
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnderstanding metabolic characteristics and molecular mechanisms of large to giant congenital melanocytic nevi: implications for melanoma risk and therapeutic targets†
Ge
Song‡
 ac,
Tao
Dai‡
b,
Yongqiang
Ren
c,
Yajie
Chang
a,
Pengfei
Guo
a,
Zhanwei
Wang
b,
Guiping
Shen
*a and
Jianghua
Feng
*a
ac,
Tao
Dai‡
b,
Yongqiang
Ren
c,
Yajie
Chang
a,
Pengfei
Guo
a,
Zhanwei
Wang
b,
Guiping
Shen
*a and
Jianghua
Feng
*a
aDepartment of Electronic Science, Fujian Provincial Key Laboratory of Plasma and Magnetic Resonance, Xiamen University, Xiamen 361005, China. E-mail: jianghua.feng@xmu.edu.cn; gpshen@xmu.edu.cn
bDepartment of Wound Repair, Tongji Hospital Affiliated Tongji University, Shanghai 200065, China
cDepartment of Plastic Surgery, First Affiliated Hospital of Henan University of Science and Technology, Luoyang 471003, China
First published on 24th March 2025
Abstract
Large to giant congenital melanocytic nevi (LGCMN) present clinical challenges due to their complex phenotypic heterogeneity and increased melanoma risk. Molecular-level research is essential for understanding the pathogenic mechanisms of LGCMN and identifying potential therapeutic targets. Tissue samples from 67 LGCMN lesions and 49 matched controls were analyzed using metabolomics and transcriptomics to identify metabolic characteristics and gene expression differences. A protein–protein interaction network and a multi-layer network of key metabolites–genes-pathways were established to explore the metabolic characteristics and gene associations with LGCMN. Metabolic analysis revealed a consistent dysregulation in amino acid metabolisms, including arginine, alanine, aspartate, glutamate, phenylalanine, and tyrosine, across LGCMN lesions and subtypes. Compared to controls, 18 upregulated metabolites and 7 downregulated metabolites were identified in LGCMN lesions. Metabolic profiles varied among LGCMN subtypes, with the trunks subtype exhibiting significant alterations in branched-chain amino acids. Network analysis identified 23 genes related to melanogenesis and amino acid metabolism, including TYR, SOX10, and MITF, which showed strong correlation with tyrosine, phenylalanine, and branched-chain amino acids (r > 0.6). High centrality values for genes (e.g., EDNRB, TYR, MITF, SOX10, and MAPK3 > 0.300) and amino acids (e.g., tyrosine at 0.397 and phenylalanine at 0.374) emphasize their pivotal roles in melanogenesis. This study reveals significant metabolic and molecular differences between LGCMN lesions, normal skin, and across LGCMN subtypes, highlighting the deregulation of amino acid metabolism and key genes involved in melanogenesis. These insights enhance our understanding of LGCMN's biological heterogeneity and provide novel avenues for therapeutic intervention.
1. Introduction
Recent medical research has improved our understanding of congenital melanocytic nevi (CMN).1,2 Large CMN (LCMN, 20–40 cm in diameter) and giant CMN (GCMN, >40 cm in diameter) are rare, neural crest-derived melanocytic lesions3,4 that tend to enlarge with age and are often characterized by a rough and irregular surface. The prevalence of large to giant CMN (LGCMN) is approximately 1 in 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 to 1 in 500000, and their complex morphology and heterogeneous structure pose significant challenges for surgical removal and increase the risk of malignant transformation into melanoma.5
000 to 1 in 500000, and their complex morphology and heterogeneous structure pose significant challenges for surgical removal and increase the risk of malignant transformation into melanoma.5
Surgical intervention, while effective in removing large skin lesions, can result in significant surgical trauma, postoperative wound healing challenges, and potential functional impairments, imposing substantial psychological and economic burdens on patients.6 Non-surgical treatments, such as laser therapy and chemical peeling, offer symptomatic relief but cannot completely remove the lesions and may increase the risks of recurrence and malignancy. These limitations highlight the importance of in-depth molecular-level research into the pathological mechanisms of LGCMN.
Multi-omics approaches have become a focal point in disease research, aiming to uncover novel therapeutic targets and develop more effective treatment strategies.7 Notably, NRASQ61R and BRAF have been identified as major somatic mutations in LGCMN, which persistently activate the MAPK signaling pathway in melanocytes.8 Studies on immortalized cell lines originating from GCMN have revealed heightened activity in PI3K/Akt and Bcl-2 pathways, suggesting their crucial role in the maintenance and survival of LGCMN. Transcriptomic studies have also identified abnormal expression of various transcription factors (especially MITF) in LGCMN. Histone deacetylase inhibitors, such as vorinostat (SAHA), can suppress MITF expression and induce cell apoptosis, providing a promising strategy for non-surgical treatment of LGCMN.9 RNA sequencing of distinct LGCMN features suggests that different regions of the lesion may be driven by diverse molecular events, resulting in phenotypic variations.8 Metabolomics studies provide new insights into the pathological mechanisms of LGCMN. Research indicates significant abnormalities in amino acid metabolism, tricarboxylic acid (TCA) cycle, and carbohydrate metabolism pathways among LGCMN patients. 1H nuclear magnetic resonance (1H NMR)-based metabolomics studies have revealed alterations in multiple metabolite levels in the serum and urine of GCMN patients, particularly in amino acid and energy metabolism, suggesting their key role in disease progression.10 Additionally, primary cutaneous melanoma exhibits metabolic reprogramming, with significant changes in tryptophan and histidine metabolism pathways.11 The associations between changes in arginine metabolism and glycerophospholipids and melanoma progression highlight the significance of metabolic regulation in lesion processes.12
In this study, we employed 1H NMR-based metabolomics to discern metabolic differences between LGCMN lesions and normal control skin, as well as among subtypes. We also employed transcriptomics (RNA-seq) to identify differentially expressed genes, integrating network pharmacology and various algorithms to determine potential targets for LGCMN. Ultimately, a gene–metabolite-pathway network was constructed for a comprehensive evaluation. The findings from this study provide a foundation for further investigation into the pathogenesis of LGCMN and offer possibilities for the development of targeted therapies for these lesions.
