
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Leaky waveguide biosensors for label-free measurement of human serum albumin†
Anil Kumar
Pal
ad,
Md Nazmul
Hossain
 be,
Saverio
Brogna
b,
Nicholas J.
Goddard
c and
Ruchi
Gupta
*a
be,
Saverio
Brogna
b,
Nicholas J.
Goddard
c and
Ruchi
Gupta
*a
aSchool of Chemistry, University of Birmingham, Birmingham, B15 2TT, UK. E-mail: r.gupta.3@bham.ac.uk
bSchool of Biosciences and Birmingham Centre of Genome Biology, University of Birmingham, B15 2TT UK
cUnaffiliated
dDepartment of Physics and Nanotechnology, College of Engineering and Technology, SRM Institute of Science and Technology, Kattankulathur, Chennai 603203, Tamil Nadu, India
eDepartment of Microbial Biotechnology, Faculty of Biotechnology and Genetic Engineering, Sylhet Agricultural University, Sylhet 3100, Bangladesh
First published on 2nd May 2025
Abstract
Early diagnosis of diseases such as kidney disease relies on the successful measurement of albumin concentration in urine. We report label-free detection of human serum albumin (HSA) using a leaky waveguide (LW) optical biosensor. The LW reported in this work comprised a few microns-thick mesoporous polyacrylamide hydrogel film deposited on a glass substrate by casting and, for the first time, copolymerized with N-(3-aminopropyl)methacrylamide (APMAA) to provide functional amine groups required to immobilise recognition elements, half-antibody fragments. Furthermore, this is an unprecedented report on the use of a high molecular weight (3700 D) poly(ethylene glycol) diacrylamide in contrast to previously reported low molecular weight bis-acrylamide crosslinkers to increase the porosity of waveguide films. Equally, other parameters such as molar ratio of APMAA to acrylamide and total weight of (monomers and crosslinker) to volume ratio were optimised to obtain hydrogel films with pore size and amine groups required to immobilise half-antibody fragments in hydrogel films. Three different strategies for immobilisation of recognition elements; two based on streptavidin biotin interactions and the third based on half fragments of antibody were studied. The third immobilisation strategy resulted in the most reproducible results and hence was used to measure the equilibrium dissociation constant of HSA and its corresponding half-antibody fragments. Using the LW-based label-free optical biosensor, HSA was successfully detected with a limit of detection of 28 ng mL−1 in buffer and the lowest concentration of HSA measured in this work was 66.5 ng mL−1. This capability of quantitation of HSA by the LW can be built upon to realise a LW biosensor for early detection of diseases including kidney disease.
1. Introduction
Biologically important human serum albumin (HSA) is the most predominant circulating protein in humans. The concentration of HSA is ∼500–800 μM in blood and <0.3 μM in urine in healthy adults. While an increase in HSA levels in urine indicates a chronic kidney disease called microalbuminuria, a much lower level of HSA in urine is indicative of kidney and liver diseases.1–5 Thus, early detection of abnormalities in the concentration of HSA is of great importance for delivering timely treatments. The state of art methods used in clinical biochemistry laboratories for measuring the concentration of HSA are based on the interaction of the protein with dyes such as bromocresol green (BCG) and bromocresol purple (BCP).6,7 However, the concentrations of HSA estimated using BCG and BCP dyes are often significantly different, resulting in clinical uncertainty. To overcome the limitation associated with the use of dyes, biosensors have been developed.8–13 For example, optical biosensors have been reported for the detection of HSA in aqueous buffers and body fluids such as serum and urine.9,14–16 More specifically, Rasouli et al.14 demonstrated optical detection of HSA in serum using a nanocurcumin–VO2+ ensemble–based optical nanoprobe by observing the colour intensity of the HSA-optical nanoprobe complex. Semeradtova et al. developed optical microchips and utilised fluorescently labelled HSA for the detection of the protein in urine.15,17 Labelling of proteins with fluorophores adds to the number of analysis steps and can often increase the probability of false positives/negatives. These limitations can be overcome by label-free optical biosensors that rely on transducing changes in refractive index (RI), which can be caused by binding of analyte to recognition elements, into an easily measurable signal (e.g., changes in wavelength, intensity, angle).18 For example, Liu et al.17 demonstrated the sensing of HSA by a surface plasmon resonance (SPR) sensor using anthraquinone dye Cibacron Blue F3G-A (CB) as a recognition element for the specific binding of HSA. The RI changes caused by binding of HSA to CB caused shifts in resonance wavelength, which were measured to determine the concentration of HSA.Another label-free optical sensor is the leaky waveguide (LW),19 which has been shown to be able to measure diverse analytes including immunoglobulin G (IgG), thrombin, average iron content of ferritin, bacteria, and organophosphorus pesticides.20–24 The simplest LWs comprise a few microns thick film with RI lower than the substrate but higher than the liquid sample on top of the film. Light is partially confined in the low RI film of LWs by Fresnel reflection at the film/substrate interface which makes the waveguide film leaky, allowing prism coupling. The RI of the waveguide can change because of binding of analytes to recognition elements immobilised in the film or and/or change in composition and/or temperature of liquid sample. Compared to LWs with non-porous waveguide films where analytes and recognition elements are only present on the surface of the waveguide films, LWs with waveguide films made of porous hydrogels offer ∼9 times higher sensitivity by increasing the immobilisation density of recognition elements and maximizing the fraction of light that can interact with analyte-recognition elements complexes.21 As reported previously,20 the refractive index sensitivity (RIS) of LWs with porous waveguide films is ∼120° RIU−1. The minimum RI resolution of LWs is ∼10−6, which is comparable to typical surface Plasmon resonance (SPR) devices with continuous metal film and prism coupling.25,26 In contrast to SPR, which only works with light of transverse magnetic (TM) polarisation, LWs can work with light of any polarisation and even with unpolarised light (as in this work). Equally, the RIS and minimum RI resolution of LWs is independent of the polarisation of light.27 Furthermore, RIS of LWs with a porous waveguide is independent of the thickness of the waveguide film.27 This is because LWs with porous waveguides are bulk sensors, and their performance is minimally influenced by variations in film thickness and/or surface roughness.
