
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Application of AQbD principles on the development of a stability-indicating HPLC method for the determination of acetylsalicylic acid, ramipril and atorvastatin in their fixed-dose polypills†
Halil
Kasem
a,
Marianna
Ntorkou
a,
Paraskevas D.
Tzanavaras
b and
Constantinos K.
Zacharis
 *a
*a
aLaboratory of Pharmaceutical Analysis, Department of Pharmacy, Aristotle University of Thessaloniki, GR-54124, Greece. E-mail: czacharis@pharm.auth.gr
bLaboratory of Analytical Chemistry, School of Chemistry, Faculty of Sciences, Aristotle University of Thessaloniki, GR-54124, Greece
First published on 28th March 2025
Abstract
In this study, an analytical quality by design approach was proposed for the development of a stability-indicating HPLC method for the determination of acetylsalicylic acid, ramipril and atorvastatin in their fixed-dose polypills. The structures, retention and the forced degradation studies of each drug served as useful prior knowledge. Using risk assessment and screen design, three critical method parameters (buffer pH, gradient slope and % CH3OH initial content) were defined and optimized using a Box–Behnken response surface methodology. The stability-indicating features of the proposed method are assessed through forced degradation studies. The chromatographic separation of the analytes was carried out with a gradient mode using 10 mM phosphate buffer (pH 2.3) and methanol on a C18 analytical column. The method operable design region was approved by the establishment of a robust zone using Monte Carlo simulation and capability analysis. The determination coefficients (R2) were higher than 0.9939. The proposed method indicated good precision (RSD < 7.7%) and the accuracy expressed as average % relative recovery ranged between 91.4–106.7%. The developed analytical scheme was successfully applied to quantify the selected APIs in the commercially available polypill Trinomia® capsules. The dosage uniformity of the drug-containing formulations was evaluated.
Introduction
Cardiovascular disease is the primary cause of death and health complications globally. Taking a polypill containing essential medications that enhance outcomes (such as aspirin, ACE inhibitors, and statins) has been suggested as an easy method for reducing the risk of cardiovascular-related deaths and complications following a heart attack.1,2 In 2001, the World Health Organization (WHO) introduced the concept of a polypill comprising blood pressure-lowering agents, a statin, and an anti-platelet agent in fixed doses as a preventive measure for cardiovascular disease.1 Such polypills were first made available in Europe and contain 100 mg of acetylsalicylic acid (ASA), 20 or 40 mg of atorvastatin (ATOR), and 2.5, 5, or 10 mg of ramipril (RAM). They are used for secondary prevention in adult patients who are effectively controlled with individual components at equivalent therapeutic doses.1The traditional way to develop and optimize an analytical method procedure is by means of the Quality by Testing approach generally by varying one factor at a time (OFAT). This results in a high number of experiments, with limited understanding, as the approach does not effectively analyze interactions between parameters.3 Additionally, the absence of robust analytical methods has long been a concern for the pharmaceutical industry. To tackle this issue, the International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use (ICH) has developed a set of guidelines. The ICH Q14 promotes a risk-based approach to developing analytical procedures, encouraging the adoption of Analytical Quality by Design (AQbD) over traditional methods.4,5
The AQbD provides a systematic approach for method development allowing the identification of variables that influence the method performance. It ensures that the analytical process is well-designed, robust, and consistently delivers the expected results throughout its lifecycle. This approach involves several key steps, including defining the Analytical Target Profile (ATP), the assessment of Critical Process Parameters, determining Critical Quality Attributes (CQAs), risk assessments, method development using chemometrics, testing robustness, establishment of Method Operable Design Region (MODR), etc. The ATP outlines the performance requirements, anticipated goals, and objectives for method development.6 The risk assessment was employed to identify method parameters that influence analytical performance, as well as risks related to variability, including sample preparation, instrument configuration, and environmental conditions.7 The MODR is determined by testing robustness to confirm that the method consistently produces reliable results within set acceptance criteria.8 Developing an analytical method using AQbD offers several advantages such as the reduction of the occurrence of out-of-specification results, making the method more cost-efficient after development and validation, as it requires no further adjustments.9 Overall, this approach aligns and serves as an extension of the existing pharmaceutical guidelines, incorporating the principles of QbD. Several scientific papers on the application of AQbD showed the advantages of this approach in method development10–12 and especially in analysis of combined pharmaceutical products.13–15
The literature review for the analytes revealed that only a few HPLC methods are reported for the analysis of ASA, RAM and ATOR in combination with other drugs in fixed-dose pharmaceutical products.16–19 Only two HPLC methods have been proposed for the simultaneous determination of the three drugs in their combined dosage forms.20–22 Abdallah et al. developed an ion-pair HPLC of the analytes in fixed-dose pharmaceutical products.23 However, ion-pair HPLC methods require long equilibration time of the stationary phase especially when gradient elution is used. More importantly, the ion-pairing reagents are typically adsorbed onto the stationary phase leading to its permanent modification and column degradation.24 These approaches require a large number of experiments involved in OFAT and lack a deep understanding of critical method parameters (CMPs) and are only partially validated. This issue results in an incomplete scientific understanding of method performance characteristics and their proper pharmaceutical application. The proposed study presents an AQbD strategy for the development of a reliable and robust stability-indicating HPLC method with adequate separation efficiency of the studied drugs and their main degradation products. To the best of our knowledge, such an approach has not been previously reported in the literature. Risk assessment combined with chemometrics (Plackett–Burman and Box–Behnken designs) is utilized for the screening, identification and optimization of the CMPs. The method's MODR was established using a robustness test via Monte Carlo simulation experiments and capability analysis. Finally, the developed method was validated to demonstrate its suitability for the intended purpose.
