
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A RP-HPLC-UV method for the dual detection of fluconazole and clobetasol propionate and application to a model dual drug delivery hydrogel†
Robyn A.
Macartney
 a,
Annabelle T. R.
Fricker
b,
Andrew M.
Smith
c,
Stefano
Fedele
d,
Ipsita
Roy
be and
Jonathan C.
Knowles
*a
a,
Annabelle T. R.
Fricker
b,
Andrew M.
Smith
c,
Stefano
Fedele
d,
Ipsita
Roy
be and
Jonathan C.
Knowles
*a
aDivision of Biomaterials and Tissue Engineering, University College London (UCL) Eastman Dental Institute, London, UK. E-mail: j.knowles@ucl.ac.uk
bSchool of Chemical, Materials & Biological Engineering, Faculty of Engineering, University of Sheffield, Sheffield, UK
cDepartment of Microbial Diseases, UCL Eastman Dental Institute, Royal Free Campus, University College London, London, UK
dUCL Eastman Dental Institute, University College London, London, UK
eInsigneo Institute for in Silico Medicine, University of Sheffield, UK
First published on 24th April 2025
Abstract
Advanced drug delivery systems have become widely investigated to improve the efficacy of treatments for several diseases. These devices offer improved efficient, sustained, and targeted delivery which improves patient compliance, quality of life and minimises potential systemic side effects. As these therapeutic devices have advanced there is a potential for the development of products which deliver multiple drugs for simultaneous treatment of diseases. Given the interest in these dual-delivery devices it follows that new analytical methods need to be developed to detect and quantify different analytes during device development and validation. Here, for the first time, a reverse-phase high performance liquid chromatography (RP-HPLC) method is validated, utilising UV detection, for the dual detection of fluconazole and clobetasol propionate. The method is tested on a dual loaded model implant material intended as mucosal patches for the direct treatment of lichen planus and associated fungal infections. The method described here exhibited specificity and robustness with accurate and precise results. Good linearity was obtained between 0.25 and 2.5 mg mL−1 for fluconazole and 5 and 50 μg mL−1 for clobetasol propionate, with an R2 value of 0.9999 for the dual detection of fluconazole and clobetasol propionate. The developed method demonstrated selectivity and the solution containing both fluconazole and clobetasol propionate remained stable over a range of storage temperatures for up to 28 days. Within this validation study, the protocol was applied to a relevant dual loaded film showing the suitability of the method in studying drug release characteristics. The method described here also has a broader applicability for analysis and quantification of in vitro and in vivo drug release studies.
1. Introduction
Oral lichen planus (OLP) is a relatively common chronic inflammatory disease mediated by T lymphocytes, which affects the mucous membranes of the oral cavity. In its most severe form, OLP is associated with mucosal erosion and ulceration, resulting in significant pain and discomfort, particularly during eating or drinking. The World Health Organisation (WHO) suggests that OLP is a potentially malignant disorder, but there has been controversy over the malignant transformation, which has been reported to range from zero to 9.52% across patients.1 Reports indicate that the worldwide prevalence of OLP is up to 1.01% of the global population.2 There are reports of higher incidences of the disease in patients over the age of 40 and some evidence of a geographical influence in disease development with increased incidences reported in regions such as Europe and South America.3 Routine treatment involves the use of corticosteroids usually applied directly to the painful area or sometimes taken systemically.4 There is no cure for this disease, but these treatments can reduce pain and improve healing potential around the affected area. However, these treatments usually require long-term regimens, which can bring adverse consequences associated with prolonged steroid use.5,6 One of the side-effects of long-term steroid use is the susceptibility of patients to fungal infections, leading to the need for a second prescription to treat the fungal infection.7 The key drawback with using topical ointments for treatment of these afflictions is the short contact time with the affected area, as well as poor transdermal bioavailability.8–10 This is especially prevalent in the oral cavity due to the effects of salivary washout. Whilst treatments such as intralesional injections and laser therapy have shown some promise as treatments for OLP these are invasive procedures which are not suitable for all patient groups.11,12The most prescribed corticosteroid for the purpose of OLP treatment is clobetasol propionate (CP).13,14 This drug works by binding to cytoplasmic glucocorticoid receptors, thereby activating receptor mediated gene expression. The downstream changes in protein syntheses associated with these gene expression changes include an increase in anti-inflammatory protein synthesis and a decrease in the production of pro-inflammatory mediators.15 Specifically, CP acts to produce phospholipase A2 inhibitory proteins that help control the release of arachidonic acid from phospholipid membranes which is an inflammatory precursor.16
Fungal infections, identified as a common secondary complication of long-term steroid treatment, are routinely treated using fluconazole (FLU). This drug shares a mechanism of action with many other fungal treatments via disruption of the fungal cell membrane.17 Disruption to the membrane is achieved by preventing the conversion of lanosterol to ergosterol, responsible for forming a crucial part of the fungal cell membrane, via interaction of the drug with 14-demethylase.
Given the fact these two drugs are commonly prescribed in unison it would be helpful to have a means of dual quantification following delivery for analysis of parameters such as efficacy in delivery and tissue targeting by respective drugs. Additionally, recently the development of new drug delivery mechanisms in this field has become of interest to improve treatment outcomes via prolonged drug contact times and sustained release mechanisms.18 Often steroidal treatments are applied as topical ointments and cannot maintain prolonged contact times with the affected areas. Currently there is expressed interest in developing in situ patch style, drug eluting type treatments for these sorts of infections.19,20Via this patch style delivery system, the prolonged delivery of multiple drugs becomes more achievable, these multi drug therapies are already the norm in cancer treatments and are extending into normal practice in both the dual targeting of primary ailments, and in the targeting of both the primary source of disease and related secondary complications. With the development of these new drug delivery systems comes the need for new analytical methods capable of detecting and quantifying the analytes used in the development of new formulations. These new drug delivery interventions require extensive in vitro drug dissolution studies during development stages. Whilst many studies have described the individual detection of either CP or FLU,21–23 none to the authors knowledge, have reported on a reliable method for the dual detection of the two drugs. The development of such an HPLC method leads to many advantages such as cost and time savings, versatility, and high sensitivity and specificity, all whilst using readily available equipment with no need for specialist or in-house modification.24 Using HPLC for this application is appropriate given that both CP and FLU are UV-active compounds and can therefore be separated and detected using a single HPLC method.25,26 To ensure the suitability of the method developed the system suitability factors such as resolution, retention time, tailing factor and capacity factor were assessed. Therefore, here we present a single-wavelength UV-HPLC quantification method for the simultaneous detection and quantification of FLU and CP which is validated following ICH guidelines.
