
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReduction of background-triggered amplification in lesion-induced DNA amplification (LIDA)†
Anantha S.
Ealeswarapu
 ,
Nahida
Akter
and
Julianne M.
Gibbs
*
,
Nahida
Akter
and
Julianne M.
Gibbs
*
Department of Chemistry, University of Alberta, Edmonton, Alberta T6G 2G2, Canada. E-mail: julianne.gibbs@ualberta.ca
First published on 6th March 2025
Abstract
Lesion-induced DNA amplification (LIDA) enables isothermal amplification of nucleic acids, and the only enzyme required is T4 DNA ligase. However, the application of LIDA for the amplification of trace amounts of nucleic acids has been hindered by the observed background-triggered amplification in the absence of the initial target due to a pseudo-blunt end ligation reaction of two of the primers. In this work, we have tested three approaches to minimize the background-triggered amplification: increasing and decreasing the concentration of salts such as NaCl and MgCl2, respectively, and increasing the concentration of ATP. All these optimizations sharply decreased the background-triggered amplification. Employing the most favourable buffer condition of 2.5 mM MgCl2 where the target-initiated amplification was least affected while reducing the background-triggered process enabled us to achieve a detection range of 14 nM–140 aM with an approximate limit of detection of 680 aM, which is five orders of magnitude more sensitive than using our standard amplification conditions. This optimization of the salt and co-factor concentrations to decrease the background and enhance the sensitivity of LIDA has demonstrated LIDA's potential for application in clinical diagnostics.
Introduction
Nucleic acid amplification is an important method for detecting low-abundance nucleic acid sequences in biological samples associated with infectious pathogens or disease states.1,2 Polymerase chain reaction (PCR) is by far the most commonly used nucleic acid amplification method owing to its sensitivity.3 However, its dependence on expensive thermal cyclers for amplifying nucleic acids and its requirement for skilled operators make PCR less accessible for usage in resource-limited settings or in point-of-care testing.4,5 The emergence of isothermal nucleic acid amplification techniques (INAATs) has improved the accessibility of NAATs in low-resource settings as these can be performed at a constant temperature using less equipment making them more convenient and cost-effective.1,6,7 INAATs like loop-mediated isothermal amplification (LAMP),8,9 strand-displacement amplification (SDA),10 and exponential-amplification reaction (EXPAR)11 can amplify target nucleic acid sequences to produce detectable amplicons. For example, within one hour LAMP can detect (∼102) copies per μL of target, but it demands extensive primer design and high temperatures between 60 and 65 °C.9 Strand-displacement amplification (SDA) has potential for amplifying DNA with a nearly ∼107 fold amplification factor under 2 hours,5,10 while the exponential amplification reaction (EXPAR) displays 106–108 amplification efficiency under 30 min.11 However, the latter two methods employ two enzymes DNA polymerase and nicking endonuclease enzyme and are performed at 37 °C (ref. 10 and 12) and 60 °C,11 respectively. Our group has developed an isothermal nucleic acid amplification process that uses only one enzyme based on a ligase chain reaction (LCR) rather than a polymerase chain reaction.13,14 Traditionally, LCR is a highly sensitive exponential amplification technique that utilizes DNA ligase and needs a thermocycler for amplification.15,16 Our approach to achieving DNA amplification isothermally involves introducing a destabilizing abasic lesion in one of the ligating primers that facilitates spontaneous turnover in the ligase chain reaction. This process is called lesion-induced DNA amplification (LIDA).17 Our previous work has shown that LIDA can operate at room temperature, alleviating the need for temperature control.17,18In LIDA, one of the primers complementary to the target sequence, DNA-I, contains a destabilizing model abasic group (1′,2′-dideoxy-5′-phosphate) at its terminus. The other primer is complementary to the target at the sequence adjacent to the destabilizing primer, leading to