Experimental
Chemicals and solutions
ASA (≥98%) was purchased from TCI Chemicals (Riga, Latvia), and RAM (≥98%) and salicylic acid (SA) (≥99%) were provided by Sigma-Aldrich (Darmstadt, Germany). ATOR (≥99%) was kindly donated by Pharmathen SA. Acetonitrile (ACN) (HPLC grade), methanol (HPLC grade), NaH2PO4 (>98%), concentrated H3PO4 (85% w/v), NaOH pellets, H2O2 (30% w/w), and HCl (37% w/v) were also provided by Sigma-Aldrich (Darmstadt, Germany). All other chemicals were of analytical grade. A B30 water purification system (Adrona SIA, Riga, Latvia) was utilized to produce high-purity water. A cellulose acetate membrane filter was purchased from ISOLAB Laborgerate GmbH (Eschau, Germany).Pharmaceutical formulations (Trinomia® caps, 100 mg ASA/20 mg (ATOR calcium trihydrate)/5 mg RAM) were obtained from Ferrer-Galenica AE (Athens, Greece) through local pharmacy stores.
Stock standard solutions of the APIs were prepared in methanol at a concentration of 1000 μg mL−1. ASA stock solutions were kept at −18 °C and the rest ones at 4 °C for a period of five days at the most. Working standards were prepared daily in water by serial dilutions in a mixture of water/methanol, 50/50 v/v (diluent). A mixture of the studied APIs at a concentration of 100 μg mL−1 was prepared in the diluent and used for peak identification.
For method development, a mixture of the three drugs and their main degradation products was used throughout this study. To undertake this task, preliminary forced degradation experiments were conducted for each drug separately. It was found that acidic conditions were favourable for the degradation of ASA (degraded to SA) and ATOR while RAM was degraded in alkaline medium. On this basis, ASA and ATOR solutions were prepared in 0.1 M HCl and RAM in 0.1 M NaOH (300 μg mL−1 each) and incubated at 60 °C for 1 h. After cooling, equal volumes of each solution were mixed and used.
System suitability test (SST) solution
The SST helps to verify that the chromatographic system meets the required performance criteria, ensuring accurate and reproducible results. Based on this, a mixture of SA, ASA, RAM, and ATOR was prepared by transferring appropriate volumes of the individual stock solutions of each compound into a 25 mL volumetric flask, followed by dilution with the diluent. The final concentrations of SA, ASA, RAM, and ATOR in the obtained solution were 25, 100, 50, and 100 μg mL−1, respectively.HPLC instrumentation and conditions
A Shimadzu HPLC quaternary solvent system 2010 (Kyoto, Japan) equipped with a UV-Vis detector was used for the method development and validation. For the forced degradation studies, a Shimadzu HPLC quaternary system coupled with an SPD-M20A photodiode array (PDA) detector was utilized. In both cases, instrument control and data acquisition were performed via LCsolutions® software (Version 1.25 SP4).All separations of the drugs were carried out on a Supelcosil C18, 250 × 4.6 mm, 5 μm, (Supelco, Bellefonte, USA) under gradient elution. The mobile phases A and B consisted of 10 mM phosphate buffer (pH = 2.3) and MeOH. Initially, the mobile phase ratio was 20% v/v of B and then linearly decreased to 75% in 40 min and returned to the initial composition and kept constant up to 55 min for column equilibration. The mobile phase was pumped constantly at 1 mL min−1. An injection volume of 25 μL was used throughout this study. The column temperature was 50 °C and the analytes were detected at 220 nm. A mixture of water/CH3OH/isopropanol (50/25/25% v/v) was utilized for the autosampler washing solution.
Analysis of pharmaceutical formulations
Each capsule contains two tablets of each ASA (50 mg per tab) and ATOR (10 mg per tab), and one tablet of RAM (5 mg per tab). Each tablet was weighed and transferred in a 100 mL volumetric flask filled with ca 80 mL of diluent, sonicated for 10 min and made up to volume with the diluent. A portion of ca 10 mL of the obtained solution was filtered through a PTFE disposable syringe filter (0.45 μm) and ca 1 mL was transferred to an HPLC vial. This solution was used for the quantitation of RAM and ATOR. An additional 10-fold dilution was required for the quantification of ASA.Placebo powder equivalent to the excipients contained in each formulation was accurately weighed and transferred into a 100 mL volumetric flask. Then, ca 80 mL of diluent was added and the suspension was sonicated for 10 min with intermittent shaking. The volume was filled up to the mark with the diluent and filtered and analysed.
Method development using AQbD
A Plackett–Burman design (PBD) was constructed where 5 factors and 6 dummies plus 3 central points resulted in a matrix of 15 experimental runs. The selected CPMs ranged between two levels as follows: buffer concentration (10–30 mM) (Factor 1), buffer pH (2–4) (Factor 2), column temperature (25–50 °C) (Factor 3), gradient slope (1.0–2.2 %B/min) (Factor 4) and %CH3OH initial content (20–60% v/v) (Factor 5). The analysis time was considered as the retention time (Rt) of the latest eluting compound. The mathematical model related to the design consists of main effects and possible two-factor interaction effects. Pareto charts were employed as the principal model selection technique identifying the independent variables that significantly affect the chosen CMAs.
Results and discussion
Preliminary experiments, ATP
One of the initial steps in the analytical QbD pathway is defining the ATP (Table S1†), where the quality requirements, expected goals, and objectives of method development are established.4,25 In this context, a rapid HPLC method is needed for quantifying the studied APIs in their fixed dose polypills and ensuring a robust experimental design space. The primary quality criteria include the method's (a) selectivity, (b) accuracy (ranging from 90% to 110%), and (c) precision (with %RSD < 10%).Preliminary experiments were conducted to establish the base for method development, as well as to identify the CMAs and CMPs. According to physicochemical data of the analytes (Table S2†), all drugs are relatively polar, with ASA being the most polar of them. According to the literature, the drugs are typically separated using the C18 stationary phase.17,21 Due to their varying physicochemical properties, separation under isocratic elution is extremely time consuming (ca > 90 min). Initial experimentation was based on using gradient elution on various analytical columns i.e. Supelcosil C8 (150 × 4.6 mm, 5 μm, Supelco), ACE Phenyl (150 × 4.6 mm, 5 μm), BDS C18 (100 × 4.6 mm, 3 μm, ThermoScientific) and Supelcosil C18 (250 × 4.6 mm, 5 μm, Supelco) using phosphate buffer (10 mM, pH 3.0) and methanol as mobile phases at a flow rate of 1 mL min−1. For the 150 mm-long columns, even the investigation of the phenyl analogous stationary phases showed insufficient resolution (Rs = 0.9–1.2) between the adjacent peaks such as the ASA/RAM-degradant (RAMD), ATOR and its degradation product (ATORD). Thus, the 250 mm-long Supelcosil C18 stationary phase was selected for its superior efficiency in separating the degradation products from the studied APIs.