2. Materials and methods
2.1. Materials
CP powder, ≥98%, was purchased from Cambridge Bioscience (Cambridge, England, UK). FLU powder, 98%, dimethylformamide (DMF), methanol (MeOH), trifluoracetic acid (TFA) and acetonitrile (ACN) were procured from fisher scientific (Loughborough, England, UK). Polyvinyl alcohol (PVA) and dimethyl sulfoxide (DMSO) were purchased from Sigma-Aldrich (Dorsett, UK).2.2. Stock solution, working standards, quality control solutions of FLU and CP
A stock solution containing 2.5 mg per mL FLU and 50 μg per mL CP was prepared in a 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30 MeOH:dH2O (v/v) solution. Six standard solutions were prepared by further dilution of the FLU–CP stock solution with the MeOH:dH2O solution, which were used for the calibration curve, giving concentrations of FLU and CP corresponding to (0.25, 0.5, 1, 1.5, 2 and 2.5 mg mL−1) and (5, 10, 20, 30, 40 and 50 μg mL−1) respectively. Quality control (QC) solutions at three concentrations were prepared from the same stock solution of FLU–CP at concentrations of 0.5 mg mL−1–10 μg mL−1, 1 mg mL−1–20 μg mL−1 and 2 mg mL−1–40 μg mL−1 which were utilised for accuracy and precision studies. These concentration ranges for the analysis of FLU and CP were chosen as they are reflective of the therapeutic dosage of these drugs when prescribed in their topical form.27,28
:30 MeOH:dH2O (v/v) solution. Six standard solutions were prepared by further dilution of the FLU–CP stock solution with the MeOH:dH2O solution, which were used for the calibration curve, giving concentrations of FLU and CP corresponding to (0.25, 0.5, 1, 1.5, 2 and 2.5 mg mL−1) and (5, 10, 20, 30, 40 and 50 μg mL−1) respectively. Quality control (QC) solutions at three concentrations were prepared from the same stock solution of FLU–CP at concentrations of 0.5 mg mL−1–10 μg mL−1, 1 mg mL−1–20 μg mL−1 and 2 mg mL−1–40 μg mL−1 which were utilised for accuracy and precision studies. These concentration ranges for the analysis of FLU and CP were chosen as they are reflective of the therapeutic dosage of these drugs when prescribed in their topical form.27,28
2.3. Chromatographic conditions
HPLC analysis was conducted using an Agilent Technologies 1100 Infinity compact LC Series (Agilent Technologies, Stockport, Cheshire, UK), comprising a degassing unit, binary pump, autosampler, and UV-detector with a Zorbax LC column (5 μm particle size, C18, 170 Å, 250 × 4.6 mm) from Agilent Technologies (Stockport, Cheshire, UK) was employed for chromatographic separation. The separation of analytes occurred at 24 °C, utilising mobile phases composed of 0.1% TFA in water and 0.1% TFA in ACN. Conditions were as follows; 35% of ACN in water for 5 minutes at a flow rate of 0.6 mL min−1, the flow rate was then increased to 1 mL min−1 between 5 and 6 minutes, finally the mobile phase composition was ramped to 100% ACN (+0.1% TFA) between 5 and 15 minutes at 1 mL min−1. The conditions were then held as such until 20 minutes. UV signal was measured using an Agilent G1315A DAD detector at 250 nm. For each concentration, single injections of 20 μL were made to obtain the peak area for constructing the calibration curve. Detection of the analytes transpired after 7.3 minutes for FLU and 17.4 minutes for CP. The UV absorption maximum of each drug was determined using a Unicam UV-500 UV-vis spectrometer (Thermospectronic, Leeds, UK) to identify the optimal wavelength necessary to validate the HPLC method. A 1 cm2 quartz cuvette filled with 70:30 MeOH:dH2O (v/v) was used as a blank for FLU and CP. Standard solutions containing each drug were scanned at room temperature between 220 and 400 nm to produce a spectrum for both drugs.
2.4. Method validation
The analytical method was validated according to ICH guidelines, ‘Validation of Analytical Procedures Q2(R2)’,29 covering aspects such as linearity, limit of detection (LOD), limit of quantification (LOQ), specificity, accuracy, precision, and robustness.:30 MeOH:dH2O (v/v)). The chromatograms were inspected for any signs of interference between the peaks for the two drugs and for any peak interaction caused by the solvents.
 | (1) |
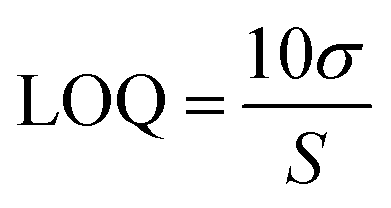 | (2) |
On the other hand, the precision evaluates the level of agreement between multiple measurements taken using various samples of the same homogenous drug solution under the predetermined conditions. Thus, assessing the presence of random error. Metrics like relative standard deviation (RSD) (eqn (3)) where σ is the standard deviation, repeatability, and intermediate precision were computed based on the estimated concentrations. For concentrations under analysis, an RSD value lower than 10% is considered acceptable.
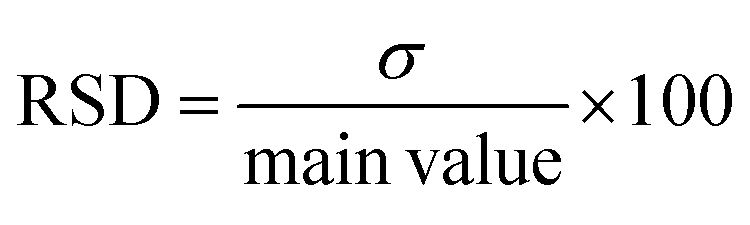 | (3) |
:dH2O after the highest concentration of the calibration standard solution of FLU–CP (2.5 mg mL−1, 50 μg mL−1 respectively). Acceptable performance is determined when the area of peak observed for each drug in the blank samples is not greater than 20% of the area of a sample solution at the LOQ concentration. This analysis is based on the chromatogram of the blank solution.
:dH2O at a concentration of double the maximum concentration from the calibration curve. Samples were initially prepared at a concentration of 5 mg per mL FLU and 100 μg per mL CP and then diluted 5 and 10 times in the MeOH:dH2O buffer to give FLU–CP solutions of final concentrations of 1 mg mL−1–20 μg mL−1 and 0.5 mg mL−1–10 μg mL−1 respectively. Accuracy and precision were determined based on three replicates for each dilution via comparison with the calibration curve.