2. Methods
2.1 Sample collection
This study was approved by the Medical Ethics Committee of Tongji Hospital affiliated to Tongji University, China (SBKT-2023-149). Written informed consent was obtained from all participants. Patients with LGCMN were diagnosed by experienced plastic surgeons based on clinical presentations and confirmed postoperatively through histopathology using the BEST classification system.13 From January 2020 to September 2023, a total of 67 LGCMN patients undergoing surgical excision were enrolled in this study. Of these, perilesional normal skin samples were successfully obtained from 49 patients due to intraoperative constraints. These 49 adjacent normal tissues were matched one-to-one with their corresponding lesion samples. Among these, five patients were diagnosed with LGCMN and concomitant other diseases during pathological examination, including three with neurofibromatosis, one with leiomyoma, and one with malignant melanoma. All collected tissues, including LGCMN lesion samples and corresponding perilesional normal skin (control, more than 5 mm away from the tumor margin), were stripped of subcutaneous fat while retaining the epidermis and dermis, then placed in liquid nitrogen and stored at −80 °C for subsequent analyses.2.2 Pathological examination of skin tissues
Tissue blocks soaked in 4% paraformaldehyde were dehydrated, cleared, waxed-embedded, and sectioned coronally at a thickness of 4 μm using a rotary microtome. The sections were dewaxed and stained with hematoxylin and eosin (HE). The stained samples were examined under an Olympus light microscope (Olympus Optical Co., Japan).2.3 1H NMR analysis and data processing
Approximately 150 mg of tissue was extracted using the Bligh–Dyer method to obtain freeze-dried powder.14 This powder was then mixed with 0.6 mL of ice-cooled extraction solution (CH3OH:CHCl3:H2O = 2:2:3). The mixture was vortexed and left on ice for 15 min, followed by centrifugation at 4 °C at 10000 g for 15 min to obtain the supernatant. The supernatant was transferred to a 1.5 mL tube and lyophilized for 24 h to remove methanol and water. The resulting freeze-dried tissue powder was dissolved in 1.5 mol L−1 phosphate buffer prepared with 500 μL of D2O (pH 7.4, containing 0.05% sodium 3-(trimethylsilyl) propionate-2,2,3,3-d4 (TSP)). Each sample was mixed and centrifuged at 4 °C at 10000 g for 10 min. The supernatant was then transferred into a 5 mm NMR tube.
1H NMR spectra were acquired on a Bruker Advance III spectrometer (Bruker Corporation, Kalsruhe, Germany) operating at 850.29 MHz and 298 K. A NOESYGPPR1D (RD-90°-t1-90°-tm-90°-Acq) was used to detect small molecule metabolites. A total of 64 transients were collected into 32 K data points over a spectral width of 17 kHz. The acquisition time was set to 1.93 s, with a relaxation delay of 3.0 s and a fixed interval t1 of 4 μs. The water resonance was irradiated during the relaxation delay, and the mixing time tm was 100 ms. NMR spectra were processed using MestReNova (Version 14.1.1, Mestrelab Research S. L., Spain), including phase and baseline corrections, window selection (δ0.6–9.8), and removal of water (δ4.68–5.18) and methanol (δ3.32–3.38) peaks. Data were normalized to the total spectral area to account for sample concentration differences and imported into MATLAB (R2014b, MathWorks, USA) for spectral alignment using the icoshift package.15
2.4 RNA sequence analysis and data processing
Total RNA was extracted from skin tissues using an RNeasy Fibrous Tissue Mini Kit (Qiagen, USA). RNA quality was assessed with a nanophotometer (IMPLEN, USA) and Agilent Bioanalyzer 2100 (Agilent, USA), retaining only samples with an RNA Integrity Number (RIN) ≥ 7.0. cDNA libraries were prepared using an NEBNext Ultra RNA Library Prep Kit for Illumina (NEB, USA) and sequenced. Clean reads were obtained by removing adapters, poly-N sequences, and low-quality reads. Reads were aligned to the reference genome using Hisat2 v2.0.5, and gene expression was quantified with Cufflinks v2.2.1. RNA-seq data were first normalized using the Trimmed Mean of M-values (TMM) method in the edgeR package to correct for sequencing depth and composition bias. Differential expression analysis was then performed using the limma R package and Student's t-test, considering genes with a fold change >2 and adjusted p < 0.05 as significantly differentially expressed.2.5 Metabolites and gene set enrichment, pathway and molecular network analysis
Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses were performed to elucidate the biological significance of differentially expressed genes. GO analysis (http://geneontology.org/) categorized target genes according to biological processes, cellular components, and molecular functions, while KEGG pathway analysis (https://www.kegg.jp/) identified relevant signaling pathways. Both analyses were conducted using the clusterProfiler packages in R, considering a p < 0.05 as indicative of statistically significant enrichment pathways associated with the candidate genes. The Human Metabolome Database (HMDB, https://www.hmdb.ca/) provides comprehensive information on metabolites. Differential genes identified from the volcano plot were used to construct a protein–protein interaction (PPI) network in the STRING database (https://string-db.org/) with a confidence score > 0.4. In this study, we employed three distinct methods, MCODE, cytoNCA, and cytoHubba plugins in Cytoscape (v3.9.1, Cytoscape Consortium, USA), to identify hub genes. Each method utilizes a unique algorithm to assess network topology. MCODE was used to identify densely connected subnetworks within the PPI network. Subnetworks with an MCODE score > 5 were selected as significant modules. cytoNCA evaluated six topological metrics (betweenness, closeness, degree, eigenvector, LAC, and network centrality), selecting hub genes that exceeded the median values for all six metrics through three iterative rounds. cytoHubba ranked genes based on five centrality metrics (closeness, betweenness, stress, radiality, and bottleneck), defining hub genes as those appearing in all five rankings. The intersection and pairwise unions of the three methods were extracted using Venn diagram analysis to determine central hub genes. Finally, gene–metabolite correlations and enriched pathways were integrated to construct a multi-layer gene–metabolite-pathway network.2.6 Statistical analyses
Multivariate statistical analysis was conducted using SIMCA (v14.1, Umetrics, Umea, Sweden). Principal component analysis (PCA) was used to assess sample distribution and identify clusters or outliers, while supervised partial least squares discriminant analysis (PLS-DA) and orthogonal PLS-DA (OPLS-DA) with Pareto scaling were utilized to highlight metabolic differences between groups. Model quality was evaluated using R2X, R2Y, and Q2 values obtained from cross-validation, and permutation tests with 200 iterations were conducted to confirm model reliability and prevent overfitting. Significant metabolites were identified based on their absolute correlation coefficient (|r|), variable importance in projection (VIP), fold change, and p-value and were visualized in a four-dimensional volcano plot, where fold change and p-value served as axes and |r| and VIP were represented by color and size.All other statistical analyses, including non-parametric tests, Student's t-tests, and chi-square (χ2) tests, were conducted using OriginPro (v2024b, OriginLab, Northampton, MA, USA). The results were expressed as mean ± standard deviation (SD). T-tests were used to assess differences in continuous variables, chi-square tests were used for categorical data, and the Wilcoxon test was applied for non-normally distributed data. Visualizations such as bubble plots, Venn diagrams, and correlation heatmaps were also generated using OriginPro. A p-value of less than 0.05 was considered statistically significant.