Several experiments were performed using different mobile phase compositions to identify the most suitable one. Since all the tested APIs contain a carboxylic group, a phosphate buffer with a pH lower than the analytes' pKa (i.e., 3.5, as shown in Table S2†) is preferable to ensure the existence of the non-ionic forms of the drugs and enhance their peak shape. Organic modifiers (acetonitrile, methanol) were evaluated and methanol proved to be the best and most cost-effective option, offering well-resolved peaks along with appropriate analysis times. Various trials were conducted with different gradient slopes (ranging from 1.0 to 2.2) and temperatures (ranging from 25 °C to 50 °C) using a NaH2PO4 aqueous solution (10–30 mM, pH 3.0) and methanol as mobile phases. In all cases the mobile phase flow rate was 1 mL min−1. The experiments confirmed that the number of eluted peaks and the resolution between them are directly dependent on the gradient slope and the %CH3OH initial content. UV spectra of the studied drugs were obtained (Fig. S1†) and a wavelength of 220 nm was selected for the monitoring of all drugs, as a single-wavelength UV detector was utilized in this study. The optimization strategy was designed to achieve satisfactory separation of the three drugs and their degradation products within the shortest possible analysis time.
Risk assessment, CMAs, and CMPs
The resolution (Rs) between each API and its adjacent peaks was designated as a CMA and required to be higher than 1.7. Additionally, the number of theoretical plates (N) for the drug peaks (N > 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) and the analysis time were also defined as CMAs.
000) and the analysis time were also defined as CMAs.
The subsequent stage focused on selecting CMPs to ensure the desired quality of the analytical method. This process involved a science-based quality risk management approach to establish the relationship between CMPs and CMAs. In this sense, an Ishikawa diagram was developed using TIBCO® Statistica v. 13.3.0 software (TIBCO Software Inc., Palo Alto, CA, USA) to facilitate brainstorming and identify all parameters that could potentially impact the CMAs (Fig. 1). Experimental parameters were categorized into low, medium, and high-risk groups based on their influence on the selected CMAs. Five high-risk parameters were identified as critical and subjected to further evaluation, while medium-risk parameters were fixed based on preliminary experiments. The instrumental parameters were considered low risk as they are dependent on the HPLC system.
 | ||
| Fig. 1 Ishikawa diagram. High-risk factors (CMPs) are marked in red. Fixed and other medium-risk parameters are green- and orange-marked, respectively. | ||
Screening design
The primary objective of the screening design was to identify the factors and their interactions that could potentially influence the separation selectivity. Compared to the OFAT approach, DoE is an efficient way to identify these factors with a minimized number of experiments.26 Five parameters were screened using PBD and the dataset is tabulated in Table S3 (ESI†).Pareto charts (Fig. 2) revealed that increasing the buffer pH led to lower resolution between ASA and SA, RAM and ATOR, and decrease the peak efficiency of ASA (negative effect). The issue is attributed to the fact that at higher pH values the carboxylic groups of the analytes are ionized leading to lower chromatographic efficiencies. As expected, higher gradient slopes resulted in lower resolutions, especially in pairs of ATOR-RAM and ATOR-ATORD and lower analysis time as well. The column temperature was statistically significant and positively affected the resolution of pairs RAM-RAMD and ATOR-ATORD, with a lesser impact on the other CMAs. Given this, we decided to exclude this factor by setting its highest value (50 °C) to minimize the number of experiments in the RSM study. Buffer concentration had no impact on the studied CMAs and therefore the lower concentration of 10 mM was adopted for further experiments.
 | ||
| Fig. 2 Pareto charts for the chromatographic parameters: (A) Rs(1), (B) Rs(2), (C) Rs(3), (D) Rs(4), (E) Rs(5), (F) NASA, (G) NRAM, (H) NATOR and (I) Rt. | ||
Optimization design
The next phase of AQbD focused on optimizing the three CMPs and defining the mathematical relationship between the CPMs and CQAs. To achieve this, a BBD was employed, demonstrating its effectiveness in identifying the MODR through both numerical and graphical optimization techniques. The coefficients for the predictive models of the evaluated CMAs were determined using multiple linear regression. According to Design Expert® (Version 22.0.8, Stat-Ease Inc, Minneapolis, MN, USA) software, appropriate model adjustments, such as eliminating non-significant parameters or choosing the appropriate transformation, lead to improved summary statistics.ANOVA was employed to statistically evaluate the models. All the models showed statistical significance, while the lack-of-fit was found to be non-significant in all cases (Table S4–S12†). All cases demonstrated good predictability and accuracy, with R2 and adjusted R2 values exceeding 0.8687. Additionally, the adequate precision values for all models were above 93.83, confirming the models' significance. As anticipated, the resolution between the drugs and their degradants was significantly affected by the studied CMPs. Analogous effects were recorded for the number of theoretical plates of all studied APIs peaks expect for N(ASA) which was only affected by buffer pH and the initial %CH3OH content. Table 1 summarizes the predicted regression models along with the statistical parameters. The predicted models were properly fitted to the experimental data which are randomly scattered across the line as shown in the plots of the residuals (Fig. S2–S4†).