2.5. Stability of analytical solution
To assess the degradation of FLU and CP in the release media (MeOH:dH2O), stability studies were conducted. A standard solution of FLU and CP was prepared in MeOH:dH2O 70:30 (v/v) and stored under various conditions: room temperature, in an incubator (37 °C), in the refrigerator (4 °C), and in the freezer (−25 °C). The concentration of each drug was quantified using the HPLC method at intervals of 7, 14, 21, and 28 days.
2.6. Applications of the validated analytical method
The validated HPLC method was then further applied for the simultaneous quantification of FLU and CP release from a model drug delivery patch dual loaded with both drugs. The percentage drug encapsulation within the film and the percentage cumulative release over a 24-hour study was performed employing the use of the HPLC method in this application.:3 (v/v) ratio of DMSO:DMF by stirring for 3 hours at 130 °C. The solution was allowed to cool whilst stirring to homogenise, the drugs were then added to the solution with FLU and CP added to a final concentration of 2.5 mg mL−1 and 150 μg mL−1 respectively. Solutions were mixed to homogenise and then cast in 6-well plates and allowed to cool naturally over 12 hours to room temperature. The solvent was evaporated overnight and then films removed for use in the drug release study.
:dH2O overnight and the supernatant collected for analysis. This solution was then used to quantify the amount of drug encapsulated in each film.
:dH2O 70:30 (v/v), and incubated at 37 °C under atmospheric humidity for up to 24-hours. At predefined timepoints 1 mL of release media was removed for analysis by the HPLC method previously described and replaced with 1 mL of prewarmed release buffer. The samples were analysed immediately, without further post processing, upon collection using the previously described HPLC method. The experiment was run in triplicate and the cumulative release calculated based on the maximum release possible calculated by the encapsulation efficiency study using eqn (4). | (4) |
The equation used to calculate cumulative release from scaffolds at time x, where releaseTx = total release at time x, releaseTx−1 = total release at time x − 1 and maximum release = full possible release from the scaffold.
2.7. Statistical analysis
Quantification of drug concentration was carried out using peak integration in OpenLab CDS (Agilent Technologies, California, USA). Means, standard deviation, relative standard deviation (RSD), and relative error were calculated using Microsoft® Excel® Version 2402 (Microsoft Corporation, Redmond, USA). GraphPad Prism® 10.1.0 (Graphpad Software, San Diego, California, USA) was used for any statistical analysis of data and for graphing purposes.3. Results and discussions
3.1. Method validation
The UV absorbance of FLU and CP were examined using a UV-vis spectrophotometer to identify at which wavelength the signal was most intense and demonstrated the best specificity. The UV-vis spectra for 2.5 mg per mL FLU and 50 μg per mL CP in the MeOH:dH2O buffer showed maximum absorption peaks at 258 and 244 nm respectively for each drug. The point at which the two spectra overlap is considered the maximum absorption for both drugs simultaneously and therefore should be used in the single wavelength detection of the mixture, here this is observed at 250 nm, as shown in S1.†
886.87 mAu obtained using 250 nm. Whilst the difference in the area under the CP peak was less affected by this change in wavelength with a difference of only 231.58 mAu. Therefore, the final wavelength selected for analysis was 250 nm.
:dH2O alongside samples of FLU–CP solution at the highest and lowest concentration used in the calibration curve. The retention times for FLU and CP under the selected chromatographic conditions were 7.3 minutes and 17.4 minutes, respectively, within a total running time of 20 minutes. The individual detection of CP has been reported up to a retention time of 18 minutes31,32 and that of FLU usually utilises methods lasting between 6 and 10 minutes.22,23 At present it would be necessary to individually detect these components using different analysis methods, the novel dual detection method presented here allows the opportunity for the rapid detection of both elements, saving considerable time when analysing large numbers of samples. Additionally, the possibility of detecting both components in a single method reduces the required sample volume for analysis, often sample volumes may be extremely limited and requirement for multiple analyses puts strain on the available resources. As shown in Fig. 1B and C, the presence of both drugs in the sample chromatogram is well-defined and the two are well separated without any interaction, indicating the method's specificity for the selected drugs. Conversely, the blank sample exhibited no peak at the retention times corresponding to those of the drugs. The data here confirms that the analytical method for dual detection of FLU and CP demonstrates good specificity and selectivity.
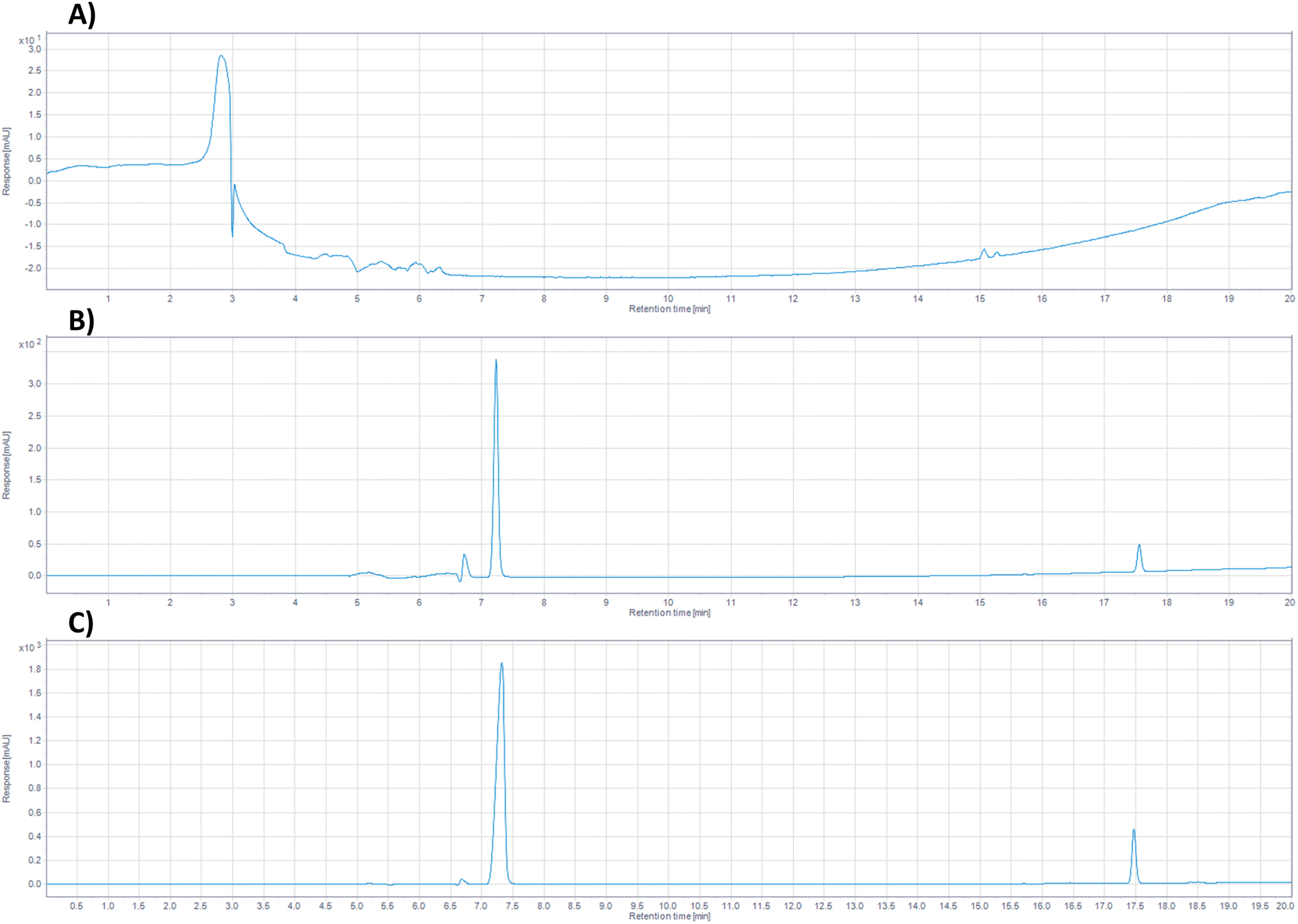 | ||
| Fig. 1 Chromatograph spectrum of (A) a blank sample using the MeOH:dH2O buffer, (B) FLU–CP in a concentration of 0.25 mg per mL FLU, 5 μg per mL CP and (C) FLU–CP in a concentration of 2.5 mg per mL FLU, 50 μg per mL CP, respectively where the x-axis represents the time in minutes and the y-axis represents the peak height in mAU, without blank subtraction. | ||
Capacity factor describes the interaction of analytes with the sorbent in the column, taking into account the void volume time of the column. Typically, capacity factors of 1–10 are considered good for chromatographic separation,33 meaning the values obtained here are acceptable for the detection and quantification of FLU and CP. Additionally, the ratio of the capacity factors for the two eluted compounds provides information on the selectivity of the method. Here, this separation factor, is calculated as 3.35, which greatly exceeds the accepted value of 1.1 which indicates good selectivity of a method.
