
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Long-term device stability for Raman spectroscopy†
Shuxia
Guo
 *ab,
Anuradha
Ramoji
ab,
Aikaterini
Pistiki
ab,
Hulya
Yilmaz
ab,
Uwe
Glaser
a,
David L.
Vasquez-Pinzon
ab,
Iwan W.
Schie
abd,
Ute
Neugebauer
abc,
Anja
Silge
ab,
Jürgen
Popp
ab and
Thomas
Bocklitz
ab
*ab,
Anuradha
Ramoji
ab,
Aikaterini
Pistiki
ab,
Hulya
Yilmaz
ab,
Uwe
Glaser
a,
David L.
Vasquez-Pinzon
ab,
Iwan W.
Schie
abd,
Ute
Neugebauer
abc,
Anja
Silge
ab,
Jürgen
Popp
ab and
Thomas
Bocklitz
ab
aLeibniz Institute of Photonic Technology, Member of Leibniz Health Technologies, Member of the Leibniz Centre for Photonics in Infection Research (LPI), Albert Einstein Strasse 9, 07745 Jena, Germany. E-mail: shuxia.guo@uni-jena.de
bInstitute of Physical Chemistry (IPC) and Abbe Center of Photonics (ACP), Friedrich Schiller University Jena, Member of the Leibniz Centre for Photonics in Infection Research (LPI), Helmholtzweg 4, 07743 Jena, Germany
cCenter for Sepsis Control and Care (CSCC), Jena University Hospital, Am Klinikum 1, 07745, Jena, Germany
dUniversity of Applied Science Jena, Department Medical Engineering and Biotechnology, Carl-Zeiss-Promenade 2, 07745 Jena, Germany
First published on 19th May 2025
Abstract
Long-term stability of Raman setups is one of the critical criteria for using Raman spectroscopy in real-world applications. Substantial differences from long-term drifts of a device can largely reduce the reliability of the technology and lead to serious consequences in scenarios such as disease diagnostics. A systematic investigation of long-term device stability is urgently needed to understand the device-related variations and to help improve the situation. In this study, 13 substances were measured as quality control references weekly for 10 months on a Raman device to investigate instrumental stability over time. The 13 substances were selected to be stable and to cover a wide range of standards, solvents, lipids, and carbohydrates. Approximately 50 Raman spectra of each substance were acquired per measurement day. A data pipeline was constructed to discover the variability (i.e., instability) of the device for the covered time window. Therein, the stability of the measurement was benchmarked from multiple perspectives, including the intensity variations, the correlation coefficients, the clustering, and the classification. The results suggested the device variability to be more random than systematic. Nonetheless, we demonstrated the possibility of decreasing the variations from the data via computational methods. In particular, we estimated the spectral variations by a network adapted from the variational autoencoder (VAE) and suppressed them from the measured data by the extensive multiplicative scattering correction (EMSC) method. This could improve the prediction of independent measurement days for three representative classification tasks.
1. Introduction
Raman spectroscopy provides the unique fingerprints of the molecular components being measured by indirectly detecting the molecular vibrations.1–5 This makes Raman spectroscopy a versatile technology for biological, medical, and clinical investigations.6–8 The power of the technology is further enhanced by machine learning techniques9 that can effectively extract the intrinsically subtle spectral variations of interest and translate them into high-level knowledge. However, the widespread application of Raman spectroscopy remains an unfulfilled and challenging task. One of the major obstacles is the substantial differences between devices, from long-term drifts of a device, or due to the amendment/replacement of an optical compartment.1 This challenge was highlighted in one of our previous studies by comparing the spectral variations of 35 instruments across the European institutes.10The consequences of the issue are two-fold. First, a trained machine learning model is disabled to predict the unknown data well if they are from a different device or, more likely, on the same device but at a long time after.11,12 This can lead to severe consequences in applications like disease diagnostics. Second, it is extremely challenging for the community to build standard databases, especially from multi-center measurements where the measurement-dependent variations are significant.13 The lack of standard databases has posed a challenge to the development of large-scale machine learning models such as deep neural networks, which require large datasets.
