
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDevelopment of a high-resolution paper-spray mass spectrometry method using street drugs for the early detection of emerging drugs in the unregulated supply†
Allie
Miskulin
a,
Bruce
Wallace
bc,
Dennis K.
Hore
 *acd and
Chris
Gill
*acef
*acd and
Chris
Gill
*acef
aDepartment of Chemistry, University of Victoria, Victoria, British Columbia V8W 3V6, Canada. E-mail: dkhore@uvic.ca
bSchool of Social Work, University of Victoria, Victoria, British Columbia V8W 2Y2, Canada
cCanadian Institute for Substance Use Research, University of Victoria, Victoria, British Columbia V8W 2Y2, Canada
dDepartment of Computer Science, University of Victoria, Victoria, British Columbia V8W 3P6, Canada
eDepartment of Chemistry, Vancouver Island University, Nanaimo, British Columbia V9R 5S5, Canada. E-mail: Chris.Gill@viu.ca
fDepartment of Environmental and Occupational Health Sciences, University of Washington, Seattle, WA 98195, USA
First published on 7th March 2025
Abstract
Adulteration of the unregulated opioid supply has contributed to increasing numbers of overdose deaths in North America. Harm-reduction drug checking has emerged as a strategy to address increasing adulteration rates by providing information about sample composition to people who use drugs. While paper-spray mass spectrometry is capable of trace detection for drug checking, the presence of newly emerging substances often goes undetected if not included in the targeted analysis method. High-resolution mass spectrometry has not been widely used in drug-checking efforts to date, but it has advanced capabilities to facilitate the detection of newly emerging substances. We present a high-resolution paper-spray mass spectrometry method developed for the detection of newly emerging compounds in the street-drug supply. The method was used to analyze a selection of opioid samples received at a drug-checking service in Victoria, British Columbia, Canada. Using this approach, newly emerging adulterants, precursors and byproducts were identified in the local street-drug supply.
1 Introduction
Over the last decade there has been a trend of increasing overdose deaths in North America, where the volatility of the unregulated drug supply has been reported as a major driving factor.1–4 The United Nations Office on Drugs and Crime has identified over 1200 unique novel psychoactive substances internationally over the past several decades, creating great concern regarding the evolving composition of the street supply for people who use drugs.5 The opioid supply in North America consists primarily of fentanyl, but there are large inconsistencies in the amount of fentanyl present, with a wide and variable range of adulterants added to opioid drug mixtures.6–8 Specifically, the opioid supply is often adulterated with other sedatives, including fentanyl analogs such as carfentanil,9 benzodiazepines,10 veterinary sedatives such as xylazine,11,12 and nitazenes.13 It is suspected that adulteration occurs for several reasons, including to enhance or produce synergistic effects of the drug, or to reduce the amount of active drug necessary to achieve the desired effect.11,14 Drug adulteration changes over time in ways that are not necessarily predictable and is considered to be a result of the various economic and law-enforcement pressures experienced by illicit drug manufacturers.14 Adulteration results in the consumption of unknown drugs, which can cause unpredictable effects on the individual, and therefore the detection of such compounds is of great importance for people who use drugs.15Harm reduction drug checking has emerged to identify drug compositions and to provide information about the drug supply to people who use drugs.16,17 Currently, one of the limitations of drug checking is the detection of newly emerging substances, especially when they are present in low concentrations.18,19 Most routine drug testing methods are able to identify only a limited number of compounds, resulting in newly emerging substances going undetected.20 Some of the most commonly used drug-checking strategies include immunoassay test strips and Fourier transform infrared (FTIR) spectroscopy. Immunoassay test strips exist for the detection of fentanyl, benzodiazepines, and xylazine, among other drug classes, but they are unable to differentiate between the various analogs present in each class.21–23 While FTIR has large libraries that enable the identification of a wide range of compounds, detection efforts become challenging with complex mixtures or when compounds are present at low concentrations.24,25
Various mass spectrometry methods have been investigated as drug-checking tools because of their ability for trace detection and quantitation. While gas chromatography-mass spectrometry (GC-MS) and liquid chromatography-tandem mass spectrometry (LC-MS/MS) are considered to be gold-standard techniques in forensic contexts, their use for drug checking is hindered by expensive equipment, the need for highly trained personnel, and long analysis times.26–28 Various other mass spectrometry systems have been implemented for point-of-care drug analysis, notably paper-spray mass spectrometry (PS-MS),29,30 portable GC-MS,31 handheld high-pressure mass spectrometry (HPMS),32 and miniature paper capillary spray ionization (PCSI) mass spectrometry.33 While mass spectrometry methods have the advantage in sensitivity and quantitation over other drug-checking instruments, targeted methods used in routine analyses are limited to specific analytes and typically use low-resolution mass analyzers.34,35 Such analyzers are not designed for the detection of newly emerging substances, as unit-mass resolution does not allow for a confident identification of new substances in the absence of chromatographic separation.35 On the other hand, targeted methods require the availability of analytical reference standards, and therefore have a limited ability to keep up with the constantly evolving street-drug supply.36
Untargeted methods for the detection of newly emerging substances are commonly developed on LC-MS/MS instruments,37 or other hyphenated techniques that give definitive results, but face limitations of long analysis times, highly optimized conditions, and extensive sample preparation.38 Ambient ionization techniques offer a way to address such limitations, with rapid measurement times, little to no sample preparation and minimal operator training requirements. Such techniques have been demonstrated for drug analysis, including direct analysis in real time mass spectrometry (DART-MS)39,40 and PS-MS.29,30,41–43 Additionally, PS-MS has the ability to quantify trace amounts of analytes in complex matrices, which is an advantage over other ambient ionization techniques that are largely considered to be semi-quantitative.44,45 PS-MS has been used in drug checking for quantitative analyses, but non-targeted screening in drug-checking methods with high-resolution PS-MS instruments has not been thoroughly explored.29
High-resolution mass spectrometry (HRMS) drug screening has emerged in response to the increasing proliferation of novel substances, and provides superior resolution and accompanying capabilities of an advanced scanning method, such as data-dependent (DDA) and data-independent (DIA) acquisition modes.35 DIA methods fragment all precursor ions within a given mass window, which provides an abundance of data.46 However, these data can be convoluted and it is therefore challenging to associate fragment ions with their corresponding precursors.47,48 In contrast, DDA methods select the highest intensity ions for fragmentation. To avoid the assumption that the highest intensity ions are the most relevant, algorithms allowing for the use of exclusion lists and dynamic exclusion are beneficial for samples with large numbers of species.49 The information obtained using HRMS allows compounds to be identified without a reference standard by obtaining the accurate mass and isotope ratio information, allowing the narrowing of possible candidates to a certain molecular formula.37In silico fragmentation algorithms can then be used to provide theoretical fragmentation patterns for molecules of interest, and the possible molecular structures associated with these fragments.37 PS-HRMS methods have been demonstrated for drug analysis in biological matrices such as urine50 and blood,51 but pre-consumption testing of street drugs is limited.