the formation of a nicked duplex (Fig. 1). The enzyme T4 DNA ligase can then catalyze the formation of a novel template through the ligation of these two primers (DNA-II, Fig. 1). Due to the presence of the abasic group on DNA-II, the original template DNA-I spontaneously dissociates. Subsequently, DNA-II functions as a template for hybridization with two additional primers that constitute the DNA-I sequence, one of which includes a fluorescent label. This results in the formation of another nicked duplex, which is subsequently ligated by T4 DNA ligase to form a fluorescently labeled copy of DNA-I. Once again, the duplex spontaneously dissociates due to the destabilization caused by the presence of the abasic group releasing DNA-I and DNA-II and allowing both to template more reactions. This cross-catalytic cycle leads to rapid, exponential self-replication of an 18 nt target and has been used to amplify a variety of sequences.14,17–19 By employing a serial-dilution strategy with low concentration of the primers in a cross-catalytic approach we were able to detect as low as 140 fM of target DNA.14 In more recent work, lesion-induced DNA amplification has been utilized to detect RNA targets by incorporating an RNA-templated step where cDNA is generated, isothermally liberated and then subsequently amplified by LIDA. Using reverse transcription (RT-) LIDA with optimized primer concentrations we achieved a detection limit of 240 amol (32 pM) at 28 °C of target RNA spiked into samples of total cellular RNA.18
 | ||
| Fig. 1 Schematic of self-replication of DNA-I by lesion-induced DNA amplification (LIDA) from four primers (Ia, IbP, IIaP, and IIb). The IIaP strand contains the destabilizing abasic group at the 5′-phosphate position. In typical LIDA, Ia is the limiting primer reagent, which is labeled with fluorescein (green star). The formation of DNA-I is measured by fluorescence imaging coupled with polyacrylamide gel electrophoresis. | ||
However, one major limitation of lesion-induced DNA amplification (LIDA) is the presence of background-triggered amplification that occurs in the absence of the target, which is observed in all the sequences that we successfully amplified by LIDA.14,17–19 This background-triggered amplification leads to: (i) false positives by incorrectly indicating the presence of the target in the biological sample20 and (ii) limited analytical sensitivity of the assay (too high of a limit of detection, LOD) leading to false negatives.21 The background triggered reaction in LIDA results from T4 DNA ligase catalyzed ligation of the primers in the absence of the initial target to form DNA-II via a blunt-end like (pseudo blunt-end) ligation (Fig. 2)14,17,18 Recently, we determined that it is specifically due to the non-templated formation of DNA-II (ligation of the 5′-phosphate abasic primer) in the first cycle, which is a slow process but results in a template that can amplify by cross-catalysis.22
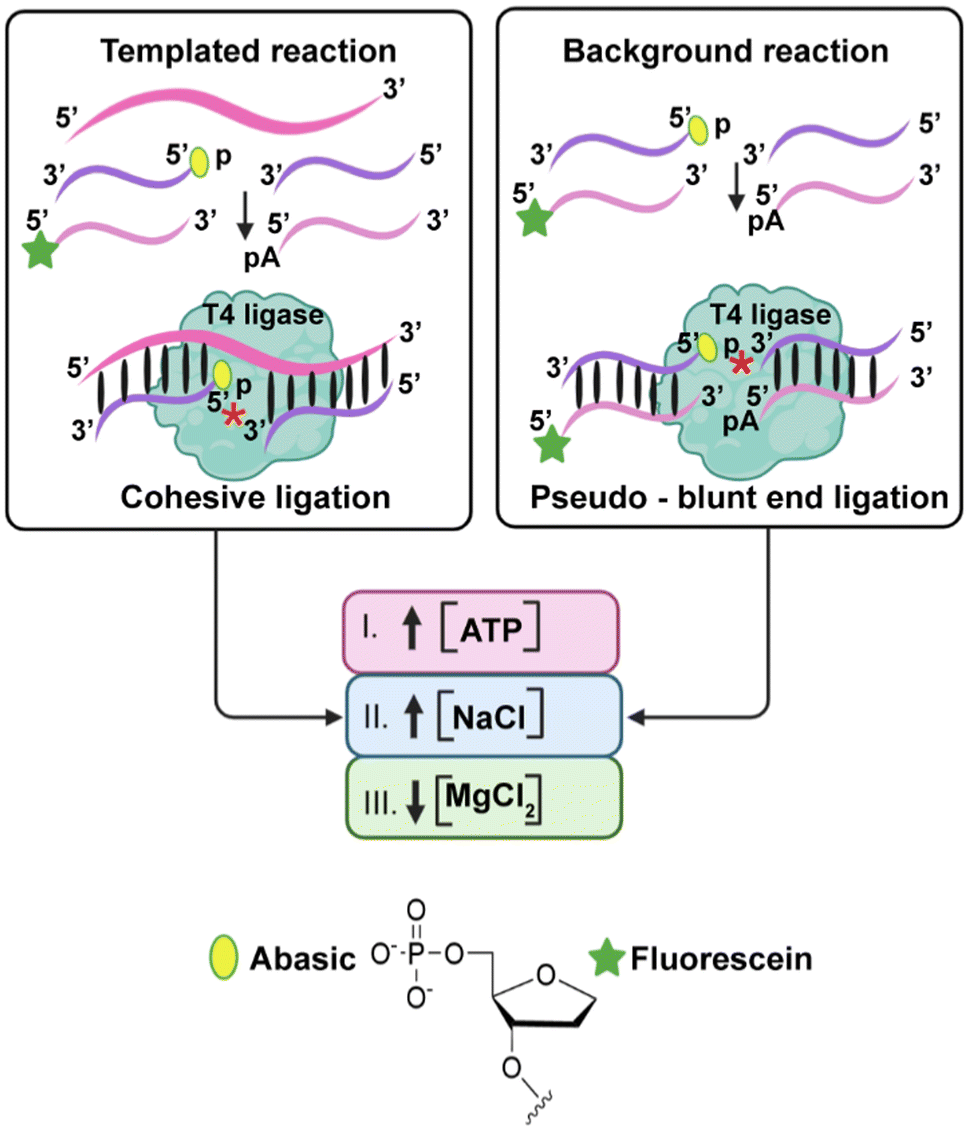 | ||
| Fig. 2 Schematic illustration of the cohesive (templated reaction) and pseudo-blunt end ligations (background reaction) by the T4 DNA ligase involved in LIDA and the reported factors that help to reduce blunt end ligation, which we hypothesize would reduce background-triggered amplification in LIDA. *Shows the site of phosphodiester formation. Created in BioRender.com. | ||
Studies of T4 DNA ligase have shown that increasing the concentration of the enzyme co-factor ATP and decreasing the concentration of Mg2+ can inhibit the joining of blunt ends with only slight reduction in the rate of the templated ligation.23 Other studies have also shown that increasing the concentration of monovalent salts like NaCl can inhibit both blunt end and cohesive ligation, albeit to different extents.24 Therefore, we hypothesized that the factors that influence blunt end ligation by T4 DNA ligase would influence the pseudo-blunt end ligation that causes background-triggered LIDA (Fig. 2). Here we show how the background triggered process can be minimized by varying the concentration of ATP, NaCl, and Mg2+ leading to a reduction in the LOD of our standard LIDA process by five orders of magnitude. This enormous improvement in sensitivity opens the door for the use of LIDA in point-of-care testing.