To suppress the device-dependent spectral variations is therefore an important task in Raman spectroscopy. Spectrometer calibration,14 for instance, brings measured peak positions and intensities to their theoretical values according to the well-defined standard materials, which helps to remove the device-related variations in a measured spectrum. The improvement, however, is limited. As is shown in ref. 11, the remaining spectral variations, both on the wavenumber and intensity axis, are still substantial. The reasons can be multiple, as have been discussed in ref. 10. An alternative method, warping,15,16 does not rely on standard materials, but attempts to align the measured spectra to a reference spectrum. This helps to reduce both instrument and sample-related spectral variations, provided a proper reference spectrum is employed. Nevertheless, warping does not guarantee a spectrum to be closer to the ‘truth’ unless a standard material is used as the reference. This makes the technique reference dependent, introducing another source of variation into multicenter studies.
To better understand the instrumental drifts and explore the possibilities of better calibration, we investigated the variations of a Raman setup over a term of ten months. The workflow is shown in Fig. 1. Raman measurements were performed weekly on 13 carefully selected pure substances, ranging from standard solids to solvents, lipids, and carbohydrates. A sequence of chemometric steps, including preprocessing and statistical analysis, was developed to verify the stability of the measurement. The variability underlined from the analysis was further estimated and suppressed with a variational autoencoder (VAE) in combination with extensive multiplicative scattering correction (EMSC).
 | ||
| Fig. 1 Workflow of the study. 13 reference substances were carefully selected and measured weekly, of which we covered in this study the data over 10 months (2022-11-02 to 2023-08-30). The measured spectra went through the same analysis pipeline. Preprocessing steps were applied including spikes removal, wavenumber calibration, baseline correction, and normalization. All preprocessed spectra were subjected for intensity analysis, clustering, and classifications to discover the potential changes in the device over the measurement period. | ||
2. Materials and methods
2.1 Raman spectrometer and measurement
The Raman measurements in this study were performed on high throughput screening RS (HTS-RS) system as was described in ref. 17 and 18 with 1 s integration time. The system was equipped with a 785 nm single-mode excitation laser (Xtra, Toptica, Germany) and a nominal output power of 400 mW. It was designed for in vitro cell diagnostics and has been used for clinical investigations, which makes its long-term stability particularly important. We conducted the measurement weekly on 13 substances included in European Pharmacopoeia (EP) or National Institute of Standards and Technology (NIST): four standard references (cyclohexane, paracetamol, polystyrene, silicon), four solvents (dimethyl sulfoxide (DMSO), benzonitrile, isopropanol, ethanol), three carbohydrates (fructose, glucose, sucrose), and two lipids (squalene, squalane). Additionally, we measured the dark current and Raman spectra from water on each measurement day. A detailed description of each substance is included in ESI.† These substances were chosen because they have Raman signals over the entire wavenumber range. Their bandwidth, intensity ratios, and distribution are similar to the biological spectra that are examined for diagnostic purposes (blood, tissues, cells, microorganisms). Particularly, the liquid substances were placed inside quartz cuvettes (Fig. S2a and b†) while the powder substances were pressed inside aluminum holders that were produced in the workshop of the Institute of Physical Chemistry at Friedrich Schiller University (Fig. S2c†). Details and pictures of the different sample holders are given in Fig. S2.†The number of spectra is shown in Fig. S1† for each substance and each measurement day. The mean spectra are visualized along with the standard deviation in Fig. S3† for each substance. Noteworthy, the ‘Paracetamol’ and ‘Paracetamol_Slide’ refer to paracetamol being measured on a substrate of slide, which demonstrated focusing instability compared with those measured on an aluminum holder (‘Paracetamol_Alum’, see Fig. S4†). Therefore, we used the former two measurements only for the wavenumber calibration but excluded them for further analysis. Moreover, spectra from cyclohexane were excluded except being used as standard of the wavenumber calibration, considering the contamination on the sample during the measurement (Fig. S5†). Squalane was excluded as well due to the instability in the spectral range below 500 cm−1 (larger standard deviation shown in Fig. S6†). Silicon was used to calibrate exposure time of the measurement, by making sure a relatively constant intensity of the Raman band at 520 cm−1. For analysis it was excluded because it contains only one single Raman band. The spectra from dark current and water were not used for the analysis, as the former does not contain Raman bands while the latter was only measured occasionally (see Fig. S1†).
