gangliosidosis, Front. Chem. React. Eng., 2022, 4, 91 Search PubMed.
- D. Foster, A. Cakley and J. Larsen, Optimizing Enzyme-Responsive Polymersomes for Protein-Based Therapies, Nanomedicine, 2024, 19(3), 213–229 CrossRef CAS PubMed . Available from: https://www.tandfonline.com/doi/full/10.2217/nnm-2023-0300.
- J. M. Kelly, A. L. Gross, D. R. Martin and M. E. Byrne, Polyethylene glycol-b-poly(lactic acid) polymersomes as vehicles for enzyme replacement therapy, Nanomedicine, 2017, 12(23), 2591–2606 CrossRef CAS PubMed . Available from: https://www.futuremedicine.com/doi/10.2217/nnm-2017-0221.
- J. M. Kelly, E. E. Pearce, D. R. Martin and M. E. Byrne, Lyoprotectants modify and stabilize self-assembly of polymersomes, Polymer, 2016, 87, 316–322 CrossRef CAS.
- L. L. Stookey, Ferrozine—a new spectrophotometric reagent for iron, Anal. Chem., 1970, 42(7), 779–781 CrossRef CAS . Available from: https://pubs.acs.org/doi/abs/10.1021/ac60289a016.
- T. M. Jeitner, Optimized ferrozine-based assay for dissolved iron, Anal. Biochem., 2014, 454(1), 36–37 CrossRef CAS PubMed.
- R. T. Pearson, N. J. Warren, A. L. Lewis, S. P. Armes and G. Battaglia, Effect of pH and temperature on PMPC-PDPA copolymer self-assembly, Macromolecules, 2013, 46(4), 1400–1407 CrossRef CAS.
- C. Fetsch, J. Gaitzsch, L. Messager, G. Battaglia and R. Luxenhofer, Self-assembly of amphiphilic block copolypeptoids - Micelles, worms and polymersomes, Sci. Rep., 2016, 6, 33491 CrossRef PubMed.
- P. Hiemenz and R. Rajagopalan, Principles of Colloid and Surface Chemistry, Taylor & Francis Group, Boca Raton, FL, 3rd edn, 1997, pp. 369–371 Search PubMed.
- A. Hellerbach, V. Schuster, A. Jansen and J. Sommer, MRI Phantoms – Are There Alternatives to Agar?, PLoS One, 2013, 8(8), e70343 CrossRef CAS PubMed.
- C. N. Hahn, M. del Pilar Martin, M. Schröder, M. T. Vanier, Y. Hara and K. Suzuki, et al., Generalized CNS disease and massive G(M1)-ganglioside accumulation in mice defective in lysosomal acid β-galactosidase, Hum. Mol. Genet., 1997, 6(2), 205–211 CrossRef CAS PubMed.
- D. R. Martin, B. A. Rigat, P. Foureman, G. S. Varadarajan, M. Hwang and B. K. Krum, et al., Molecular consequences of the pathogenic mutation in feline GM1gangliosidosis, Mol. Genet. Metab., 2008, 94(2), 212–221 CrossRef CAS PubMed.
- D. Foster, L. Williams, N. Arnold and J. Larsen, Therapeutic developments for neurodegenerative GM1 gangliosidosis, in Frontiers in Neuroscience, Frontiers Media SA, 2024, vol. 18 Search PubMed.
- E. Rideau, R. Dimova, P. Schwille, F. R. Wurm and K. Landfester, Liposomes and polymersomes: a comparative review towards cell mimicking, Chem. Soc. Rev., 2018, 47(23), 8572–8610 RSC.
- D. E. Discher, V. Ortiz, G. Srinivas, M. L. Klein, Y. Kim and D. Christian, et al., Emerging applications of polymersomes in delivery: From molecular dynamics to shrinkage of tumors, Prog. Polym. Sci., 2007, 32(8–9), 838–857 CrossRef CAS PubMed.