Experimental section
Materials and instrumentation
Chemicals were purchased from Fisher Scientific or Sigma-Aldrich and used as received. T4 DNA ligase enzyme (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000000 cohesive end units per mL, catalog #M0202T) and ATP (10 mM, catalog #9804S) were purchased from New England Biolabs. For making 15% polyacrylamide denaturing gels, 40% acrylamide/bis-solution 19:1 was purchased from Bio-Rad (California, USA). The ligation master mix was prepared immediately before the experiment from separate solutions of TRIS-HCl (pH 7.5) (containing an appropriate amount of MgCl2) and ATP and NaCl. Ultrapure water was obtained from a Milli-Q-Ultrapure Water System, which was autoclaved and used for the experiments.
000000 cohesive end units per mL, catalog #M0202T) and ATP (10 mM, catalog #9804S) were purchased from New England Biolabs. For making 15% polyacrylamide denaturing gels, 40% acrylamide/bis-solution 19:1 was purchased from Bio-Rad (California, USA). The ligation master mix was prepared immediately before the experiment from separate solutions of TRIS-HCl (pH 7.5) (containing an appropriate amount of MgCl2) and ATP and NaCl. Ultrapure water was obtained from a Milli-Q-Ultrapure Water System, which was autoclaved and used for the experiments.
The oligonucleotide synthesis was performed on an Applied Biosystems Model 392 DNA/RNA Synthesizer. A Torrey Pines Scientific Echotherm Chilling/Heating Plate (Model IC22) was used to maintain temperature for the experiments. 15% denaturing gels were made using 8 M urea. The gels were imaged using an Image Quant RT ECL Imager from GE Healthcare Life Science by UV transillumination. Igor Pro v7.08A (Wavemetrics, Oswego, OR) was used to fit the data with a sigmoidal growth function. We employed Axygen 0.6 mL Maxymum Recovery Snaplock Polypropylene Microcentrifuge tubes (MCT-060-L-C) for our experiments.
Oligonucleotide preparation and characterization
All the oligonucleotides listed in Table S1† were synthesized on an Applied Biosystems Model 392 automated solid-phase synthesizer using Glen Research reagents. All synthesized DNA was purified with Glen-Pak Purification cartridges (catalog # 60-5200-01) following the DMT-ON protocol and freeze-dried afterward.Some of the DNA strands had special modifications with compounds such as 5′-phosphate, fluorescein, and the abasic group. The abasic group was incorporated using dSpacer CE phosphoramidite (catalog #10-1914-90) and fluorescein using 5′-fluorescein-dT phosphoramidite (catalog #10-1056-95). Solid phosphorylation reagent II (catalog #10-1902-90) was used to synthesize 5′-phosphate strands.
The synthesized strands were characterized by MALDI-TOF in a linear negative mode using a Voyager Elite time of flight-mass spectrometer (Applied Biosystems, Foster City, CA) following our earlier work.22 Purity was assessed by visualizing the oligonucleotides with Stains-All reagent after it was run on a 15% polyacrylamide denaturing gel and analyzed using the Image Quant RT ECL Imager from GE healthcare Life Science (with the fluorescein filter).