3. Results
3.1 Demographic data and clinical characteristics of the patients
A total of 67 patients were enrolled, providing 67 samples of LGCMN tissue and 49 matched samples of perilesional normal tissue. Table 1 details the patients and tissue characteristics, which predominantly consist of intradermal nevi and compound nevi. The pathological tissue images are shown in Fig. 1a. HE staining revealed the structural variations of different types of nevi with distinct patterns and depths of melanocyte distribution.| # Subjects | LGCMN tissues | Matched control tissues | p-value |
|---|---|---|---|
| Total (N = 67) | Total (N = 49) | ||
| Gender | 67 | 49 | 0.263 |
| Male | 26 (38.81%) | 25 (51.02%) | — |
| Female | 41 (61.19%) | 24 (48.98%) | — |
| Age at diagnosis (years) | 5.17 ± 7.19 | 4.38 ± 8.09 | 0.330 |
| Distribution pattern | 0.699 | ||
| Bonce | 13 (19.40%) | 14 (28.57%) | — |
| Extremity | 8 (11.94%) | 6 (12.24%) | — |
| Shawl | 20 (29.85%) | 13 (26.53%) | — |
| Trunks | 26 (38.81%) | 16 (32.65%) | — |
| Pathological types | 0.540 | ||
| Intradermal nevus | 31 (46.27%) | 27(55.10%) | — |
| Compound nevus | 35 (52.24%) | 22(44.90%) | — |
| Junctional nevus | 1 (1.49%) | 0 | — |
| Comorbidities | — | — | |
| Neurofibromatosis | 3 (4.48%) | — | — |
| Leiomyoma | 1 (1.49%) | — | — |
| Malignant melanoma | 1 (1.49%) | — | — |
 | ||
| Fig. 1 Histological and metabolomic differences between LGCMN lesions and control skin. (a) HE staining of intradermal, junctional, and compound nevi (×100, ×200). (b) PCA, OPLS-DA plots, and model validation by permutation analysis. (c) Volcano plot of significant differential metabolites (|r| > 0.6, VIP > 1, and p < 0.01). (d) Bubble plot of disturbed metabolic pathways. | ||
3.2 Metabolomics analysis and identification of key metabolic pathways in LGCMN tissues
To investigate the potential pathogenesis of LGCMN, metabolomics analysis was conducted on the 1H NMR data of the tissues. Metabolites were identified from NMR spectra of the LGCMN tissues with references to the existing literature16,17 and confirmed through the HMDB. Spectral details of the identified metabolites are provided in supplementary Table S1.† The PCA score plot revealed a significant separation between the two groups of samples (Fig. 1b, left), reflecting a substantial metabolic difference between LGCMN and perilesional control skins. To enhance the resolution and effectiveness of the model, OPLS-DA was conducted on the NMR data. The score plot (Fig. 1b, middle) and model parameters (R2Y and Q2) indicated significant differences between LGCMN and control groups. The permutation test (Fig. 1b, right) confirmed the validity of the model. Twenty-five metabolites were identified to be differentially expressed between the two groups using |r| > 0.6, VIP > 1, and p < 0.01 as the critical criteria. Among them, 18 metabolites were upregulated in LGCMN skin, including citrulline, alanine, phenylalanine, serine, glycine, hypoxanthine, tyrosine, choline, phosphorylcholine (PC), aspartate, trimethylamine N-oxide (TMAO), glutamate, lysine, valine, isoleucine, leucine, phosphocreatine (PCr), and glycerophosphorylcholine (GPC), while 7 metabolites were downregulated, including 3-hydroxybutyrate, N-acetylglutamate (NAG), methylmalonate, α-ketoisovalerate, asparagine, pyruvate, and methionine. The corresponding volcano plot displayed the metabolites contributing to classification (Fig. 1c and ESI Table S2†).To explore the potential metabolic mechanisms of LGCMN, KEGG pathway analysis was performed on the differential metabolites. The resultant bubble plot illustrated the significance and impact of various metabolic pathways (Fig. 1d). The metabolic differences between LGCMN and control tissues are primarily influenced by several amino acid metabolism pathways. Notably, arginine biosynthesis, alanine, aspartate and glutamate metabolism, glycine, serine and threonine metabolism, and phenylalanine, tyrosine and tryptophan biosynthesis exhibited the highest significance, indicating their critical roles in the metabolic network. Other important pathways include valine, leucine and isoleucine degradation and phenylalanine metabolism, highlighting their relevance in the overall metabolic landscape.
3.3 Specific metabolic features of different LGCMN subtypes
To investigate the heterogeneity within LGCMN and link it to metabolic diversity, metabolomic analysis was conducted on tissue samples from different phenotypic LGCMN lesions. However, PCA and PLS-DA score plots did not effectively distinguish between different LGCMN phenotypes (Fig. S1†). To enhance the resolution of subtype differences, OPLS-DA was performed on the pair-wise groups including bonce LGCMN vs. control-bonce, extremity LGCMN vs. control-extremity, shawl LGCMN vs. control-shawl, and trunks LGCMN vs. control-trunks. The permutation test results confirmed the model's strong explanatory and predictive power, underscoring significant metabolic differences between LGCMN lesions and control skin. Volcano plots (Fig. 2a–d), integrating the correlation coefficients, VIP values, fold changes, and t-test P values (ESI Table S2†), were used to identify potential biomarkers for each subtype. | ||
| Fig. 2 OPLS-DA score plots (left), permutation tests (middle), and volcano plots (right) for 1H NMR data of LGCMN lesions and peri-lesion skin in four subtypes: bonce (a), extremity (b), shawl (c), and trunks (d). Differential metabolites (|r| > 0.6, VIP > 1, and p < 0.01) are shown, with key metabolites listed in Table S2.† | ||
A Venn diagram illustrated significant metabolic differences among the four LGCMN subtypes (Fig. 3a, left). For example, the trunks subtype uniquely contained 6 metabolites (aspartate, choline, leucine, lysine, pyruvate, and α-ketoisovalerate), while also sharing 5 metabolites with the shawl subtype (3-hydroxybutyrate, aspartate, isoleucine, NAG, and tyrosine), suggesting a certain degree of similarity in their metabolic profiles. Notably, only the trunks subtype included all branched chain amino acids (BCAAs: leucine, isoleucine, and valine). Among the 30 differential metabolites, 21 demonstrated significant differences in their relative concentrations across the four LGCMN subtypes as shown in the box plots (Fig. 3a, right). These findings highlight distinct metabolic differences among subtypes, providing a comprehensive view of metabolic characteristics in each LGCMN subtype.
 | ||
| Fig. 3 Potential biomarkers and differential metabolic pathways in LGCMN subtypes (bonce, extremity, shawl, and trunks). (a) Venn diagram of biomarkers between lesions and adjacent tissues (left) and their relative concentrations analyzed by the Wilcoxon test (right, ***p < 0.001, **p < 0.01, *p < 0.05, and ns: not significant). (b) Bubble plot of disrupted pathways, with deeper reds indicating higher significance (p < 0.05). | ||
Pathway enrichment analysis of these potential biomarkers was conducted to further understand the metabolic dysregulation among different subtypes of LGCMN. Based on a criterion of p < 0.05, eight, three, ten, and seven disrupted metabolic pathways were identified from the bonce, extremity, shawl, and trunks subtypes, respectively (Fig. 3b and Table S3†). Notably, the shawl and trunks subtypes exhibited pronounced disruptions in amino acid metabolism (for example, arginine biosynthesis, alanine, aspartate and glutamate metabolism, glycine, serine and threonine metabolism, and phenylalanine, tyrosine, and tryptophan biosynthesis) and energy metabolism (for example, pantothenate and CoA biosynthesis). This suggests distinct metabolic characteristics between these subtypes. Furthermore, significant metabolic disruption in valine, leucine, and isoleucine degradation was exclusively observed in the trunks subtype.