- D. Furtado, M. Björnmalm, S. Ayton, A. I. Bush, K. Kempe and F. Caruso, Overcoming the Blood-Brain Barrier: The Role of Nanomaterials in Treating Neurological Diseases, Adv. Mater., 2018, 1801362 CrossRef PubMed . Available from: https://doi.wiley.com/10.1002/adma.201801362.
- R. Toy, L. Bauer, C. Hoimes, K. B. Ghaghada and E. Karathanasis, Targeted nanotechnology for cancer imaging, in Advanced Drug Delivery Reviews, Elsevier B.V., 2014, vol. 76, pp. 79–97 Search PubMed.
- D. Vorselen, M. Marchetti, C. López-Iglesias, P. J. Peters, W. H. Roos and G. J. L. Wuite, Multilamellar nanovesicles show distinct mechanical properties depending on their degree of lamellarity, Nanoscale, 2018, 10(11), 5318–5324 RSC.
- Z. Song, H. Kim, X. Ba, R. Baumgartner, J. S. Lee and H. Tang, et al., Polypeptide vesicles with densely packed multilayer membranes, Soft Matter, 2015, 11(20), 4091–4098 RSC.
- J. Habel, A. Ogbonna, N. Larsen, S. Cherré, S. Kynde and S. R. Midtgaard, et al., Selecting analytical tools for characterization of polymersomes in aqueous solution, RSC Adv., 2015, 5(97), 79924–79946 RSC.
- M. A. Chaves, P. L. Oseliero Filho, C. G. Jange, R. Sinigaglia-Coimbra, C. L. P. Oliveira and S. C. Pinho, Structural characterization of multilamellar liposomes coencapsulating curcumin and vitamin D3, Colloids Surf., A, 2018, 549, 112–121 CrossRef CAS.
- X. Zhang, Y. Liu, Y. J. Kim, J. Mac, R. Zhuang and P. Wang, Co-delivery of carboplatin and paclitaxel via cross-linked multilamellar liposomes for ovarian cancer treatment, RSC Adv., 2017, 7(32), 19685–19693 RSC.
- S. M. Gruner, R. P. Lenk, A. S. Janoff and M. J. Ostro, Downloaded via CLEMSON UNIV on [Internet], in Biochemistry, UTC, 1985, vol. 24. Available from: https://pubs.acs.org/sharingguidelines Search PubMed.
- K. I. Joo, L. Xiao, S. Liu, Y. Liu, C. L. Lee and P. S. Conti, et al., Crosslinked multilamellar liposomes for controlled delivery of anticancer drugs, Biomaterials, 2013, 34(12), 3098–3109 CrossRef CAS PubMed . Available from: https://www.ncbi.nlm.nih.gov/pubmed/23375392.
- Y. Liu, J. Fang, Y. J. Kim, M. K. Wong and P. Wang, Codelivery of doxorubicin and paclitaxel by cross-linked multilamellar liposome enables synergistic antitumor activity, Mol. Pharm., 2014, 11(5), 1651–1661 CrossRef CAS PubMed.
- S. Allen, O. Osorio, Y. G. Liu and E. Scott, Facile assembly and loading of theranostic polymersomes via multi-impingement flash nanoprecipitation, J. Controlled Release, 2017, 262, 91–103 CrossRef CAS PubMed.
- M. Krack, H. Hohenberg, A. Kornowski, P. Lindner, H. Weller and S. Förster, Nanoparticle-loaded magnetophoretic vesicles, J. Am. Chem. Soc., 2008, 130(23), 7315–7320 CrossRef CAS PubMed.
- D. Chandler, Interfaces and the driving force of hydrophobic assembly, Nature, 2005, 437(7059), 640–647 CrossRef CAS PubMed.