LIDA protocol
The isothermal self-replication experiments were performed as described in our earlier work13,14 with some modifications. For 15 μL total reaction volume, we used 1 μL of T4 DNA ligase (2000000 CEU mL−1). The final concentration of the three primers (Ibp, IIap, and IIb) was 2.8 μM and that of the limiting fluorescent primer (Ia) was 1.4 μM. The reaction mixtures were prepared by mixing four primers with the template (template-initiated reaction) and without the template (background-triggered reaction) to reach a volume of 10 μL. The DNA solution was incubated for 10 minutes at 30 °C. Each component of the ligation buffer and the enzyme were then added to different spots near the top of the Eppendorf tube containing the DNA. The sample was then spun down and vortexed, thereby initiating the reaction. 1.5 μL aliquots of the reaction mixture were taken at various time points and placed in a microcentrifuge tube containing 2 μL of a mixture of bromophenol blue dye (0.011 g mL−1) and EDTA (0.5 M). The samples were analyzed using a 15% PAGE denaturing gel. The target concentration variation experiments were performed in the same way using the same primer ratios as mentioned above, but with various lower concentrations of the DNA target (14 nM, 140 pM, 14 pM, 14 fM, and 140 aM). A master mix containing each component of the ligation buffer and the enzyme was prepared and incubated for 10 minutes at 30 °C. To minimize any potential error or variation arising from different enzyme bottles, we combined 10 μL from each of the two T4 DNA ligase enzyme (2000000CEU mL−1) bottles. This pooled enzyme solution was then used to perform two replicate target concentration variation experiments to ensure consistency and reliability in the enzymatic activity across both experimental replicates.
Results and discussion
T4 DNA ligase catalyzes the formation of a phosphodiester bond between the 5′-phosphate and 3′-hydroxyl groups of two adjacent primers hybridized to a template strand.25 This enzyme can also perform this bond sealing with blunt end duplex substrates. T4 DNA ligase requires divalent metal cations such as Mg2+ or Mn2+ and is dependent on ATP as an adenylate-donating co-factor that provides energy for ligation.26 Ligation of nicked DNA involves three catalytic steps: (1) adenylation of the ligase at a lysine, which releases pyrophosphate (PPi) and results in the formation of a covalent ligase-adenosine monophosphate (AMP) complex; (2) transfer of AMP onto the 5′-phosphate of the nicked DNA, activating it for nucleophilic attack by the 3′-hydroxy group from the adjacent DNA strand; and (3) formation of the phosphodiester bond releasing the AMP.26,27 For blunt-end ligation, the mechanism is virtually the same except that the enzyme can adenylate the 5′-phosphate on one duplex prior to the binding of the duplex containing the 3′-hydroxy group.28In the presence of the target, LIDA proceeds by cohesive ligation of the destabilizing primer strands hybridized to the target template to generate DNA-II (Fig. 2). In the absence of the target, however, ligation can occur because of the single-base overhang between the 5′-phosphate abasic primer and the 5′-phosphate adenosine primer, which also results in the formation of DNA-II. We refer to this mechanism of ligation to form DNA-II as a pseudo-blunt end ligation as there is no hydrogen-bonding in this single A:Abasic overhang (Fig. 2). We found that it occurs for LIDA-based amplification of all sequences that we have explored using T4 DNA ligase.14,17,18
Effect of increasing ATP on target-initiated and background-triggered LIDA