3.4 Systematic screening and analysis of differential genes and core networks in LGCMN
In the transcriptomic analysis of 10 matched pairs of LGCMN lesions and adjacent control tissues (details provided in ESI Table S4†), the OPLS-DA score plot revealed their distinct transcriptomic profiles. The model's robustness and predictive strength were confirmed with R2Y = 0.943 and Q2 = 0.893 (Fig. 4a). A volcano plot revealed 991 differentially expressed genes (DEGs) with at least twofold changes (P < 0.05), of which 395 were upregulated and 596 were downregulated (Fig. 4a, right). The DEGs were imported into the STRING database for topological analysis. After removing isolated nodes, a protein–protein interaction (PPI) network comprising 612 nodes and 921 edges was obtained. This initial PPI network was further analyzed using Cytoscape 3.9.1, where the MCODE plugin identified 22 functional modules. Six clusters with scores greater than 5 were highlighted (Fig. 4b), with details provided in Table 2.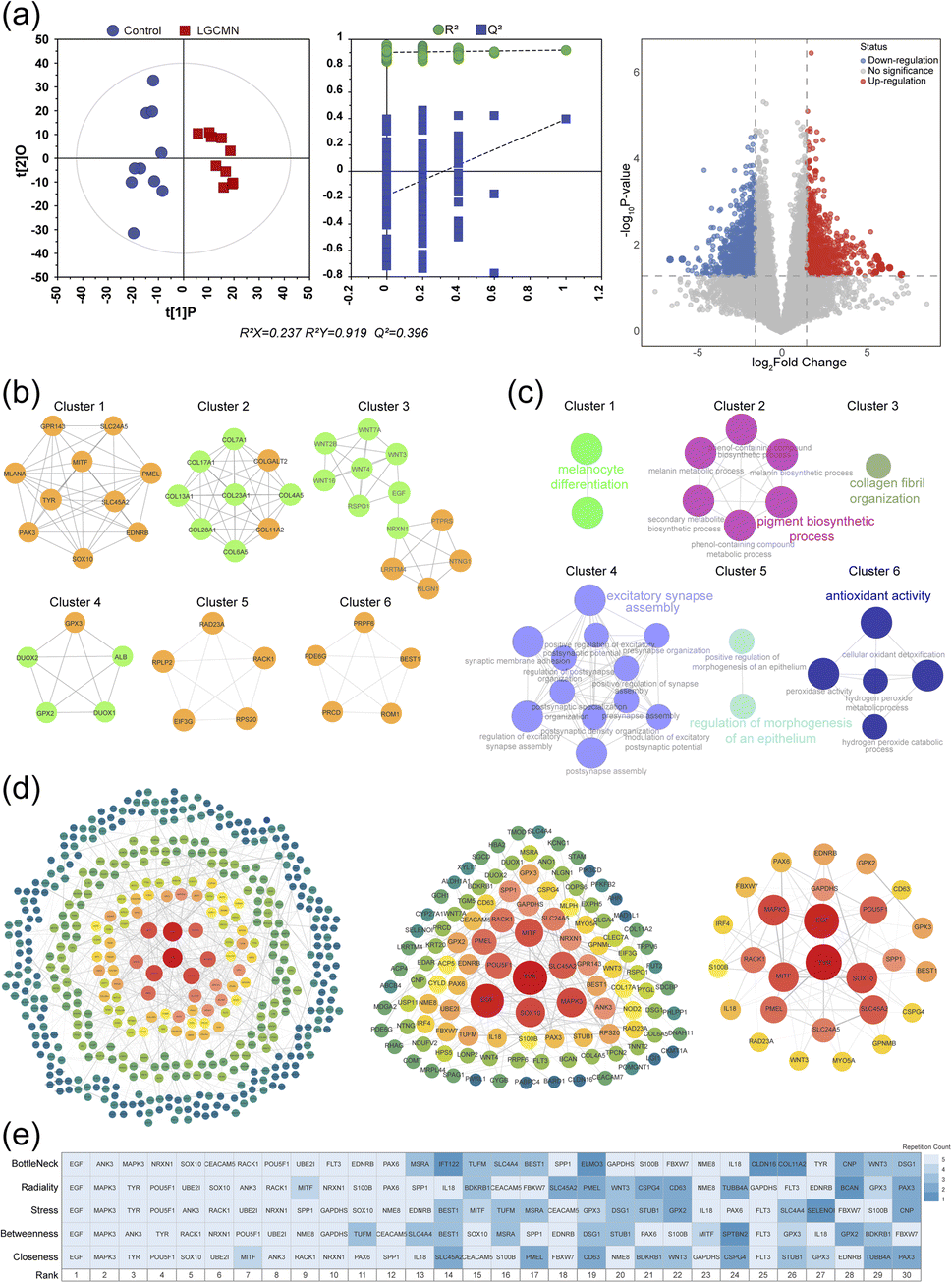 | ||
| Fig. 4 Comprehensive analysis of DEGs in LGCMN. (a) OPLS-DA plots and volcano plots highlight DEGs (|log2FC| > 2 and padj < 0.05). (b) Protein–protein interaction network (MCODE) identifies six gene clusters. (c) ClueGO enrichment analysis for clusters. (d) Three-layer hub gene network (cytoNCA) with centrality-based nodes. (e) Heatmap of top 30 hub genes ranked by cytoHubba algorithms. | ||
| Cluster | MCODE score | Nodes | Edges | Gene |
|---|---|---|---|---|
| 1 | 9.111 | 10 | 41 | PAX3, MITF, MLANA, PMEL, EDNRB, GPR143, SLC24A5, SLC45A2, TYR, and SOX10 |
| 2 | 9 | 9 | 36 | COL7A1, COL4A5, COL6A5, COL11A2, COL28A1, COL17A1, COL23A1, COL13A1, and COLGALT2 |
| 3 | 5.091 | 12 | 28 | NLGN1, WNT2B, NRXN1, WNT7A, EGF, WNT16, LRRTM4, PTPRS, WNT3, RSPO1, NTNG1, and WNT4 |
| 4 | 5 | 5 | 10 | DUOX1, GPX2, DUOX2, ALB, and GPX3 |
| 5 | 5 | 5 | 10 | RPLP2, RPS20, RACK1, EIF3G, and RAD23A |
| 6 | 5 | 5 | 10 | PDE6G, ROM1, PRPF6, PRCD, and BEST1 |
Enrichment analyses of the six clusters showed the key processes such as melanocyte differentiation in cluster 1, the pigment biosynthetic process in cluster 2, collagen fibril organization in cluster 3, excitatory synapse assembly in cluster 4, regulation of morphogenesis of an epithelium in cluster 5, and antioxidant activity in cluster 6 (Fig. 4c). These findings suggest that the hub genes within these functional modules are meaningful and align with the pathological mechanisms associated with LGCMN.