This is an unprecedented report on LWs with waveguide films made by polymerising acrylamide and N-(3-aminopropyl)methacrylamide (APMAA) monomers with crosslinker, poly(ethylene glycol) diacrylamide (PEGDAm-3700). While APMAA provided the amine groups required for immobilisation of recognition elements, the high molecular weight of PEGDAm-3700 in comparison to previously used bis-acrylamide crosslinker, resulted in films with high porosity. This porosity allowed immobilisation of recognition elements in the waveguide films, improving sensitivity and limit of detection. The films of APMAA, acrylamide and PEGDAm-3700 were deposited on glass substrates by casting where the precursor solution was sandwiched between two glass substrates separated by polystyrene beads. In this case, the initial thickness of the hydrogel films was determined by the diameter of the polystyrene beads, and one of the glass substrates was chemically treated to allow covalent immobilisation of the hydrogel film to the glass. Although the approach of using beads to make films of controlled thickness has been previously reported,28 it has not been used to make waveguide films for sensing as is the case in this work. As the films were covalently bound to the glass substrate, they were robust and could not be damaged by flow of solutions over long periods of time. Equally, for the first time, we showed immobilisation of half fragments of antibodies in the waveguide films of LWs. We studied the affinity between HSA and its corresponding half-antibody fragments immobilised in waveguide films of LWs and determined the equilibrium dissociation constant (KD). Finally, LW biosensors with optimised concentration of half-antibody fragments were for the first time used for label-free quantitation of HSA with a limit of detection (LOD) of 28 ng mL−1 in buffer and the lowest concentration of HSA measured in this work was 66.5 ng mL−1.
2. Experimental
2.1. Materials and methods
Ethanol, toluene, polystyrene beads (mean diameter ∼1 μm), chloro(dimethyl)vinylsilane (97%) (CDVS), trimethoxy(3,3,3-trifluoropropyl)silane (99%) (TMTFS), N-(3-aminopropyl)methacrylamide hydrochloride (98%) (APMAA), 40% acrylamide solution, poly(ethylene glycol) diacrylamide with average Mn of 3700 (PEGDAm-3700), ammonium persulphate (APS), N,N,N′,N′-tetramethylethylenediamine (TEMED), sodium phosphate monobasic monohydrate, sodium phosphate dibasic dodecahydrate, ethylenediaminetetraacetic acid (EDTA), bromophenol blue, sodium chloride (99%), glycerol (Mw: 92), poly(ethylene glycol) (PEG) (Mw: 10, 100 and 300 kDa), biotin–protein A (P2165), IgG (I5131), anti-IgG (B3773), human serum albumin (A9511, ≥96%) (HSA), and centrifugal filter units (Amicon Ultra-0.5 mL with molecular weight cut-off (MWCO) of 30 kDa) were purchased from Sigma Aldrich (UK). Decon 90, tris-HCl, sodium dodecyl sulfate (SDS) and 2-mercaptoethylamine–HCl (2-MEA) were purchased from Fisher Scientific (UK). NHS–PEG2–maleimide and NHS–PEG12–biotin (where NHS is N-hydroxysuccinimide) were purchased from Broadpharm (USA). Streptavidin (2-0203-100) was purchased from IBA Lifesciences (Germany). Anti-human albumin antibody from goat (A80-129A) was purchased from Cambridge Bioscience (UK). 1 mm thick glass microscope slides were purchased from VWR (Leicestershire, UK). Colour pre-stained protein standards (10–250 kDa) were bought from New England Biolabs (USA).2.2. Fabrication of aminopropyl co-polymerised polyacrylamide LW films
Hydrogel films of APMAA copolymerised with acrylamide and crosslinked with PEGDAm-3700 were deposited on ∼25 × 25 mm2 glass substrates by casting where the thickness of the films was determined by the diameter of polystyrene beads used as spacers. The use of beads to make films of controlled thickness by casting has been previously reported.28 Glass substrates were cleaned in an ultrasonic bath using the procedure described previously.24 For chemical attachment of hydrogel films to glass surfaces, a few cleaned substrates were treated with 1% (v/v) CDVS solution in toluene for 30 min. The remaining few substrates were treated with 5% (v/v) TMTFS solution in toluene for 30 min to make the glass surface hydrophobic. All substrates were then washed with toluene and dried before use. Subsequently, a spacer layer was formed by drop casting 0.5 μL of a colloidal solution of 1% (w![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :v) polystyrene beads at four corners of TMTFS treated glass substrates and dried under ambient condition.
:v) polystyrene beads at four corners of TMTFS treated glass substrates and dried under ambient condition.
100 μL of hydrogel precursor solution was prepared by mixing 9 μL of 40% (w/v) acrylamide, 1.25 μL of TEMED, 0.79 mg of PEGDAm-3700, 0.89 mg of APMAA, and 1.25 mg of APS in N2-degassed de-ionised water. The total concentration of monomers in the precursor solution was 4.5% (w/v) with 10% (mol:mol) of APMAA:acrylamide. This precursor solution was cast between CDVS and TMTFS treated glass substrates with a spacer layer of polystyrene beads. After the solution was polymerised, the TMTFS treated glass substrate was peeled off and the hydrogel film was left behind on the CDVS treated glass substrate because of chemical linkages between the film and CDVS treated glass. The resulting LWs were stored in 100 mM phosphate buffer, pH 7.4 until use.