2.2 Spectral preprocessing and wavenumber calibration
All spectra were preprocessed following a routine procedure, including despiking, wavenumber calibration, baseline correction, and l2 normalization.9 More details of each step are given in ESI.† The wavenumber calibration was conducted following the procedure described in our previous studies.14 Particularly, we performed the wavenumber calibration using different standards including cyclohexane, paracetamol, polystyrene, and a combination of all three standards. The results were compared according to three metrics: the mean absolute deviation (MAD) and the Pearson's correlation coefficient (PCC). To calculate MAD, we located on each spectrum the positions of well-defined Raman bands and calculated their absolute deviations from the values provided in standard database or literature. The MAD was obtained as the average over the deviations of multiple Raman bands within each spectrum. The PCC was calculated between the mean spectrum of each measurement day and the overall mean spectrum for each substance. All three metrics were calculated on spectra without wavenumber calibration and those calibrated using different standards. The results were compared to find the optimal standard for wavenumber calibration.2.3 Benchmarking stability
As our aim is to investigate the stability of Raman setups over a long period, we describe in this section a data pipeline to discover the potential spectral variations across the measurement days. The pipeline starts from correlation analysis as the very basic approach and goes further to classification as a more advanced process. We employed different approaches to incorporate the different perspective each approach brings, which helps to reach conclusions without being biased by a certain approach. Details will be given in the next subsections. | ||
| Fig. 2 The workflow of clustering analysis. (a) k-Means clustering was performed on each substance with the k value varying from 3 to 15. (b) For each k value and each substance, the ratio between the within- and cross-cluster distance was calculated based on the distance metrics of correlation coefficients, cosine distance, Euclidean distance, and Mahalanobis distance. (c) The ratios in (b) were input into sum-ranking difference (SRD), according to which the optimal k value of the corresponding substance is determined. (d) Steps (a–c) were repeated for all substances, of which the optimal k values were collected to find the k values of the top frequencies, i.e., the common-k. (e) k-Means clustering was performed for each substance again based on each k value from the common-k. (f) Each clustering result in (e) is translated into a co-clustering matrix, indicating if spectra from one measurement day (d1, d2, …, dn) are clustered into the same cluster as those from the other day. (g) The co-clustering matrices calculated in (f) were averaged for each substance. (h) The final co-clustering matrix was calculated by averaging results in (f) from all substances. | ||
 | ||
| Fig. 3 Illustration of the move-window strategy. Here we fixed the window size to be 3, i.e., to cover one before and one after the active day, except the cases where the active day is the first or the last day point. During the computation, the spectra are split into two parts: inside and outside of the active window. The RMSE is calculated between the two parts. In case of classification, a model is always trained on those outside of the window and used to predict those inside the window. The output, i.e., the RMSE or accuracy will be assigned as the result of the active day. | ||
Similarly, we performed one-class classification tasks based on the move-window strategy. The model was trained with spectra outside of the window and used to predict those inside the window. The accuracy of the prediction was considered as the result of the current active day. The one-class classification resulted in a vector of (nday, 1) for each substance. We collected results of all substances to form an accuracy matrix of dimension (nday, nsub), from which the SRD was calculated to summarize the results from different substances. A lower SRD value, which means a better prediction accuracy, represents a higher similarity of the day compared to the other days.
2.4 Verification of spectral variations
The potential patterns of the device variations unraveled from the previous analysis were verified as follows. We separated the measurement days into n segments, with lower spectral variations within each group compared to those across different segments. We made sure successive measurement days were in one segment, as the device drift is supposed to occur continuously over time. The spectral differences between the n segments were verified with the three representative classification tasks in Table 1. The three tasks were designed according to their chemical/biological information, the similar ones assigned into one task. To do so, we considered the n segments as n batches and performed the three classifications with a leave-one-batch-out cross-validation (LOBOCV). The results were compared to those from a random n-fold cross-validation, in which the measurement days were split randomly into n folds. The assumption was that the classification results will be worse for the LOBOCV if the differences between the segments are significant.| Task | Substances |
|---|---|
| Alcohol | Isopropanol, ethanol |
| Sugar | Fructose, glucose, sucrose |
| Other | Benzonitrile, dimethyl sulfoxide |
2.5 Suppression of spectral variations
The analysis described above helps to reveal the variability of the measurement. A more important but extremely challenging question is how to suppress the spectral variations. As a first attempt, we established a network by adapting a variational autoencoder (VAE) to estimate the representative component of the spectral variations, which were used as interference in the extensive multiplicative scattering correction (EMSC). This will suppress the contributions of the estimated variations from the measured data. Detailed description of the VAE architecture and EMSC method is given in ESI (see Fig. S10 and eqn S(1)†). Briefly, the VAE takes a spectral pair (s1, s2) as input and outputs the difference spectrum between s1 and s2. It consists of an Encoder and a Decoder. The Encoder transforms the spectral pair into two vectors representing the mean and variance of the distribution of the latent space, out of which a latent vector was resampled and used for the Decoder for reconstruction. Noteworthy, we kept the Decoder to be linear without using any activation functions. This could remove not only the nonlinear effects for the reconstruction but also lead to a more powerful Encoder and less powerful Decoder. In this way we expect the Encoder to better extract the hidden spectral variations that suffice for reconstruction.To make the situation easier, the patterns of the spectral variations were assumed not to significantly change over time. To start, we mean-centered and scaled against standard deviation for all spectra of each substance independently and constructed spectral pairs with two spectra (sbn, sbn+1) randomly selected from the same substance but two successive batches (bn, bn+1). The spectral pairs from different substances were concatenated together and used to train the VAE, in which the difference between the paired spectra was used as ground truth. In this way, the latent space is expected to embed the spectral variability from the measurement.