As the sensitivity of LIDA is impacted by the background amplification that arises from the in situ synthesis of a catalytic amount of DNA-II by pseudo-blunt end ligation, our goal was to identify experimental conditions that decreased background-triggered amplification, thereby enhancing the sensitivity of LIDA. Studies have shown that an increase in ATP concentration decreases blunt-end ligation.23 Accordingly, we hypothesized that increasing the ATP concentration beyond our standard LIDA conditions (1 mM ATP) would adversely affect the background-triggered LIDA, leading to a shift to the right of the sigmoidal amplification profile corresponding to LIDA without the initial target.The effect of increasing ATP concentration on LIDA in the absence and presence of the initial DNA-I target is presented in Fig. 3. For simplicity, we utilized standard primers that operate best at 30 °C where only one primer contains a destabilizing (abasic) group rather than our dual-destabilization primers that can be tuned to operate at lower temperatures.17 We also used a temperature controller to ensure that any changes in the rate of amplification could be attributed to changes in the buffer conditions. First, we evaluated the impact of ATP concentration on the target-initiated LIDA. Accordingly, a 14 nM DNA-I target was incubated with an excess of primers at varying ATP concentrations in the presence of T4 DNA ligase and the conversion of the fluorescent primer to a DNA-I product was monitored by gel electrophoresis. Increasing the ATP concentration from 1 mM to 7.5 mM did not have a significant effect on the amplification rate for the LIDA reactions initiated by 14 nM DNA-I (Fig. 3A). In contrast, the onset of amplification for the background-triggered LIDA in the absence of the initial DNA-I target slowed down with an increase in ATP concentration, supporting our hypothesis (Fig. 3B). The observed trends in the template-initiated and background-triggered LIDA are consistent with the observations by Ferretti and co-workers where they reported a significant decrease in the extent of blunt-end joining with an increase in ATP concentration with only minimal reduction in the joining of the cohesive-ends by T4 DNA ligase.23 However, at an ATP concentration of 10 mM, complete inhibition of LIDA was observed in both the absence and presence of the initial DNA-I target. Such a strong nonlinear effect from varying the ATP concentration as shown in Fig. 3A has been observed in T4DNA ligase catalyzed cohesive end ligation in the percent yield,23 as well as in the enzyme turnover rate.26 Moreover, in these reports, 10 mM ATP either completely shut down cohesive end ligation23 or inhibited it significantly,26 consistent with our observations. This hindering of ligation by excessive ATP has been attributed to the dissociation of the ligase from the DNA complex after the transfer of AMP. In the presence of excess ATP, the ligase can be readenylated prior to rebinding to the DNA complex disabling its ability to catalyze the ligation of the adenylated DNA intermediate (App DNA), leading to the accumulation of the latter.23,26
 | ||
| Fig. 3 Effect of increased ATP concentration on the kinetics of cross-catalytic formation of F-DNA-I initiated by (A) 14 nM and (B) 0 nM DNA-I in the presence of different ATP concentrations. % conversion refers to the amount of fluorescent primer converted to DNA-I. Standard experimental conditions: 14 nM DNA-I, 1.4 μM Ia, 2.8 μM IIaP, 2.8 μM IbP, 2.8 μM IIb, 50 mM TRIS-HCl (pH 7.5), 10 mM MgCl2, 1 mM ATP, 2000 CEU T4 DNA ligase, and 30 °C. | ||
Effect of increasing monovalent ion concentration on target-initiated and background-triggered LIDA