A common approach for development of a non-targeted method is to optimize and evaluate the method using analytical standards, or standards spiked into a laboratory-developed matrix. Analytical standards have high levels of purity, and are most often obtained in solution form. Street drugs largely consist of chemicals synthesized in clandestine laboratories, where incomplete purification is common.43,52 Additionally, street drugs have complex matrices, often with a variety of cutting agents and adulterants.53 Essentially, there are any number of unknown factors that contribute to the complexities of street-drug samples that cannot be replicated in a laboratory setting. The use of street drugs in analytical method development has been proven to be successful, as it accounts for the complexities in the samples that the developed method is being aimed at.54 Therefore, method development using street-drug samples will be more directly applicable for the detection of newly emerging adulterants in street drugs.
We present a PSMS-HRMS DDA method for the detection of newly emerging components of street drugs without the use of reference standards. To ensure that the method will be directly applicable to street-drug matrices, drug samples collected from the Substance Drug Checking Project55 were used in the development of this method. Several newly emerging substances were detected throughout the work, highlighting the need for regular untargeted screening of the street-drug supply.
2 Methods
2.1 Materials
Acetonitrile and Optima high performance liquid chromatography (HPLC)-grade formic acid and methanol were acquired from Fisher Scientific (Ottawa, ON, Canada). Deionized water was prepared utilizing a water purification system (18.4 MΩ cm Facility Scale Reverse Osmosis/Ion Exchange Water Purification System from Applied Membranes Inc., Vista, California, USA). VeriSpray™ sample plates for PS-MS measurements were obtained from Thermo Fisher Scientific (San Jose, CA, USA).2.2 Sample preparation and analysis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 was used over a scan range of 50–600 m/z. The four highest intensity precursor ions were selected in each scan, and these precursors were then dynamically excluded for 20 s to allow for the fragmentation of lower intensity ions. A ±5 ppm window was implemented for the dynamic exclusion of these compounds, and isotope peaks were also excluded. An exclusion list (provided in the ESI†) of 1942 contaminant peaks was included in the method, with a 10 ppm tolerance window. For the data-dependent MS/MS analysis, a precursor isolation window of 2 m/z was used, with a stepped high-energy collisional dissociation (HCD) of 30, 50, and 70%, normalized to the precursor mass. The resolution was set to 30000, and data were collected in centroid mode. Further details of the instrument parameters can be found in Table S2.† The spray solvent used was a mixture of acetonitrile, deionized water, and formic acid (90/9.9/0.1 v/v% acetonitrile/water/formic acid). Isopropanol was also investigated as a spray solvent in an attempt to improve detection of poorly ionizing compounds. Further details of the method and results for the spray solvent optimization can be found in the ESI.†
000 was used over a scan range of 50–600 m/z. The four highest intensity precursor ions were selected in each scan, and these precursors were then dynamically excluded for 20 s to allow for the fragmentation of lower intensity ions. A ±5 ppm window was implemented for the dynamic exclusion of these compounds, and isotope peaks were also excluded. An exclusion list (provided in the ESI†) of 1942 contaminant peaks was included in the method, with a 10 ppm tolerance window. For the data-dependent MS/MS analysis, a precursor isolation window of 2 m/z was used, with a stepped high-energy collisional dissociation (HCD) of 30, 50, and 70%, normalized to the precursor mass. The resolution was set to 30000, and data were collected in centroid mode. Further details of the instrument parameters can be found in Table S2.† The spray solvent used was a mixture of acetonitrile, deionized water, and formic acid (90/9.9/0.1 v/v% acetonitrile/water/formic acid). Isopropanol was also investigated as a spray solvent in an attempt to improve detection of poorly ionizing compounds. Further details of the method and results for the spray solvent optimization can be found in the ESI.†
2.3 Exclusion list development
An exclusion list consisting of m/z values to be omitted by the method was developed to improve the detection of drug-sample components. Methanolic blank samples that were deposited on a paper strip were analyzed in full-scan mode in triplicate, and the masses of peaks with an intensity above 100000 were added to the exclusion list (n = 2148). Illicit drug samples of five different types, namely 3,4-methylenedioxymethamphetamine (MDMA), ketamine, cocaine, a benzodiazepine (bromazolam), and a fentanyl mixture consisting of fentanyl, caffeine, and erythritol, were analyzed to identify peaks that may present as a result of sample storage or preparation. The components in these drug samples had been previously verified by analysis on a QqQ instrument. These drug samples were analyzed using the developed DDA method, which included the exclusion list of peaks from the blank sample. The aim of this strategy was to identify additional contaminants present in drug samples, where precursors selected for DDA in at least 3 of the 5 analyzed drug samples were excluded. This strategy was repeated three times, resulting in an additional 196 m/z values being added to the exclusion list. Following the removal of duplicate values, and those that shared precursor masses close to values in a developed drug precursor database, the final exclusion list contained 1942 m/z values.
2.4 DDA peak search algorithm
A database of 132 common and emerging drugs was created that included precursor masses and 1–3 fragments for a given precursor to confirm identity. Three fragments were used in most cases, and one or two fragments were used for molecules that did not have 3 major fragments in their MS/MS spectrum. Masses in this database are listed in Table S3.† It should be noted that we have assumed the presence of specific isomers for some compounds, including ortho-methyl fentanyl and para-fluoro-phenethyl-ANPP, as they have been previously detected and reported. However, this method is incapable of distinguishing between ring substitution isomers. Fragments were selected based on their relative intensities in reference spectra from mzCloud (HighChem, Slovakia), which also correspond to those collected with an Orbitrap instrument using HCD 50%. For cases where the required data did not exist in mzCloud, reference spectra were obtained from the Center for Forensic Science Research and Education (CFSRE) monographs.56 These spectra were collected using an LC-quadrupole time-of-flight-MS instrument at a range of 100–510 Da with a collision energy spread of 35 ± 15 eV.56Instrument RAW files were parsed to the mzML format using the ThermoRawFile parser.57 A Python script using the pymzML package was then created to parse the mzML files to obtain the various spectra and scan types. The extracted precursors and their corresponding MS/MS spectra could then be compared to values within the developed database. A tolerance window of 0.001 m/z for the selected precursors was used to tentatively identify compounds. The presence of all corresponding fragment ions within a tolerance window of 0.001 m/z in the MS/MS spectrum was used to tentatively confirm the identity of the compound. For compounds that did not have reference spectra available, in silico fragmentation was performed using the Mass Frontier™ software (Thermo Fisher Scientific). Briefly, the molecular structure for the compound of interest was imported into the software, and the three most abundant fragments generated as a result of in silico fragmentation were used to confirm the presence of the compound.