After being trained, the VAE was used to extract the representative components of the spectral variations. To do so, we calculated the mean spectrum of each day for each substance. Spectral pairs were constructed with two mean spectra randomly selected from two successive batches of the same substance. We randomly constructed 200 spectral pairs and fed into the encoder of the VAE. The latent vectors resulted from the encoder were supposed to encode the spectral variations between successive measurement days. To extract the representative components of the spectral variations, we performed a 5-means clustering on the latent vectors. Out of each cluster, we selected the latent vector that is closest to the centroid according to the Pearson's correlation coefficient. This led to five latent vectors which were fed into the trained decoder to obtain the representative components of the spectral variations.
The representative components for the spectral variations were used in the EMSC model (eqn (S1)†) as interference (pk(![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) )) to remove their influence on the measured spectra. We verified the effect of such variation suppression based on the three classification tasks in Table 1. The difference between the LOBOCV and random CV were compared to those without the EMSC correction.
)) to remove their influence on the measured spectra. We verified the effect of such variation suppression based on the three classification tasks in Table 1. The difference between the LOBOCV and random CV were compared to those without the EMSC correction.
3. Results and discussion
3.1 Benchmark of variability
 | ||
| Fig. 4 Comparison of different approaches for wavenumber calibration: no calibration, calibration based on paracetamol, cyclohexane, polystyrene, or a combination of the three standards. (a) Mean absolute deviation (MAD) of measured peak positions from their theoretical values. (b) Pearson's correlation coefficients (PCC) between day-wise mean spectrum and the overall mean spectrum for different substances. It is demonstrated that the wavenumber calibration largely decreased the spectral variations, with smaller MAD and increased PCC. It is also indicated that the cyclohexane provides inferior calibration compared to the other standards. However, we did not see clear differences among the other three cases of standards. Nonetheless, we chose to use a combination of the three standards for the calibration in the subsequent analysis, which helps to reduce the influence from standard-relevant variations on the calibration. | ||
 | ||
| Fig. 5 Results of correlation coefficients between the mean spectrum of different days from the same substance before (a and b) and after (c and d) wavenumber calibration. (a and c) Example results from the substance benzonitrile; (b and d) results after averaging over all substances. The measurement days of 2023-06-16 and 2022-12-02 are highlighted in panels (c and d), where the correlation coefficients changed quite obviously on these two days. | ||
 | ||
| Fig. 6 Results of k-means clustering with Paracetamol-based wavenumber calibration. (a) The scattering plot of PCA on benzonitrile, legends are shown for every five dates. (b) The ratio between the within- and cross-cluster distance with different k-values calculated from benzonitrile as an example. (c) SRD calculated from the ratios results and the automatically detected optimal k value. (d) Histogram of optimal k values for all substances. In this case, 6 was used as the common k value. (e) The cluster-matrix for benzonitrile, of which the value Mij = 1 demonstrating data from measurement days i and j are clustered into one cluster. (f) The final cluster-matrix averaged over all substances, where the arrows demonstrate the dates with obvious variations. | ||
 | ||
| Fig. 7 Comparison of optimal k values (a1–e1) and the cluster matrix (a2–e2) in case of different wavenumber calibrations. Accordingly, 4, 6, 6, 4, 6 are used as the optimal k-values in each case of wavenumber calibration, respectively. The distribution of the optimal k values varied across different wavenumber calibrations, most likely suggested the standard-dependence of wavenumber calibration or certain instabilities within the standard substances themselves. By combining all three standards during the calibration, nonetheless, we expected to reduce the influence from such standard-relevant variations. Therefore, the results from the combined calibration are considered more ‘reliable’. | ||
Before moving on to the next step, we compared the distribution of the optimal k values among different substances for the different strategies of the wavenumber calibration. The histograms of the optimal k values and the co-cluster matrices are visualized in Fig. 7(a1–e1), which varies among different standards for wavenumber calibration. This suggested the standard-dependence of calibration as one of the sources for spectral variations. By combining all three standards during the calibration, we expected to reduce such standard-dependent variations. Therefore, further analysis will be performed with spectra from the combined calibration.