2.3. Instrumentation
The instrument used to study LWs has been described in our previous report24 and a schematic is provided in Fig. 1. Briefly, the instrument comprised a point source red LED (TL-6, iC-Haus, 640 nm). Light from the LED was passed through optical lenses (40 mm focal length achromatic doublet and 63 mm focal length cylindrical lens) to obtain a wedge-shaped unpolarised beam. Light was focussed onto a LW mounted on a BK7 equilateral prism (Qioptic Photonics, UK). Light reflected from LWs was collected using a 20 Mpixel (MER-2000-19U3M-L, Daheng Imaging) CMOS camera. A dual channel flow cell containing through holes for fluidic connections (inlets and outlets) was mounted on top of the LW using a clamping plate. One of the fluidic channels served as a sensor and the other as a reference. Analyte, immobilisation reagents, antibody and buffer solutions were pumped into sample and/or reference channels as required using a peristaltic pump (Minipuls® 3, Gilson, UK) at a flow rate of 200 μl min−1. Shifts in resonance angles of LWs corresponding to the reference channel were subtracted from the sensor channel to measure analyte response while removing baseline drifts and other common-mode effects. | ||
| Fig. 1 Schematic of the leaky waveguide instrumentation. | ||
2.4. Preparation of half antibody fragments
In immobilisation strategy 3 (discussed in sub-section 2.6), half-antibody fragments were used. The reduction of antibody dithiol bonds was carried out using 2-MEA prior to immobilisation29 in hydrogel films of LWs. 1 mg ml−1 antibody stock solution was prepared in 100 mM phosphate buffer, pH 7.4 containing 150 mM NaCl and 5 mM EDTA. 50 μL of 500 mM 2-MEA solution was added to 500 μL of antibody solution. The resulting solution was incubated at 37 °C for 90 min. The solution was then cooled to room temperature and 2-MEA was separated from the antibody and half-antibody fragments using 30 kDa MWCO filter unit where the filtrate contained 2-MEA, and the residue contained antibody and the fragments.2.5. SDS-PAGE
The formation of antibody half fragments was investigated by performing sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE) (Mini-Protean precast gels, BioRad, UK). The residue retained on the membrane of 30 kDa MWCO filter was analysed by SDS-PAGE. The SDS-PAGE was performed under non-reducing conditions. 10 ml of 10% (w:v) resolving gel was prepared using 4.1 ml of de-ionised water, 3.3 ml of 30% (w:v) acrylamide:bisacrylamide, 2.5 ml of 1.5 M Tris HCl (pH 8.8), 100 μl of 10% SDS, 100 μl of 10% (w:v) APS and 10 μl of TEMED. Similarly, 10 ml of 4% (w:v) stacking gel was prepared using 6.1 ml of de-ionised water, 1.3 ml of 30% (w:v) acrylamide:bisacrylamide, 2.5 ml of 1.5 M Tris HCl (pH 6.8), 100 μl of 10% SDS, 100 μl of 10% (w:v) of APS and 10 μl of TEMED. The residue was mixed with non-reducing loading buffer (60 mM Tris-HCl pH 6.8, 4% (w/v) SDS, 20% (v/v) glycerol, and 0.04% (w/v) bromophenol blue). A solution of pre-stained protein standard was used for molecular weight sizing. 20 μL of each solution was placed in different wells of the SDS-PAGE gel, and a constant voltage of 100 V was applied for ∼2.5 h. Protein bands were visualised by Coomassie blue staining.30,31
2.6. Immobilisation of recognition elements
The experimental conditions for immobilisation of recognition elements in LW films using three different strategies are described below. In strategies 1 and 2, biotin–protein A and IgG were used as exemplar recognition element and analyte, respectively. In strategy 3, half fragments of either anti-IgG or anti-HSA were used as recognition elements while either IgG or HSA were used as analytes.• Immobilisation strategy 1: 30 μl of 2.5 mg mL−1 NHS–PEG12–biotin solution was pipetted in the sensor flow channel of the flow cell placed on top of the LW. The NHS–PEG12–biotin was allowed to react with the –NH2 groups in LW films for 30 min. A buffer wash was then performed by flowing phosphate buffer pH 7.4 for 12 min. Streptavidin solution of 0.05 mg mL−1 was introduced into the sensor flow channel for 30 min followed by flowing of buffer for 12 min. Then, 0.07 mg mL−1 biotin–protein A was passed through the sensor flow channel for 30 min followed by washing with buffer for 12 min. Throughout these steps, buffer was flowed in the reference channel. Subsequently, 0.1 mg mL−1 of IgG solution was passed through for 30 min in both sensor and reference flow channels. Finally, buffer was passed through for 12 min in both sensor and reference flow channels.
• Immobilisation strategy 2: NHS–PEG12–biotin was attached to the LW film using the procedure described above. A solution containing streptavidin and biotin–protein A in the molar ratio of 1:0.75, with the concentration of streptavidin being 0.05 mg mL−1, was prepared. The solution was passed through the sensor flow channel for 30 min followed by washing with buffer for 12 min. Throughout these steps, buffer was flowed in the reference channel. Subsequently, 0.1 mg mL−1 of IgG solution was passed through for 30 min in both sensor and reference flow channels. Finally, buffer was passed through for 12 min in both sensor and reference flow channels.
• Immobilisation strategy 3: To immobilise half-antibody fragments, the amine groups in the hydrogel film of a LW were reacted with 8.3 mg mL−1 NHS–PEG2–maleimide for 30 min by pipetting 30 μl of the linker solution in the sensor flow channel of the flow cell. This was followed by flowing of phosphate buffer pH 7.4 for 12 min. Subsequently, 30 μl of half-antibody fragment solution was flowed for 30 min in the sensor channel and then phosphate buffer pH 7.4 for 12 min. Throughout these steps, buffer was flowed in the reference channel. Finally, analyte solution (either anti-IgG or HSA) was passed through both the sensor and reference channels for 30 min followed by washing with phosphate buffer pH 7.4 for 20 min.
3. Results and discussion
3.1. SDS-PAGE analysis
SDS-PAGE analysis was performed to confirm the formation of antibody fragments by treatment of antibodies with 2-MEA. 2-MEA is a mild reducing agent, reducing disulphide bonds preferentially at the hinge region of the antibody to form two identical half-antibody fragments with expected molecular weight of 72–90 kDa.29 After 1 h of reaction between antibody and 2-MEA, the residue retained on 30 kDa MWCO membrane was expected to contain a mixture of antibody and its half-antibody fragments, and this was confirmed by SDS-PAGE analysis. Fig. S1 in the ESI† clearly shows that lane 5, which was used to analyse untreated antibody solutions, contained a major band around 150 kDa. In contrast, lanes 3–4 that were used to analyse antibody solution after treatment with 2-MEA contained bands at 75 kDa, suggesting the formation of half-antibody fragments.3.2. Studies on LW biosensors
To increase the porosity of the hydrogel LW films, we used PEGDAm-3700 as a cross-linker instead of previously reported low molecular weight bis-acrylamide crosslinker. The porosity is also dependent on the concentration of total monomer and the percentage of crosslinker used. Besides making a porous LW film, the quantity of –NH2 groups in the hydrogel films is also important. The films should have a higher –NH2 concentration to bind more recognition elements and hence analytes. The –NH2 concentration is dependent on the concentration of APMAA. Therefore, LW films were fabricated using precursor solutions containing different concentrations of total monomer (4, 4.5 and 5% (w/v)) and different mole percentages of APMAA with respect to acrylamide (5, 10, and 15%). The concentration of the crosslinker PEGDAm-3700 was 0.79% (w/v).