To ensure robust identification of hub genes, MCODE, cytoNCA, and cytoHubba were combined to capture network centrality and connectivity. As shown in Fig. 4d, a three-layer hub gene network was constructed and analyzed with cytoNCA, finally identifying 27 hub genes (Fig. 4d, right). Additionally, a heatmap was generated to rank the top 30 hub genes based on five different centrality algorithms (closeness, betweenness, stress, radiality, and bottleneck) in cytoHubba, from which 19 core genes were selected for further analysis (Fig. 4e and Table S5†). This multi-dimensional approach ensures the robustness of the selection, highlighting key genes consistently identified across different analytical methods.
To refine the selection of hub genes, a Venn diagram was used to integrate the results from MCODE, cytoNCA, and cytoHubba. The final set comprised five genes identified by all three methods including EDNRB, TYR, SOX10, EGF, and RACK1 and an additional 18 genes from pairwise intersections, resulting in 23 key genes (Fig. 5a and Table S6†). These hub genes were subjected to GO and KEGG pathway analyses, revealing significant enrichment in biological processes such as pigmentation, melanocyte differentiation, and melanin biosynthesis; cellular components such as the melanosome membrane and pigment granules; and molecular functions including protein binding and oxidative activities (Fig. 5b and Table S7†). The KEGG pathway analysis highlighted pathways associated with melanogenesis, stem cell pluripotency signaling, and cancer-related pathways, including tyrosine metabolism, MAPK signaling, and PD-1/PD-L1 immune interaction. This functional annotation highlights the central roles of these hub genes in key biological processes and pathways, suggesting their potential as therapeutic targets in LGCMN.
 | ||
| Fig. 5 Analysis of key hub genes and their correlations with metabolites and pathways. (a) Venn diagram of hub genes identified using MCODE, cytoNCA, and cytoHubba. (b) GO and KEGG enrichment analysis with corresponding bubble plot. (c and d) Correlation between hub genes and metabolites (*p < 0.05). (e) Cytoscape network of metabolites, genes, and pathways, with correlations assessed using the Spearman coefficient. | ||
Spearman correlation analysis revealed significant associations among the 23 hub genes (Fig. 5c), suggesting co-regulation in shared biological processes. EDNRB showed strong positive correlations with TYR, SOX10, MITF, SLC24A5, and SLC45A2 (r > 0.6), indicating their joint involvement in melanogenesis. Similarly, TYR correlated positively with SOX10 and MITF (r > 0.6), supporting their cooperative roles in melanocyte function. Moderate correlations between EGF, RACK1, and MAPK3 (0.4 < r < 0.6) suggest their interactions in cell proliferation and signaling.
Fig. 5d shows correlations between 23 hub genes and various metabolites, emphasizing links between gene expression and metabolic processes. EDNRB, TYR, SOX10, MITF, PMEL, SLC24A5, and SLC45A2 showed strong positive correlations with tyrosine and phenylalanine (melanin precursors) (r > 0.6) and BCAA, indicating that these genes and amino acids may have mutual regulatory roles in melanogenesis. MAPK3 was positively correlated with PC, tyrosine, and phenylalanine, and negatively with 3-hydroxybutyrate, suggesting regulatory roles in signaling and energy metabolism.
The gene–metabolite-pathway interaction network offers a more holistic and integrated perspective (Fig. 5e). It reveals significant disruption in amino acid metabolism and melanogenesis pathways in LGCMN, with particular involvement of genes such as TYR, MITF, MAPK3, SLC45A2, PMEL, SLC24A5, EDNRB, SOX10, and RACK1, all of which exhibit a closeness centrality above 0.300. Tyrosine and phenylalanine also exhibited high closeness centrality values (0.397 and 0.374, respectively), further emphasizing their centrality in the melanogenic process. The interactions of 3-hydroxybutyrate, leucine, valine, and isoleucine with genes such as TYR, MITF, and SOX10 suggest a role of BCAA in melanocyte metabolism and proliferation. Although melanogenesis and tyrosine metabolism pathways may have fewer direct connections, they play central roles through their associations with key genes and metabolites. Overall, the interaction between melanogenesis and amino acid metabolism is particularly pronounced in the network analysis, underscoring the critical importance of these pathways in the pathophysiology of LGCMN.
4. Discussion
4.1 Metabolic abnormalities and potential pathogenic mechanisms of LGCMN
LGCMN is characterized not only by abnormal phenotypic and pathological features but also by various metabolic abnormalities. CMN syndrome is associated with significant increases in BMI, reductions in sub-nevus fat and muscle mass, and endocrine abnormalities such as the premature thelarche variant (3%), persistent cryptorchidism (6%), and insulin resistance.18 Although endocrine disorders may influence metabolic status, studies on endocrine changes in LGCMN are limited, and their specific role remains unclear. Hormonal imbalances could potentially impact metabolic pathways by modulating cellular signaling networks and enzyme activities.18 Building on our previous findings of systemic metabolic changes in GCMN, this study shifts the focus to tissue-specific metabolic alterations in LGCMN lesions. While our earlier work revealed significant changes in serum and urine metabolomes, the present study offers direct insights into lesion-site metabolism rather than systemic indicators in biofluids. We identified significant disruptions in amino acid and energy metabolism in LGCMN tissues, hallmarks of metabolic reprogramming closely linked to tumor onset and progression.19 These tissue-specific findings provide novel insights into the interactions between metabolic pathways and key melanogenesis-related genes, extending our understanding of LGCMN pathophysiology beyond the systemic changes previously reported. Approximately 33% of melanomas originate from benign melanocytic nevi, with some studies considering melanocytoma as an intermediate genetic stage between nevi and melanoma. As a benign melanocytic tumor, LGCMN may share genetic and metabolic traits with melanoma.20,21The amino acid alterations in LGCMN tissues are similar to characteristics observed in melanoma. For example, in the BRAFV600E melanoma model, the same types of amino acids observed in our results are upregulated. During activated BCAA catabolism, the enzymes branched-chain aminotransferases 1 and 2 (BCAT1/2) transfer nitrogen to α-ketoglutarate to produce glutamine, supporting cell proliferation. The BCAT inhibitor gabapentin effectively blocks BCAA transamination, thereby suppressing melanocyte proliferation and invasion.22,23 While LGCMN is not malignant, its metabolic profile shares certain features with neoplastic conditions, suggesting that BCAA metabolism may play a role in lesion progression. Leucine, a key BCAA, activates mTORC1 signaling, which regulates protein synthesis, cellular growth, and metabolic adaptation. Moreover, α-ketoisovalerate, a BCAA-derived keto acid, has been implicated in metabolic signaling and redox balance, potentially influencing cellular homeostasis in LGCMN lesions.23
In parallel, deficiencies in amino acids such as citrulline, alanine, and aspartate can significantly inhibit melanoma cell proliferation.24,25 Inducing citrulline deficiency, for instance, can lead to the death of up to 80% of cancer cells in vitro, providing a theoretical basis for citrulline-deficient therapies.26 Glutamate, serine, and glycine provide precursors for protein, nucleic acid, and lipid synthesis, supporting the metabolic demands of tumor cells.27,28 Lysine influences the tumor microenvironment and genomic stability through epigenetic regulation, such as via KMTs and KDMs.29,30 These findings suggest that alterations in amino acid availability and metabolism could impact the growth dynamics and clinical behavior of LGCMN lesions.