2.5 Method evaluation and identifying emerging compounds
Opioid samples (n = 65) were analyzed using the developed method to evaluate the relative detection threshold, and to investigate the detection of components of the street-drug supply outside of those detected with routine targeted analysis methods. The majority of the samples (n = 40) were collected from the Victoria, British Columbia area, while the remainder (n = 25) were collected from various locations around the province of British Columbia. Previously obtained quantitative results by PS-MS with a QqQ instrument were used to provide a relative detection threshold for the compounds analyzed.3 Results and discussion
3.1 Evaluation of the developed method
000. However, upon preliminary analysis of a subset of street-drug samples (n = 18) using this method, no compounds were detected at levels below 1% w/w. These results suggested that additional m/z values should be added to the exclusion list to avoid DDA being performed on background or matrix components, allowing for the detection of lower intensity drug m/z signals. To select additional m/z values to add to the exclusion list, street-drug samples from various drug classes were analyzed to determine if any additional peaks were present from sample preparation, storage, or other sources present in street-drug samples. Street-drug samples of MDMA, ketamine, cocaine, bromazolam, and a fentanyl mixture were analyzed, and peaks selected for DDA fragmentation were compared across all five classes. Five different drug classes were analyzed to avoid the elimination of specific precursors or byproducts belonging to each drug class. The m/z values selected for fragmentation in the majority (3 of 5) of samples were added to the exclusion list. This process was repeated three times, using an updated exclusion list of the selected peaks each time.
Compounds that had quantitative information available from previous analyses were used to assess the relative method detection threshold. Using the updated exclusion list, sample components could now be detected at below 1% in the majority of cases for the analyzed samples (n = 47). Table 1 compares the detection rates for compounds above and below the detection threshold that was determined to be 1% w/w. Only compounds with previous quantitative information from routine analysis are included in Table 1. Out of the 128 sample components present across 47 samples, only four were not detected at a concentration over 1% w/w, and were therefore considered to be outliers. Two of these four components were bromazolam, where the undetected instances were at a concentration of approximately 3% w/w. The other instances included fentanyl at 2% and methamphetamine at 6%. Therefore, 1% was considered to be an appropriate approximate detection threshold for this work, with many compounds being detected below this limit consistently. Several of the studied components, including 4-anilino-N-phenethylpiperidine (ANPP) (n = 4), fluorofentanyl (n = 18), furanyl UF-17 (n = 1), medetomidine (n = 8), ortho-methylfentanyl (n = 19), and phenacetin (n = 1) were detected at all studied concentrations. Notably, no benzodiazepines were detected at below 1% w/w. However, the median concentration of bromazolam observed for samples analyzed by the Substance Drug Checking Project in 2023 was 3.6%,58 suggesting that the 1% detection threshold is sufficient for a drug-checking context.
| Compound | Detection rate | ||
|---|---|---|---|
| >1% | <1% | Overall | |
| Acetylcodeine | N/A | 0 (1) | 0 (1) |
| Acetylmorphine | 1.0 (1) | 0 (1) | 0.5 (2) |
| ANPP | 1.0 (3) | 1.0 (1) | 1.0 (4) |
| Bromazolam | 0.83 (12) | 0 (2) | 0.71 (14) |
| Desalkygidazepam | 1.0 (1) | 0 (1) | 0.50 (2) |
| Fentanyl | 0.97 (32) | 0.40 (5) | 0.89 (37) |
| Flubromazepam | 1.0 (2) | 0 (1) | 0.67 (3) |
| Fluorofentanyl | 1.0 (10) | 1.0 (8) | 1.0 (18) |
| Furanyl UF-17 | 1.0 (1) | N/A | 1.0 (1) |
| Medetomidine | 1.0 (4) | 1.0 (4) | 1.0 (8) |
| Methamphetamine | 0.67 (3) | N/A | 0.67 (3) |
| ortho-Methylfentanyl | 1.0 (17) | 1.0 (2) | 1.0 (19) |
| Phenacetin | 1.0 (1) | N/A | 1.0 (1) |
| Xylazine | 1.0 (8) | 1.0 (7) | 1.0 (15) |
The limited ability to detect benzodiazepines at low concentrations is suspected to be due to competitive ionization. Competitive ionization results in sample components with high proton affinities consuming most of the available charge in the ionization step, hindering the ionization of components with lower proton affinities.53 The proton affinity of a compound refers to its ability to accept protons, which speaks to its ability to form protonated cations for positive ion mode mass spectrometry. Fig. 1 highlights the complex nature of the analyzed samples, where the majority of samples contain four or more active components, suggesting that there are multiple compounds competing for the available charge. Competitive ionization has previously been documented in drug mixtures using thermal desorption-DART-MS.59 The proton affinities for fentanyl and several analogs has been previously reported to range from 1018 to 1078 kJ mol−1.60 While this study did not include fluorofentanyl or ortho-methylfentanyl, we expect new analogs to be close to or within the reported range due to their structural similarities. The proton affinities for several benzodiazepine drugs have been reported to range from 554 to 856 kJ mol−1.61 However, that study was performed prior to the emergence of many of the designer benzodiazepines that are common in the street-drug supply. While there is a larger range of proton affinity values reported for benzodiazepines due to their diverse structures, the values are lower than fentanyl analogs, and thus we expect a similar trend for the newly emerging benzodiazepines. The lower proton affinities for benzodiazepines suggest that they are subject to more competitive ionization effects, and could explain why we see lower signal intensities and a lower rate of detection for these compounds.
 | ||
| Fig. 1 A breakdown of the number of sample components for 47 samples, and the proportional makeup of drugs from each category. The number of components represents detection by either the routine quantitative analysis method at Substance Drug Checking, the developed PS-HRMS DDA method, or both. The “other” category includes methamphetamine, primidone, BTMPS, and phenacetin. | ||
In an attempt to improve the signal intensity observed for benzodiazepines, isopropanol was also investigated as a spray solvent for PS-MS. While previous studies involving PS-MS for drug detection have used a mixture of acetonitrile, water, and formic acid for various drugs and metabolites in biological samples, spray-solvent optimization studies have not been demonstrated for street-drug samples prepared in methanol.44,62,63 It has been reported that binary solvent mixtures could create challenges for PS-MS, where solvents with different evaporation rates could cause changes in solvent composition over the course of the ionization.64 There is support in the literature for the improvement of signal intensity using isopropanol alone, which prompted the comparison of this solvent to that of the acetonitrile mixture.64–66 However, our results showed that the acetonitrile solution resulted in superior signal intensity and repeatability for all six components in the analyzed street-drug mixture (Fig. S1†). This is likely because of improved protonation in positive ion PS-MS for acetonitrile, and the addition of formic acid for signal stability.63,67 Therefore, the acetonitrile solution continues to be used for all analyses in this work.
| Compound | Theoretical m/z | Experimental m/z | Error (ppm) | Ion 1 | Ion 2 | Ion 3 |
|---|---|---|---|---|---|---|
| Caffeine | 195.0877 | 195.0878 | 0.5126 | Y | Y | N/A |
| Fentanyl | 337.2275 | 337.2275 | 0.0000 | Y | Y | Y |
| Medetomidine | 201.1386 | 201.1388 | 0.9943 | Y | Y | N/A |
| ortho-Methyl-ANPP | 295.2169 | 295.2168 | −0.3387 | Y | Y | Y |
| ortho-Methylfentanyl | 351.2431 | 351.2432 | 0.2847 | Y | Y | Y |
| Xylazine | 221.1107 | 221.1107 | 0.0000 | Y | Y | Y |
While spectral libraries are widely used for the identification of compounds from MS/MS spectra, they are typically developed for MS/MS spectra for chromatographically separated compounds. This can create challenges when attempting to match direct mass spectrometry spectral data against reference library spectra. Additional peaks present in the spectra resulting from the lack of chromatographic separation cause library match scores to be low, and the likelihood of identification in this manner is unreliable. Another challenge with relying on spectral libraries is that the identification of compounds is only possible if an entry exists in the database.53 Spectra for newly emerging substances are often available online or can be predicted by in silico fragmentation prediction software before spectral libraries are updated. Therefore, the developed spectral matching algorithm can aid in compound identification efforts for PS-MS spectra through using fragment ion matching instead of using the entire MS/MS spectra.
Another advantage of this method is that the mass database is simple to update, and data can be reprocessed to identify compounds retrospectively. Therefore, in our approach, the aim was to optimize the instrumental method to be able to select relevant peaks, and eliminate the need for an inclusion list, allowing retrospective analysis of the data for newly emerging substances in the illicit supply when required.
3.2 Detection of substances outside of targeted methods
| Compound | Number of detections |
|---|---|
| BTMPS | 7 |
| F-ANPP | 10 |
| Fluoro-phenethyl-ANPP | 5 |
| ortho-Methyl-ANPP | 7 |
| Phenethyl-ANPP | 1 |
| Primidone | 4 |
| Protonitazepyne/isotonitazepyne | 1 |
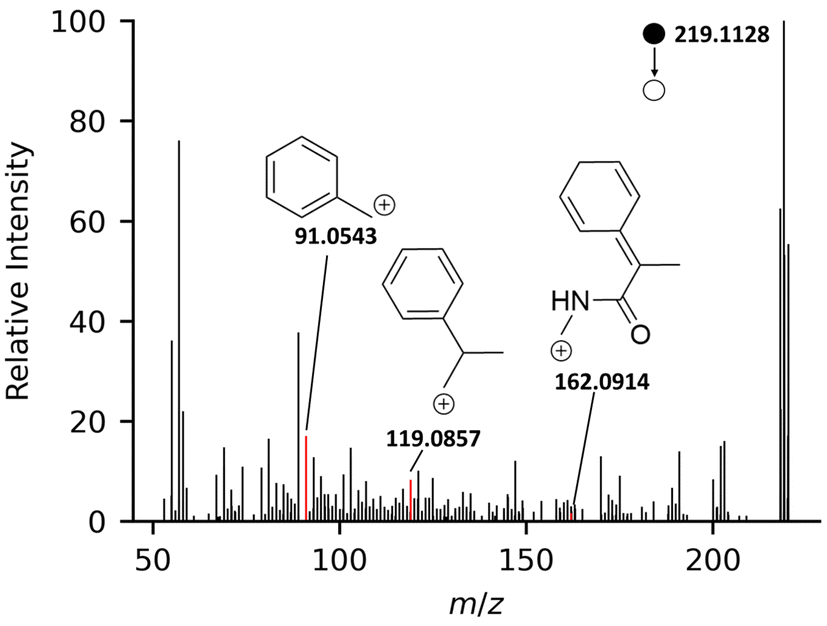 | ||
| Fig. 2 High-resolution PS-MS/MS [M + H]+ spectrum of primidone, with the three selected confirming ions highlighted. | ||
BTMPS was also detected in several samples and has been identified in the unregulated drug supply in various locations across the United States.73 BTMPS is pharmacologically active, and is widely used as a light stabilizer for plastics and in pharmaceutical products.74,75 While this compound has been recently identified in the street-drug supply, it is difficult to identify the origin or purpose of the compound with regard to street drugs. Due to its use as a pharmaceutical stabilizer, there is a possibility that it was introduced into the street-drug supply to play a similar role.74 BTMPS has also been studied in relation to its potential in decreasing withdrawal symptoms in rats, but such effects of the drug have not been studied on humans, and other related health harms are largely unknown.76 The presence of BTMPS was confirmed with the PS-HRMS [M + 2H]2+ MS/MS spectrum, as shown in Fig. 3, and is in agreement with previously published data on BTMPS.73 The noise in the spectrum can be attributed to other ions present within the precursor isolation window, which is shown in Fig. S4.†
 | ||
| Fig. 3 High-resolution MS/MS [M + 2H]2+ spectrum of BTMPS, with the three selected confirming ions highlighted. | ||
The compositions of samples with BTMPS and primidone were investigated to determine if there were any trends associated with the makeup of the samples where these components are present. Table 4 lists a breakdown of the components detected for each sample in addition to the newly emerging substances. While no significant trends are apparent, all samples are complex in nature, containing drugs from 3–5 different drug classes, with 4–7 components present (Fig. 4). This highlights the importance of regular drug screening, as newly emerging substances can be present in samples that contain multiple known active components, and are not necessarily replacing known active components. Additionally, the sample compositions identified for primidone are in agreement with previous reports that indicate primidone was detected in the presence of fentanyl and/or an analog, along with medetomidine, a benzodiazepine, and/or xylazine.69
 | ||
| Fig. 4 Components detected in addition to primidone (Pn) and BTMPS (Bn) in street-drug samples. | ||
| Original results | Unknown identification |
|---|---|
| Benzodiazepine (unknown type), microcrystalline cellulose | Phenazolam |
| Benzodiazepine (unknown type), caffeine, erythritol, fentanyl, | |
| ortho-methylfentanyl, xylazine | Desalkylgidazepam |
| Microcrystalline cellulose, unknown | Protonitazepyne, isotonitazepyne |
| Caffeine, erythritol, fluorofentanyl, precursor (unknown type) | F-ANPP, fluoro-phenethyl-ANPP |
| Caffeine, erythritol, fluorofentanyl, precursor (unknown type) | F-ANPP, fluoro-phenethyl-ANPP |
| Fentanyl, precursor (unknown type) | F-ANPP, phenethyl-ANPP |
Methoxyacetyl fentanyl, which has been identified as both a synthesis byproduct and an active analog, was detected in 14 samples in this work.78 This drug has been identified as a synthesis byproduct in both the Gupta79 and Siegfried synthesis80 methods,81 which are commonly used in clandestine laboratories as a result of their relative simplicity over the patented Janssen method.82 All sources for detection of this compound were samples where fentanyl was also present, which suggests its presence as a synthesis byproduct over being an added adulterant.