| CMA | Regression modela | Adjusted R2 | Predicted R2 | % C.V | Adeq. Precision |
|---|---|---|---|---|---|
| a Significant coefficients (p < 0.05) are only included. Factors in the coded level. b A: buffer pH. c B: initial %CH3PH content. d C: gradient slope (%CH3OH/min). | |||||
| R s(1) | 55.74 − 33.28 Ab + 0.465 Bc − 7.94 Cd + 3.15 AC + 4.26 A2 − 0.006 B2 | 0.8687 | 0.7330 | 24.68 | 13.43 |
| R s(2) | 8.27 + 3.82 A − 0.154 B − 4.83 C − 0.067 AB + 0.117 BC | 0.8761 | 0.5630 | 25.48 | 12.32 |
| R s(3) | −13.45 + 9.31 A + 0.084B + 5.24 C − 0.063 AB − 2.004 AC | 0.9281 | 0.7398 | 12.58 | 15.70 |
| logRs(4) |
+4.18–1.67 A + 0.007 B − 0.439 C + 0.046 AC + 0.003 BC + 0.227 A2 − 0.0002 B2 | 0.9975 | 0.9942 | 1.26 | 93.83 |
| logRs(5) |
−1.13 + 0.563 A − 0.010 B + 0.982 C − 0.186 AC + 0.0076 BC − 0.295 C2 | 0.9551 | 0.8841 | 10.82 | 22.06 |
| N ASA | 2.07 × 105 − 40014 Α − 4933 B + 683 AB + 26.14 B2 |
0.9314 | 0.8224 | 25.27 | 23.63 |
| logNRAM |
22.97 − 6.69 A − 0.028 B − 2.63 C + 0.557 AC + 0.027 BC +0.899 A2 − 0.0009 B2 | 0.9499 | 0.8350 | 2.78 | 22.25 |

|
1156 − 21.69 B − 2.11 C2 + 7.26 BC − 123.1 C2 | 0.9665 | 0.8849 | 7.66 | 30.24 |
| R t | 113.8 − 1.65 B − 39.47 C + 0.655 BC | 0.8999 | 0.8238 | 14.61 | 20.09 |
The 3D plots of the studied CMAs are portrayed in Fig. 3 and S5 (ESI†). Curvature was observed in most of the resolution plots. The maximum values of Rs(1) and Rs(4) were recorded at lower buffer pH while the rest of the resolutions were maximized at pH 4. Acidic mobile phases (pH 2) in combination with lower gradient slopes were beneficial leading to a more symmetric peak and higher numbers of theoretical plates (Fig. S5A–C†).
 | ||
| Fig. 3 3D plots demonstrating the effects of selected CMPs on (A) Rs(1), (B) Rs(2), (C) Rs(3), (D) Rs(4) and (E) Rs(5). | ||
The optimization of the individual CMPs was carried out using the desirability function. All CMAs were adjusted to maximize their values while minimizing Rt. During this process, each parameter was assigned an equal weighting factor (w = 1). As a result of numerical optimization, a global desirability score of 0.537 was obtained (Fig. S6†). The optimum values of CMPs after rounding were 2.3, 20% and 1.4 for the buffer pH, initial %CH3OH content and gradient slope, respectively.
The design space, also referred to as MODR, represents a range where changes in method parameters do not compromise performance.27 As shown in Fig. 4, the purple region highlights the area where all responses meet the specified criteria. These criteria are defined by acceptance limits for resolution (Rs(1), Rs(2) > 3.0; Rs(3), Rs(4) > 5.0; Rs(5) > 1.7), peak efficiency (NASA > 20k, NRAM > 15k; NATOR > 300k), and analysis time (Rt < 45 min). During routine analysis, variations in the specified method parameters may lead to one or more CMAs exceeding the boundaries of the grey area. To prevent this, simulation experiments were conducted to identify a robust MODR and ensure the reliability of the analytical method.
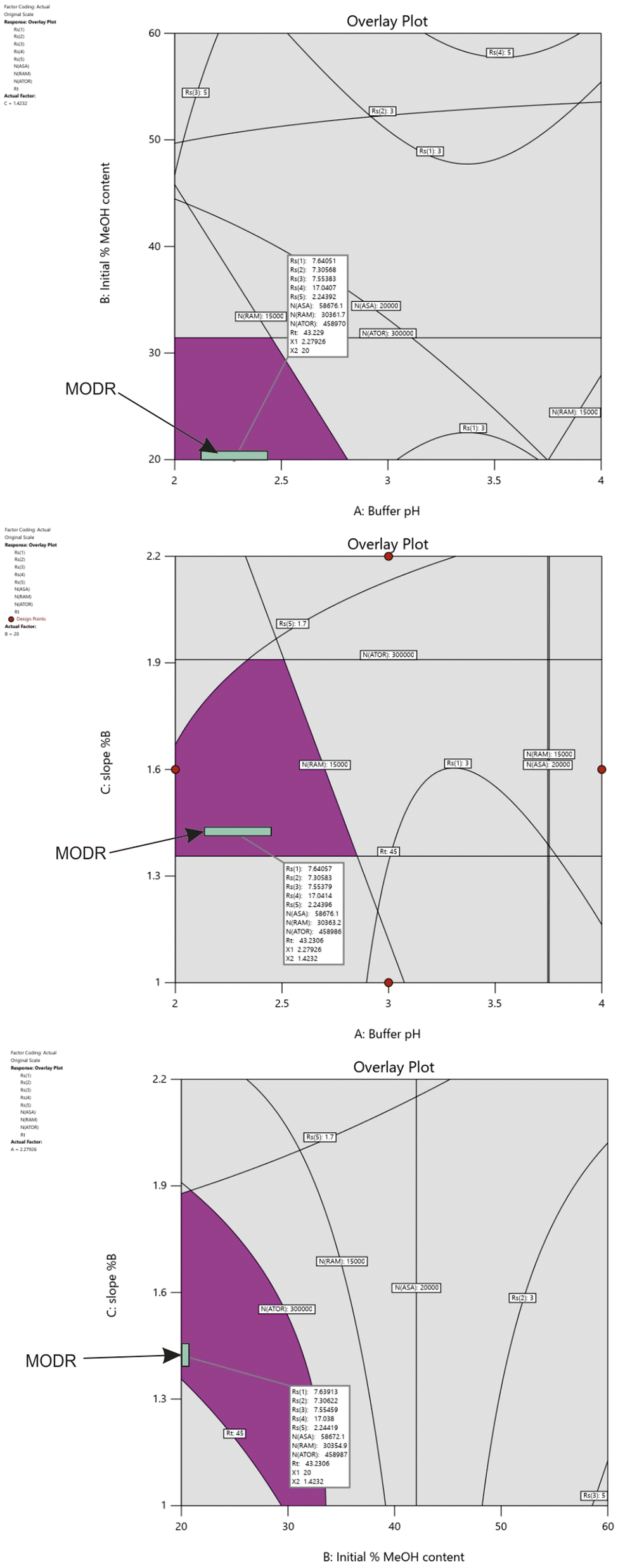 | ||
| Fig. 4 Design space established by overlaying contour plots of chromatographic responses. | ||
Computational determination of MODR
Monte Carlo simulations combined with capability analysis were utilized to determine the MODR. Predicted response variations were generated by randomly altering the CMPs within predefined limits. The capability index (CpK) was calculated with a threshold value of 1.33, indicating that the process variation occupies 75% of the specification limits range. All calculations were performed through the Minitab Workspace 1.3.1 (trial version) software.Initial simulation experiments (100k iterations) were conducted using mean values of 2.3 for buffer pH, 20% for initial %CH3OH content, and 1.4 for the gradient slope with standard deviation (SD) values of 0.3, 1 and 0.1, respectively. Only Rs(1), NASA and NATOR achieved higher CpK values than 1.33 concluding that there is a need to reduce SD values. These results indicate