The energy metabolism pathways in LGCMN also show notable alterations, including pyruvate dehydrogenase kinase (PDK) inhibiting the conversion of pyruvate to acetyl-CoA to maintain high glycolytic levels and support rapid tumor proliferation.31 PCr, as a quick energy reserve, helps mitigate oxidative stress during tumor cell proliferation, while 3-hydroxybutyrate and α-ketoisovalerate regulate tumor energy balance, aiding adaptation to oxidative stress.32 In lipid metabolism, choline metabolites such as PC and GPC are closely linked to melanoma progression; the former marks tumor malignancy, while the accumulation of the latter inhibits apoptosis and promotes tumor cell spread.33,34 Thus, the alterations in amino acid, energy, and lipid metabolism observed in LGCMN are similar to those in melanoma, suggesting that these metabolic pathways may play a significant role in LGCMN pathology.
4.2 Metabolic differences and clinical significance of LGCMN subtypes
Some researchers have identified potential genetic and molecular differences among LGCMN classifications such as bolero, bathing trunk, and satellite nevi, but these efforts have yielded limited correlations.8 Similarly, our metabolomic analysis across subtypes revealed only limited differentiation. The metabolic differences observed across different LGCMN subtypes reflect their biological heterogeneity. The shawl and trunks subtypes, in particular, exhibit significant variations in amino acid metabolism, including arginine, alanine, and phenylalanine. An abnormal valine, leucine and isoleucine degradation pathway is especially prominent in the trunks subtype, potentially associated with its unique clinical features or growth pattern. These lesions often cover larger areas and are more likely to exhibit complex features such as satellite nevi and nodules. Some cases in the trunks subtype also display a distinctive “butterfly” distribution13,18 and a higher risk of malignancy. In this study, one patient with the trunks subtype developed melanoma and ultimately succumbed to the disease. This phenomenon has also been observed in previous literature and recent cases of bathing trunk patterns of malignancy.4 Case reports have shown that patients with the trunks subtype of LGCMN may experience rapid progression to melanoma following immunotherapy.35 Elevated BCAA levels in melanoma are closely associated with tumor proliferation and malignancy, reflecting its metabolic state and serving as a potential biomarker for cancer progression and prognosis.36,37 Such characteristics further highlight the clinical distinctiveness and malignancy risk of the trunks subtype.4.3 Hub gene-metabolite-pathway network and potential regulatory mechanisms in LGCMN
The multi-omics based multi-layered gene–metabolite-pathway network for LGCMN revealed significant differences in melanin production, cell proliferation, and signal regulation. The core regulatory axis consists of genes such as EDNRB, SOX10, TYR, and MITF, which directly influence melanin production and melanocyte development.38 TYR, as a rate-limiting enzyme, catalyzes the conversion of tyrosine to dopaquinone, initiating melanin synthesis.39 SOX10, as a key regulator of neural crest cell differentiation, supports melanocyte proliferation by regulating MITF, TYR, and PMEL. MITF, a central transcription factor, promotes melanogenesis and cell proliferation.40 High expression of SOX10 and MITF in LGCMN and melanoma, along with their inhibition reducing stem cell characteristics and suppressing tumor formation, indicates their critical role in LGCMN malignant transformation.9,41 Additionally, genes such as PMEL, SLC24A5, and SLC45A2 stabilize the melanosome structure and regulate calcium balance and pH to support the stability of the melanogenesis network.42–44In terms of metabolic regulation, tyrosine and BCAA metabolism play particularly important roles. The association of BCAA with genes such as SOX10 and MITF underscores its significance in melanocyte proliferation and metabolic regulation, suggesting that intervening in amino acid metabolism could potentially aid tumor therapy by targeting specific metabolic pathways or combining dietary regulation.45 Previous studies have shown that BCAA application in B16F0 melanocytes can inhibit melanin production.46 In melanoma cells with BRAF mutations and hyperactive MAPK pathways, limiting leucine availability could suppress melanoma growth.47 The amino acid enrichment characteristics of LGCMN also suggest new intervention possibilities for targeting MAPK and PI3K/AKT signaling pathways.
The MAPK pathway regulates transcription factors such as Elk-1, Myc, and CREB, promoting the expression of MITF, TYR, and SOX10 and enhancing melanocyte proliferation and malignant potential.48 Furthermore, the MAPK pathway forms a synergistic network with genes such as EGF, RACK1, and IL-18. EGF activates the MAPK pathway to stimulate cell proliferation, with observed downregulation in melanoma consistent with our findings.49 RACK1 regulates signaling correlating with melanoma malignancy, and IL-18 regulates melanin production, potentially influencing local inflammatory responses within LGCMN.50 Inhibitors such as trametinib and alpelisib show promise in LGCMN treatment, significantly controlling lesion progression by inhibiting MAPK and PI3K pathways.2,51
It should be pointed out that this study has several limitations. Firstly, due to the small sample size, validation in larger cohorts is required to establish the reliability and generalizability of disease-specific biomarkers. Secondly, the lack of hormone level assessments in LGCMN patients in clinical reports limits the analysis of the relationship between metabolic findings and hormonal status, affecting our understanding of LGCMN pathogenic mechanisms. Finally, due to the rarity of malignant melanomas arising from giant nevi, it has not been possible to collect sufficient malignant samples for comparative analysis with LGCMN, limiting a comprehensive understanding of the potential pathogenic mechanisms underlying LGCMN malignancy.