Peak asymmetries of less than 2 are generally acceptable when the asymmetry factor is calculated at 10% of the peak height, with values of less than 1.05 being described as excellent.30 The asymmetry for the peaks of both eluted compounds in this method are therefore considered excellent regarding asymmetric properties.
Results presented in Table 1 summarise the values for slope, y-intercept, coefficient of correlation, LOD and LOQ achieved from this method. The correlation coefficient (R2) was 0.99 for both FLU and CP, confirming a robust linear response across the concentration ranges investigated. The LOD and LOQ were calculated as 0.003 mg mL−1 and 0.008 mg mL−1 respectively for FLU, and 0.004 μg mL−1 and 0.012 μg mL−1 respectively for CP. Previous studies have described lower values for the limit of quantification of FLU using HPLC methods within the nanogram range.34–36 These studies dealt primarily with lower concentration ranges for quantification due to the proposed application of the methods for quantification being detection of FLU in human plasma. A study by Singh et al. demonstrated that these factors are media dependent with LOD ranging from 0.2–0.39 μg mL−1 and LOQ ranging from 0.61–1.19 μg mL−1 depending on the media in which the FLU was contained.37 However, another study with an application closer to that described here, into the detection of FLU in solid dosage forms gave similar LOD and LOQ to those found in our validation study.38 Therefore, it is confirmed that the ranges for FLU quantification investigated here are appropriate for the intended application. The LOD and LOQ reported in the current study are improved over those described previously in the literature for the detection of CP using RP-HPLC methods. In the past LOQs of 0.64–9.72 μg mL−1 for CP have been reported, suggesting an enhanced performance of the method investigated here.39–42 Fontana et al. reported values of 0.45 μg mL−1 and 1.38 μg mL−1 for LOD and LOQ respectively during their validation of a single detection method of CP using HPLC.40 Similarly, Bagad et al. reported much greater values of LOD and LOQ (3.20 and 9.72 μg mL−1 respectively) than those obtained in our method.43 In a method for the dual detection of nadifloxacin and CP, one group have obtained LOD and LOQ of 0.21 μg mL−1 and 0.64 μg mL−1 respectively.41 Whilst these values lie closer to those obtained in our study, still the values obtained in our method suggest that the new method validated here offers reliable detection and quantification of the analyte at lower concentrations than those previously described. Given the intended application of the method under validation the chosen range for linearity studies within the milligram range for FLU and microgram range for CP is appropriate and ensures relevance of the method validation for future research.
| FLU | CP | |
|---|---|---|
| Calibration standards range | 0.25–2.5 mg mL−1 | 5–50 μg mL−1 |
| Slope | 6730.10 | 38.99 |
| y-Intercept | −10.64 | 0.38 |
| R 2 | 0.9999 | 0.9999 |
| LOD | 0.003 mg mL−1 | 0.004 μg mL−1 |
| LOQ | 0.008 mg mL−1 | 0.012 μg mL−1 |
Samples were prepared and injected in triplicate over three consecutive days. To calculate the percentage recovery the nominal concentration of quality control samples was compared with those obtained using the developed method. Results are presented in Tables 2 and 3. Across the concentrations used for this experiment, the mean percentage recovery falls within the range of 98–101.35% is within the acceptable standards of 98–102% as described the FDA.44
| Quality control concentration (mg mL−1) | Percentage recovery (%) | Mean recovery ± SD (%) | |
|---|---|---|---|
| 0.5 | Day 1 | 101.92 | 101.05 ± 1.54 |
| Day 2 | 101.96 | ||
| Day 3 | 99.27 | ||
| 1 | Day 1 | 101.71 | 101.49 ± 0.63 |
| Day 2 | 101.98 | ||
| Day 3 | 100.78 | ||
| 2 | Day 1 | 100.48 | 99.11 ± 1.19 |
| Day 2 | 98.48 | ||
| Day 3 | 98.37 | ||
| Quality control concentration (μg mL−1) | Percentage recovery (%) | Mean recovery ± SD (%) | |
|---|---|---|---|
| 10 | Day 1 | 99.33 | 99.26 ± 0.73 |
| Day 2 | 99.09 | ||
| Day 3 | 99.37 | ||
| 20 | Day 1 | 101.35 | 99.31 ± 1.78 |
| Day 2 | 98.17 | ||
| Day 3 | 98.40 | ||
| 40 | Day 1 | 101.13 | 101.34 ± 0.49 |
| Day 2 | 98.40 | ||
| Day 3 | 101.48 | ||
Additionally, to ensure the reliability and repeatability of the method for analysing FLU and CP within the same concentration ranges described previously, the precision of the method was assessed. For this assessment, samples of the three different concentrations were quantified in triplicate on a single day (intra-day) and across three consecutive days (inter-day). The results are presented in Tables 4 and 5, mean and relative standard deviation are described, indicative of precision. Again, the calculated results remained within acceptable limits, as the variability (RSD) for all concentrations was below 2%.45 This study aimed to develop a suitably sensitive and accurate approach for the detecting and quantifying FLU and CP simultaneously. The chosen analytical technique for dual detection of FLU and CP was HPLC due to its capability to handle complex molecules with diverse polarity and molecular mass. The results presented here show that the HPLC method for FLU and CP has been successfully developed and validated following guidelines put in place by the ICH, via testing for specificity, linearity, range LOD and LOQ, accuracy, and precision. The results obtained show that this method is suitable for in vitro quantification of both FLU and CP.