that some responses are highly sensitive to variations in the defined parameters. When SD values were set to 0.1 for buffer pH, and 0.05 for initial %CH3OH content, and 0.05 for the gradient slope, adequate CpK values (>1.33) were achieved for all CMAs (Fig. S7†). Based on the results, the MODR was determined within the design space, as illustrated in Fig. 4 with a predefined probability of 95% confidence interval. The finalized HPLC conditions were established as follows: buffer pH at 2.3, initial %CH3OH content at 20%, and a gradient slope of 1.4 %B/min. The buffer concentration and the column temperature were set to 10 mM and 50 °C, respectively.
Method validation
The developed method was validated according to the ICH Q2(R2) and the USP chapter 1225 Validation of compendial methods.28,29System suitability
The SST solution was analysed under the specified method conditions to evaluate the system suitability parameters. The following criteria were established for system suitability: the USP resolution (Rs(1) between ASA and SA) should be ≥ 3.0. The theoretical plate count for ASA should be greater than 20k, while for ATOR, it should exceed 300k. The %RSD of peak areas for the analytes from six replicate injections should be less than 2.0%. These criteria confirm that the developed HPLC method is appropriate for assessing method validation parameters.Specificity
To investigate the specificity of the method, forced degradation studies were performed. For this purpose, separate API standard solutions (100 μg mL−1) were employed to assess the degradation pathway and impurity profiling of each drug. According to the literature, the studied drugs are prone to acidic, basic, oxidative and thermal hydrolysis, and photodegradation and therefore we decided to investigate them under the above conditions.30–32 The degradation study was conducted for each API separately under the following conditions: acidic (0.1 M HCl for 1 h at 60 °C), alkaline (0.1 M NaOH for 1 h at 60 °C), oxidative (3% w/v H2O2 solution for 1 h at 60 °C and protected from light) degradation, thermal degradation in the solid state (24 h at 60 °C) and aqueous solution (24 h at 60 °C) and photolytic degradation (24 h exposure to daylight). The acidic and basic degradation samples were neutralized to prevent further degradation. The possible degradation was screened by comparing the recorded chromatograms with the standard control sample (Fig. 5). | ||
| Fig. 5 Overlaid chromatograms of the analysis of (A) the standard solution of each drug (100 μg mL−1) and under (B) acidic, (C) alkaline, (D) oxidative, (E) thermal hydrolysis, (F) thermal degradation in the solid state, and (G) photolytic degradation stressed conditions. Insets: peak purity of each drug. For experimental conditions see the main text. | ||
Under acidic conditions, ASA and ATOR were partially degraded to SA (ca 38%) and ATORD (ca 29.7%), respectively. SA was the primary degradation product derived from ASA33 and it was also identified by injecting the SA standard. ATORD was likely the lactone derivative (Impurity H as specified in the monograph) of ATOR, typically formed during acidic hydrolysis of the drug.34,35 Under alkaline conditions, ASA and RAM were almost completely degraded to SA and RAMD at higher than 96%. RAMD was likely Impurity E, formed due to alkaline hydrolysis of the ester bond.36,37 Simultaneous exposure to oxidative stress and elevated temperature had no effect on the stability of the drugs except for ASA which showed significant degradation as only ca 15% of ASA was recovered. The evaluation of the rest stress conditions (thermal hydrolysis, photolytic degradation) revealed no significant degradation of the tested drugs. HPLC-PDA analysis of the peak purity of the APIs across all stressed samples confirmed their complete separation from degradation products. The peak purity index exceeded 0.99999 (threshold 0.999), indicating thorough resolution of the analyte peak from its degradation products.