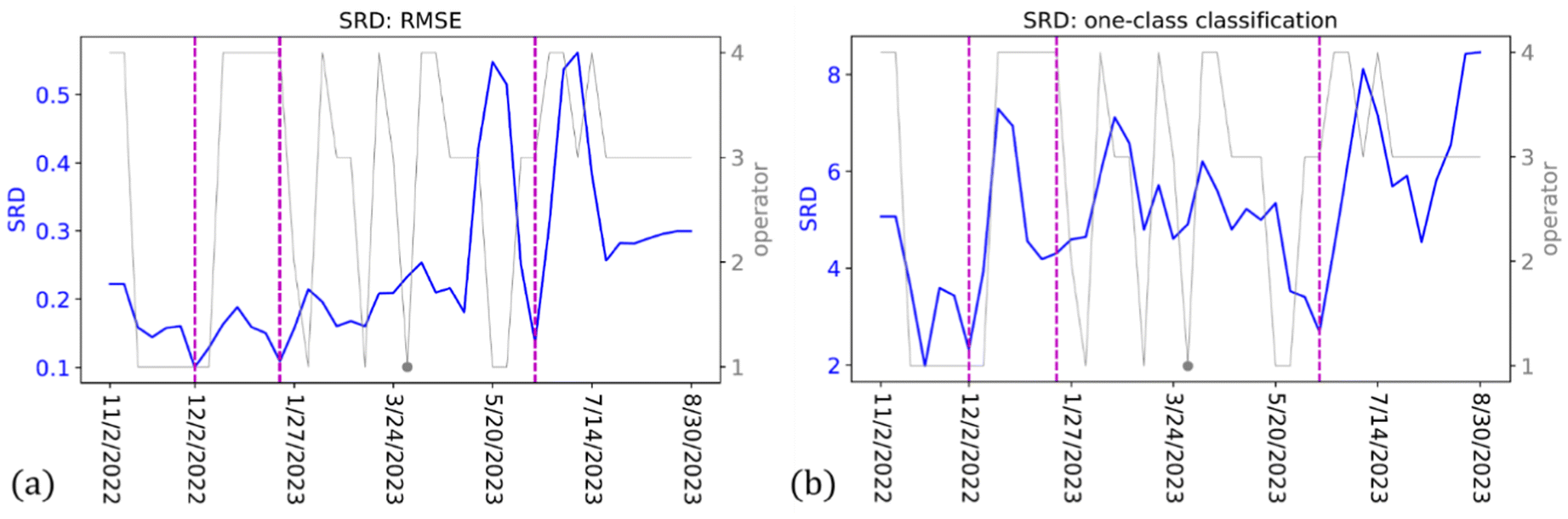 | ||
| Fig. 8 Results of moving-window calculations, where the key measurement dates are marked as dashed lines, the information of the operator is given as dual axis in gray. (a) Sum-ranking difference (SRD) calculated from the RMSE of different measurement days. (b) SRD calculated from the accuracy of the one-class classification. It is clear to see different changing patterns of the SRD within different segments split by the key dates. For both sets of the results, we could see correlation between the changes in operators and the spectral variations. | ||
3.2 Verification of variability
The verification of the measurement variability was performed via three classification tasks given in Table 1. We employed two cross-validation schemes: leave-one-batch (segment)-out cross-validation (LOBOCV) and random cross-validation. Spectra from each substance were mean-centered and scaled against the standard deviation of the same substance. This largely reduced the separability between the substances and made the classification more challenging. Otherwise, the ‘batch-wise’ changes are hidden as the substance-wise differences are much higher. The balanced accuracy based on three classification models are shown in Fig. 9 named ‘None’. Accordingly, the prediction of the LOBOCV was generally inferior to that of the randomized cross-validation. This means that differences between batches (segments) are larger than those between randomly separated folds. This is in line with the results given in Fig. 6. Noteworthy, the linear models, including LDA and linear-kernel SVM, did not provide satisfied results in all three classification tasks. This suggests that the classification tasks were not linearly solvable. | ||
| Fig. 9 Balanced accuracy of the three classification tasks under the framework of LOBOCV as well as the randomized CV for different cases of variation suppression: no suppression, EMSC based on average difference spectrum (diff-EMSC), and EMSC based on VAE estimated variations (VAE-EMSC). Different models, including linear models LDA and linear SVM, nonlinear models from ‘rbf’ SVM were employed for the classification. In comparison, the randomized CV (r) and LOBOCV (p) tend to be more similar after the EMSC in comparison to those without EMSC, especially for SVM with radial kernel. The VAE-EMSC outperforms diff-EMSC with higher balanced accuracy. | ||
3.3 Suppression of variability