Two analogs of ANPP were also detected. Fluoro-ANPP (F-ANPP), a fluorofentanyl precursor, was detected in 10 samples, though notably not always alongside fluorofentanyl. F-ANPP was confirmed by the presence of the selected fragments at m/z 105.0700, 134.0965, and 188.143, which are all common fragment ions for fentanyl-related compounds. The PS-HRMS/MS product spectrum for this detection is shown in Fig. S5.†ortho-Methyl-ANPP, an ortho-methylfentanyl precursor, was detected in 7 samples. This precursor was most frequently detected alongside ortho-methylfentanyl, except in one instance. The spectrum for ortho-methyl-ANPP is shown in Fig. 5. The confirming fragments for this compound are of high intensity in the spectrum, which is related to the high intensity of the ortho-methyl-ANPP ion in the precursor selection window (Fig. S6†).
 | ||
| Fig. 5 High-resolution PS-MS/MS spectra for ortho-methyl-ANPP (m/z, 295.2169), with the three target fragment ions highlighted. | ||
Other emerging precursors, namely phenethyl-ANPP and fluoro-phenethyl-ANPP, were tentatively identified based on their similar structure and fragmentation pattern to ANPP and F-ANPP respectively. Phenethyl-ANPP was detected in 1 sample, and fluoro-phenethyl-ANPP was detected in 5 samples. These compounds have been suspected to result from different synthesis routes in the manufacturing of fentanyl.83 The in vitro μ-opioid receptor activity of phenethyl-ANPP has been reported to be negligible, especially when compared to that of fentanyl.83 There have been no reports on the μ-opioid receptor activity of fluoro-phenethyl-ANPP at this time, but it is suspected to be of similar activity to phenethyl-ANPP as a result of their structural similarity. While there have been several previous reports of the detection of phenethyl-ANPP in the United States,14,83 there are limited reports regarding the detection of fluoro-phenethyl-ANPP. Fig. 6 demonstrates the similarity between the major fragments for fluoro-phenethyl-ANPP and F-ANPP, specifically at m/z 105.0700, 134.0967, and 188.1436. These fragments are shared among many 4-anilinopiperidine-type fentanyl analogs and precursors, and were identified by in silico fragmentation of this compound.84 Therefore, we were able to tentatively identify the presence of this precursor in 5 samples.
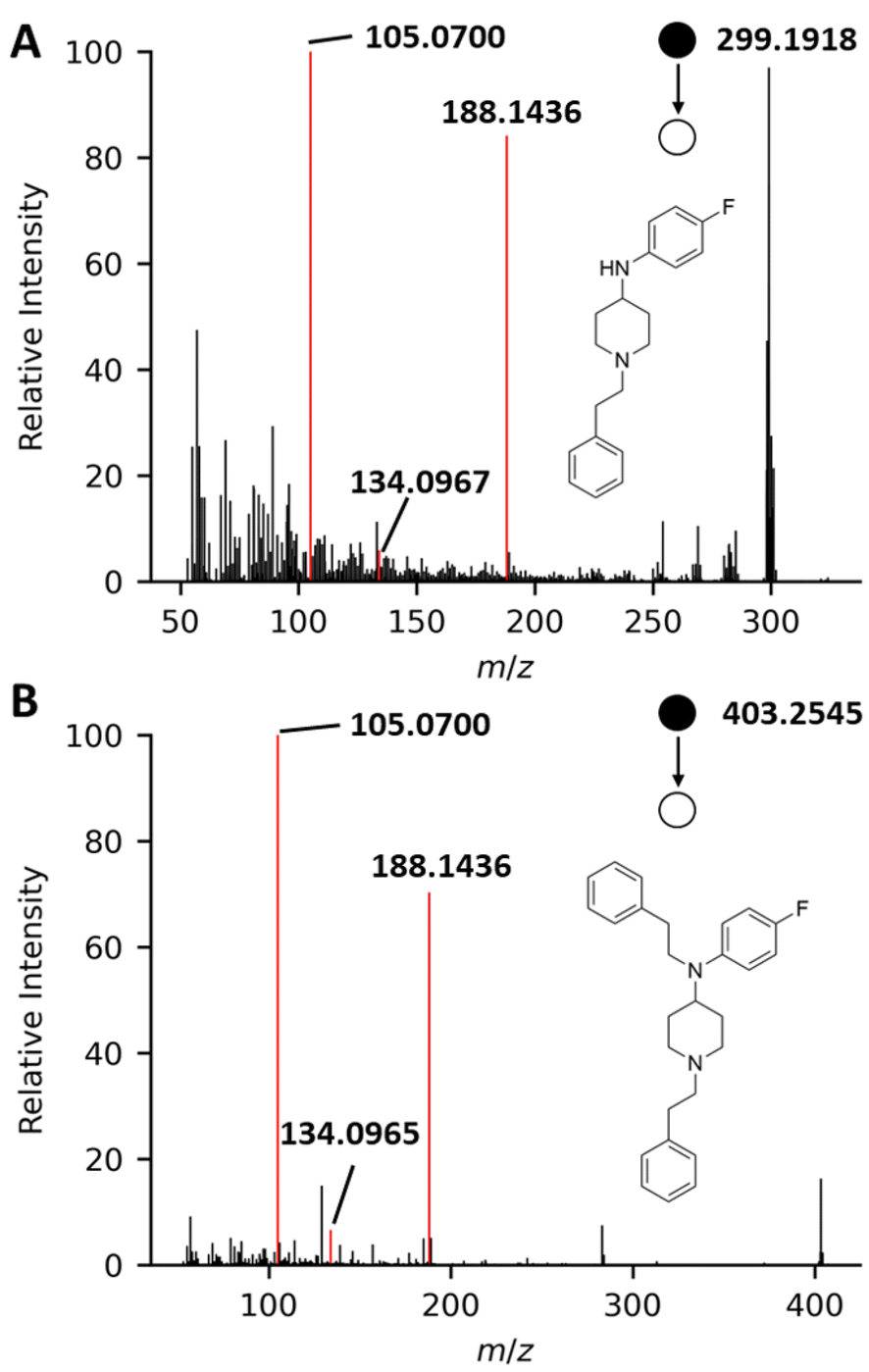 | ||
| Fig. 6 High-resolution PS-MS/MS [M + H]+ spectrum of (A) F-ANPP with the three selected confirming ions highlighted, and (B) fluoro-phenethyl-ANPP with three selected confirming ions highlighted. | ||
The detection of these compounds highlights the benefits of using multiple instruments for drug checking, and how together, they allow for a more complete picture of the composition of the street-drug supply, the necessity of which has been previously reported.34,85 Samples labeled as containing “Benzodiazepine (unknown type)” had a positive result with an immunoassay test strip, with none of the benzodiazepines in the targeted PS-MS method detected. Using the non-targeted PS-HRMS DDA method in this case allowed for the detection of benzodiazepines that were not yet included in the targeted method: phenazolam and desalkylgidazepam. Additionally, a pill with no active components detected was found to contain either of protonitazepyne or isotonitazepyne, which are isobaric and cannot be distinguished using mass spectrometry alone. There have been reports of isotonitazepyne appearing in expected oxycodone pills in Australia,86 and protonitazepyne in drug powders in Ireland.87 A study investigating the μ-opioid receptor activity of protonitazepyne and several other nitazenes suggests that these compounds are more potent than fentanyl.88 However, in-depth studies of the potencies of these drugs are limited. In other cases, FTIR signals indicated a precursor is present based on partial library matching to similar precursor compounds but were unable to be confirmed as spectra for the specific precursors, F-ANPP, phenethyl-ANPP, and fluoro-phenethyl-ANPP, which are not present in the libraries. Confidence in identification can also be increased with the agreement of results from different instrumental analyses.