Linearity, precision and accuracy
The method was validated at seven concentration levels ranging from 50 to 150% of the specification limit of each drug. Since ASA is easily hydrolyzed to SA, the individual calibration curve of SA was constructed in the range of 4.0–48.5 μg mL−1 to determine its quantity in the calibration standards and unknown samples and correct the concentration of ASA. Unweighted linear regression models were applied to the experimental data within the specified calibration range to streamline future calculations. Linear calibration plots were achieved for the studied APIs within the tested calibration range, with coefficients of determination (r2) exceeding 0.9939 (Table 2). The residuals' distribution closely aligns with a normal distribution, as indicated by the p-values (p > 0.05) from the Shapiro–Wilk normality test. Triplicate analysis was performed in all cases.| Compound | Linear range (μg mL−1) | r 2 | Slope ± SDa | Intercept ± SD | Normality of residuals (SW test)b | |
|---|---|---|---|---|---|---|
| P | W | |||||
| a SD: standard deviation. b Normal distribution of residuals using the Shapiro–Wilk test. | ||||||
| SA | 4.04–48.5 | 1.0000 | 61040 ± 152 |
20043 ± 4253 |
0.6422 | 0.9379 |
| ASA | 51.4–154.2 | 0.9946 | 51734 ± 1697 |
182022 ± 188832 |
0.6935 | 0.9460 |
| RAM | 24.8–74.4 | 0.9939 | 27974 ± 504 |
−138720 ± 27061 |
0.1350 | 0.9296 |
| ATOR | 100–300 | 0.9943 | 51654 ± 898 |
155952 ± 194554 |
0.2333 | 0.8909 |
The intra-day and intermediate precisions were evaluated at three concentration levels of SA (4.0, 25.4 and 48.5 μg mL−1) and 50%, 100 and 150% for the specification limit of each API. For intra-day experiments, the relative recoveries were reported as average % recovery of the three levels tested for all analytes (Fig. 6) while the precision was less than 7.3%. Adequate values for intermediate precision were obtained to be less than 7.7% with recoveries being in the range of 91.4–106.7% for all analytes, respectively (Table S13†).
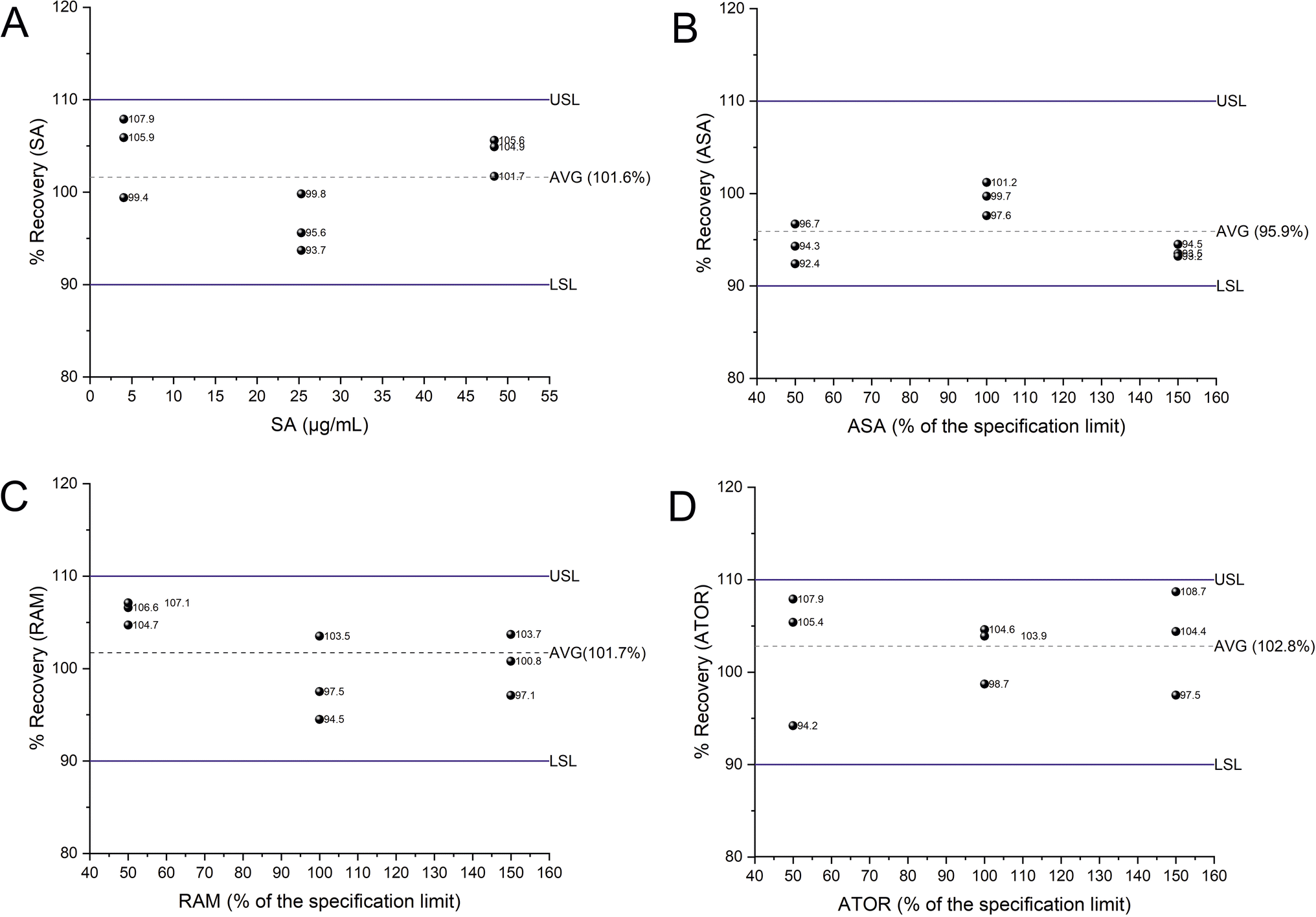 | ||
| Fig. 6 Plots of the % recovery for the studied APIs (A–C) and SA (D) across the working range. The horizontal dotted lines indicate the average (Avg) % recovery of the three levels. The blue horizonal lines indicate the upper (USL) and lower specification limits (LSL) targeting atp criteria of ± 10% for relative accuracy. | ||
Taking together, the above validation results demonstrate that the developed method is fit-for-purpose relative to ATP criteria.