Another approach to inhibit the activity of T4 DNA ligase in a manner that affects blunt end vs. cohesive ligation more is to introduce K+ or Na+ into the ligation mixture. Studies have shown that increasing monovalent ion concentrations to ionic strengths much higher than those provided by the presence of Mg2+ (vide infra) can inhibit the activity of the T4 DNA ligase albeit to varying extents on cohesive and blunt end substrates.24 Therefore, we hypothesized that introducing NaCl would decrease the background-triggered amplification more so than the template-initiated LIDA reaction owing to its deleterious effect on the pseudo-blunt end ligation of the primers. LIDA was performed under our standard conditions with the addition of NaCl at various concentrations (Fig. 4). | ||
| Fig. 4 Effect of NaCl concentration on the kinetics of LIDA initiated by (A) 14 nM and (B) 0 nM DNA-I using otherwise standard conditions. % conversion refers to the amount of fluorescent primer converted to DNA-I. | ||
With 100 mM NaCl, we observed faster target initiated and background-triggered amplification compared to the reaction without any added NaCl. This increase in activity is attributed to the shielding by the monovalent sodium cations that stabilizes the hybridized DNA complexes via mitigation of the negative charge on the sugar-phosphate backbone.29 However, T4 DNA ligase activity started to decline at salt concentrations above 200 mM, consistent with previous T4 DNA activity studies.30,31 We observed that at a 200 mM concentration both the target-initiated and background-triggered LIDA declined with a greater impact on the latter, suggesting that pseudo-blunt end ligation was more compromised compared with cohesive ligation. This trend of slower amplification with increasing salt concentration continued at 300 mM, and by 400 mM no amplification was observed within two hours. The inhibitory effect of monovalent salts at higher concentration on T4 DNA ligase has been attributed to a decreased affinity of the enzyme-AMP towards the DNA resulting in a higher effective Km.30,31 This proposed decrease in ligase affinity for the DNA substrate is thought to stem from neutralization of the negative charges on the phosphate group that play an important role in binding of the DNA to the T4 ligase.29,30
Effect of increasing divalent ion concentration on target-initiated and background-triggered LIDA
In addition to ATP, the ligase requires a divalent metal cation such as Mg2+ or Mn2+. Studies have shown that a decrease in Mg2+ concentration results in the decreased joining or full inhibition of ligation of blunt ends, while only exhibiting a minimal decrease in the joining of the cohesive ends.23 Accordingly, we hypothesized that decreasing the Mg2+ concentration below our standard LIDA conditions (10 mM MgCl2) would adversely affect background-triggered LIDA. The effect of decreasing Mg2+ concentration on the target-initiated and background-triggered reactions is presented in Fig. 5.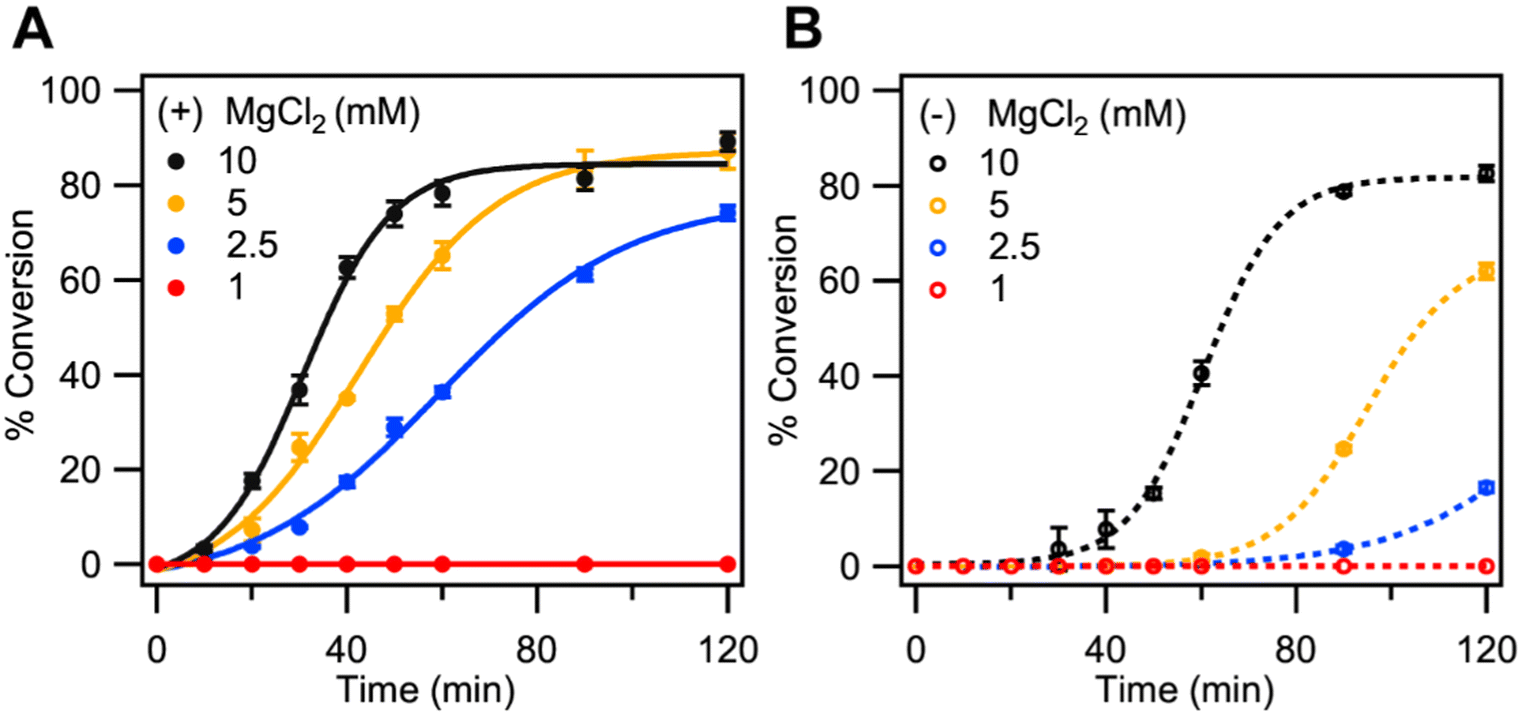 | ||
| Fig. 5 Effect of decreasing MgCl2 concentration on the kinetics of cross-catalytic formation of F-DNA-I initiated by (A) 14 nM and (B) 0 nM DNA-I under otherwise standard conditions. % conversion refers to the amount of fluorescent primer converted to DNA-I. | ||
Decreasing the Mg2+ concentration from 10 mM to 2.5 mM did not have a significant effect on the amplification rate for the target-initiated LIDA reactions (Fig. 5A) while a more significant effect was observed for the background-triggered process (Fig. 5B). A further decrease in Mg2+ concentration to 1.0 mM caused complete inhibition of both cohesive and blunt ends by the T4 DNA ligase. The observed trends in the templated and background reactions are consistent with the observations by Ferretti and co-workers, where they reported a significant decrease in the extent of blunt end joining with a decrease in Mg2+ concentration with only minimal reduction in the joining of the cohesive-ends by T4 DNA ligase.23 The decrease in blunt end joining in the presence of low Mg2+ could be attributed to the abortive ligation that occurs at lower Mg2+ concentrations also leading to the accumulation of the App DNA.26,32