The PS-HRMS method developed here was able to identify newly emerging substances in samples selected at random, in addition to samples selected because an unknown component was present. This demonstrates the utility of both analyzing samples where there is an obvious unidentified component, and random selection of samples for additional trace components that may be present. The identification of newly emerging substance by the PS-HRMS method can also guide the inclusion of compounds on targeted mass spectrometry methods, allowing these substances to be routinely detected by drug-checking services. Therefore, the importance of multi-instrument drug checking is demonstrated, where the combined strengths of multiple instruments allow for a more complete understanding of the unregulated drug supply.
4 Conclusions
The developed PS-HRMS DDA method provides a solution to address a current limitation in harm-reduction drug checking regarding the detection of newly emerging substances in the street-drug supply using PS-HRMS. The simplicity and speed of such a method provides a rapid screening tool to monitor the rapidly evolving drug supply, and therefore allows further insight into the complexities of street drugs. The presented work demonstrates the usefulness of combined quantitative and qualitative mass spectrometry methods in the context of harm-reduction drug checking.Author contributions
Conceptualization: A. M., C. G.; data collection: A. M.; methodology: A. M.; analysis and interpretation: A. M.; writing: A. M., D. K. H., C. G.; funding acquisition: B. W., D. K. H., and C. G.Data availability
Data will be made available from the corresponding author upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was supported by a New Frontiers in Research Fund (NFRFE-2022-00886), NSERC Discovery Grant (RGPIN-2021-02981), and a Canadian Foundation for Innovation (CFI) John R. Evans Leaders Fund (40274). Additional support and contributions were made by the Vancouver Foundation (F0120-5607), BC Ministry of Health, and the Island Health Authority. High performance computing support and server resources were provided by the University of Victoria and the Digital Research Alliance of Canada. We thank the community members for making this work possible through the donation of their drug samples. We also acknowledge that the laws and regulations in British Columbia regarding illicit substances have facilitated this study.References
- H. Hedegaard, A. M. Miniño, M. R. Spencer and M. Warner, Drug Overdose Deaths in the United States, 1999–2020, U.S. department of health and human services, centers for disease control and prevention, national center for health statistics technical report, 2021.
- S. Imtiaz, K. Shield, B. Fischer, T. Elton-Marshall, B. Sornpaisarn, C. Probst and J. Rehm, Subst. Abuse Treat., Prev., Policy, 2020, 15, 1–9 CrossRef PubMed.
- BC Coroners Service, BC Coroners Service Death Review Panel: An Urgent Response to a Continuing Crisis, BC coroners service technical report, 2023.
- A. Ospina-Escobar and C. A. D. Cervantes, Int. J. Drug Policy, 2024, 129, 104464 Search PubMed.
- UNODC, World Drug Report 2024, United nations technical report, 2024.
- A. Larnder, A. Saatchi, S. A. Borden, B. Moa, C. G. Gill, B. Wallace and D. Hore, Drug Alcohol Depend., 2022, 235, 109427 CrossRef PubMed.
- M. Zoorob, Int. J. Drug Policy, 2019, 70, 40–46 Search PubMed.
- P. Reuter, B. Pardo and J. Taylor, Int. J. Drug Policy, 2021, 94, 103086 CrossRef PubMed.
- Z. Dai, M. Abate, G. Smith, J. Kraner and A. R. Mock, Drug Alcohol Depend., 2019, 196, 1–8 CrossRef PubMed.
- C. Russell, J. Law, M. Bonn, J. Rehm and F. Ali, Int. J. Drug Policy, 2023, 111, 103933 CrossRef PubMed.
- S. Marshall and L. Nelson, J. Pharm. Pract., 2024, 38(2), 264–269 Search PubMed.
- J. Palamar and A. Krotulski, J. Am. Med. Assoc., 2024, 332, 1425 Search PubMed.
- M. M. Vandeputte, A. J. Krotulski, D. Walther, G. Glatfelter, D. Papsun, S. E. Walton, B. K. Logan, M. H. Baumann and C. P. Stove, Arch. Toxicol., 2022, 96, 1845–1863 CrossRef PubMed.
- H. Galust, J. Seltzer, J. Hardin, N. Friedman, J. Salamat, R. Clark and J. Harmon, BMC Public Health, 2024, 24, 923 Search PubMed.
- C. G. Rose, A. S. Pickard, V. Kulbokas, S. Hoferka, K. Friedman, J. Epstein and T. A. Lee, Harm Reduct. J., 2023, 20, 151 CrossRef PubMed.
- B. Wallace, L. Gozdzialski, A. Qbaich, S. Azam, P. Burek, A. Hutchison, T. Teal, R. Louw, C. Kielty, D. Robinson, B. Moa, M.-A. Storey, C. Gill and D. Hore, Drugs, Habits Soc. Policy, 2022, 23, 220–231 CrossRef.
- N. Maghsoudi, J. Tanguay, K. Scarfone, I. Rammohan, C. Ziegler, D. Werb and A. I. Scheim, Addiction, 2022, 117, 532–544 Search PubMed.