All results presented so far suggest a variability over the 10 months of measurement, despite the wavenumber calibration. It is therefore beneficial to explore methods of suppressing such variability. To do so, we developed a VAE neural network to estimate the variations, with an assumption that the variations feature a relatively stable pattern. The network was trained with substances of benzonitrile, dimethyl sulfoxide, isopropanol, and polystyrene, which are chemically more stable than the others and were measured over the full period. After VAE was trained, we fed it with spectral pairs constructed as follows. First, we calculated the mean spectrum of each day for each substance. 200 spectral pairs were constructed with two mean spectra randomly selected from two successive batches of the same substance. We fed these spectral pairs into the encoder of the VAE, of which the output is plotted along with the ground truth in Fig. S11(a and b†). The resulting latent vectors are visualized in Fig. S12.† To extract the representative spectral variations, we performed 5-means clustering on the latent vectors (Fig. S11(c)†) and used the centroids as the input of the decoder, leading to 5 representative components of the spectral variation (Fig. S11(d)†).To suppress the spectral variations from the measurement, we employed the obtained representative components as ‘interference’ in EMSC model (eqn (S1a)†) for correction. The resulting spectra were expected to contain less variability from day to day. To verify this point, we performed again the three classification tasks of Table 1 with different classification models. As is shown in Fig. 9 (‘VAE-EMSC’), the differences between the LOBOCV and the randomized CV were generally smaller, especially for nonlinear SVM.
To verify the capability of VAE for estimating the spectral variability, we calculated the average difference spectrum between spectral pairs used for VAE training and used it as interference in EMSC. The corrected spectra were used again for the classification tasks. The balanced accuracy of all three models and two cross-validation strategies are shown in Fig. 9 (‘diff-EMSC’). Similar to VAE-EMSC, the difference between the two cross-validations was reduced by diff-EMSC comparing with those without EMSC (‘None’). Nonetheless, we also see that VAE-EMSC outperformed diff-EMSC with higher balanced accuracies, particularly for the nonlinear SVM. This highly suggested the benefit of VAE for variability estimation than the simple difference spectrum. As the difference spectrum still contains fingerprints of substances, using the average difference spectrum in EMSC can lose useful spectral information except the measurement variability. On the other hand, the VAE model aims to learn a ‘general’ measurement variability that is substance independent, making it more beneficial than the difference spectrum.