5. Conclusions
This study presents a systematical exploration of unique gene and metabolic characteristics of LGCMN through metabolomic and transcriptomic analyses. A multi-layered hub gene–metabolite-pathway network provides novel insights into the pathogenic mechanisms of LGCMN. Key genes such as TYR, SOX10, and MITF play significant roles in melanogenesis and cell proliferation, and their associations with tyrosine, phenylalanine, and BCAA underscore the importance of amino acid metabolism in LGCMN. Notably, the potential for BCAA metabolism intervention to inhibit melanin synthesis introduces a promising avenue for non-surgical LGCMN treatment. Furthermore, the activation of the MAPK and PI3K/AKT pathways is closely correlated with LGCMN proliferation and potential malignancy. The integration of gene regulation and metabolic associations offers valuable insights into LGCMN pathogenesis and guides development of targeted therapies.Ethical statement
This study was performed in line with the principles of the Declaration of Helsinki. Approval was granted by the Institutional Ethics Committee of Tongji Hospital affiliated to Tongji University, China (SBKT-2023-149). The patient's parents/guardians in this manuscript have given written informed consent to the publication of their case details.Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request. The RNA sequencing data used in this study are available in the National Center for Biotechnology Information (NCBI) SRA under accession PRJNA730598.Author contributions
Conceptualization, J. F. and T. D.; data curation, G. Song; formal analysis, J. F.; G. Song and T. D.; funding acquisition, G. Shen and T. D.; investigation, G. Song; methodology, J. F.; project administration, G. Shen; resources, G. Song; T. D.; Y. R. and Z. W.; software, G. Song and P. G.; supervision, J. F. and G. Shen; validation, T. D.; Y. R. and Z. W.; Y. R.; P. G. and Y. C.; visualization, G. Song; writing—original draft preparation, G. Song; writing—review and editing, J. F.; G. Shen; T. D.; Y. R.; Y. C.; P. G. and Z. W.; all authors have read and agreed to the published version of the manuscript.Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 82072015), the Natural Science Foundation of Fujian Province of China (No. 2022J01062), and Jointly Constructed Project in Henan Province of China (No. LHGJ20220674). We would like to thank Dr Yinan Ma, Huile Pei, and Fangfang Jiao of the Department of Dermatology, The Second Affiliated Hospital of the Henan University of Science and Technology, and Xialin Cheng from the School of Medicine, Xiamen University, for their assistance in collecting clinical case data.References
- Y. S. Choi, T. H. Erlich, M. von Franque, I. Rachmin, J. L. Flesher, E. B. Schiferle, Y. Zhang, M. Pereira da Silva, A. Jiang, A. S. Dobry, M. Su, S. Germana, S. Lacher, O. Freund, E. Feder, J. L. Cortez, S. Ryu, T. Babila Propp, Y. L. Samuels, L. R. Zakka, M. Azin, C. E. Burd, N. E. Sharpless, X. S. Liu, C. Meyer, W. G. Austen Jr, B. Bojovic, C. L. Cetrulo Jr, M. C. Mihm, D. S. Hoon, S. Demehri, E. B. Hawryluk and D. E. Fisher, Cell, 2022, 185, 2071–2085.e12 CAS.
- A. Tomás-Velázquez, J. C. López-Gutiérrez, C. de Andrea, M. Reyes-Múgica, C. M. Salgado and P. Redondo, J. Eur. Acad. Dermatol. Venereol., 2024, 38, 1147–1151 CrossRef PubMed.
- S. Krengel, A. Scope, S. W. Dusza, R. Vonthein and A. A. Marghoob, J. Am. Acad. Dermatol., 2013, 68, 441–451 CrossRef.
- V. P. Martins da Silva, A. Marghoob, R. Pigem, C. Carrera, P. Aguilera, J. A. Puig-Butillé, S. Puig and J. Malvehy, J. Am. Acad. Dermatol., 2017, 76, 689–694 CrossRef.
- A. Alikhan, O. A. Ibrahimi and D. B. Eisen, J. Am. Acad. Dermatol., 2012, 67, 495.e1–495.e17 CrossRef PubMed.
- M. Carmen Ceballos-Rodríguez, P. Redondo, A. Tomás-Velázquez, D. Cieza-Díaz and J. Carlos López-Gutiérrez, J. Pediatr. Surg., 2021, 56, 2113–2117 Search PubMed.
- K. J. Karczewski and M. P. Snyder, Nat. Rev. Genet., 2018, 19, 299–310 CAS.
- V. Martins da Silva, E. Martinez-Barrios, G. Tell-Martí, M. Dabad, C. Carrera, P. Aguilera, D. Brualla, A. Esteve-Codina, A. Vicente, S. Puig, J. A. Puig-Butillé and J. Malvehy, J. Invest. Dermatol., 2019, 139, 900–908 CAS.
- D. Basu, C. M. Salgado, B. Bauer, R. M. Hoehl, C. N. Moscinski, L. Schmitt and M. Reyes-Múgica, Melanoma Res., 2021, 31, 319–327 CrossRef CAS PubMed.
- Y. Chang, T. Dai, G. Song, S. Wang, H. Pei, G. Shen and J. Feng, J. Pharm. Biomed. Anal., 2024, 242, 116060 CAS.
- N. J. Taylor, I. Gaynanova, S. A. Eschrich, E. A. Welsh, T. J. Garrett, C. Beecher, R. Sharma, J. M. Koomen, K. S. M. Smalley, J. L. Messina and P. A. Kanetsky, PLoS One, 2020, 15, e0240849 CAS.
- M. Kertys, M. Grendar, V. Horak, N. Zidekova, H. Kupcova Skalnikova, J. Mokry, E. Halasova and J. Strnadel, Melanoma Res., 2021, 31, 140–151 CAS.
- G. Song, T. Dai, Y. Chang, H. Pei, W. Liu, P. Guo, Y. Ren, G. Shen and J. Feng, J. Eur. Acad. Dermatol., 2024, 39, 171–180 Search PubMed.
- P. Manirakiza, A. Covaci and P. Schepens, J. Food Compos. Anal., 2001, 14, 93–100 CAS.
- F. Savorani, G. Tomasi and S. B. Engelsen, J. Magn. Reson., 2010, 202, 190–202 CAS.
- J. M. Fonville, A. D. Maher, M. Coen, E. Holmes, J. C. Lindon and J. K. Nicholson, Anal. Chem., 2010, 82, 1811–1821 CAS.
- P. Guo, T. Teng, W. Liu, Y. Fang, B. Wei, J. Feng and H. Huang, Int. J. Cancer, 2022, 151, 1835–1846 CAS.
- R. Waelchli, J. Williams, T. Cole, M. Dattani, P. Hindmarsh, H. Kennedy, A. Martinez, S. Khan, R. K. Semple, A. White, N. Sebire, E. Healy, G. Moore and V. A. Kinsler, Br. J. Dermatol., 2015, 173, 1471–1478 CrossRef CAS.