- E. D. Smolensky, H. Y. E. Park, Y. Zhou, G. A. Rolla, M. Marjańska and M. Botta, et al., Scaling laws at the nanosize: The effect of particle size and shape on the magnetism and relaxivity of iron oxide nanoparticle contrast agents, J. Mater. Chem. B, 2013, 1(22), 2818–2828 RSC.
- M. Cho, J. Villanova, D. M. Ines, J. Chen, S. S. Lee and Z. Xiao, et al., Sensitive T2 MRI Contrast Agents from the Rational Design of Iron Oxide Nanoparticle Surface Coatings, J. Phys. Chem. C, 2023, 127(2), 1057–1070 CrossRef CAS.
- G. Knobloch, T. Colgan, C. N. Wiens, X. Wang, T. Schubert and D. Hernando, et al., Relaxivity of Ferumoxytol at 1.5 T and 3.0 T, Invest. Radiol., 2018, 53(5), 257–263 CrossRef CAS PubMed.
- Q. Liu, L. Song, S. Chen, J. Gao, P. Zhao and J. Du, A superparamagnetic polymersome with extremely high T2 relaxivity for MRI and cancer-targeted drug delivery, Biomaterials, 2017, 114, 23–33 CrossRef CAS PubMed.
- M. Dovedytis, Z. J. Liu and S. Bartlett, Hyaluronic acid and its biomedical applications: A review, in Engineered Regeneration, KeAi Communications Co., 2020, vol. 1, pp. 102–113 Search PubMed.
- P. Snetkov, K. Zakharova, S. Morozkina, R. Olekhnovich and M. Uspenskaya, Hyaluronic Acid: The Influence of Molecular Weight on Structural, Physical, Physico-Chemical, and Degradable Properties of Biopolymer, Polymers (Basel), 2020, 12(8), 1800 CrossRef CAS PubMed.
- J. K. Van Zandwijk, F. F. J. Simonis, F. G. Heslinga, E. I. S. Hofmeijer, R. H. Geelkerken and B. Haken, Comparing the signal enhancement of a gadolinium based and an iron-oxide based contrast agent in low-field MRI, PLoS One, 2021, 16(8), e0256252 CrossRef CAS PubMed.
- E. Scarpa, J. L. Bailey, A. A. Janeczek, P. S. Stumpf, A. H. Johnston and R. O. C. Oreffo, et al., Quantification of intracellular payload release from polymersome nanoparticles, Sci. Rep., 2016, 6, 29460 CrossRef PubMed.
- K. Trumbull, S. Fetten, N. Arnold, V. Marahrens, D. Montgomery, O. Myers, J. L. Twiss and J. Larsen, Targeted Polymersomes Enable Enhanced Delivery to Peripheral Nerves Post-Injury, Bioconjugate Chem., 2025, 36(4), 823–837 CrossRef CAS PubMed.
- N. Brunetti-Pierri and F. Scaglia, GM1 gangliosidosis: review of clinical, molecular, and therapeutic aspects, Mol. Genet. Metab., 2008, 94(4), 391–396 CrossRef CAS PubMed . Available from: https://www.ncbi.nlm.nih.gov/pubmed/18524657.
- H. L. Gray-Edwards, D. S. Regier, J. L. Shirley, A. N. Randle, N. Salibi and S. E. Thomas, et al., Novel Biomarkers of Human GM1 Gangliosidosis Reflect the Clinical Efficacy of Gene Therapy in a Feline Model, Mol. Ther., 2017, 25(4), 892–903 CrossRef CAS PubMed.
- V. J. McCurdy, A. K. Johnson, H. L. Gray-Edwards, A. N. Randle, B. L. Brunson and N. E. Morrison, et al., Sustained Normalization of Neurological Disease after Intracranial Gene Therapy in a Feline Model, Sci. Transl. Med., 2014, 6(231), 231ra48 Search PubMed . Available from: https://www.ncbi.nlm.nih.gov/pubmed/25792328.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4bm01583e |
| This journal is © The Royal Society of Chemistry 2025 |