3.4 Discussion
We have demonstrated that the variability over time in Raman spectroscopy to be present but show no systematic pattern, which however can be suppressed via computational approaches. As the most straightforward manner, a wavenumber calibration based on multiple reference standards could be considered in Raman spectroscopy to improve the spectral reproducibility. Moreover, it is good practice to track the device drift overtime regularly based on the measurement on standard references to extract the hidden variability and correct it. Further, we recommend recording the environmental parameters of the measurement, such as humidity, temperature, operator, etc. to allow for better understanding of the source of potential variability. Last but not the least, we would like to briefly link to our previous studies on device-to-device variability and the approaches to handling it.11,12,20 Therein, the issue was handled via model transfer approaches, which are task- and sample-dependent. In this study, however, we aim to understand the measurement variability more systematically and estimate it in a sample- and task-anonymous manner, which is supposed to act as a ‘general calibration’ method that can suit the correction of different samples. Despite its use in structured time-series data, noteworthy, the VAE-EMSC method is potentially useful for data from multiple devices or replicates.4. Conclusions
In this study, we investigated the long-term stability of a Raman setup and constructed a data pipeline to benchmark, verify, and suppress the variability based on machine learning and statistical analysis. We based our analysis on the spectra of 13 substances measured weekly over a 10-month time window. The substances covered a wide range of standard materials, solvents, carbohydrates, and lipids. To start, the performance of the wavenumber calibration was compared across different standards: cyclohexane, polystyrene and paracetamol (Acetaminophen). A combined wavenumber calibration with all standards was eventually employed to reduce the possible standard dependence of the calibration. Further, we benchmarked the variability of the measurement from different perspectives, including correlation analysis, clustering, and moving window analysis. Therein, we could spot three dates when a clear change occurred for the measurement. This observation was further verified with three classification tasks, which were significantly inferior when predicting data from a different time segment split by the three days. In addition, we were able to estimate the spectral variations with VAE-based neural network and suppress them via EMSC method, which obviously improved the results of three classification tasks.Nevertheless, we did not see a strong correlation between the variability and the known facts like operator and element adjustment. This largely suggests a multi-source origin of the measurement variability. To systematically investigate such origins requires an extremely carefully controlled experimental condition. However, our results showed the promise of suppressing such variability with computational methods, which can be particularly beneficial for applications such as measurements on biological samples where the variability can heavily overwhelm spectral changes of interest. Particularly, the method of VAE-EMSC demonstrated the promise of estimating the measurement variability and reducing its influence from the measured data while maintaining useful spectral information. This makes it a potential ‘general calibration’ method for better reproducibility of long-term measurements. Its applicability on biological samples such as cells, tissues, and biofluid will be verified in our future study.
Data availability
All data needed to evaluate the conclusions in the paper is available on Zenodo via DOI: 10.5281/zenodo.15471666.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
We highly acknowledge the project TELEGRAFT (101057673) with the funding from the European Union's 9th Framework Program Horizon Europe. Views and opinions expressed are however those of the author(s) only and do not necessarily reflect those of the European Union or the Health and Digital Executive Agency. Neither the European Union nor the granting authority can be held responsible for them. This work is additionally supported by the BMBF, funding program Photonics Research Germany (LPI-BT1-FSU, FKZ: 13N15466; LPI-BT3-FSU, FKZ: 13N15710; LPI-BT4-Leibniz IPHT, FKZ: 13N15713) and is integrated into the Leibniz Center for Photonics in Infection Research (LPI). We thank support from Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) via the project TOOLs (528591139-FIP-31/1).References
- R. L. McCreery, Raman Spectroscopy for Chemical Analysis, John Wiley & Sons, 2005 Search PubMed.
- C.-S. Ho, N. Jean, C. A. Hogan, L. Blackmon, S. S. Jeffrey, M. Holodniy, N. Banaei, A. A. E. Saleh, S. Ermon and J. Dionne, Rapid Identification of Pathogenic Bacteria Using Raman Spectroscopy and Deep Learning, Nat. Commun., 2019, 10(1), 4927, DOI:10.1038/s41467-019-12898-9.
- C. V. Raman and K. S. Krishnan, A New Type of Secondary Radiation, Nature, 1928, 121(3048), 501–502, DOI:10.1038/121501c0.
- T.-Y. Huang and J. C. C. Yu, Development of Crime Scene Intelligence Using a Hand-Held Raman Spectrometer and Transfer Learning, Anal. Chem., 2021, 93(25), 8889–8896, DOI:10.1021/acs.analchem.1c01099.
- F. Lussier, V. Thibault, B. Charron, G. Q. Wallace and J.-F. Masson, Deep Learning and Artificial Intelligence Methods for Raman and Surface-Enhanced Raman Scattering, TrAC, Trends Anal. Chem., 2020, 124, 115796, DOI:10.1016/j.trac.2019.115796.
- G. Pezzotti, Raman Spectroscopy in Cell Biology and Microbiology, J. Raman Spectrosc., 2021, 52(12), 2348–2443, DOI:10.1002/jrs.6204.
- K. C. Doty and I. K. Lednev, Raman Spectroscopy for Forensic Purposes: Recent Applications for Serology and Gunshot Residue Analysis, TrAC, Trends Anal. Chem., 2018, 103, 215–222, DOI:10.1016/j.trac.2017.12.003.