Diffusion studies were performed to determine the porosity of the hydrogel films to species of different molecular weights. The shifts in resonance angles of LW with hydrogel films of different compositions were monitored for glycerol (92 D) and PEG (35, 100, and 300 kDa) solutions of similar RI. The resonance angle is a function of the RI of the waveguide and the solution above. If a species can diffuse in the hydrogel waveguide, the RI of the waveguide will change more in comparison with the case if the species cannot diffuse in the waveguide film. Since the solutions used here have similar RI, the shift in the resonance angle will depend on the porosity of the hydrogel as it will determine if a species can enter the film. Based on diffusion studies, we concluded that the porosity of hydrogel films increased as (1) APMAA concentration increased because of electrostatic interactions between the charged amines causing the film to swell and (2) the total monomer concentration decreased. However, the precursor solution containing 4% (w/v) of total monomers failed to gel reliably. The porosity to 300 kDa PEG was highest for 4.5% (w/v) hydrogel containing 0.89% (w/v) (or, 10 mol%) of APMAA and 0.79% (w/v) of PEGDAm-3700. Thus, subsequent work was undertaken using LWs with hydrogel films of this composition.
Typical two- and one-dimensional reflectivity curves of a LW with a hydrogel waveguide of optimised composition is shown in Fig. 2(a). A comparison of shifts in resonance angles of the optimised LW to glycerol and PEG solutions of similar RI (see Fig. 2(b)) shows that the waveguide film excluded more PEG as the molecular weight increased.
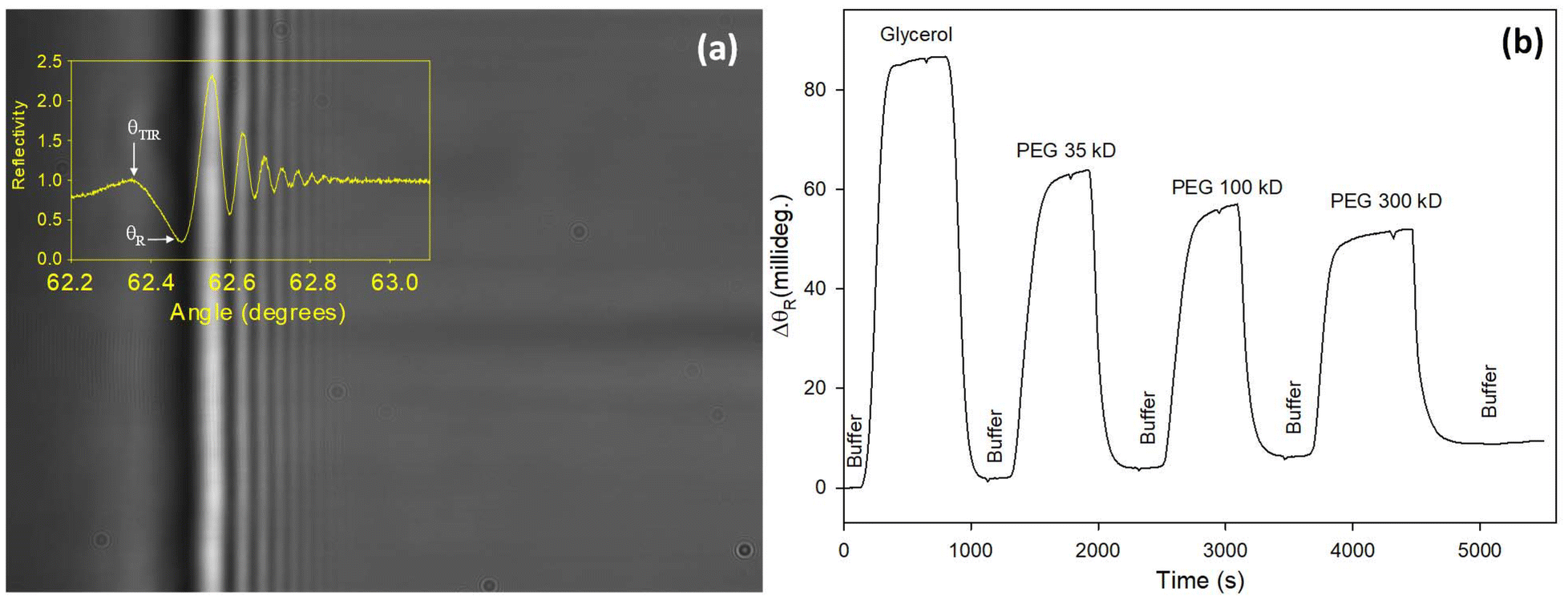 | ||
| Fig. 2 (a) Two- and one-dimensional reflectivity curves and (b) shift in resonance angle (ΔθR) for 0.5% (v:v) glycerol and 0.5% (w:v) PEG solutions of an optimised LW. | ||
:v) LW device after immersion in buffer but before immobilisation, along with the best fit to theory. The best fit parameters were for a waveguide 2.886 μm thick with a RI increment above the sample of 0.00342. The best fit thickness is more than twice the diameter of the spacer beads, which is most likely caused by swelling of the film as a result of electrostatic interactions between the protonated amines of APMAA.
 | ||
| Fig. 3 One-dimensional experimental and fitted reflectivity profiles of a LW with a cast polyacrylamide waveguide layer. | ||
We measured two-dimensional reflectivity curves of 42 LWs made using this fabrication method. Each image was divided into 153 rectangles 24 pixels (57.6 μm) high and 5496 pixels wide and the one-dimensional reflectivity profile extracted for each rectangle. These reflectivity profiles were then analysed to extract the slope and non-uniformities in the position of resonance angle across the imaged width of the LW. First, a linear regression was used to determine the slope of the resonance angle with distance across the LW device. After this, the standard deviation of the residuals from the best fite line was used to determine the non-uniformity of the LW. The resulting plot is provided in Fig. 4. Devices with non-uniformity of below 50 millideg. and slope of below ±20 millideg. mm−1 in their resonance angle position were considered to pass the required quality. Considering this criteria, ∼57% LW devices passed the quality check. The two-dimensional reflectivity profiles of a passed and failed LW device are shown in the inset in Fig. 4.