- A. Peacock, R. Bruno, N. Gisev, L. Degenhardt, W. Hall, R. Sedefov, J. White, K. Thomas, M. Farrell and P. Griffiths, Lancet, 2019, 394, 1668–1684 CrossRef PubMed.
- C. Awuchi, M. Aja, N. Mitaki, S. Morya, I. Amagwula, C. Echeta and V. Igwe, J. Chem., 2023, 1, 1–36 Search PubMed.
- V. Singh, T. Browne and J. Montgomery, Public Health Rep., 2020, 135, 6–10 CrossRef PubMed.
- N. C. Peiper, S. D. Clarke, L. B. Vincent, D. Ciccarone, A. H. Kral and J. E. Zibbell, Int. J. Drug Policy, 2019, 63, 122–128 CrossRef PubMed.
- J. E. Goldman, K. M. Waye, K. A. Periera, M. S. Krieger, J. L. Yedinak and B. D. L. Marshall, Harm Reduct. J., 2019, 16, 3 Search PubMed.
- S. A. Shuda and H. Y. Lam, Characterization of Xylazine Test Strips for Use in Drug Checking, The center for forensic science research & education technical report, 2022.
- A. A. Bunaciu, H. Y. Aboul-Enein and S. Fleschin, Appl. Spectrosc. Rev., 2010, 45, 206–219 Search PubMed.
- S. Tobias, A. M. Shapiro, C. J. Grant, P. Patel, M. Lysychyn and L. Ti, Drug Alcohol Depend., 2021, 218, 108300 CrossRef CAS.
- R. Gonzales, K. Titier, V. Latour, A. Peyre, N. Castaing, A. Daveluy and M. Molimard, Int. J. Drug Policy, 2021, 88, 103037 CrossRef PubMed.
- M. Fregonese, A. Albino, C. Covino, A. Gili, M. Bacci, A. Nicoletti and C. Gambelunghe, Front. Pyschiatry, 2021, 12, 596895 CrossRef.
- N. Maghsoudi, K. McDonald, C. Stefan, D. R. Beriault, K. Mason, L. Barnaby, J. Altenbert, R. D. MacDonald, J. Caldwell, R. Nisenbaum, P. Leece, T. M. Watson, K. W. Tupper, L. Kufner, A. I. Scheim and D. Werb, Harm Reduct. J., 2020, 17, 9 CrossRef PubMed.
- S. A. Borden, A. Saatchi, G. W. Vandergrift, J. Palaty, M. Lyshyshyn and C. G. Gill, Drug Alcohol Rev., 2022, 41, 410–418 Search PubMed.
- S. A. Borden, A. Saatchi, E. T. Krogh and C. G. Gill, Anal. Sci. Adv., 2020, 1, 97–108 Search PubMed.
- L. Gozdzialski, J. Aasen, A. Larnder, M. Ramsay, S. A. Borden, A. Saatchi, C. G. Gill, B. Wallace and D. K. Hore, Int. J. Drug Policy, 2021, 97, 103409 CrossRef PubMed.
- L. Karch, S. Tobias, C. Schmidt, M. Doe-Simkins, N. Carter, E. Salisbury-Afshar and S. Carlberg-Racich, Drug Alcohol Depend., 2021, 228, 108976 Search PubMed.
- J.-C. Laxton, J. Monaghan, B. Wallace, D. Hore, N. Wang and C. Gill, Int. J. Mass Spectrom., 2023, 484, 116976 Search PubMed.
- L. Gozdzialski, B. Wallace and D. Hore, Harm Reduct. J., 2023, 20, 39 Search PubMed.
- S. A. Borden, J. Palaty, V. Termopoli, G. Famiglini, A. Cappiello, C. G. Gill and P. Palma, Mass Spectrom. Rev., 2020, 39, 703–744 Search PubMed.
- J. Yu, K. Diekhans, A. Tsang and L. Rodda, J. Anal. Toxicol., 2024, 48, 573–581 CrossRef PubMed.
- C. Mollerup, P. Dalsgaard, M. Mardal and K. Linnet, Drug Test. Anal., 2017, 9, 1052–1061 Search PubMed.
- E. Drug, D. Marder, I. Binyamin, D. Yeffet, E. Gershonov and S. Dagan, J. Mass Spectrom., 2024, 59, e4994 CrossRef PubMed.
- T. Cooman, C. Ott, K. Dalzell, A. Burns, E. Sisco and L. Arroyo, Forensic Chem., 2021, 25, 100352 Search PubMed.
- A. Grange and G. Sovocool, Rapid Commun. Mass Spectrom., 2011, 25, 1271–1281 Search PubMed.
- J. Mckenna, R. Jett, K. Shanks and N. Manicke, J. Anal. Toxicol., 2018, 42, 300–310 CrossRef PubMed.
- S. Prunty, D. Carmany, E. Dhummakupt and N. Manicke, Analyst, 2023, 148, 3274 Search PubMed.
- S. Borden, S. R. Mercer, A. Saatchi, E. Wong, C. M. Stefan, H. Wiebe, D. K. Hore, B. Wallace and C. G. Gill, Drug Test. Anal., 2023, 15, 484–494 CrossRef.
- Y. Su, H. Wang, J. Liu, P. Wei, R. Cooks and Z. Ouyang, Analyst, 2013, 138, 4443 RSC.
- G. W. Vandergrift and C. G. Gill, J. Mass Spectrom., 2019, 54, 729–737 CrossRef PubMed.
- D. Amodei, J. Egertson, B. MacLean, R. Johnson, G. Merrihew, A. Keller, D. Marsh, O. Vitek, P. Mallick and M. MacCoss, J. Am. Soc. Mass Spectrom., 2019, 30, 669–684 CAS.
- F. Sun, H. Tan, Y. Li, M. D. Boevre, H. Zhang, J. Zhou, Y. Li and S. Yang, J. Hazard. Mater., 2021, 401, 123266 CAS.
- Y. Yang, L. Yang, M. Zheng, D. Cao and G. Liu, Trends Anal. Chem., 2023, 160, 116966 CAS.
- H. Tan, F. Sun, M. Abdallah, J. Li, J. Zhou, Y. Li and S. Yang, Food Chem., 2022, 385, 132669 CAS.
- J. A. Michely, M. Meyer and H. Maurer, Anal. Chem., 2017, 89, 11779–11786 CAS.
- H. Zimmerman-Federle, G. Ren, S. Dowling, C. Warren, D. Rusyniak, R. Avera and N. E. Manicke, Drug Test. Anal., 2024, 17(1), 138–151 Search PubMed.
- K. Norman, A. Ciesielski and J. Wagner, WIREs Forensic Sci., 2020, 3, e1393 Search PubMed.
- M. Joshi and E. Sisco, WIREs Forensic Sci., 2023, 5, e1486 CrossRef.