Application to pharmaceutical formulations
The developed HPLC-UV method was successfully applied for the determination of ASA, RAM and ATOR in commercially available fixed-dose formulations (Trinomia® caps). Each capsule was individually treated as described in the “Analysis of pharmaceutical formulations” section and processed following the USP guidelines for dosage uniformity testing.38The experimental results are tabulated in Table 3. The samples complied with the pharmacopoeial specifications and limits for individual dosage uniformity. The assay values of the formulation, based on the mean of ten capsules, were calculated as 102.0% for ASA, 102.9% for RAM, and 105.5% for ATOR, all of which fall within the specified limits. Representative chromatograms of the analysis of the standard and sample are depicted in Fig. S8.†
| Sample | Content uniformity (%) (labeled value 100 mg ASA, 5 mg RAM, 20 mg ATOR per capsule) | ||
|---|---|---|---|
| ASA | RAM | ATOR | |
| CAP-1 | 103.6 | 101.6 | 109.3 |
| CAP-2 | 94.1 | 108.6 | 104.5 |
| CAP-3 | 101.0 | 104.8 | 104.9 |
| CAP-4 | 105.1 | 103.1 | 96.8 |
| CAP-5 | 109.3 | 104.5 | 101.6 |
| CAP-6 | 102.7 | 105.4 | 98.6 |
| CAP-7 | 108.4 | 107.2 | 107.2 |
| CAP-8 | 88.9 | 99.5 | 110.9 |
| CAP-9 | 106.5 | 103.1 | 110.5 |
| CAP-10 | 103.4 | 99.6 | 108.8 |
| Mean of individual contents (% of the label claim) (X) | 102.0 | 102.9 | 105.5 |
| Reference value (M) | 101.5 | 101.5 | 101.5 |
| Standard deviation (s) | 5.8 | 3.3 | 4.5 |
| Acceptance value (AV) | 12.1 | 8.1 | 13.1 |
| Maximum allowed acceptance value (L1) | 15 | 15 | 15 |
| Result | Pass (AV < L1) | Pass (AV < L1) | Pass (AV < L1) |
Conclusions
In the present study, an HPLC-UV method was developed, validated, and applied for the simultaneous determination of ASA, RAM and ATOR in their fixed-dose polypill formulation. An AQbD approach was implemented, encompassing defining the analytical target profile, method scouting, risk assessment, and the identification of CMAs and CMPs. Optimal experimental conditions were determined using a chemometric approach, with all models demonstrating strong predictability and fit. The MODR comprised a set of CPM conditions that provided acceptable values for CMAs. Forced degradation experiments were conducted to prove the selectivity of the proposed approach. Capability analysis, combined with Monte Carlo simulations, was employed to assess the method's robustness. Method validation was conducted, focusing on selectivity, precision, and accuracy. The method was successfully applied to quantify the drug content in commercially available Trinomia® capsules.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Halil Kasem: formal analysis, data curation, investigation, validation. Marianna Ntorkou: data curation, investigation validation, writing – original draft. Paraskevas D. Tzanavaras: methodology, writing – review & editing. Constantinos K. Zacharis: conceptualization, methodology, investigation, methodology, software, validation, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Pharmathen SA company is kindly acknowledged for the donation of the atorvastatin reference standard.References
- J. R. González-Juanatey, L. Masana, R. Dalmau and A. Cordero, Int. J. Cardiol., 2025, 418, 132578 CrossRef PubMed.
- J. M. Castellano, S. J. Pocock, D. L. Bhatt, A. J. Quesada, R. Owen, A. Fernandez-Ortiz, P. L. Sanchez, F. Marin Ortuño, J. M. Vazquez Rodriguez, A. Domingo-Fernández, I. Lozano, M. C. Roncaglioni, M. Baviera, A. Foresta, L. Ojeda-Fernandez, F. Colivicchi, S. A. Di Fusco, W. Doehner, A. Meyer, F. Schiele, F. Ecarnot, A. Linhart, J.-C. Lubanda, G. Barczi, B. Merkely, P. Ponikowski, M. Kasprzak, J. M. Fernandez Alvira, V. Andres, H. Bueno, T. Collier, F. Van de Werf, P. Perel, M. Rodriguez-Manero, A. Alonso Garcia, M. Proietti, M. M. Schoos, T. Simon, J. Fernandez Ferro, N. Lopez, E. Beghi, Y. Bejot, D. Vivas, A. Cordero, B. Ibañez and V. Fuster, N. Engl. J. Med., 2022, 387, 967–977 CrossRef CAS PubMed.
- D. T. Xuan, H. M. T. Nguyen and V. D. Hoang, Chemom. Intell. Lab. Syst., 2024, 254, 105243 CrossRef CAS.
- L. Chiarentin, C. Gonçalves, C. Augusto, M. Miranda, C. Cardoso and C. Vitorino, Crit. Rev. Anal. Chem., 2023, 54(8), 3478–3519 CrossRef PubMed.
- Analytical Procedure Development Q14, https://database.ich.org/sites/default/files/ICH_Q14_Guideline_2023_1116.pdf, (accessed 17 February 2025).
- K. H. Kim, J. E. Lee, J. C. Lee, R. Maharjan, H. Oh, K. Lee, N. A. Kim and S. H. Jeong, J. Chromatogr. A, 2023, 1709, 464375 CrossRef CAS PubMed.
- M. K. Parr and A. H. Schmidt, J. Pharm. Biomed. Anal., 2018, 147, 506–517 CrossRef CAS PubMed.
- R. Deidda, S. Orlandini, P. Hubert and C. Hubert, J. Pharm. Biomed. Anal., 2018, 161, 110–121 CrossRef CAS PubMed.
- S. M. Aboelghar, M. A. Hegazy and H. A. Wagdy, Microchem. J., 2024, 200, 110292 CrossRef CAS.
- Salwa and L. Kumar, Microchem. J., 2023, 193, 108944 CrossRef CAS.