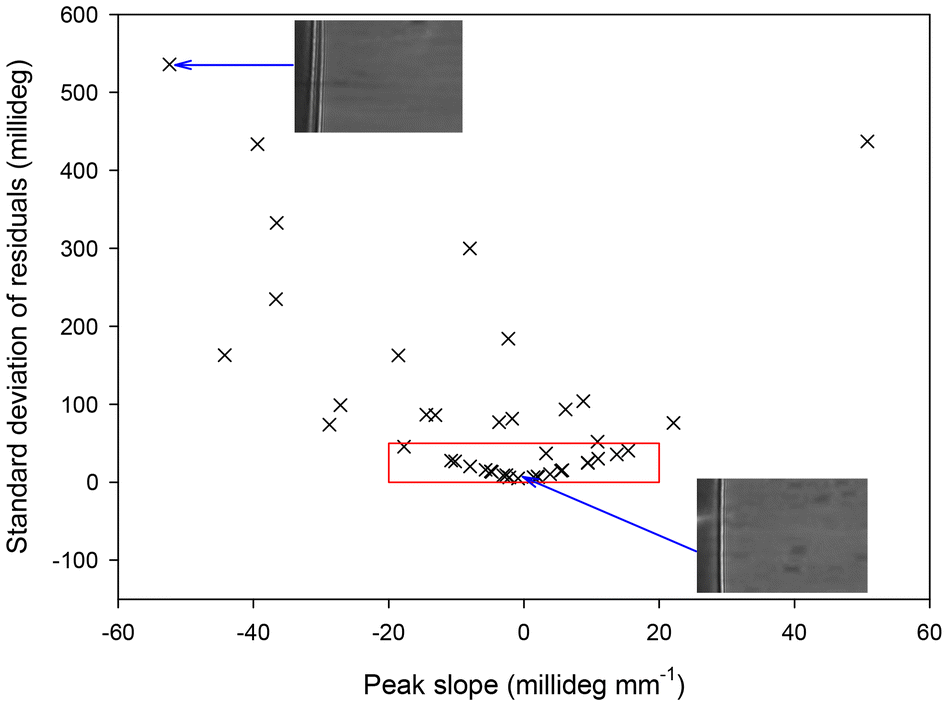 | ||
| Fig. 4 Plot of non-uniformity versus tilt in the position of resonance angle for hydrogels films of 42 LW devices fabricated using casting where scatter points in the red box are the devices that passed the quality check. | ||
The effectiveness of the immobilisation strategies was determined by the detection of analytes with the results discussed below.
• Immobilisation strategy 1: A schematic showing the attachment of biotin–protein A (recognition element) using strategy 1 is provided in Fig. 5(a). As different solutions were introduced in the sensor and reference channels of the flow cell mounted on top of the LW, shifts in resonance angle (ΔθR) of sensor and reference channels were recorded with time as shown in Fig. 6. The large shift in ΔθR in the sensor channel for NHS–PEG12–biotin was because of the presence of dimethyl sulfoxide (DMSO), which has a RI of 1.479 compared to 1.333 for water. The immobilisation of streptavidin and biotin–protein A in the sensor channel was confirmed by an increase in ΔθR from the baseline at t = 0 to 52.4 ± 0.4 millideg. and 63.1 ± 0.1 millideg., respectively. Finally, IgG was flowed in both sensor and reference channels, but only ΔθR in the sensor channel changed to any significant level (79.1 ± 0.1 millideg.), suggesting specific binding. Although the analyte (IgG) was successfully detected using strategy 1, ΔθR in the sensor channel for the recognition element was relatively small. This can be explained by considering that biotin binding sites of streptavidin might have been used up by the biotin of NHS–PEG12–biotin, leaving only a few sites for the binding of biotin–protein A. Additionally, Fig. 5 shows that ΔθR in the sensor channel decreases on a buffer wash, suggesting that IgG–protein A complex dissociates. This is not surprising considering that analyte and recognition elements (here, IgG and protein A) are involved in dynamic equilibrium where the analyte can bind to recognition elements and then dissociate.34 This dynamic equilibrium between analyte and recognition elements including the on-rate (binding) and off-rate (dissociation) has been studied by other label-free optical biosensors.35,36
 | ||
| Fig. 5 Schematic showing immobilisation of recognition element using (a) strategy 1 and strategy 2, and (b) strategy 3 in waveguide films of LWs. | ||
 | ||
| Fig. 6 Shifts in resonance angle (ΔθR) as different solutions used in immobilisation strategy 1 were flowed in the sensor and reference flow channels of the flow cell on LW. | ||
• Immobilisation strategy 2: Strategies 1 and 2 were similar, but to ensure that ΔθR in the sensor channel for the recognition element was large, streptavidin and biotin–protein A were mixed in a relative molar ratio of 1:0.75 and then introduced in the sensor channel of the waveguide film treated with NHS–PEG12–biotin. As different solutions were introduced in the sensor and reference channels of the flow cell mounted on top of the LW, ΔθR of the sensor and reference channels were recorded with time as shown in Fig. 7. In this case, ΔθR in the sensor channel from the baseline at t = 0 changed to 417.3 ± 0.8 millideg. And 594.7 ± 1.0 millideg. for streptavidin + biotin–protein A and IgG, respectively.
 | ||
| Fig. 7 Shifts in resonance angle (ΔθR) as different solutions used in immobilisation strategy 2 were flowed in the sensor and reference flow channels of the flow cell on LW. | ||
A comparison of shifts in resonance angles because of IgG binding for strategy 2 was higher than strategy 1 (see Table 1). Despite this improvement, strategy 2 was not ideal because reproducibility was poor. The poor reproducibility of strategy 2 might be because of uncontrolled binding of biotin–protein A to streptavidin during their mixing. Similar to Fig. 6, Fig. 7 shows that ΔθR in the sensor channel decreases on a buffer wash, suggesting that IgG–protein A complex dissociates because IgG and protein A are involved in dynamic equilibrium.
| Immobilisation strategy | Shift in resonance angle (ΔθR) from the preceding buffer baseline because of IgG binding (millideg.) (shown as gray arrow in Fig. 6–8) | ||
|---|---|---|---|
| Sensor | Reference | Sensor–reference | |
| Strategy 1 | 16.0 ± 0.1 | 1.3 ± 0.2 | 14.7 ± 0.2 |
| Strategy 2 | 178.3 ± 1.0 | −4.9 ± 0.3 | 183.2 ± 1.1 |
| Strategy 3 | 103.1 ± 0.4 | −30.3 ± 0.3 | 133.5 ± 0.5 |
• Immobilisation strategy 3: As shown in Fig. 5(b), half fragments of antibodies were immobilised in waveguide films using NHS–PEG2–malemide linker. ΔθR of the sensor and reference channels versus time as different solutions were introduced is shown in Fig. 8. In this case, ΔθR in the sensor channel changed from the baseline at t = 0 to 74.2 ± 0.1 millideg. and 177.3 ± 0.4 millideg. for half fragment of anti-IgG and IgG, respectively. A comparison of shifts in resonance angles because of IgG binding for strategy 3 was significantly higher than strategy 1 but lower than strategy 2 (see Table 1). Furthermore, 3 LW biosensors were made using immobilisation strategy 3 and shift in resonance angle because of IgG binding was recorded for each LW biosensor. These experiments showed that variance in IgG binding signal was ∼25% from sensor to sensor created using immobilisation strategy 3. As this variance was significantly lower than the other two strategies (∼25% versus >50%), immobilisation strategy 3 was used for the remainder of this work. In comparison to strategies 1 and 2, strategy 3 was also beneficial because fewer number of steps were involved.
 | ||
| Fig. 8 Shifts in resonance angle (ΔθR) as different solutions used in immobilisation strategy 3 were flowed in the sensor and reference flow channels of the flow cell on LW. | ||
As discussed in section 1, LWs with porous waveguide films offer higher sensitivity than those with non-porous films. Thus, the porosity of waveguide films before and after attachment of half fragments of antibodies immobilised using strategy 3 was determined. As discussed in sub-section 3.2.1, porosity of waveguide films was determined by diffusion studies where ΔθR was measured as glycerol and PEGs of different molecular weights were flowed over the LW. Fig. 9 shows that ΔθR for LWs comprising waveguide films without and with immobilised half antibody fragments was comparable. Thus, the waveguide films with immobilised half antibody fragments were expected to be largely porous to the analyte of interest, HSA, with a molecular weight of ∼66 kDa.
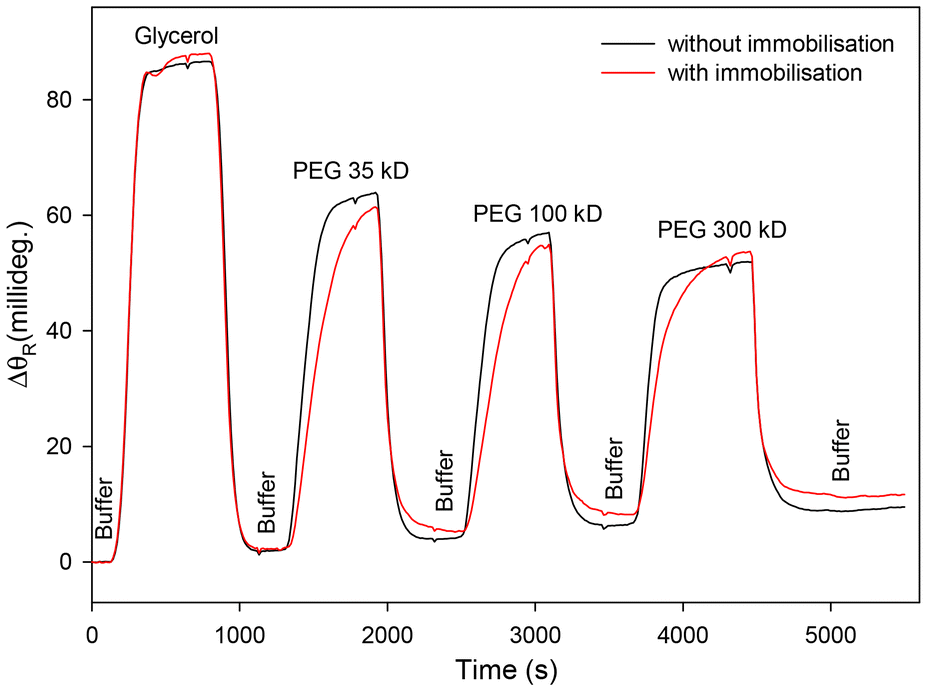 | ||
| Fig. 9 Shifts in resonance angle (ΔθR) of LWs comprising waveguide films without and with immobilised half antibody fragments in response to solutions of PEGs of different molecular weights. | ||
 | (1) |
 | ||
| Fig. 10 (a) Real-time measurement of shift in resonance angle (ΔθR) (sensor–reference) of the LW with increase in concentration of HSA from 66.5 ng ml−1 (or 1 nM) to 6650 ng ml−1 (or 100 nM) and (b) shift in resonance angle versus concentration of HSA where the error bars for each concentration of the protein were calculated using 3 replicates on a LW biosensor and error bars on blank were calculated using 11 LWs. | ||
The Langmuirian adsorption model is non-linear except where [A] ≪ KD, when eqn (1) reduces to:
 | (2) |
Since only one concentration used (66.5 ng mL−1) was significantly below the KD, the linear part of the Langmuir adsorption model was not applicable and could not be used to determine the limit of detection. Instead, the slope of the response at [A] = 0 was derived by differentiating eqn (1) and substituting [A] = 0.
 | (3) |
At [A] = 0:
 | (4) |
The slope expressed in degrees is given by:
 | (5) |
The standard deviation of the intercept (Sa) was estimated from the root mean square of the standard deviations of the responses of 10 LW biosensors for 0 ng mL−1 HSA, which was 2.31 millideg. The LOD was calculated as:
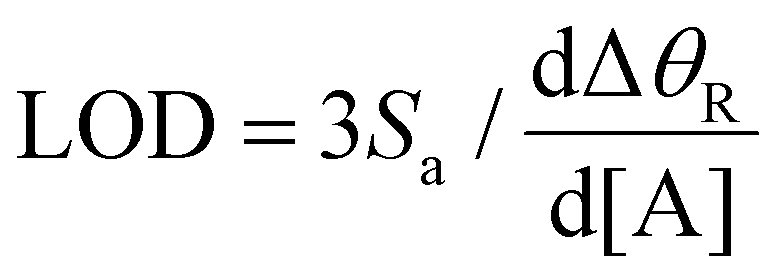 | (6) |
This gave a LOD of 28 ng mL−1. Fig. 10(a) shows that detection time is long at low concentrations of HSA. This can be explained by considering that detection time is determined by the time taken by the protein to diffuse from solution to the surface of and then in the volume of the waveguide film. The detection time can be reduced by active transport of proteins using for example, electric fields as previously shown by the authors.37
At the time of writing, as summarised in Table 2, there is one electrochemical sensor reported with lower LOD. Attar et al. reported an electrochemical sensor with a LOD of 8.6 ng ml−1 that could also distinguish glycated and non-glycated HSA.38 Most other reported sensors have much higher LODs, ranging from 650 ng ml−1 to 2.5 mg ml−1. As far as label-free optical sensors for HSA are concerned, Liu et al. reported a SPR based label-free optical biosensor for HSA detection with LOD of 4000 ng mL−1 in aqueous buffer.17 This work has improved that LOD to 28 ng ml−1 using a porous LW with a high density of immobilised recognition elements. The LOD of the LW biosensor is well below the physiological range, which is normally 35–50 mg mL−1 in serum and <20 μg mL−1 in urine of healthy persons.39,40
| Type of biosensor | Sensing strategy/platform | LOD of HSA (ng ml−1) | Sample matrix | Ref. |
|---|---|---|---|---|
| Electrochemical | Electrodes coated with a thin layer of a composite containing M13 virus particles | 6600 | Urine | 11 |
| Screen-printed carbon electrodes immobilized with AHA | 1550 | Buffer | 8 | |
| Electrode surface immobilized with albumin ligand, ∝-HSA | 8.6 | Buffer | 38 | |
| Optical | Chemiluminescence lateral flow immunoassay cartridge with integrated amorphous silicon photosensors | 2.5 × 106 | Urine | 9 |
| SPR sensor, anthraquinone dye Cibacron Blue F3G-A (CB) immobilised on gold film | 4000 | Buffer | 17 | |
| Absorbance of NCur–VO2+ optical nanoprobe forming complex with HSA | 731.5 | Serum | 14 | |
| Optical microchips containing recombinant protein binders to sense fluorescently labelled HSA | 650 | Urine | 15 | |
| Fluorescence measurement of AHA-(CdSe/ZnS) quantum dot complex | 32 | Buffer | 12 | |
| Optical | LW sensor, half fragment of anti-HSA immobilised in polyacrylamide mesoporous hydrogel film | 28 | Buffer | This work |
4. Conclusions
This work demonstrated label-free detection of human serum albumin (HSA) by leaky waveguide (LW) optical biosensor via covalent attachment of half fragments of HSA antibody into the 3D polymer network of the waveguide. The LW was fabricated by depositing a microns-thick film of polyacrylamide copolymerised with aminopropyl that provided amine functional groups for the immobilisation of recognition elements, half fragments of anti-HSA. The molar concentration of N-(3-aminopropyl)methacrylamide (APMAA) monomer relative to acrylamide and the total monomer concentration were varied, resulting in an optimal waveguide film with the highest porosity to poly(ethylene glycols) (PEGs) of molecular weights 35, 100, and 300 kDa. The waveguide film with the highest porosity contained 10 mol% of APMAA with respect to acrylamide and 4.5% (w/v) total monomer. The LW porosity was improved by using a longer cross-linking agent poly(ethylene glycol) diacrylamide with average molecular weight of 3700 D (PEGDAm-3700) as compared to our previous work where a low molecular weight crosslinker, bis-acrylamide, was used. The antibody fragments were successfully synthesized using the reducing agent 2-MEA, as confirmed by SDS-PAGE analysis. Among immobilisation strategies demonstrated for recognition elements, the detection by immobilisation of half-antibody fragments involved fewer steps and was shown to have better repeatability but slightly lower sensitivity compared to immobilisation involving streptavidin–biotin interactions. The equilibrium dissociation constant was found to be 266 ± 65 ng mL−1 or 4.03 ± 0.98 nM between HSA and half fragments of anti-HSA immobilised in waveguide films of LWs. Finally, HSA was successfully quantified using the LW biosensor with a limit of detection of 28 ng mL−1 and the lowest concentration of HSA measured in this work was 66.5 ng mL−1.Author contributions
AKP: Validation, formal analysis, investigation, writing – original draft, writing – review and editing; MNH: Investigation, writing – review and editing; SB: Supervision, writing – review and editing, NJG: Software, validation, formal analysis, writing – review and editing; RG: Conceptualization, methodology, validation, formal analysis, resources, writing – review and editing, visualization, supervision, project administration, funding acquisition.Data availability
The datasets supporting this article are available upon request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
RG acknowledges funding support from the Engineering and Physical Sciences Research Council (Grants EP/N02074X/1 and EP/N02074X/2).References
- S. M. Figueroa, P. Araos, J. Reyes, B. Gravez, J. Barrera-Chimal and C. A. Amador, Antioxidants, 2021, 10, 404 CrossRef CAS PubMed.
- P. A. Sarafidis and G. L. Bakris, Nephrol., Dial., Transplant., 2006, 21, 2366–2374 CrossRef PubMed.
- S. Koulouris, I. Lekatsas, I. Karabinos, G. Ioannidis, T. Katostaras, A. Kranidis, K. Triantafillou, N. Thalassinos and L. Anthopoulos, Am. Heart J., 2005, 149, 840–845 CrossRef CAS PubMed.
- J. Anguizola, R. Matsuda, O. S. Barnaby, K. S. Hoy, C. Wa, E. DeBolt, M. Koke and D. S. Hage, Clin. Chim. Acta, 2013, 425, 64–76 CrossRef CAS PubMed.
- D. Kumar, P. Dutta, R. Ramachandran, R. Bhattacharyya and D. Banerjee, Clin. Chim. Acta, 2025, 565, 119947 CrossRef CAS PubMed.
- V. Garcia Moreira, N. Beridze Vaktangova, M. D. Martinez Gago, B. Laborda Gonzalez, S. Garcia Alonso and E. Fernandez Rodriguez, Lab. Med., 2018, 49, 355–361 Search PubMed.
- D. Kumar and D. Banerjee, Clin. Chim. Acta, 2017, 469, 150–160 CrossRef CAS PubMed.
- V. Stanković, S. Đurđić, M. Ognjanović, B. Antić, K. Kalcher, J. Mutić and D. M. Stanković, J. Electroanal. Chem., 2020, 860, 113928 CrossRef.
- M. Zangheri, F. Di Nardo, M. Mirasoli, L. Anfossi, A. Nascetti, D. Caputo, G. De Cesare, M. Guardigli, C. Baggiani and A. Roda, Anal. Bioanal. Chem., 2016, 408, 8869–8879 CrossRef CAS PubMed.
- W. Lee, J.-H. Lee, B.-K. Oh and J.-W. Choi, Ultramicroscopy, 2010, 110, 723–728 CrossRef CAS PubMed.
- A. F. Ogata, J. M. Edgar, S. Majumdar, J. S. Briggs, S. V. Patterson, M. X. Tan, S. T. Kudlacek, C. A. Schneider, G. A. Weiss and R. M. Penner, Anal. Chem., 2017, 89, 1373–1381 CrossRef CAS PubMed.
- M.-C. Tu, Y.-T. Chang, Y.-T. Kang, H.-Y. Chang, P. Chang and T.-R. Yew, Biosens. Bioelectron., 2012, 34, 286–290 CrossRef CAS PubMed.
- B. Charron, A. Delorme, C. Dubois, M. H. Jodaylami and J.-F. Masson, Analyst, 2023, 148, 5525–5533 RSC.
- Z. Rasouli and R. Ghavami, Microchim. Acta, 2020, 187, 208 CrossRef CAS PubMed.
- A. Semeradtova, M. Stofik, L. Vankova, P. Maly, O. Stanek and J. Maly, Sens. Actuators, B, 2018, 272, 441–447 CrossRef CAS.
- X. Chen, M. Li, Z. Wang, K. Zhao, J. Gu, Q. Li and J.-J. He, Sensors, 2024, 24, 677 CrossRef PubMed.
- J.-T. Liu, P.-S. Lin, Y.-M. Hsin, J.-Z. Tsai and W.-Y. Chen, J. Taiwan Inst. Chem. Eng., 2011, 42, 696–700 CrossRef CAS.
- C. Chen and J. Wang, Analyst, 2020, 145, 1605–1628 RSC.
- N. J. Goddard and R. Gupta, Sens. Actuators, B, 2020, 309, 127776 CrossRef CAS.
- N. A. Alamrani, G. M. Greenway, N. Pamme, N. J. Goddard and R. Gupta, Analyst, 2019, 144, 6048–6054 RSC.
- R. Gupta, N. A. Alamrani, G. M. Greenway, N. Pamme and N. J. Goddard, Anal. Chem., 2019, 91, 7366–7372 CrossRef CAS PubMed.
- M. Zourob, S. Mohr, B. J. Treves Brown, P. R. Fielden, M. B. McDonnell and N. J. Goddard, Anal. Chem., 2005, 77, 232–242 CrossRef CAS PubMed.
- M. Zourob, A. Simonian, J. Wild, S. Mohr, X. Fan, I. Abdulhalim and N. J. Goddard, Analyst, 2007, 132, 114–120 RSC.
- A. K. Pal, N. J. Goddard, H. J. Dixon and R. Gupta, Biosensors, 2020, 10, 134 CrossRef CAS PubMed.
- R. Gupta and N. J. Goddard, Analyst, 2021, 146, 4964–4971 RSC.
- X. Wang, M. Jefferson, P. C. D. Hobbs, W. P. Risk, B. E. Feller, R. D. Miller and A. Knoesen, Opt. Express, 2011, 19, 107–117 CrossRef PubMed.
- R. Gupta and N. J. Goddard, Sens. Actuators, B, 2020, 322, 128628 CrossRef CAS.
- J. Xia, Z.-Y. Liu, Z.-Y. Han, Y. Yuan, Y. Shao, X.-Q. Feng and D. A. Weitz, Acta Biomater., 2022, 141, 178–189 CrossRef CAS PubMed.
- A. Makaraviciute, C. D. Jackson, P. A. Millner and A. Ramanaviciene, J. Immunol. Methods, 2016, 429, 50–56 CrossRef CAS PubMed.
- A. Aitken, Genet. Res., 2003, 81, 243–244 CrossRef.
- M. R. Green and J. Sambrook, Molecular Cloning: A Laboratory Manual, Cold Spring Harbor Laboratory Press, New York, 4th edn, 2012 Search PubMed.
- G. G. Nenninger, J. B. Clendenning, C. E. Furlong and S. S. Yee, Sens. Actuators, B, 1998, 51, 38–45 CrossRef CAS.
- C. W. Wu, C. Y. Chiang, C. H. S. Chen, C. S. Chiang, C. T. Wang and L. K. Chau, Talanta, 2016, 146, 291–298 CrossRef CAS PubMed.
- S. de Picciotto, B. Imperiali, L. G. Griffith and K. D. Wittrup, Anal. Biochem., 2014, 460, 9–15 CrossRef CAS PubMed.
- D. Capelli, V. Scognamiglio and R. Montanari, TrAC, Trends Anal. Chem., 2023, 163, 117079 CrossRef CAS.
- T. Gjetting, M. Gad, C. Fröhlich, T. Lindsted, M. C. Melander, V. K. Bhatia, M. M. Grandal, N. Dietrich, F. Uhlenbrock, G. R. Galler, M. Strandh, J. Lantto, T. Bouquin, I. D. Horak, M. Kragh, M. W. Pedersen and K. Koefoed, mAbs, 2019, 11, 666–680 CrossRef CAS PubMed.
- N. J. Goddard and R. Gupta, Sens. Actuators, B, 2019, 301, 127063 CrossRef CAS.
- A. M. Attar, M. B. Richardson, G. Speciale, S. Majumdar, R. P. Dyer, E. C. Sanders, R. M. Penner and G. A. Weiss, ACS Appl. Mater. Interfaces, 2019, 11, 4757–4765 CrossRef CAS PubMed.
- R. Choudhury, S. Rajeshbhai Patel and A. Ghosh, J. Photochem. Photobiol., A, 2019, 376, 100–107 CrossRef CAS PubMed.
- Z. Stojanovic, J. Erdőssy, K. Keltai, F. W. Scheller and R. E. Gyurcsányi, Anal. Chim. Acta, 2017, 977, 1–9 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5an00108k |
| This journal is © The Royal Society of Chemistry 2025 |