- R. R. Martens, L. Gozdzialski, E. Newman, C. Gill, B. Wallace and D. K. Hore, Anal. Chem., 2024, 96, 12277–12285 CAS.
- Substance: Vancouver Island Drug Checking Project, https://substance.uvic.ca.
- Monographs, https://www.cfsre.org/nps-discovery/monographs, accessed: 2025-01-13.
- N. Hulstaert, J. Shofstahl, T. Sachsenberg, M. Walzer, H. Barsnes, L. Martens and Y. Perez-Riverol, J. Proteome Res., 2020, 19, 537–542 Search PubMed.
- Substance Drug Checking: Annual Review 2023, https://substance.uvic.ca/files/reports/SubstanceDrugChecking2023AnnualReport.pdf, accessed: 2025-01-17.
- E. Sisco, J. Verkouteren, J. Staymates and J. Lawrence, Forensic Chem., 2017, 4, 108–115 CrossRef CAS PubMed.
- E. Denis, J. Bade, R. Renslow, K. Morrison, M. Nims, N. Govind and R. Ewing, J. Am. Soc. Mass Spectrom., 2022, 33, 482–490 Search PubMed.
- L. Schove, J. Perez and G. Loew, Bioorg. Med. Chem., 1994, 2, 1029–1049 Search PubMed.
- C. Skaggs, L. Kirkpatrick, W. R. A. Wichert, N. Skaggs and N. Manicke, Rapid Commun. Mass Spectrom., 2020, 34, e8601 CrossRef CAS PubMed.
- Q. Yang, N. Manicke, H. Wang, C. Petucci, R. Cooks and Z. Ouyang, Anal. Bioanal. Chem., 2012, 404, 1389–1397 Search PubMed.
- C.-W. Tsai, C. Tipple and R. Yost, Rapid Commun. Mass Spectrom., 2017, 31, 1565–1572 Search PubMed.
- Z.-P. Zhang, X. N. Liu and Y. J. Zheng, Chin. J. Anal. Chem., 2014, 42, 145–152 Search PubMed.
- Z. Zhang, W. Xu, N. Manicke, R. Cooks and Z. Ouyang, Anal. Chem., 2012, 84, 931–938 CAS.
- C. Vega, C. Spence, C. Zhang, B. Bills and N. Manicke, J. Am. Soc. Mass Spectrom., 2015, 27, 726–734 CrossRef PubMed.
- S. I. Johannessen and C. J. Landmark, in NeuroPsychoparmacotherapy, ed. P. Riederer, et al., Springer Nature Switzerland, Cham, Switzerland, 2022, pp. 3537–3546 Search PubMed.
- R. Booker , Primidone in the Unregulated Drug Supply in Canada, Criminal intelligence service alberta technical report, 2024.
- W. M. A. Neissen, Mass Spectrom. Rev., 2012, 31, 626–665 Search PubMed.
- R. Zhu, Y. Dong, X. Cai and C. Huang, Molecules, 2019, 24, 1494 CAS.
- L. Høj, C. Mollerup, B. Rasmussen, S. Johansen, K. Linnet and P. Dalsgaard, Drug Test. Anal., 2019, 11, 1258–1263 Search PubMed.
- S. E. Walton, E. Tracy, J. Shinefeld, D. Teixeira da Silva, M. T. Denn, A. D. Quinter, J. S. DeBord, B. K. Logan and A. J. Krotulski, NPS Discovery – New Drug Monograph: BTMPS, Center for forensic science research and education technical report, 2024.
- P. Sótonyi, A. Kovács, G. Volk, J. Járay and A. Benko, J. Chromatogr. Sci., 2004, 42, 49–53 Search PubMed.
- M. Gill, M. Garber, Y. Hua and D. Jenke, J. Chromatogr. Sci., 2010, 48, 200–207 CAS.
- B. Hall, L. Pearson and A. V. Terry Jr., Neuropharmacology, 2011, 61, 798–806 CAS.
- T. Green, R. Olsen, C. Jarczyk, E. Erowid, F. Erowid, S. Thyssen, R. Wightman, B. del Pozo, L. Michelson, A. Consigli, B. Reilly and S. Ruiz, J. Public Health Manage. Pract., 2022, 28, S347–S354 Search PubMed.
- M. Mardal, S. S. Johansen, A. B. Davidsen, R. Telving, J. R. Jornil, P. W. Dalsgaard, J. B. Hasselstrøm, Å. M. Øiestad, K. Linnet and M. F. Andreasen, Forensic Sci. Int., 2018, 290, 310–317 CAS.
- P. Gupta, K. Ganesan, A. Pande and R. Malhotra, J. Chem. Res., 2005, 2005, 452–453 Search PubMed.
- Siegfried, Synthesis of Fentanyl, https://www.erowid.org/archive/rhodium/chemistry/fentanyl.html, accessed: 2025-01-14.
- M. de Bruin-Hoegèe, D. Kleigwg, D. Noort and A. van Asten, Forensic Chem., 2021, 24, 100330 Search PubMed.
- L. Mörén, J. Qvarnström, M. Engqvist, R. Afshin-Sander, X. Wu, J. Dahlén, C. Löfberg, A. Larsson and A. Östin, Talanta, 2019, 203, 122–130 Search PubMed.
- M. M. Vandeputte, A. Krotulski, F. Hulpia, S. Van Calenbergh and C. P. Stove, J. Anal. Toxicol., 2022, 46, 350–357 Search PubMed.
- Y. Fan, X. Lin, X. Chen, H. Wu, J. Xu and Y. Xu, Forensic Chem., 2023, 33, 100485 CrossRef CAS.
- F. Giulini, E. Keenan, N. Killeen and J.-H. Ivers, J. Psychoact. Drugs, 2022, 55, 85–93 CrossRef PubMed.
- B. Curtis, D. J. Lawes, D. Caldicott and M. D. McLeod, ChemRxiv, 2024, preprint, DOI:10.26434/chemrxiv-2024-0mdvs.
- S. Killoran, S. McNamara, P. Kavanagh, J. O’Brien and R. Lakes, Drug Test. Anal., 2024, 17(3), 350–357 Search PubMed.
- L. M. D. Vrieze, S. E. Walton, E. Pottie, D. Papsun, B. Logan, A. Krotulski, C. Stove and M. Vandeputte, Arch. Toxicol., 2024, 98, 2999–3018 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Instrument parameters for the triple quadrupole and Orbitrap mass spectrometers; compounds included in the DDA peak search algorithm; spray solvent optimization details; PS-HRMS precursor isolation windows for primidone, BTMPS, and ortho-methyl-ANPP; annotated spectrum for F-ANPP; the exclusion m/z list for the high-resolution measurements. See DOI: https://doi.org/10.1039/d5an00086f |
| This journal is © The Royal Society of Chemistry 2025 |