| Quality control concentration (mg mL−1) | Intra-day precision | Inter-day precision | ||
|---|---|---|---|---|
| Mean concentration found ± SD (mg mL−1) | Relative standard deviation (%) | Mean concentration found ± SD (mg mL−1) | Relative standard deviation (%) | |
| 0.5 | 0.53 ± 0.01 | 1.60 | 0.51 ± 0.01 | 1.55 |
| 1 | 1.04 ± 0.01 | 0.41 | 1.01 ± 0.01 | 1.20 |
| 2 | 2.02 ± 0.02 | 0.89 | 1.98 ± 0.04 | 1.77 |
| Quality control concentration (μg mL−1) | Intra-day precision | Inter-day precision | ||
|---|---|---|---|---|
| Mean concentration found ± SD (μg mL−1) | Relative standard deviation (%) | Mean concentration found ± SD (μg mL−1) | Relative standard deviation (%) | |
| 10 | 9.86 ± 0.06 | 0.58 | 9.93 ± 0.12 | 1.26 |
| 20 | 20.35 ± 0.12 | 0.59 | 19.86 ± 0.33 | 1.68 |
| 40 | 38.25 ± 0.35 | 0.92 | 40.42 ± 0.79 | 1.96 |
:dH2O sample after injecting FLU–CP at the highest concentration of the calibration curve. As shown in Fig. 2B chromatograms of the blank samples revealed no evidence of peaks around 7.3 minutes or 17.4 minutes, corresponding to the retention times of FLU and CP, respectively. This indicates the absence of any carry-over effect after injecting the high-concentration drug sample.
 | ||
| Fig. 2 Chromatograph spectra of (A) a sample of the highest sample concentration used in the calibration curve (FLU–CP at 2.5 mg mL−1 and 50 μg mL−1 respectively) (B) a blank sample (MeOH:dH2O buffer) injected immediately after the highest concentration sample, without blank subtraction. | ||
:dH2O buffer. In this experiment dilution factors of 5 and 10 were used. Percentage recovery for FLU was calculated as 99.11 ± 4.48% and 101.29 ± 0.29% for the 1 in 10 and 1 in 5 dilutions respectively. The CP solutions showed percentage recoveries of 103.94 ± 3.94% and 101.06 ± 4.68% for the 1 in 10 and 1 in 5 dilutions respectively. Results obtained here lie within the acceptable precision range of ±15% outlined in the ICH guidelines.46
| Parameter | Condition | FLU | CP | ||||
|---|---|---|---|---|---|---|---|
| Recovery (%) | Retention time (min) | Peak area (mAU) | Recovery (%) | Retention time (min) | Peak area (mAU) | ||
| Column temperature (°C) | 20.5 | 99.94 | 7.3 | 16980.8 |
100.17 | 17.5 | 2032.0 |
| 21 | 100 | 7.3 | 16991.3 |
100 | 17.5 | 2028.6 | |
| 21.5 | 99.65 | 7.3 | 16931.9 |
99.76 | 17.5 | 2023.7 | |
| Flow rate, 0–5, 5–20 min (mL min−1) | 0.59, 0.99 | 99.57 | 7.4 | 16918.4 |
100.97 | 17.5 | 2048.3 |
| 0.6, 1 | 100 | 7.3 | 16991.3 |
100 | 17.5 | 2028.6 | |
| 0.61, 1.01 | 99.64 | 7.2 | 16929.7 |
99.64 | 17.4 | 2021.2 | |
| Mobile phase, 0–5, ramping to 20 min (dH2O:MeOH) |
64.8:35.2, 0.2:99.8 |
99.43 | 7.3 | 16894.3 |
100.65 | 17.4 | 2041.7 |
| 65:35, 0:100 |
100 | 7.3 | 16991.3 |
100 | 17.5 | 2028.6 | |
| 65.2:34.8, 0:100 |
99.08 | 7.3 | 16834.8 |
100.43 | 17.4 | 2037.4 | |
3.2. Stability of analytical solution
Stability of the analytical solution has an important role in estimating the shelf life of samples when stored under controlled environmental conditions taking into consideration factors such as temperature, humidity, and light exposure. Additionally, in order to perform successful drug release or dissolution experiments, it is necessary for the FLU and CP to remain stable in solution for an extended duration to enable accurate drug quantification. In this study a solution containing 2.5 mg per mL FLU, and 50 μg per mL CP was stored under various conditions: −20 °C, 4 °C, 21 °C and 37 °C. Sampling was conducted at predetermined time points up to 28 days and drug concentration quantified using the HPLC method. Fig. 3 shows the stability of the solution over the 28-day period.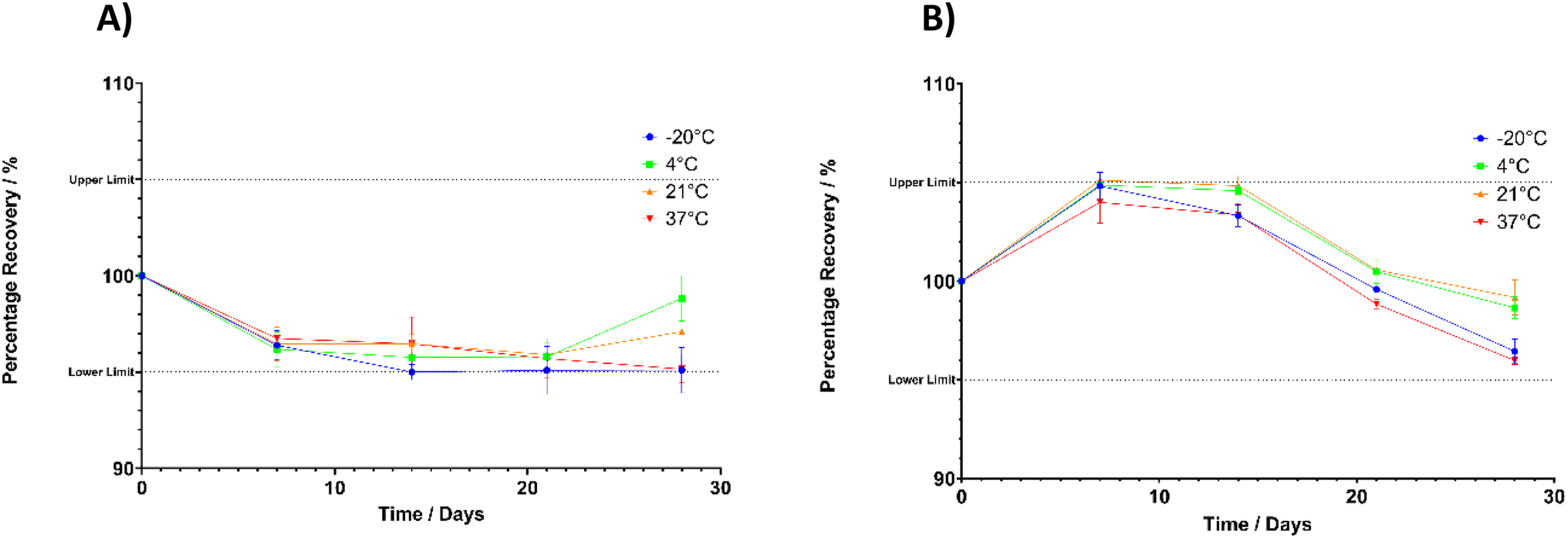 | ||
| Fig. 3 Stability of the FLU–CP solution at the highest standard concentration used, containing 2.5 mg mL−1 and 50 μg mL−1 respectively (A) FLU stability (B) CP stability over 28 days in vitro at a range of storage temperatures. | ||
Whilst previous reports have been made on the stability of FLU when in solution, many have not investigated the stability over the temperature ranges described here. Nevertheless, the results obtained in the present study reflect those obtained by other studies. The preparation of FLU in a buffer for oral delivery was shown to be stable at room temperature for up to 77 days, demonstrating percentage recovery of over 90% at all timepoints throughout the study period.48 Another stability study using FLU in solution at temperatures of −20 °C and – 80 °C for up to 4 months showed high levels of recovery of 97.7 and 98.1% respectively at 4 months of storage.49 Most closely related to the current study, Bin Liew et al. reported on the stability of FLU in a methanol solution at room temperature, giving a percentage recovery of 104.83% at 8 hours.36 No studies to date report on the stability of FLU in solution at 37 °C, however if this drug is to be incorporated into drug delivery devices for prolonged delivery the compound stability under post-administration conditions need to be evaluated. The results shown in Fig. 3A confirm the drug stability under such conditions demonstrating percentage recoveries between 95.15% and 96.73% for FLU throughout the experiment at 37 °C.
Investigating the stability of CP in solution showed promising stability over 28-days across all storage temperatures. This aligns with results reported elsewhere describing the stability of CP in solution at temperatures below 80 °C whereas storage temperatures between 80 and 100 °C showed evidence of degradation products eluting in corresponding chromatograms.50 Therefore, as the highest storage temperature here was 37 °C it is unsurprising that the solution remained stable despite the prolonged incubation duration. Furthermore, elsewhere stability studies at 4 °C and room temperature report on the stability of CP in a number of different solvents for up to 7 days.51Fig. 3B shows that the current stability study demonstrated percentage recoveries between 95.97% and 104.87% for CP.
3.3. Drug release study
An in vitro drug release study was conducted to demonstrate the applicability of the method to relevant experiments. The cumulative release was calculated with Fig. 4B showing the release profile of FLU and CP from the PVA hydrogel. Both drugs demonstrated burst release with the majority of FLU and CP released within the initial 6 hours of release. At 6 hours 92% of FLU was released whilst 68% CP release was observed. The release data was fitted to a number of release models to assess the release kinetics with the release profile of both drugs best fitting the Korsmeyer-Peppas model of release kinetics demonstrating R squared values of 0.98 and 0.99 for FLU and CP respectively.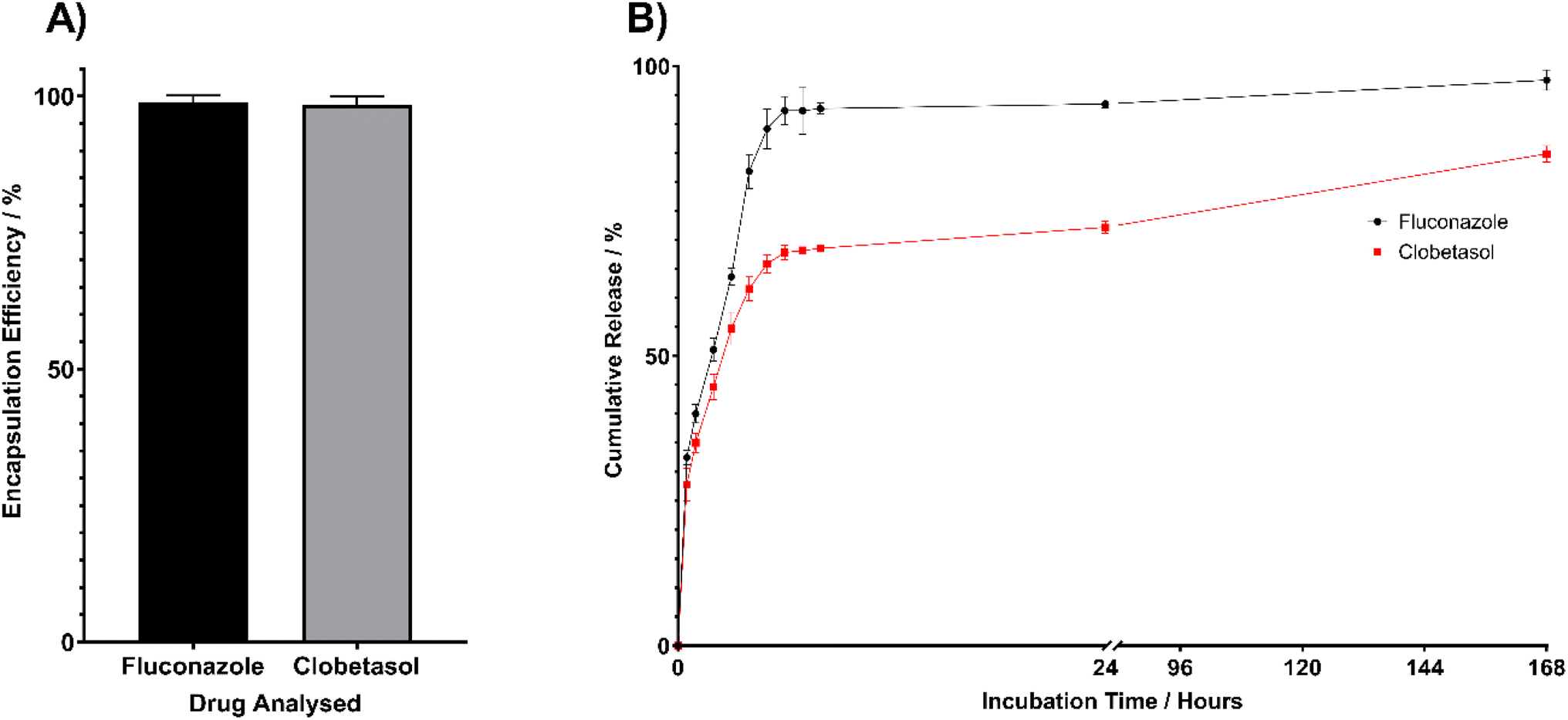 | ||
| Fig. 4 (A) Mean encapsulation efficiency of FLU and CP in the PVA films used to calculate the cumulative release during the drug release study. (B) Cumulative release profile of FLU and CP from a model biomaterial, PVA hydrogel, over a 24-hour period demonstrating the potential applications of the dual detection and quantification method described here. | ||
The chromatographic peaks remained well resolved and drug release was successfully quantified using the validated RP-HPLC method. From a quantification perspective, it can be concluded that the method developed and validated here is capable enough of detection and quantification of both FLU and CP simultaneously. The method described here is therefore appropriate for the investigation of in vitro physicochemical properties.
4. Conclusions
This study introduced a novel, sensitive and reliable RP-HPLC method with UV detection for the simultaneous detection and quantification of FLU and CP. The method underwent rigorous validation in alignment with the guidelines set out in the ICH for validation of analytical procedures. Results confirmed the efficacy, specificity, and high precision of the method for the quantification of both FLU and CP. A hallmark indicator of the high sensitivity of this method was the low values obtained for LOQ, these were calculated as 0.008 mg mL−1 and 0.012 μg mL−1 for FLU and CP respectively. Accordingly, the validated HPLC method was successfully applied to investigate the drug release from a PVA hydrogel over a 7-day period in vitro. The analytical method is therefore valid, fit for use and can be used for regular, routine analysis such as stability studies, drug encapsulation and drug release investigations. This dual detection analysis method will also provide advantages in the study of oral lichen planus and the development of new dual delivery therapeutic devices. The method provides the capability of rapid testing of the efficacy of dual drug diffusion across mucous membranes, allowing analysis of the spatial distribution of delivered drugs across different tissues. In terms of future applications, via this method, it can be determined if therapeutics are being delivered systemically or to the target tissues to predict the potential for secondary complications because of long-term treatment regimens if steroids such as CP.Data availability
The authors confirm that the data supporting the findings of this study are available within the article [and/or] its ESI.† Additional raw data is available from the corresponding author, upon reasonable request.Author contributions
Robyn A. Macartney: conceptualization, data curation, formal analysis, investigation, methodology, validation, writing – original draft, writing – review and editing. Annabelle T. R. Fricker: writing – review and editing. Andrew M. Smith: writing – review and editing. Stefano Fedele: writing – review and editing. Ipsita Roy: writing – review and editing, funding acquisition. Jonathan C. Knowles: conceptualization, writing – review and editing, funding acquisition, resources, supervision.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the Engineering and Physical Science Research Council (EPSRC) [grant number EP/X022196/1, EP/X026108/1].References
- Y.-S. L. Cheng, A. Gould, Z. Kurago, J. Fantasia and S. Muller, J. Oral Maxillofac. Pathol., 2016, 122, 332–354 Search PubMed.
- M. Á. González-Moles, S. Warnakulasuriya, I. González-Ruiz, L. González-Ruiz, Á. Ayén, D. Lenouvel, I. Ruiz-Ávila and P. Ramos-García, Oral Dis., 2021, 27, 813–828 CrossRef PubMed.
- C. Li, X. Tang, X. Zheng, S. Ge, H. Wen, X. Lin, Z. Chen and L. Lu, JAMA Dermatol., 2020, 156, 172–181 CrossRef PubMed.
- N. Lavanya, P. Jayanthi, U. Rao and K. Ranganathan, J. Oral Maxillofac. Pathol., 2011, 15, 127–132 CrossRef CAS PubMed.
- D. Wojciechowski and A. Wiseman, Clin. J. Am. Soc. Nephrol., 2021, 16, 1264–1271 CrossRef CAS PubMed.
- S. K. Stacey and M. Mceleney, Am. Fam. Physician, 2021, 103(6), 337–343 Search PubMed.
- H. Akan, V. P. Antia, M. Kouba, J. Nos Sinkó, A. Daniela, A. D. Tănase, R. Vrhovac and R. Herbrecht, J. Antimicrob. Chemother., 2013, 68, 5–15 CrossRef PubMed.
- R. Hosseinpour-Moghadam, F. Mehryab, M. Torshabi and A. Haeri, Clin. Ther., 2021, 43, e377–e402 CrossRef CAS PubMed.
- S. Ghosalkar, P. Singh and P. Ravikumar, J. Cosmet. Dermatol., 2022, 21, 536–549 CrossRef PubMed.
- S. Kwon, R. K. Singh, R. A. Perez, E. A. A. Neel, H. W. Kim and W. Chrzanowski, J. Tissue Eng., 2013, 4, 1–18 Search PubMed.
- N. Gholizadeh, M. S. Sadrzadeh-Afshar and N. Sheykhbahaei, J. Pharmaceut. Sci., 2020, 56, 1–15 Search PubMed.
- O. Mahdavi, N. Boostani, H. Jajarm, F. Falaki and A. Tabesh, J. Dent., 2013, 14, 201–204 CAS.
- M. Carbone, P. G. Arduino, M. Carrozzo, G. Caiazzo, R. Broccoletti, D. Conrotto, C. Bezzo and S. Gandolfo, J. Oral Pathol. Med., 2009, 38, 227–233 CrossRef CAS PubMed.
- P. Yuan, X. Qiu, L. Ye, F. Hou, Y. Liang, H. Jiang, Y. Zhang, Y. Xu, Y. Sun, X. Deng, H. Xu and L. Jiang, Oral Dis., 2022, 28, 670–681 CrossRef PubMed.
- P. J. Barnes, Eur. Respir. J., 2006, 27, 413–426 CrossRef CAS PubMed.
- A. Zampetti, C. Feliciani, A. Tulli and P. Amerio, Clin. Med. Insights Ther., 2010, 2, 523–537 CAS.
- A. Khodavandi, F. Alizadeh, N. A. Vanda, G. Karimi and P. P. Chong, Pharm. Biol., 2014, 52, 1505–1509 CrossRef CAS PubMed.
- A. B. Nair, S. Kumar, P. Dalal, C. Nagpal, S. Dalal, R. Rao, N. Sreeharsha and S. Jacob, Pharmaceutics, 2022, 14, 383 CrossRef CAS PubMed.
- A. Ghalayani Esfahani, L. Altomare, E. M. Varoni, S. Bertoldi, S. Farè and L. De Nardo, J. Mater. Sci. Mater. Med., 2019, 30, 40 CrossRef PubMed.
- H. E. Colley, Z. Said, M. E. Santocildes-Romero, S. R. Baker, K. D'Apice, J. Hansen, L. S. Madsen, M. H. Thornhill, P. V. Hatton and C. Murdoch, Biomaterials, 2018, 178, 134–146 CrossRef CAS PubMed.
- F. Q. Hu, H. Yuan, H. H. Zhang and M. Fang, Int. J. Pharm., 2002, 239, 121–128 CrossRef CAS PubMed.
- J. C. R. Corrêa, C. Duarte Vianna-Soares and H. R. N. Salgado, Chromatogr. Res. Int., 2012, 2012, 1–8 CrossRef.
- D. A. Santos, K. Marques Queiroz, M.-L. Martins Silva, N. Duque Prado, P. Marcelo Andrade Lima, R. Dias Lopes Diniz, I. Costa César, G. Antônio Pianetti and D. Assis Santos, J. Pharmaceut. Sci., 2009, 45(4) DOI:10.1590/S1984-82502009000400012.
- A. Pawar, J. Drug Deliv. Ther., 2024, 1(1) DOI:10.61920/jddb.v1i01.140.
- S. Sharma and N. Khare, Adv. Powder Technol., 2018, 29, 3336–3347 CrossRef CAS.
- D. B. Kamdi, H. M. Yerne and S. R. Patil, Am. J. PharmTech Res., 2020, 10(1), 83–92 CrossRef CAS.
- National Institute for Health and Care Excellence, Dosage and Formulations, British National Fomulary, Clobetasol Propionate Search PubMed.
- Eris Oaknet Healthcare and Oaknet Healthcare, Fluconazole Gel Search PubMed.
- ICH Q2(R2), Validation of Analytical Procedures - Scientific Guideline Search PubMed.
- P. Ravisankar, S. Anusha, K. Supriya and U. Ajith Kumar, Int. J. Pharm. Sci. Rev. Res., 2019, 55, 46–50 CAS.
- Md. Bhuyian, Dr H. Rashid, A. Islam and Md. Tareque, J. Pharm. Res., 2015, 7, 375–385 Search PubMed.
- M. T. Beebeejaun, M. B. Brown, V. Hutter, L. Kravitz and W. J. McAuley, JEADV Clin. Pract., 2023, 2, 718–726 CrossRef.
- M. Wadie, S. Mahmoud Mostafa, S. Mohamed ElAdl, M. Saleh Elgawish and C. Author, Int. J. Pharm. Biol. Sci., 2017, 12, 60–68 Search PubMed.
- T. Wattananat and W. Akarawtu, Biomed. Chromatogr., 2006, 20, 1–3 CrossRef CAS PubMed.
- P. Sadasivudu, N. Shastri and M. Sadanandam, Int. J. Chemtech. Res., 2009, 1, 1131–1136 CAS.
- K. Bin Liew, G. Onn Kit Loh, Y. T. F. Tan and K. Khiang Peh, Int. J. Res. Pharm. Sci., 2012, 4, 107–111 Search PubMed.
- A. Singh, P. K. Sharma and D. K. Majumdar, J. Liq. Chromatogr. Relat. Technol., 2014, 37, 594–607 CrossRef CAS.
- A. Singh, P. Kumar Sharma and D. Kant Majumdar, Indian J. Chem. Technol., 2011, 18, 357–362 CAS.
- A. Demurtas, S. Pescina, S. Nicoli, P. Santi, D. Ribeiro de Araujo and C. Padula, J. Chromatogr. B, 2021, 1164, 122512 CrossRef CAS PubMed.
- M. C. Fontana, M. O. Bastos and R. C. R. Beck, J. Chromatogr. Sci., 2010, 48, 637–640 CAS.
- N. Patel, P. Patel and D. Meshram, Am. J. PharmTech Res., 2016, 6(5), 289–304 CAS.
- L. Antônio, D. Silva, S. Fleury Taveira, E. M. Lima and R. N. Marreto, J. Pharmaceut. Sci., 2012, 48(4), 811–817 Search PubMed.
- K. S. Bagad, K. Bacchao, S. B. Bagade, R. D. Amrutkar and D. D. Patil, Biosci. Biotechnol. Res. Asia, 2024, 21, 283–294 Search PubMed.
- G. Shabir, J. Chromatogr., A, 2003, 987, 57–66 CrossRef CAS PubMed.
- S. Del Río-Sancho, V. Merino, A. López-Castellano and Y. N. Kalia, J. Chromatogr. Sci., 2016, 54, 790–795 Search PubMed.
- European Medicines Agency and Committee for Medicinal Products for Human Use, ICH Q2(R2) Guideline on Validation of Analytical Procedures, 2023 Search PubMed.
- Y. Vander Heyden, A. Nijhuis, J. Smeyers-Verbeke, B. G. M. Vandeginste and D. L. Massart, J. Pharm. Biomed. Anal., 2001, 24, 723–753 CrossRef CAS PubMed.
- P. J. Dentinger and C. F. Swenson, Infect. Dis., 2009, 43, 485–489 CAS.
- J. Saito, A. Tanzawa, Y. Kojo, H. Maruyama, T. Isayama, K. Shoji, Y. Ito and A. Yamatani, J. Pharm. Health Care Sci., 2020, 6, 14 CrossRef PubMed.
- A. F. B. Fauzee and R. B. Walker, J. Sep. Sci., 2013, 36, 849–856 CrossRef CAS PubMed.
- D. Kamalakannan, J. Munusamy, A. Thangadurai Subramaniam, A. S. Thangadurai, P. M. Banu, V. J. Rashini and H. D. Siva Ganga Lakshmi, Pharm. Lett., 2014, 6, 52–57 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ay02219j |
| This journal is © The Royal Society of Chemistry 2025 |