- S.-M. Jeon and N. Hay, Exp. Mol. Med., 2018, 50, 1–3 Search PubMed.
- W. E. Damsky and M. Bosenberg, Oncogene, 2017, 36, 5771–5792 CrossRef CAS.
- I. Yeh, Mod. Pathol., 2020, 33, 1–14 CrossRef PubMed.
- F. J. Naser, M. M. Jackstadt, R. Fowle-Grider, J. L. Spalding, K. Cho, E. Stancliffe, S. R. Doonan, E. T. Kramer, L. Yao, B. Krasnick, L. Ding, R. C. Fields, C. K. Kaufman, L. P. Shriver, S. L. Johnson and G. J. Patti, Cell Metab., 2021, 33, 1493–1504.e5 CrossRef CAS PubMed.
- W. Guo, H. Wang and C. Li, Signal Transduct. Targeted Ther., 2021, 6, 424 Search PubMed.
- C. Wasinger, A. Hofer, O. Spadiut and M. Hohenegger, Sci. Rep., 2018, 8, 6245 Search PubMed.
- Y.-M. Fu and G. G. Meadows, J. Nutr., 2007, 137, 1591S–1596S Search PubMed.
- D. N. Wheatley and E. Campbell, Br. J. Cancer, 2003, 89, 573–576 Search PubMed.
- I. Amelio, F. Cutruzzolá, A. Antonov, M. Agostini and G. Melino, Trends Biochem. Sci., 2014, 39, 191–198 Search PubMed.
- J. Jin, J.-K. Byun, Y.-K. Choi and K.-G. Park, Exp. Mol. Med., 2023, 55, 706–715 Search PubMed.
- R. S. de Oliveira Filho, D. A. de Oliveira, M. M. Nisimoto and L. C. Marti, Cancers, 2023, 15, 5751 CAS.
- G. Punnia-Moorthy, P. Hersey, A. A. Emran and J. Tiffen, Front. Genet., 2021, 12, 680633 CAS.
- J. F. Tiersma, B. Evers, B. M. Bakker, M. Jalving and S. de Jong, Int. J. Mol. Sci., 2022, 23, 3745 CAS.
- K. Glunde, Z. M. Bhujwalla and S. M. Ronen, Nat. Rev. Cancer, 2011, 11, 835–848 Search PubMed.
- D. Morvan, A. Demidem, J. Papon, M. De Latour and J. C. Madelmont, Cancer Res., 2002, 62, 1890–1897 Search PubMed.
- K. Sonkar, V. Ayyappan, C. M. Tressler, O. Adelaja, R. Cai, M. Cheng and K. Glunde, NMR Biomed., 2019, 32, e4112 Search PubMed.
- T. Gambichler, K. Noldes, Y. Arafat, M. Neid, A. Rütten and S. Boms, Dermato, 2023, 3, 51–55 Search PubMed.
- J. Wang, W. Wang, F. Zhu and Q. Duan, Biomed. Pharmacother., 2022, 153, 113390 CAS.
- Y. Tian, J. Ma, H. Wang, X. Yi, H. Wang, H. Zhang, S. Guo, Y. Yang, B. Zhang, J. Du, Q. Shi, T. Gao, W. Guo and C. Li, Cell. Mol. Life Sci., 2023, 80, 315 Search PubMed.
- M. K. Shin, J. M. Levorse, R. S. Ingram and S. M. Tilghman, Nature, 1999, 402, 496–501 CAS.
- S. Alonso, N. Izagirre, I. Smith-Zubiaga, J. Gardeazabal, J. L. Díaz-Ramón, J. L. Díaz-Pérez, D. Zelenika, M. D. Boyano, N. Smit and C. de la Rúa, BMC Evol. Biol., 2008, 8, 74 Search PubMed.
- D. Basu, C. M. Salgado, J. R. Patel, J. Zabec, R. M. Hoehl, B. Bauer and M. Reyes-Múgica, Biomark. Res., 2019, 7, 2 CrossRef.
- O. Shakhova, D. Zingg, S. M. Schaefer, L. Hari, G. Civenni, J. Blunschi, S. Claudinot, M. Okoniewski, F. Beermann, D. Mihic-Probst, H. Moch, M. Wegner, R. Dummer, Y. Barrandon, P. Cinelli and L. Sommer, Nat. Cell Biol., 2012, 14, 882–890 CrossRef CAS.
- C. Bissig, L. Rochin and G. van Niel, Int. J. Mol. Sci., 2016, 17, 1438 Search PubMed.
- L. Le, I. E. Escobar, T. Ho, A. J. Lefkovith, E. Latteri, K. D. Haltaufderhyde, M. K. Dennis, L. Plowright, E. V. Sviderskaya, D. C. Bennett, E. Oancea and M. S. Marks, Mol. Biol. Cell, 2020, 31, 2687–2702 CrossRef CAS.
- L. B. Reis, R. M. Bakos, F. S. L. Vianna, G. S. Macedo, V. C. Jacovas, A. M. Ribeiro-dos-Santos, S. Santos, L. Bakos and P. Ashton-Prolla, BMC Cancer, 2020, 20, 1069 CrossRef CAS.
- M. B. Ishak Gabra, Y. Yang, H. Li, P. Senapati, E. A. Hanse, X. H. Lowman, T. Q. Tran, L. Zhang, L. T. Doan, X. Xu, D. E. Schones, D. A. Fruman and M. Kong, Nat. Commun., 2020, 11, 3326 CrossRef CAS.
- J. Y. Cha, H. J. Yang, H. I. Moon and Y. S. Cho, Immunopharmacol. Immunotoxicol., 2012, 34, 256–264 CrossRef CAS PubMed.
- T. Ko, R. Sharma and S. Li, Oncogene, 2020, 39, 723–738 CrossRef CAS PubMed.
- L. Y. Ballester, P. P. Aung and C.-C. R. Lee, in Genetics of Melanoma, ed. C. A. Torres-Cabala and J. L. Curry, Springer New York, New York, NY, 2016, pp. 151–163, DOI:10.1007/978-1-4939-3554-3_6.
- V. N. Ivanov and T. K. Hei, Oncogene, 2005, 24, 616–626 Search PubMed.
- C. Fu, J. Chen, J. Lu, L. Yi, X. Tong, L. Kang, S. Pei, Y. Ouyang, L. Jiang, Y. Ding, X. Zhao, S. Li, Y. Yang, J. Huang and Q. Zeng, Mol. Med. Rep., 2020, 21, 1421–1430 Search PubMed.
- A. Mir, N. G. Agim, A. A. Kane, S. C. Josephs, J. Y. Park and K. Ludwig, Pediatrics, 2019, 143, e20182469 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ay00122f |
| ‡ These authors contributed equally to this study. |
| This journal is © The Royal Society of Chemistry 2025 |