- D. A. Ahmed, O. G. Hussein, M. R. Rezk, M. Abdelkawy and Y. Rostom, Microchem. J., 2024, 196, 109717 CrossRef CAS.
- I. de Jesus do Nascimento Lopes, S. Kazumasa Fujimori, T. de Carvalho Mendes, R. Adrielle Dias de Almeida, F. Furtado de Mendonça de Sousa, C. Areias de Oliveira, D. Dibo do Nascimento, F. Rebello Lourenço, M. Isabel Rodrigues and L. Deris Prado, Microchem. J., 2024, 199, 109939 CrossRef.
- R. Patel, J. Shah and M. Patel, Sep. Sci. plus, 2025, 8, e202400229 CrossRef CAS.
- O. G. Hussein, D. A. Ahmed, M. R. Rezk, M. Abdelkawy and Y. Rostom, J. Pharm. Biomed. Anal., 2023, 235, 115598 CrossRef CAS PubMed.
- J. A. Afonso Urich, V. Marko, K. Boehm, R. A. Lara Garcia, A. Fedorko, S. Salar-Behzadi and D. Jeremic, Sci. Pharm., 2024, 92, 22 CrossRef CAS.
- S. S. Yadav and J. R. Rao, Int. J. Pharm. Pharm. Sci., 2014, 6, 443–448 Search PubMed.
- S. K. Shetty, R. M. Borkar, P. S. Devrukhakar, K. V. Surendranath, P. Radhakrishnanand, J. Satish, N. Shastri, J. Jogul and U. M. Tripathi, J. Liq. Chromatogr. Relat. Technol., 2012, 35, 662–676 CrossRef CAS.
- S. N. Ramalakshmi, B. Bhavani, R. Nagabathula, B. Chandra, K. Saipriyanaka and B. V. Renuka Sai Sri, Lat. Am. J. Pharm., 2022, 41, 1951–1954 CAS.
- S. Rao, B. Balaswami, P. Venkata Ramana, P. Sanjeeva and G. Srenivasulareddy, Int. J. Appl. Pharm., 2018, 10, 90–96 CrossRef CAS.
- N. A. Abdallah, A. M. El-Brashy, F. A. Ibrahim and M. I. El-Awady, BMC Chem., 2023, 17, 1–11 CrossRef PubMed.
- B. Żuromska-Witek, M. Stolarczyk, M. Szlósarczyk, S. Kielar and U. Hubicka, Chemosensors, 2023, 11, 25 CrossRef.
- B. Żuromska-Witek, M. Stolarczyk, M. Szlósarczyk, S. Kielar and U. Hubicka, Chemosensors, 2022, 11, 25 CrossRef.
- N. A. Abdallah, A. M. El-Brashy, F. A. Ibrahim and M. I. El-Awady, BMC Chem., 2023, 17, 19 CrossRef CAS PubMed.
- J. W. Dolan, Ion Pairing — Blessing or Curse?, https://www.chromatographyonline.com/view/ion-pairing-blessing-or-curse, (accessed 22 October 2024).
- L. Volta e Sousa, R. Gonçalves, J. C. Menezes and A. Ramos, AAPS PharmSciTech, 2021, 22, 128 CrossRef PubMed.
- T. Tome, N. Žigart, Z. Časar and A. Obreza, Org. Process Res. Dev., 2019, 23, 1784–1802 CrossRef CAS.
- T. Bastogne, F. Caputo, A. Prina-Mello, S. Borgos and M. Barberi-Heyob, J. Pharm. Biomed. Anal., 2022, 219, 114911 CrossRef CAS PubMed.
- Step, Committee for Medicinal Products for Human Use ICH Q2(R2) Guideline on validation of analytical procedures, https://www.ema.europa.eu/contact, (accessed 27 December 2024).
- General Chapters: <1225> VALIDATION OF COMPENDIAL METHODS, http://ftp.uspbpep.com/v29240/usp29nf24s0_c1225.html, (accessed 27 December 2024).
- P. Yenda, N. K. Katari, S. K. Ettaboina, B. Satheesh, S. K. Muchakayala and R. Gundla, Biomed. Chromatogr., 2023, 37, e5585 CrossRef CAS PubMed.
- M. El Kacemi, A. El Orche, K. El Bourakadi, Y. H. Benchekroun, F. Echerfaoui, K. Karrouchi, M. Bouatia and M. El Karbane, J. AOAC Int., 2024, 107, 735–748 CrossRef PubMed.
- L. Hanyšová, M. Václavková, J. Dohnal and J. Klimeš, J. Pharm. Biomed. Anal., 2005, 37, 1179–1183 CrossRef PubMed.
- J. A. A. Urich, V. Marko, K. Boehm, J. Karrer, M. Koeberle and S. Salar-Behzadi, J. Anal. Chem., 2023, 78, 1760–1769 CrossRef CAS.
- B. Ramesha, K. R. Venugopala Reddy, B. V. Kishore, M. K. Amith Kumar, L. P. Raju and B. N. Thara, J. Liq. Chromatogr. Relat. Technol., 2014, 37, 275–297 CrossRef CAS.
- E. T. Kovachovska, J. Acevska, K. Brezovska, S. Trajkovic, I. Mitrevska, S. Ugarkovic and A. Dimitrovska, Maced. J. Chem. Chem. Eng., 2022, 41, 65–76 CrossRef CAS.
- L. Hanyšová, M. Václavková, J. Dohnal and J. Klimeš, J. Pharm. Biomed. Anal., 2005, 37, 1179–1183 CrossRef PubMed.
- K. Regulska, J. Musiał and B. J. Stanisz, Pharmaceutics, 2021, 13, 1600 CrossRef CAS PubMed.
- Stage 6 Harmonization Official, https://www.usp.org/sites/default/files/usp/document/harmonization/excipients/m99694.pdf, (accessed 30 December 2024).
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ay00021a |
| This journal is © The Royal Society of Chemistry 2025 |