- K. S. Lee, Z. Landry, F. C. Pereira, M. Wagner, D. Berry, W. E. Huang, G. T. Taylor, J. Kneipp, J. Popp, M. Zhang, J.-X. Cheng and R. Stocker, Raman Microspectroscopy for Microbiology, Nat. Rev. Methods Primers, 2021, 1(1), 1–25, DOI:10.1038/s43586-021-00075-6.
- S. Guo, J. Popp and T. Bocklitz, Chemometric Analysis in Raman Spectroscopy from Experimental Design to Machine Learning–Based Modeling, Nat. Protoc., 2021, 16(12), 5426–5459, DOI:10.1038/s41596-021-00620-3.
- S. Guo, C. Beleites and U. Neugebauer, et al., Comparability of Raman Spectroscopic Configurations: A Large Scale Cross-Laboratory Study, Anal. Chem., 2020, 92(24), 15745–15756, DOI:10.1021/acs.analchem.0c02696.
- S. Guo, R. Heinke, S. Stöckel, P. Rösch, T. Bocklitz and J. Popp, Towards an Improvement of Model Transferability for Raman Spectroscopy in Biological Applications, Vib. Spectrosc., 2017, 91, 111–118 CrossRef CAS.
- S. Guo, R. Heinke, S. Stöckel, P. Rösch, J. Popp and T. Bocklitz, Model Transfer for Raman-Spectroscopy-Based Bacterial Classification, J. Raman Spectrosc., 2018, 49(4), 627–637, DOI:10.1002/jrs.5343.
- O. Ryabchykov, S. Guo and T. Bocklitz, Photonic Data Analysis in 2050, Vib. Spectrosc., 2024, 132, 103685, DOI:10.1016/j.vibspec.2024.103685.
- T. W. Bocklitz, T. Dörfer, R. Heinke, M. Schmitt and J. Popp, Spectrometer Calibration Protocol for Raman Spectra Recorded with Different Excitation Wavelengths, Spectrochim. Acta, Part A, 2015, 149, 544–549, DOI:10.1016/j.saa.2015.04.079.
- J. Sjöblom, O. Svensson, M. Josefson, H. Kullberg and S. Wold, An Evaluation of Orthogonal Signal Correction Applied to Calibration Transfer of near Infrared Spectra, Chemom. Intell. Lab. Syst., 1998, 44(1), 229–244, DOI:10.1016/S0169-7439(98)00112-9.
- J. A. Fernández Pierna, A. Boix Sanfeliu, B. Slowikowski, C. von Holst, O. Maute, L. Han, G. Amato, B. d. l. Roza Delgado, D. Perez Marin, G. Lilley, P. Dardenne and V. Baeten, Standardization of NIR Microscopy Spectra Obtained from Inter-Laboratory Studies by Using a Standardization Cell, Biotechnol., Agron., Soc. Environ., 2013, 17(4), 547–555 Search PubMed.
- I. W. Schie, J. Rüger, A. S. Mondol, A. Ramoji, U. Neugebauer, C. Krafft and J. Popp, High-Throughput Screening Raman Spectroscopy Platform for Label-Free Cellomics, Anal. Chem., 2018, 90(3), 2023–2030, DOI:10.1021/acs.analchem.7b04127.
- A. Pistiki, F. Hornung, A. Silge, A. Ramoji, O. Ryabchykov, T. W. Bocklitz, K. Weber, B. Löffler, J. Popp and S. Deinhardt-Emmer, Raman Spectroscopic Cellomics for the Detection of SARS-CoV-2-Associated Neutrophil Activation after TNF-α Stimulation, Clin. Transl. Med., 2022, 12(12), e1139, DOI:10.1002/ctm2.1139.
- K. Héberger, Sum of Ranking Differences Compares Methods or Models Fairly, TrAC, Trends Anal. Chem., 2010, 29(1), 101–109, DOI:10.1016/j.trac.2009.09.009.
- S. Guo, A. Kohler, B. Zimmermann, R. Heinke, S. Stöckel, P. Rösch, J. Popp and T. Bocklitz, Extended Multiplicative Signal Correction Based Model Transfer for Raman Spectroscopy in Biological Applications, Anal. Chem., 2018, 90(16), 9787–9795, DOI:10.1021/acs.analchem.8b01536.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5an00255a |
| This journal is © The Royal Society of Chemistry 2025 |