Sensitivity of LIDA at 2.5 mM Mg2+
From the previous studies we found that the most optimal conditions for significantly mitigating the background-triggered process while still facilitating rapid LIDA in the presence of the initial target were by decreasing the concentration of MgCl2 to 2.5 mM. To determine if these conditions with reduced Mg2+ that favor target-initiated LIDA improved the sensitivity of our assay, we performed LIDA at 2.5 mM MgCl2 for a wide range of the initial DNA-I target covering eight orders of magnitude (Fig. 6A). Remarkably, by using this simple change in ligation mixture composition, we were able to distinguish as little as 140 aM of DNA-I versus the background-triggered process (Fig. 6A). In comparison, using the same primer concentrations and buffer but with 10 mM Mg2+ we can only distinguish down to 140 pM (Fig. S4†).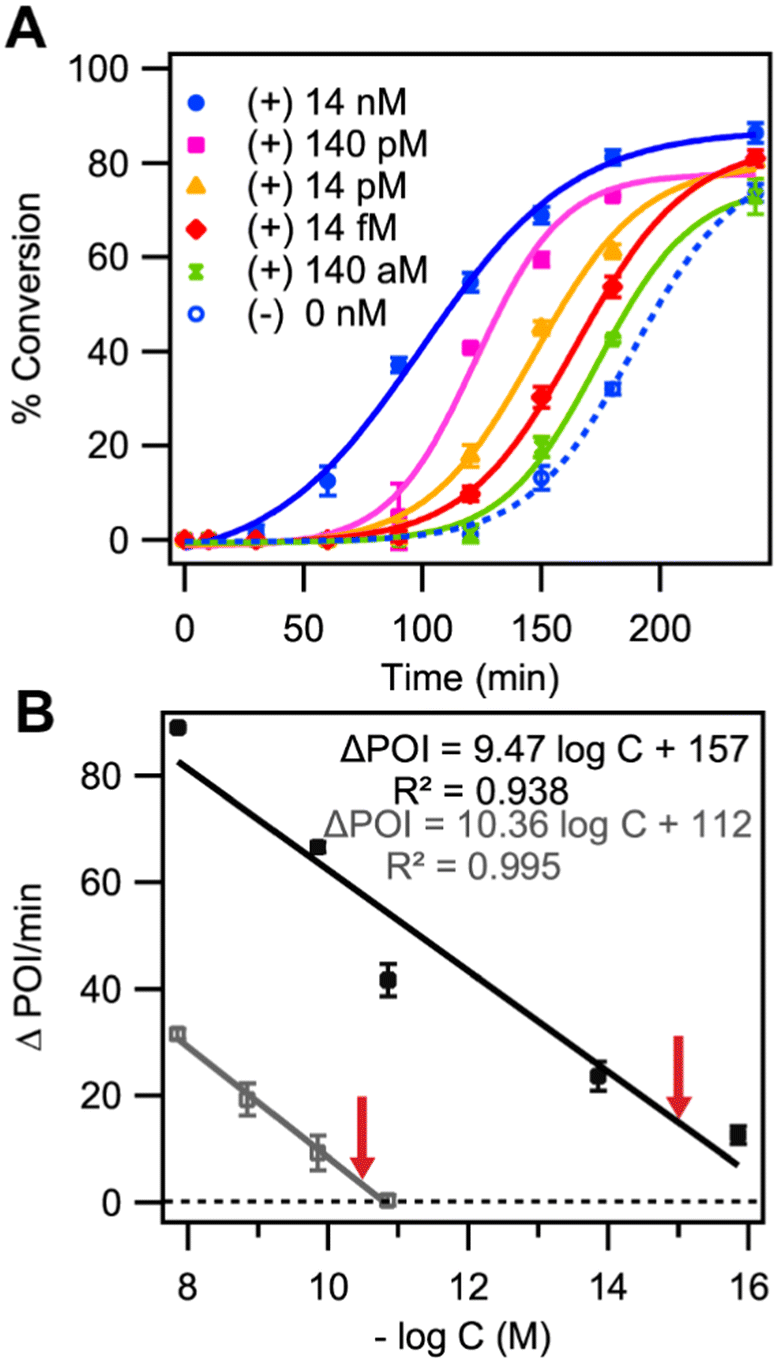 | ||
| Fig. 6 (A) Representative kinetic traces of cross-catalytic formation of DNA-I at 30 °C initiated by different concentrations of the DNA-I target with 2.5 mM Mg2+ (B) The difference in point-of-inflection for kinetic traces with and without initial target DNA-I (ΔPOI) as a function of the −log of the initial DNA-I concentration at both Mg2+ concentrations, standard 10 mM (grey) and optimized 2.5 mM (black) Mg2+. The red arrows indicate the approximate LOD for both Mg2+ concentrations, with grey and black lines representing 10 mM and 2.5 mM Mg2+ concentrations, respectively. | ||
We next considered how to quantify the differences in kinetic traces as a function of the initial DNA-I target. The utilization of time-to-threshold is a common parameter in isothermal amplification assays to determine the presence of a target.33–35 Using a related approach reported in our previous work,18 here we identified the point-of-inflection (POI) for each kinetic trace by fitting a sigmoidal curve to the data (Fig. 6A) and determining the difference in POI between the target-initiated and background-triggered amplification measured with the same enzyme master mix (Fig. 6B, black trace). The average ΔPOI from two replicates versus the –logarithm of initial DNA concentration (−logC) gave a linear trend with a line-of-best fit of ΔPOI = −9.47 ± 1.41 (−logC) + 157 ± 17 (r2-value = 0.93) (Fig. 6B, black trace). From the line of best fit, we calculated the x-intercept to be 16.58 ± 1.41, which corresponds to the −logC value when ΔPOI is equal to 0. The red arrow over the black trace in Fig. 6B marks one σ greater than the x-intercept value (−logC of 15.17), which is equal to a concentration of 680 aM, and represents a conservative estimate of the limit of detection. However, we could discriminate as low as 140 aM (−logC of 15.85) initial DNA target from the background-triggered process based on a discernible ΔPOI for both replicates (Fig. 6B-black trace). Similar analysis for the target-dependent LIDA amplification using standard (10 mM) Mg2+ concentrations prevented us from discriminating the 14 pM target from the background-triggered reaction, resulting in an approximate LOD of 55 pM (−logC of 10.26 Fig. 6B-grey trace). Thus, reducing the Mg2+ concentration led to an improvement of at least five orders of magnitude in sensitivity. These data reveal that optimizing the buffer conditions to suppress the background-triggered pseudo-blunt end process in LIDA significantly improves the assay sensitivity, underscoring the substantial impact of buffer conditions on the assay.
Conclusions
Lesion-induced DNA amplification (LIDA) represents a simple isothermal method for self-replicating 18-nucleotide DNA target sequences with an uncomplicated primer design that can be tuned to operate at room temperature making it well suited for point-of-care. Until now, this method has had a major drawback, which is the presence of a background-triggered process that limited the sensitivity of LIDA. Here we explored conditions known to minimize the rate of blunt end ligation, examining the effects of mono (Na+) and divalent (Mg2+) salt concentration as well as ATP concentration with the goal of delaying the onset of the LIDA background.The results showed that increasing ATP concentration above 1 mM and NaCl concentration above 100 mM greatly diminished the background-triggered process. However, decreasing the concentration of MgCl2 had the most significant effect on the background-triggered process while still allowing for fast target-initiated amplification. Using the optimized conditions of 2.5 mM MgCl2 enabled us to achieve a detection range of 14 nM to 140 aM, with a limit of detection of approximately 680 aM. This represents an improvement of five orders of magnitude in comparison with that of LIDA under our standard conditions. With this improvement, we have achieved detection down to clinically relevant concentrations (140 aM is 1200 copies in 15 μL) paving the way to the application of LIDA to clinical diagnostics. Although the assay demonstrates high sensitivity, the longer reaction times remain a limitation. Future work will involve testing the optimized assay at room temperature with complex, real-world samples.
Author contributions
Anantha S. Ealeswarapu: conceptualization, methodology, investigation, resources, and writing – original draft. Nahida Akter: validation of experimental data. Julianne M. Gibbs: supervision, conceptualization, and writing – review & editing.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
J. M. G. and A. S. E. gratefully acknowledge the Natural Sciences and Engineering Research Council of Canada (NSERC) for a Discovery Grant (RGPIN-2020-05976). A. S. E. gratefully acknowledges Alberta/Technical University of Munich Graduate School (ATUMS-NSERC CREATE) program for the funding. We would like to thank Ben Cobb from Ready Go Diagnostics for the valuable discussions and insights he provided during this work. We thank Gareth Lambkin for his guidance, and the University of Alberta, Department of Chemistry, Biological Services Facility for use of equipment. We thank BioRender for providing the platform used to create the figures (TOC, Fig. 2) licensed under BioRender Publication Licenses – BioRender.com/p76i257 & BioRender.com/c11b412.References
- Y. Zhao, F. Chen, Q. Li, L. Wang and C. Fan, Chem. Rev., 2015, 115, 12491–12545 CrossRef CAS PubMed.
- P. T. Monis and S. Giglio, Infect., Genet. Evol., 2006, 6, 2–12 CrossRef CAS PubMed.
- L. Garibyan and N. Avashia, J. Invest. Dermatol., 2013, 133, 1–4 CrossRef PubMed.
- M. Fakruddin, K. S. B. Mannan, A. Chowdhury, R. M. Mazumdar, Md. N. Hossain, S. Islam and Md. A. Chowdhury, J. Pharm. BioAllied Sci., 2013, 5, 245–252 CrossRef PubMed.
- P. Srivastava and D. Prasad, 3 Biotech, 2023, 13, 200 CrossRef PubMed.
- P. Craw and W. Balachandran, Lab Chip, 2012, 12, 2469–2486 RSC.
- M. M. Islam and D. Koirala, Anal. Chim. Acta, 2022, 1209, 339338 CrossRef CAS PubMed.
- M. Soroka, B. Wasowicz and A. Rymaszewska, Cells, 2021, 10, 1931 CrossRef CAS PubMed.
- V. J. Gadkar, D. M. Goldfarb, S. Gantt and P. A. G. Tilley, Sci. Rep., 2018, 8, 5548 CrossRef PubMed.
- G. T. Walker, M. S. Fraiser, J. L. Schram, M. C. Little, J. G. Nadeau and D. P. Malinowski, Nucleic Acids Res., 1992, 20, 1691–1696 CrossRef CAS PubMed.
- M. S. Reid, R. E. Paliwoda, H. Zhang and X. C. Le, Anal. Chem., 2018, 90, 11033–11039 CrossRef CAS PubMed.
- J. G. Nadeau, J. B. Pitner, C. P. Linn, J. L. Schram, C. H. Dean and C. M. Nycz, Anal. Biochem., 1999, 276, 177–187 CrossRef CAS PubMed.
- A. Kausar, R. D. McKay, J. Lam, R. S. Bhogal, A. Y. Tang and J. M. Gibbs-Davis, Angew. Chem., Int. Ed., 2011, 50, 8922–8926 CrossRef CAS PubMed.
- A. Kausar, C. J. Mitran, Y. Li and J. M. Gibbs-Davis, Angew. Chem., Int. Ed., 2013, 52, 10577–10581 CrossRef CAS PubMed.
- F. Barany, Genome Res., 1991, 1, 5–16 CrossRef CAS PubMed.
- Y. Li, X. Wang, M. Wang, M. Liu, H. Wang, W. Xia and L. Liu, Mol. Cell. Biochem., 2023, 478, 1621–1631 CrossRef CAS PubMed.
- B. S. Alladin-Mustan, C. J. Mitran and J. M. