
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInvestigation of the binding kinetics and electrochemical properties of in situ reconstructed apo-GOx using electrodes with electrodeposited FAD cofactor†
M.
Koch
a,
N.
Korkmaz
b and
Y. E.
Silina
 *c
*c
aHTW saar – University of Applied Sciences, Saarbrücken, Germany
bKIST Europe – AI Convergence Cluster, Korea Institute of Science and Technology, Saarbrücken, Germany
cSaarland University, Institute of Biochemistry, Campus B 2.2, room 317, Saarbrücken, Germany. E-mail: yuliya.silina@gmx.de; yuliya.silina@uni-saarland.de
First published on 25th June 2025
Abstract
Herein, the electroanalytical properties of in situ reconstructed apo-glucose oxidase (apo-GOx) on electrodes modified with electrodeposited cofactor flavin adenine dinucleotide (FAD) were studied in the absence of any diffusional electron mediators. In the electrodeposited sensing layer, the order of enzyme assembly using the same building blocks (e.g., spatially separated apo-GOx with FAD cofactor or native holo-GOx with entrapped cofactor) significantly affected the electrodes’ electroanalytical performance. These electrodes exhibited a pronounced real time current difference (i), and the work of in situ reconstructed apo-GOx, compared with a native electrodeposited holo-GOx analogue, was not limited by oxygen depletion (ii). This knowledge was subsequently used to profile several types of apo-GOx.
1. Introduction
Semi-artificial and reconstructed enzyme engineering is a promising research area in biotechnology and enzymology.1 A large variety of synthetic or natural cofactors can be used for reconstituting enzymes, thus generating enzymes with enhanced or entirely new functionality.2–5Despite substantial progress in reconstructed redox enzymes6–8 in the fields of enzymology, bioelectrocatalysis, and fuel cell development, essential theoretical and practical investigations remain necessary to support applications in biosensors.9,10 Amperometric biosensor applications require the electrical connection of the protein to an external current source via fast electron-relaying redox couples anchored to electrodes.5,11 To this end, a wide spectrum of methods to electrically wire redox holo-enzymes,12 or separated apo-enzymes and cofactors,9 with electrodes have been proposed, including modification of enzymes with redox-relays, and immobilization of apo-enzymes and cofactors in redox polymers.5,13,14 However, random distributions of dense cofactor-containing relay layers on electrodes often lead to insufficient electrical contact, poor electron transfer, and low electroanalytical merit.15
Fundamentally, the immobilized cofactor (i.e., flavin adenine dinucleotide, or FAD) is considered responsible for the advanced electroanalytical performance of electrodes exclusively because of a “relay-like” effect inducing a cascade of biochemical reactions supported by the FAD/FADH2 route.13 Meanwhile, the influence of the electrochemical route, i.e., oxidation of a product of biochemical transformation (e.g., hydrogen peroxide) and a dynamic oxygen supply on the efficiency of the reconstitution process of apo-enzymes, cannot be excluded.16
An approach enabling in situ reconstruction of glucose oxidase (GOx) by interaction with a thin electrodeposited cofactor layer on screen printed electrodes (SPEs) has been proposed.9 Although this approach enables the synthesis of a homogeneously distributed nano-dimensional FAD-containing layer on the SPE surface, the mechanistic aspects, operating principles, and applications of these sensing systems remain to be understood.
Notably, GOx is a FAD-dependent enzyme with unique catalytic properties. It efficiently catalyzes the oxidation of glucose using oxygen (O2), producing gluconolactone (which then spontaneously hydrolyzes to gluconic acid) and hydrogen peroxide (H2O2).17 The biochemistry of GOx offers numerous opportunities for the development of novel microanalytical systems with enhanced sensing capabilities. However, recent advances in glucose biosensing have predominantly focused on optimizing the properties of electrocatalysts (inorganic components), while comparatively little attention has been given to engineering the biocomponent itself.18 Meanwhile, modification of the GOx structure (e.g., through protein reconstruction) can enable the design of mediator-free, self-powered glucose biosensors,19 offering substantial improvements in performance and device autonomy.
The present study aims to investigate the electrochemical features of in situ reconstructed apo-GOx after its interaction with electrodeposited FAD on the SPE surface, compare its electroanalytical performance with the native holo-GOx, and elucidate the mechanistic aspects underlying their binding kinetics. During the investigations, it was revealed that the proposed mediator-free system with in situ reconstructed enzyme exhibited enhanced oxygen overproduction performance.
2. Experimental part
2.1. Chemicals and materials
(NH4)2HPO4, H2PdCl4, Na2HPO4·12H2O and 25% NH4OH from Merck (Darmstadt, Germany) were used for preparation of a palladium electrolyte (pH 9.3) and electrodeposition of sensing layers containing palladium nanoparticles (Pd-NPs). D-Glucose, Nafion™ 117 (Naf) containing solution (∼5% in a mixture of lower aliphatic alcohols and water), glucose oxidase, GOx, from Aspergillus niger (EC 1.1.3.4, Type VII, Type X-S, GOx, Aspergillus niger Recombinant, Calbiochem), flavin adenine dinucleotide (FAD) disodium salt hydrate (≥95% (HPLC), powder) were received from Merck (Darmstadt, Germany). Deionized (DI) water was generated by an Elga PureLab (Celle, Germany) purification system.Screen printed electrodes (SPEs) modified by graphene oxide (GO, DRP-110DGPHOX) were obtained from DropSens (Metrohm, Germany).
2.2. Preparation of apo-GOx
FAD is non-covalently bound to the enzyme and can readily be removed under acidic conditions.20 To prepare apo-GOx, the intact FAD cofactor was removed from native holo-GOx (type X-S, 5 μg μL−1 stock unless otherwise stated) through acidic hydrolysis in 25% (NH4)2SO4 solution, then performed centrifugation at 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 r.p.m for 15 min.9,21 The procedure was repeatedly applied three times. The obtained apo-GOx protein was next reconstituted in phosphate buffer with pH 6.98 ± 0.2 and used for subsequent studies.
000 r.p.m for 15 min.9,21 The procedure was repeatedly applied three times. The obtained apo-GOx protein was next reconstituted in phosphate buffer with pH 6.98 ± 0.2 and used for subsequent studies.
The absence of FAD in the structure of apo-GOx according to the protocol used was confirmed by UV-Vis (section 2.3) and oxygen mini-sensors (section 2.6) studies.
2.3. UV-Vis studies
To verify the completeness of FAD removal after the acidic hydrolysis of holo-GOx, UV-Vis spectra of the obtained product were measured using a QuickDrop UV-Vis spectro-photometer (SpectraMax GmbH, Germany). After repeating three steps (see section 2.2), FAD was completely removed from the holo-GOx, see ESI, Fig. S1.†2.4. Synthesis of functional layers of electrodes
Functional sensing layers of the electrodes were produced using a one-step electrodeposition methodology.9,12 This approach was specifically optimized for multicomponent electrolytes containing no more than three compounds: an inorganic precursor, polymer binding agent, and a biocomponent (either an individual cofactor or an enzyme with a deeply embedded cofactor). To preserve the integrity of the optimized electrodeposition parameters (specifically, the number and ratio of components in the multiple electrolyte), several distinct functional sensing layer designs were employed in this study.
Electrode A was obtained by electrodeposition of a Pd-NPs/holo-GOx/Naf layer (native/intact GOx) at −2.5 mA for 30 s from multiple electrolyte solution containing Pd precursors (Pd-electrolyte) at 3 g L−1, holo-GOx at 5 μg μL−1, and 2% Naf (mixed in a ratio of 1:1:1 v/v/v) according to a previously optimized procedure.16
Electrode B was obtained by electrodeposition of a Pd-NPs/apo-GOx/Naf layer at −2.5 mA for 30 s from apo-GOx containing electrolyte in the absence of FAD cofactor (Pd precursors at 3 g L−1, apo-GOx at 5 μg μL−1, and 2% Naf mixed in a ratio of 1:1:1 v/v/v). During experiments, FAD was exogenously added (in aqueous form) to droplets of glucose solution.
The preparation of FAD-modified electrodes (Electrode C and Electrode D) was performed according to the following protocol: FAD (3 mg mL−1 unless otherwise stated) was mixed with 2% Naf solution and Pd-electrolyte (pH 9.3) in a ratio of 1/1/1 v/v/v. The final multiple electrolyte solution contained FAD at 1 mg mL−1. Next, 10 μL as-prepared multiple electrolyte solution was placed over the working electrode of SPE/GO, and electrodeposition of the Pd-NPs/FAD/Naf layer was performed at −2.5 mA for 90 s.9,22
Electrode C was examined in glucose solution in the absence of apo-GOx, whereas Electrode D was examined in the presence of aqueous apo-GOx.
The differences between Electrode A and Electrode D included the spatially separated aqueous enzyme (apo-GOx) and immobilized cofactor FAD (Electrode D) vs. immobilized holo-GOx (Electrode A), see ESI, Fig. S2.†Electrode D will be further referred to as reconstructed apo-GOx or Pd-NPs/(FAD + apo-GOx)rec/Naf (rec = reconstructed).
After formation of functional sensing layers, all electrodes were carefully washed with DI water and used directly, avoiding storage.
2.5. SEM and AFM characterization of the electrodes
An FEI Quanta 400 FEG Scanning Electron Microscope (SEM) was used in high vacuum mode to visualize the as-prepared electrodes. Back-scattered electron (BSE) images were acquired at 10 kV accelerating voltage using a Solid State Detector (SDD). X-Ray spectral analysis was performed using an EDAX Genesis V 6.04 detector.A PARK NX 10 AFM system (Park Systems, South Korea) was used for AFM topography measurements of the electrode surfaces. All measurements were performed in non-contact scanning mode at room temperature under ambient conditions in air. Mounted AFM ACTA (NCHR) probes compatible with Park NX 10 were purchased from Schaefer Technologie GmbH (Germany). Images were recorded with a scan size of 25 μm × 25 μm and with a resolution of 256 pxl × 256 pxl. Average roughness (Ra) and surface root mean square roughness (Rq) values were extracted from four AFM images for each electrode using the Gwyddion image processing software (https://gwyddion.net/) and estimated as the mean roughness ± SD.
2.6. Oxygen mini-sensor studies
| Glucose + GOx/FAD(ox) → GOx/FAD(red) + gluconolactone | (1) |
| GOx/FAD(red) + O2 → GOx/FAD(ox) + H2O2 | (2) |
Notably, these biochemical reactions occur even in the absence of electrode polarization. Therefore, by analyzing oxygen consumption during these biochemical reactions, it is possible to examine the biochemical activity of holo-GOx and it's in situ reconstructed apo-GOx analogue.
For evaluation of the intensity of oxygen consumption in the presence of GOx, the oxygen concentration (μmol L−1) in a buffer and glucose containing solutions was monitored with an OXR430 needle oxygen mini-sensor (Pyro Science GmbH, Aachen, Germany).24 Experiments were conducted under ambient conditions at 22 ± 2 °C.
An oxygen mini-sensor was also employed to confirm whether the native cofactor had been completely removed from the apo-enzyme via acidic hydrolysis. As a result of cofactor exclusion, the obtained apo-GOx became biochemically inactive. Thus, inactive GOx does not consume oxygen upon exposure to the glucose solution (ESI, Fig. S3A,†see line a vs. line b; measured in a 150 μL droplet placed on a Petri dish). However, this process was shown to be reversible: based on the returned intensive oxygen consumption (ESI, Fig. S3B,†line c, measured in a 150 μL droplet placed on Pd-NPs/FAD/Naf modified electrode), the inactive apo-GOx regained its biochemically active state directly on the surface of FAD-modified electrodes. In other words, the reconstruction of apo-GOx indeed occurred on the surfaces of FAD-modified electrodes.
| H2O2 → O2 + 2H+ + 2e−, | (3) |
2.7. Electrochemical studies
The performance of electrodes with electrodeposited holo-GOx (type X-S), apo-GOx, and FAD was evaluated in a 150 μL droplet of buffer and glucose solutions with a one-channel PalmSens4 potentiostat (PalmSens, Utrecht, the Netherlands) in cyclic voltammetry (CV) mode in the range from −0.4 V to 0.4 V at scan rates of 50 mV s−1.If the conventional biochemical pathway, as described by equilibria (1) and (2) (see section 2.6) occurs, hydrogen peroxide (H2O2) is generated as a product of the enzymatic reaction. Subsequently, under applied polarization, H2O2 undergoes electrooxidation via an electrochemical pathway (see formula (3)) on the surface of Pd-NPs.16
Previously, the role of palladium surface oxides (PdO) and adsorbed oxygen (in the cathodic potential range) on the sensitivity and selectivity toward H2O2 detection using Pd-NPs-based electrodes was established.16 Therefore, a novel multi-step amperometric (MAM) read-out mode was developed, enabling faster reduction of PdO and facilitating reliable, sensitive and selective quantitative detection of hydrogen peroxide. Briefly, the MAM protocol involves two key steps: (1) cathodic polarization at −0.20 V for 60 s to reduce PdO, followed by (2) anodic polarization at 0.20 V for 30 s to detect the current corresponding to H2O2 oxidation.16
For dynamic chronoamperometric (AM) experiments, a potential of 0.20 V, corresponding to oxidation of H2O2 (a product of GOx enzymatic activity) was applied on Pd-NPs.16 The dynamic AM responses of electrodes were recorded for 2000 s, thereby providing valuable insights into apo-GOx adsorption on the surfaces of electrodes with electrodeposited functional sensing layers (Electrodes A–D). An extended duration of investigation is of limited relevance for screening application of microbial enzymes and biosensing.
2.8. Quartz microbalance (QCM) studies
In all electrochemical and oxygen mini-sensor related studies (sections 3.1–3.5), holo-GOx and its corresponding apo-form of type X-S, obtained from Aspergillus niger (EC 1.1.3.4, without added oxygen), were used.To assess the differences between GOx of type X-S (section 3.6) and several analogues, e.g., recombinant GOx and GOx of type VII, the SRS QCM 200 system (Scientific Instruments GmbH, Gilching, Germany) was used. Changes in protein quality, purity, and structure can readily be detected by QCM. Briefly, pronounced differences in frequency can be observed for molecules with similar adsorbed masses. For the experiment, 1 μL of intact holo-GOx of various type was dropped onto the surface of an Au-modified crystal (5 MHz), followed by signal recording (change in frequency, Hz).
Apo-GOx from recombinant GOx and GOx of type VII were prepared according to protocol described for apo-GOx (X-S) in section 2.2.
3. Results and discussion
3.1. Assembly of the enzymatic constructor
Electrodeposition supports direct enzyme–electrode communication regardless of the design of sensing layers, i.e. “capsule-like” or “adsorption-like” structures.22,25 For example, during electrodeposition of holo-GOx (already containing the embedded FAD cofactor), the enzyme (bioreagent) is located in a “capsule-like” structure (10–15 nm thickness) between Pd-NPs and Naf.22,25 In the case of FAD electrodeposition together with Pd-NPs and Naf (forming “adsorption-like” structures 10–15 nm thick), the cofactor (bioreagent) is located on top of the functional layer. The addition of aqueous apo-GOx results in direct contact between the enzyme and FAD-modified electrode.9,12 The same synthesis process (electrodeposition) of functional films might initially be expected to result in the formation of biosensors with similar sensing characteristics (if the same bioreagents are used) regardless of the order of assembly; however, this expectation appears to be not entirely true.In situ reconstruction of apo-GOx after interaction with solid electrodeposited FAD-doped nanoparticles (Pd-NPs/FAD/Naf) can lead to enhanced coverage of the surface of electrodes and advanced substrate-enzyme contact.26,27 This assumption was confirmed during testing of Electrodes A–D in CV mode.
Regardless of the electrode tested, the cyclic voltammograms recorded in buffer solution (Fig. 1, line a) from Pd-based electrodes exhibited characteristic profiles, with a cathodic peak observed at approximately −0.2 to −0.3 V. This peak corresponds to the oxygen reduction reaction (ORR) on Pd-NPs/PdO.16 The electrooxidation of H2O2 formed as a product of the conventional enzymatic reaction between glucose and holo-GOx is expected to be accompanied by an increase in anodic current at around 0.2 V (Fig. 1, line b).
 | ||
| Fig. 1 CV plots (second scans shown) recorded at 50 mV s−1 from: Electrode A (A) – electrodeposited Pd-NPs/holo-GOx/Naf layer; Electrode B (B) – electrodeposited Pd-NPs/apo-GOx/Naf layer explored in the presence of aqueous FAD cofactor; Electrode C (C) – electrodeposited Pd-NPs/FAD/Naf layer explored in the absence of enzyme, Electrode D (D) – reconstructed Pd-NPs/(FAD + apo-GOx)rec/Naf layer explored in buffer (a) and 25 mM glucose (b) solutions. The pH of the test solutions was maintained at 6.98 ± 0.2. The anodic range marked by a dashed square illustrates the read-out at 0.2 V on Pd-NPs.16 The blue arrows indicate an increase in anodic current corresponding to the electrooxidation of hydrogen peroxide. In contrast, the red arrows highlight the absence of a signal attributable to hydrogen peroxide electrooxidation relative to the buffer. Note: the hydrogen peroxide read-out platform employing electrodeposited Pd-NPs was optimized in previous studies.9,12,16 | ||
Unexpectedly, not all the tested electrodes exhibited this increase in anodic current upon exposure to the glucose solution. Briefly, Electrode A (electrodeposited Pd-NPs/holo-GOx/Naf layer) and Electrode D (Pd-NPs/(FAD + apo-GOx)rec/Naf) responded to the presence of glucose (Fig. 1A and Dline b). This result highlights the interaction of GOx with a physiological substrate (glucose) according to the conventional biochemical route, thereby also confirming the in situ reconstruction of apo-GOx (Electrode D, Fig. 1D, line b).
Electrode B containing an electrodeposited Pd-NPs/apo-GOx/Naf layer and examined in the presence of aqueous cofactor FAD, and Electrode C containing an electrodeposited Pd-NPs/FAD/Naf layer (examined in the absence of the aqueous apo-enzyme) did not respond to the presence of glucose (no increase in anodic current at 0.2 V compared to the buffer (line a), as indicated by the red arrows), see Fig. 1B and C, line b.
Remarkably, Electrode D (Fig. 1D, line b), containing electrodeposited cofactor FAD in the sensing layer, exhibited an advanced current response to glucose in the presence of apo-GOx in comparison to Electrode A. A similarly enhanced bioelectrocatalyzed effect has been reported in the presence of ferrocene during glucose oxidation by reconstructed apo-GOx for 24 h.13 The advanced analytical merit was explained in terms of the electron relay-FAD process (ferrocene-FAD induced relay), however, this explanation cannot apply to the present study, given that no diffusional electron mediators were applied.
Measurements of decomposed hydrogen peroxide in MAM mode16 by Pd-NPs present in the design of all tested electrodes (Electrodes A–D) showed the same trends observed in CV (ESI, Fig. S4†). Briefly, Electrode A containing electrodeposited native holo-GOx in the functional sensing layer fully retained biochemical activity toward glucose. Simultaneously, no response to glucose was recorded from Electrode B. These findings indicated that no reconstruction of the electrodeposited apo-GOx by interaction with the added aqueous FAD had occurred.
The electrode with an electrodeposited Pd-NPs/FAD/Naf layer (Electrode C) examined in the absence of aqueous apo-GOx also did not exhibit any response to glucose (e.g., no enzyme → no binding of a substrate → no biochemical reaction → no electroactive product (H2O2) formation → no electrooxidation → no current).
In contrast, the advanced current in MAM mode was recorded from the same electrode in the presence of glucose and added apo-GOx (Electrode D). The reason for the advanced electroanalytical performance of Electrode D regardless of the electrochemical readout mode used (e.g., CV (Fig. 1) or MAM (ESI, Fig. S4†)) is investigated in detail in section 3.3.
In summary, the conducted experiments highlighted the importance of the order of enzyme assembly on electrodes, even from the same electrodeposited building blocks (i.e. the same protein (GOx) and the same cofactor (FAD)).
3.2. Comparison of properties of reconstructed apo-GOx vs. native holo-GOx
Due to the pronounced electroanalytical signals recorded from Electrode A and Electrode D in the presence of glucose, the performance and properties of these systems were investigated in greater detail.SEM and AFM studies did not reveal any significant morphological differences between the sensing layers of Electrode A and Electrode D, Fig. 2. The roughness measured by AFM was almost identical for both layers (ESI, Fig. S5†). In brief, the surface of the sensing layers was represented by hybrid organic–inorganic nanoparticles. Thus, apart from the Pd peak corresponding to Pd-NPs, EDX analysis revealed the presence of Nafion (S and F peaks) and the biocomponent (characteristic C and O peaks in the EDX spectra), ESI, Fig. S6.†
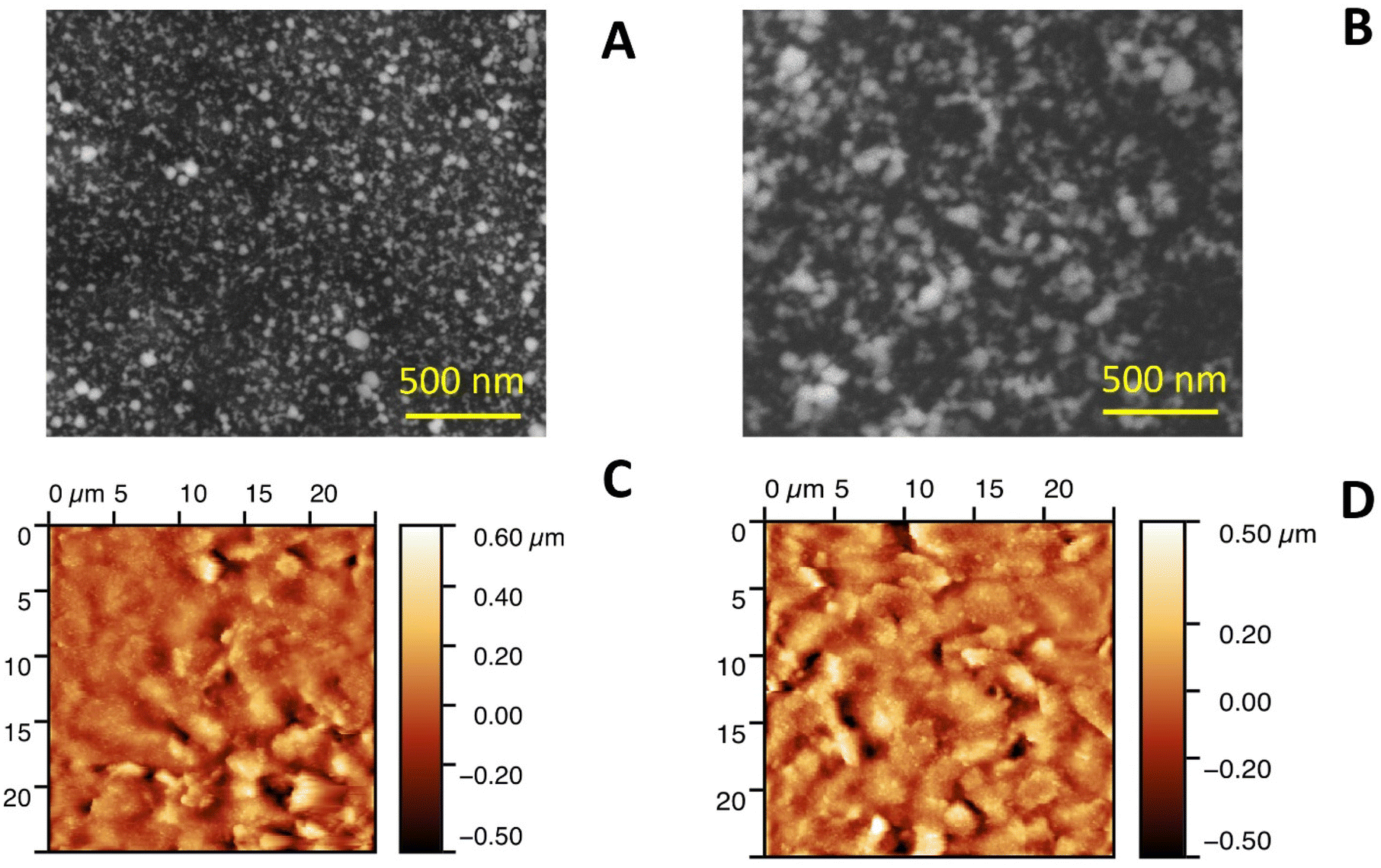 | ||
| Fig. 2 SEM (A and B) and AFM (C and D) images obtained from the sensing layers of Electrode A (A and C) and Electrode D (B and D). | ||
However, similar morphology and roughness cannot guarantee similar biochemical efficacy of the reconstructed enzyme. Therefore, next, it was necessary to clarify whether the biochemical efficacy of the reconstructed apo-GOx (Electrode D) remained identical to that of the original holo-GOx (Electrode A).
To this end, the properties of apo-GOx after interaction with the cofactor FAD immobilized on the electrode were studied in dynamic AM mode under constant potential of 0.2 V applied for 2000 s. Briefly, in the presence of apo-GOx a gradual time-dependent anodic current increase was recorded from the FAD-modified electrode (Electrode D) regardless of glucose concentration, Fig. 3 (line a and b, shown for 5 mM and 25 mM as a case study). Although a gradual, time-dependent increase in anodic current was also recorded the same FAD-modified electrode in the presence of aqueous holo-GOx and glucose (Fig. 3, line c), both the magnitude of the current and the shape of the curves differ noticeably. This suggests that the interaction mechanisms in these systems may not be entirely equivalent.
 | ||
| Fig. 3 Time-dependent AM responses recorded from: a and b – FAD-modified electrode (Electrode D) examined in a droplet of 5 mM (a) and 25 mM glucose (b) with 1 μg μL−1 of apo-GOx (in situ reconstructed apo-GOx) added; c – Electrode D tested in a droplet of 25 mM glucose with 1 μg μL−1 of holo-GOx added; d – Electrode A in a droplet of buffer (0 mM glucose); e – Electrode A in a droplet of 5 mM glucose; f – Electrode A in a droplet of 25 mM glucose. | ||
In addition, under a constant glucose concentration, and constant amount and type of protein in the droplet, the time-dependent current increase recorded from Electrode D was highly reproducible across runs and batches, ESI, Fig. S7 and S8† (shown for apo-GOx). This effect occurred exclusively in the presence of glucose – a physiological substrate of GOx. Thus, no current response was obtained from the same Electrode D examined in the presence of fructose and added apo-GOx, ESI, Fig. S9.†
By comparison, the electrode modified with Pd-NPs/holo-GOx/Naf (Electrode A, Fig. 3, lines d–f) showed no gradual, time-dependent current increase in the presence of glucose, unlike Electrode D (Fig. 3, lines a and b). The behavior of Electrode A, in contrast to Electrode D, is consistent with previously reported observations.28–31
In summary, the properties of electrodes containing electrodeposited native holo-GOx (Pd-NPs/holo-GOx/Naf, Electrode A) in the sensing functional layer differ from electrodes with in situ reconstructed apo-GOx (Pd-NPs/(apo-GOx + FAD)rec/Naf, Electrode D). More specifically, these two electrodes exhibited a significant real time difference in current behavior (Fig. 3, line a and b vs. line d and e).
3.3. Effects of dissolved oxygen on the electroanalytical performance of electrodes modified with electrodeposited native holo-GOx and reconstructed apo-GOx
Differences in the kinetics of the enzymatic reaction (as described above) result in the differences in the oxidation rates of hydrogen peroxide (a product of the enzymatic reaction) on the surfaces of electrocatalytic Pd-NPs (part of the electrodeposited sensing layers), as well as differences in the amounts of released oxygen, see formula (3).9,12,16,22Simultaneously, the amount of oxygen produced further influences the catalytic performance of reconstructed apo-GOx, e.g., catalytic turnover rates.32 For example, when the reaction mixture is saturated with oxygen, the activity of GOx can increase by up to 100%.33
To verify this assumption, next, the electroanalytical performance of both electrodes, e.g., Electrode A and Electrode D was examined in glucose solutions using a previously developed MAM protocol. This protocol enables accurate detection of H2O2 in the presence of oxygen using Pd-NPs-based electrodes.12,16 The response profile obtained during ten continuous cycles in the same droplet of glucose solution (modeling of dynamic AM conditions) differed markedly between Electrode A and Electrode D. Specifically, Electrode A showed no time-related changes in electrocatalytic behavior (Fig. 4A, line a) or in oxygen concentration (Fig. 4B, line a).
 | ||
| Fig. 4 Time-dependent MAM plots (A) and synchronized oxygen mini sensor dynamic responses (B) recorded at 22 ± 2 °C in the presence of 25 mM glucose from Electrode A (Pd-NPs/(holo-GOx)/Naf), line a and Electrode D (Pd-NPs/(apo-GOx + FAD)rec/Naf), line b during ten cycles. Note: the black dashed line shows the continuous increase in the response of Electrode D from cycle to cycle. | ||
In contrast, for Electrode D with electrodeposited FAD and explored in the presence of apo-GOx a time-dependent anodic current increase was observed starting at approximately 500 ± 50 s. This increase corresponded to the electrooxidation of H2O2 at Pd-NPs (Fig. 4A, line b) and was accompanied by dynamic oxygen release (formula (3)), Fig. 4B, line b.
According to the literature, the upper limit of the turnover rate for native GOx at 25 °C is approximately 600 ± 100 s−1.33 Therefore, it can be assumed that after in situ apo-GOx reconstruction, bioelectrocatalysis would continue on the surface of Electrode D due to the absence of oxygen depletion. This enhanced oxygen supply could explain the superior electroanalytical performance of Electrode D vs. Electrode A (see also Fig. 1 and ESI, Fig. S2†).
To determine the reason underlying the advanced oxygen production/supply from electrodes with electrodeposited FAD cofactor, next, the electrooxidation of H2O2 as a product of the enzymatic reaction was studied on Electrode D. Notably, regardless of the applied concentration of H2O2, no time-dependent current increase was recorded, Fig. 5A. The oxygen released during the decomposition of a constant amount of H2O2 on Electrode D under the synchronized applied polarization in MAM mode remained constant (Fig. 5B), in contrast to experiments performed in the presence of glucose and aqueous apo-GOx (Fig. 4B, line b). These model experiments indicate continuous H2O2 production on the surface of Electrode D in the presence of glucose and aqueous apo-GOx, further supporting a dynamic oxygen supply/release in this system.
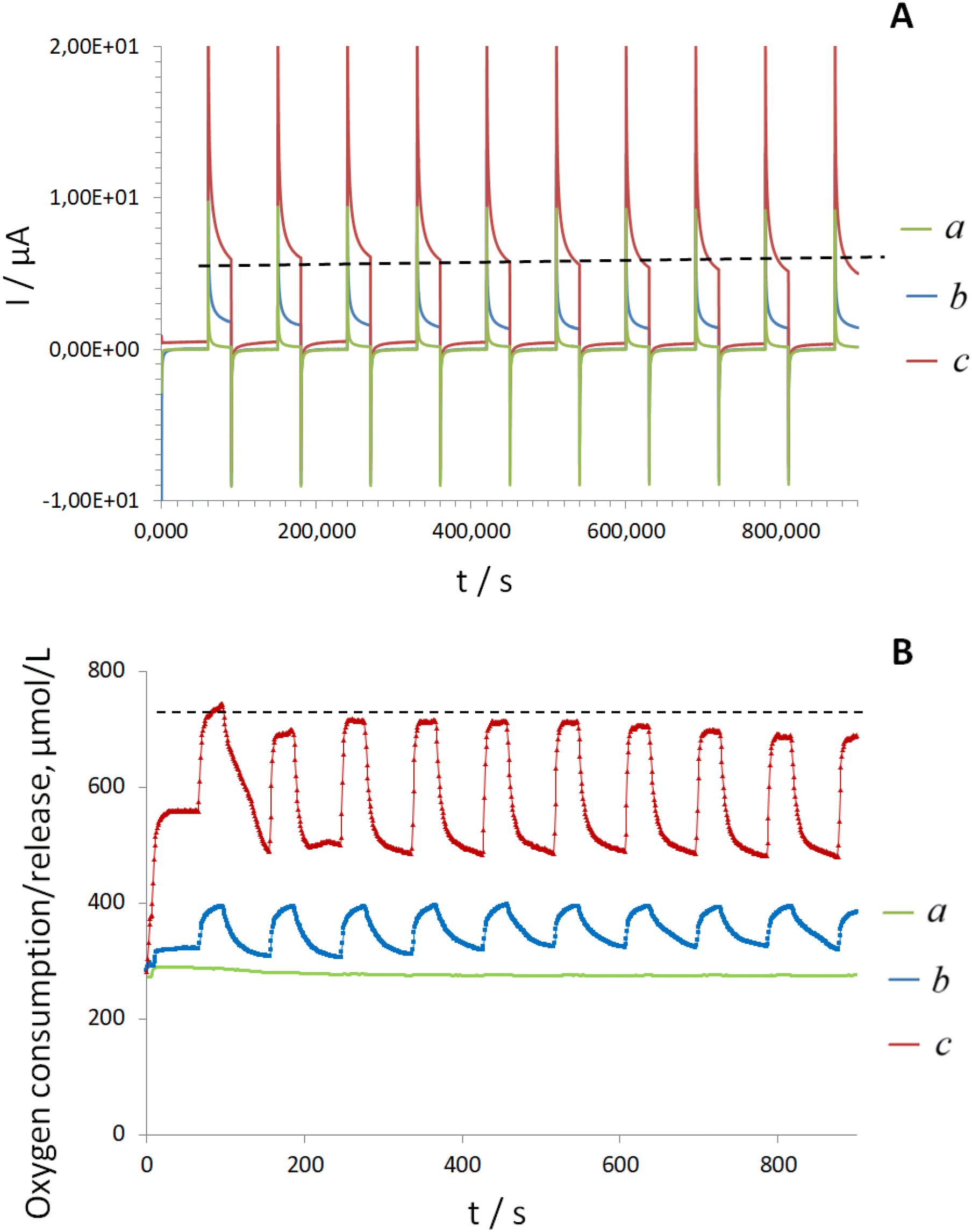 | ||
| Fig. 5 Time-dependent MAM plots (A) and synchronized oxygen mini-sensor responses, μmol L−1 (B) recorded at 22 ± 2 °C in the presence of H2O2 from Electrode D during ten cycles in: a – buffer; b – 10 mM H2O2; c – 25 mM H2O2. Note: the black dashed line shows the stable trend. The pH of all solutions was 6.98 ± 0.2. | ||
Another interesting observation from this set of experiments was that the resulting anodic current (corresponding to H2O2 oxidation) recorded in the presence of 25 mM glucose and aqueous apo-GOx was significantly higher than the currents obtained during the electrooxidation of individual 25 mM H2O2 (Fig. 4Avs. Fig. 5A). Simultaneously, the oxygen production in the presence of individual H2O2 was at least twice the oxygen release observed in the presence of aqueous apo-GOx and glucose (Fig. 4Bvs. Fig. 5B) on the surface of the same electrode. The excess oxygen (due to decomposition of high amounts of individual H2O2) likely leads to the intensive formation of palladium oxides on the surfaces of Pd-NPs, reducing their electrocatalytic ability for the electrooxidation of pure H2O2 and suppressing the anodic current at 0.2 V.16
Finally, the effects of in situ generated dissolved oxygen on the behavior of GOx on the surfaces of FAD-modified electrodes were demonstrated by tandem of MAM measurements and oxygen mini-sensor studies, performed in a droplet of glucose and holo-GOx. The results revealed trends consistent with those observed for Electrode D in the presence of aqueous apo-GOx, but with more pronounced signal intensities, Fig. 6 (line a), see also Fig. 4B (line b).
 | ||
| Fig. 6 Time-dependent oxygen mini-sensor dynamic responses, μmol L−1, recorded under applied polarization in MAM mode at 22 ± 2 °C from: (a) FAD-modified Electrode D and (b) an electrode modified with Pd-NPs in the absence of electrodeposited FAD. Measurements were performed in a droplet of test solution containing holo-GOx (type X-S, 5 μg μL−1) and 25 mM glucose. | ||
In contrast, for electrodes prepared without FAD and modified solely with Pd-NPs, no oxygen release was detected. Instead, this system exhibited strong oxygen consumption within the first 30–90 s, followed by an absence of detectable oxygen during the subsequent ten cycles (see Fig. 6, line b).
This model experiment highlighted the role of electrodeposited FAD in promoting dynamic oxygen generation and enhancing electron transfer within the reconstructed system (Electrode D). These findings confirm that the presence of electrodeposited cofactor FAD in the sensing layer design leads to oxygen overproduction.
3.4. Effects of applied polarization on the efficiency of in situ reconstitution of apo-GOx
Previously, the kinetic binding between the apo-GOx and FAD conjugated to a substrate (progesterone) was studied using fluorescence assays.20 In the present experimental set up, a slow binding between apo-GOx and the substrate (glucose), followed by interaction with the electrodeposited cofactor FAD on the electrode, can be assumed. According to eqn (1)–(3), this slow binding process leads to the gradual formation and subsequent decomposition of hydrogen peroxide under applied polarization on Pd-NPs, with slow/delayed dynamic oxygen release.To validate this assumption, experiments using apo-GOx and the FAD-modified electrode (Electrode D) were conducted under stationary current applied for 2000 s, as well as in the absence of continuous polarization. To model the absence of continuous polarization, a droplet of glucose containing apo-GOx remained on the electrode for 2000 s, however, the current was applied for only a short duration, e.g., 200 s (to record the signal corresponding to H2O2 oxidation), followed by a relaxation period (absence of current) for 5 min. This pulse-like regime was repeated four times in the same droplet of test solution.
Interestingly, in the pulse-like mode compared to stationary polarization, a nearly identical gradual, time-dependent current increase was recorded from Electrode D in a droplet containing glucose and aqueous apo-GOx, Fig. 7A (lines 1–4 vs. with line 5). The obtained results visualized the binding and biochemical transformation of glucose by in situ reconstructed apo-GOx occurring regardless of the applied current. Notably, this model experiment confirms continuous/dynamic H2O2 formation as a result of in situ apo-GOx reconstruction on the FAD-modified electrode.
 | ||
| Fig. 7 (A) Time-dependent AM plots recorded under pulse-like polarization for 200 s (1–4 curves) and stationary polarization for 2000 s (curve 5) at 0.2 V from Electrode D in the presence of apo-GOx and 25 mM glucose. (B) AM plots recorded from Electrode D in the presence of 25 mM H2O2 under pulse-like (1–4 curves) and stationary polarization (curve 5). | ||
Electrode D tested in a droplet of hydrogen peroxide solution (in pulse-like applied polarization or conventional stationary polarization) showed a different dependency from that observed with glucose solutions, Fig. 7B. Thus, regardless of the polarization mode (stationary or pulse-like), a nearly constant anodic current corresponding to H2O2 electrooxidation on Pd-NPs was detected. It means that a constant amount of H2O2 was oxidized at Pd-NPs at every time-event, thus resulting in the formation of a constant amount of oxygen in the system and the appearance of a constant anodic current.
These findings confirm the dynamic H2O2 production seen in Fig. 7A (see also Fig. 4A, line a) as a result of dynamic binding of glucose by in situ reconstructed apo-GOx, leading to dynamic oxygen release and a corresponding increase in current.
Notably, reconstructed enzyme engineering generally lacks specific applications. However, the time-dependent effect observed in this study could be readily applied in enzymology and biotechnology for real-time supervision and control of substrate binding and oxidation by in situ reconstructed apo-GOx on electrode surfaces modified with electrodeposited FAD cofactor (see section 3.6).
3.5. Controlled design of electrodes with electrodeposited FAD cofactor
Before the development of practical applications of the proposed electrodes with the electrodeposited cofactor FAD, it was important to study the impact of several experimental parameters, viz. substrate/glucose concentration, the concentration of aqueous apo-GOx and the amount of electrodeposited cofactor on time-dependent changes in the anodic current.Notably, an increase in glucose concentration, while maintaining a constant amount of protein (apo-GOx), was accompanied by a linear increase in anodic current (ESI, Fig. S10†). Since Electrode D with electrodeposited FAD cofactor linear responds to the increase in analyte concentration, the correct work of the reconstructed apo-GOx in the examined glucose concentration range was confirmed.
The effects of the amount of added aqueous apo-GOx, while maintaining a constant glucose concentration on current dependencies are summarized in ESI, Fig. S11A.† An increase in aqueous apo-GOx concentration from 1 μg μL−1 to 5 μg μL−1 was accompanied by a corresponding anodic current increase. Interestingly, AM plots recorded from Electrode D in the presence of varying amounts of apo-GOx differed in terms of not only the intensities at the end of the read-out mode (2000 s) but also showed distinct form at the beginning of the read-out region below 500 ± 50 s (close to the turnover rate of native holo-GOx). These results highlight the impact of the amount of aqueous apo-protein on its binding properties.
In the absence of electrodeposited FAD in the sensing layer (no FAD in the multiple electrolyte solution or in the electrodeposited layer), no current increase was recorded from this electrode in a droplet of glucose and aqueous apo-GOx, ESI, Fig. S11B† (line a). Increasing the FAD concentration in the multiple electrolyte solution from 0.5 mg mL−1 to 1 mg mL−1 was accompanied by a doubling of current intensity (signal recorded at 2000 s; ESI, Fig. S11B†, see lines b and c). Further increases in FAD concentration in the multiple electrolyte solution to 3–5 mg mL−1 did not result in a significant improvement in analytical performance of electrodes with electrodeposited cofactors. This effect could be explained by an increased ratio of the electrodeposited inorganic component (Pd-NPs) relative to the bioorganic component (FAD/Naf) under these conditions.23
In summary, the sensing layer design and the electroanalytical performance of electrodes with electrodeposited cofactors can readily be tuned. For the applications considered in this study (described below) the concentration of glucose in each droplet was maintained at 25 mM, the concentration of apo-GOx and holo-GOx were set at 5 μg μL−1, and the FAD concentration in multiple electrolyte solution was fixed at 1 mg mL−1.
3.6. Ranking of reconstructed apo-GOx samples
Molecular interactions, such as protein–protein, small molecule/fragment–protein interactions, ligand–protein, or protein–substrate interactions, can usually be visualized in a time-dependent manner with a Biacore platform34,35 or UV-visible spectroscopy.36 Unfortunately, few attempts have been made to develop advanced simpler, faster inexpensive electrochemical techniques for enzyme–electrode interface investigations,37 with similar technical capabilities, to enable rapid screening of native or reconstructed enzymes, and provide essential bioanalytical information on individual enzymes and their binding to physiological substrates.38 However, electrochemical investigations of enzymes and their interactions with substrates could substantially enhance understanding of enzymatic activity, structure, and function.The time-dependent dynamic current changes observed from FAD-modified electrodes during in situ reconstruction of apo-GOx were used for profiling of apo-enzymes based on their type.
All tested enzymes (GOx) were obtained from the same genetically modified strain Aspergillus niger (EC 1.1.3.4, without added oxygen) and exhibited similar activity levels (100–250 KU g−1 solid) as specified by the manufacturer. However, they differed in type: i.e. GOx type X-S, GOx type VII, and recombinant GOx. The protein purity was ≥65% for GOx type X-S and ≥60% for GOx type VII (nearly equal). The protein content of recombinant GOx was not specified. However, based on foreign activity values changing in a route: ≤5 units per mg (GOx type X-S) ≤ 10 units per mg (GOx type VII), ≤20 units per mg (recombinant GOx), it can be assumed that recombinant GOx had the lowest protein purity. The differences in GOx type, as specified by manufactures, were further confirmed by QCM studies. QCM responses recorded for GOx type VII and GOx type X-S were very similar, ESI, Fig. S12.† In contrast, the QCM signal recorded for recombinant GOx differed significantly from both.
As a negative control in this set of experiments, GOx (type X-S) heated at 70 °C for 30 min was used. Upon heating, the enzyme denatures, and the initially biochemically active monomers assemble into dimers, trimers, and tetramers.39
Next, the tested GOx of various types (type VII, type X-S and recombinant) underwent acidic hydrolysis (see Experiment) enabling their conversion to apo-GOx forms (microbial enzymes often expressed in apo-form40,41). The resulting apo-GOx were then applied to FAD-modified electrodes. The ranking results for the apo-GOx enzymes recorded from Electrode D are summarized in Fig. 8.
 | ||
| Fig. 8 Time-dependent AM plots recorded from FAD-modified electrode (Electrode D) in the presence of: a – a pure glucose solution; b – glucose with added recombinant apo-GOx; c – glucose with added apo-GOx type X-S; d – glucose with added apo-GOx type VII; e – glucose with added apo-GOx type X-S heated at 70 °C. Note: pH of all solutions was 7.01 ± 0.2; experiments were conducted in triplicate and yielded the same results. | ||
Notably, a pronounced difference in the behavior of in situ reconstructed apo-GOx enzymes was observed between 500 s (range after turnover) and 2000 s. This effect can be attributed to variations in glucose entrapment or binding by the apo-proteins. These differences likely reflect variations in protein quality or purity, thereby enabling the ranking of apo-GOx enzymes using FAD-modified electrodes.
The obtained AM plots (Fig. 8), including their shapes and current intensities, were influenced by the purity of the tested enzymes and followed the trend: apo-GOx type VII ≤ apo-GOx type X-S < recombinant apo-GOx. This trend reflects increasing efficiency of the reconstitution process. The obtained AM plots were in line with QCM curves recorded for the corresponding intact GOx variants, ESI, Fig. S12.† This indicates that the efficiency of apo-GOx reconstitution was affected by variations in protein purity and quality.
A similar observation has been reported by Chmayssem et al., where the authors have investigated the stability and activity of wild-type and recombinant GOx entrapped in a chitosan matrix.42 They have reported that recombinant form of GOx exhibited improved stability and electrochemical performance towards glucose oxidation. This effect was explained in terms of presence of catalase in the recombinant (foreign activity). In a control experiment, it was shown that when native GOx is applied together with catalase, partial decomposition of H2O2 by catalase prevents deactivation of GOx due to formation of H2O2 and enhances the catalytic activity of native GOx. To sum it up, the improvements in time-dependent reconstruction/efficiency observed for recombinant GOx can be explained by the synergistic decomposition of H2O2 (formed during enzymatic reaction) driven both by Pd-NPs (via an electrocatalytic route) and by catalase (via biochemical route) simultaneously.
Remarkably, as expected no reconstruction or time-dependent current changes were recorded for apo-GOx heated at 70 °C (denatured protein, negative control), Fig. 8.
It is believed that the established time and shape dependent differences in the dynamic electrochemical behavior of reconstructed apo-GOx on FAD-modified electrodes could serve as a valuable platform for studying the kinetic rates of microbial enzymes, conducting kinetic rate analysis, or determining turnover rates.
Furthermore, the proposed methodology could be extended to other types of oxidoreductases and apo-enzyme/cofactor pairs, facilitating the design of next-generation biosensors and biocatalytic interfaces.
4. Conclusions
Herein, the properties of in situ reconstructed apo-GOx on the surfaces of FAD-modified electrodes were studied. The role of enzyme assembly order from the same electrodeposited building units was highlighted (i). Depending on the nature of electrodeposited blocks (e.g., spatially separated FAD and apo-GOx, or complete holo-GOx) of the sensing layers, the electroanalytical performance of the electrodes exhibited real time kinetic differences (ii). Briefly, the electrode with in situ reconstructed apo-GOx, compared with an analogue with electrodeposited native holo-GOx, achieved advanced electroanalytical performance because of interactions with the electrodeposited cofactor FAD (iii).The advanced electrochemical behavior of electrodes with in situ reconstructed apo-GOx was attributed to enhanced oxygen supply resulting from dynamic substrate binding, hydrogen peroxide production, and dynamic oxygen release, thereby supporting the advanced biochemical route (iv).
The observed current differences recorded in dynamic amperometric mode from FAD-modified electrodes in the presence of aqueous apo-GOx were used for profiling the following apo-enzymes based on their type (determined by protein quality): GOx type X-S, GOx type VII, and recombinant GOx (v).
The obtained dependencies were fully validated by electrochemical and oxygen mini-sensor studies and could be used in the future for rapid electrochemical ranking, classification, and quantitative profiling of enzymes.
Author contributions
M. Koch: Formal analysis, investigation, methodology, review and editing; N. Korkmaz: Formal analysis, investigation, review and editing; Y. E. Silina: Conceptualization, investigation, data acquisition, funding acquisition, methodology, validation, writing – original draft.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the ESI.†Acknowledgements
This study was a part of the research program of Y. E. S. funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation, project 427949628).References
- L. Fruk, C.-H. Kuo, E. Torres and C. M. Niemeyer, Angew. Chem., Int. Ed., 2009, 48, 1550–1574 CrossRef CAS.
- J. S. Schultz, S. Mansouri and I. J. Goldstein, Diabetes Care, 1982, 5, 245–253 CrossRef CAS.
- S. Sitaula, S. D. Branch and M. F. Ali, Chem. Commun., 2012, 48, 9284–9286 RSC.
- S. D'Auria, P. Herman, M. Rossi and J. R. Lakowicz, Biochem. Biophys. Res. Commun., 1999, 263, 550–553 CrossRef.
- B. Willner, E. Katz and I. Willner, Curr. Opin. Biotechnol., 2006, 17, 589–596 CrossRef CAS.
- J. Yi and Z. Li, Curr. Opin. Biotechnol., 2022, 78, 102831 CrossRef CAS.
- F. Liu, L. He, S. Dong, J. Xuan, Q. Cui and Y. Feng, Molecules, 2023, 28(15), 5850 CrossRef CAS.
- U. Vyu, V. V. Shumyantseva, E. A. Bykhovskaya, L. N. Kolyada and A. I. Archakov, Biochem. Biophys. Res. Commun., 1994, 200, 722–725 CrossRef.
- N. Apushkinskaya, E. V. Zolotukhina, E. V. Butyrskaya and Y. E. Silina, Comput. Struct. Biotechnol. J., 2022, 20, 3824–3832 CrossRef CAS.
- H. Chen, O. Simoska, K. Lim, M. Grattieri, M. Yuan, F. Dong, Y. S. Lee, K. Beaver, S. Weliwatte, E. M. Gaffney and S. D. Minteer, Chem. Rev., 2020, 120, 12903–12993 CrossRef CAS.
- A. Heller, Electrical Wiring of Redox Enzymes BT, in Photochemical Conversion and Storage of Solar Energy, ed. E. Pelizzetti and M. Schiavello, SpringerNetherlands, Dordrecht, 1991, pp. 67–87 Search PubMed.
- E. V. Butyrskaya, N. Korkmaz, E. V. Zolotukhina, V. Krasiukova and Y. E. Silina, Analyst, 2021, 146, 2172–2185 RSC.
- E. Katz, A. Riklin, V. Heleg-Shabtai, I. Willner and A. F. Bückmann, Anal. Chim. Acta, 1999, 385, 45–58 CrossRef CAS.
- O. Yehezkeli, M. Moshe, R. Tel-Vered, Y. Feng, Y. Li, H. Tian and I. Willner, Analyst, 2010, 135, 474–476 RSC.
- C. Davis, S. X. Wang and L. Sepunaru, Curr. Opin. Electrochem., 2021, 25, 100643 CrossRef CAS.
- Y. E. Silina, N. Apushkinskaya, N. V. Talagaeva, M. G. Levchenko and E. V. Zolotukhina, Analyst, 2021, 146, 4873–4882 RSC.
- R. Zhao, Y. Ke, H. Sun, C. Quan, Q. Xu, J. Li, J. Guan and Y. Zhang, Microbiol. Res., 2025, 297, 128149 CrossRef CAS.
- G. Guoqiang, Q. Liang, Z. Yani, W. Pengyun, K. Fanzhuo, Z. Yuyang, L. Zhiyuan, N. Xing, Z. Xue, L. Qiongya and Z. Bin, Front. Chem., 2025, 13, 1–17 Search PubMed.
- S. Hao, H. Zhang, X. Sun, J. Zhai and S. Dong, Nano Res., 2021, 14, 707–714 CrossRef CAS.
- G. A. Posthuma-Trumpie, W. A. M. van den Berg, D. F. M. van de Wiel, W. M. M. Schaaper, J. Korf and W. J. H. van Berkel, Biochim. Biophys. Acta, Proteins Proteomics, 2007, 1774, 803–812 CrossRef CAS.
- S. D'Auria, P. Herman, M. Rossi and J. R. Lakowicz, Biochem. Biophys. Res. Commun., 1999, 263, 550–553 CrossRef.
- D. Semenova, K. V. Gernaey, B. Morgan and Y. E. Silina, Analyst, 2020, 145, 1014–1024 RSC.
- M. Koch, N. Apushkinskaya, E. V. Zolotukhina and Y. E. Silina, Biochem. Eng. J., 2021, 175, 108132 CrossRef CAS.
- D. Semenova, Y. E. Silina, M. Koch, L. Micheli, A. Zubov and K. V. Gernaey, Analyst, 2019, 144, 2511–2522 RSC.
- Y. E. Silina, Anal. Methods, 2024, 16, 2424–2443 RSC.
- A. Riklin, E. Katz, I. Wiliner, A. Stocker and A. F. Bückmann, Nature, 1995, 376, 672–675 CrossRef CAS.
- E. Katz and I. Willner, ChemPhysChem, 2004, 5, 1084–1104 CrossRef CAS.
- R. Cohen, Y. Cohen, D. Mukha and O. Yehezkeli, Electrochim. Acta, 2021, 367, 137477 CrossRef CAS.
- B. B. Collier and M. J. McShane, Proc. SPIE Int. Soc. Opt. Eng., 2015, 8591, 859104 CrossRef.
- A. Heller and B. Feldman, Chem. Rev., 2008, 108, 2482–2505 CrossRef CAS.
- B. M. Dixon, J. P. Lowry and R. D. O'Neill, J. Neurosci. Methods, 2002, 119, 135–142 CrossRef CAS.
- R. Venugopal and B. A. Saviue, Can. J. Chem. Eng., 1993, 71, 917–924 CrossRef CAS.
- C. Bourdillon, C. Demaille, J. Gueris, J. Moiroux and J. M. Saveant, J. Am. Chem. Soc., 1993, 115, 12264–12269 CrossRef CAS.
- S. Hashimoto, Principles of BIACORE, in Real-Time Analysis of Biomolecular Interactions, ed. K. Nagata and H. Handa, Springer, Tokyo, 2000, DOI:10.1007/978-4-431-66970-8_3.
- C. Davis, S. X. Wang and L. Sepunaru, Curr. Opin. Electrochem., 2021, 25, 100643 CrossRef CAS.
- N. B. Ugulava, C. J. Sacanell and J. T. Jarrett, Biochemistry, 2001, 40, 8352–8358 CrossRef CAS.
- N. Kornienko, K. H. Ly, W. E. Robinson, N. Heidary, J. Z. Zhang and E. Reisner, Acc. Chem. Res., 2019, 52, 1439–1448 CrossRef CAS.
- C. Lin, L. Sepunaru, E. Kätelhön and R. G. Compton, J. Phys. Chem. Lett., 2018, 9, 2814–2817 CrossRef CAS.
- W. N. Ye and D. Combes, Biochim. Biophys. Acta, 1989, 999, 86–93 CrossRef CAS.
- N. Mano, Bioelectrochemistry, 2019, 128, 218–240 CrossRef CAS.
- M. K. Dubey, A. Zehra, M. Aamir, M. Meena, L. Ahirwal, S. Singh, S. Shukla, R. S. Upadhyay, R. Bueno-Mari and V. K. Bajpai, Front. Microbiol., 2017, 8, 1032 CrossRef.
- A. Chmayssem, I. Shalayel, S. Marinesco and A. Zebda, Sensors, 2023, 23(1), 465 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5an00337g |
| This journal is © The Royal Society of Chemistry 2025 |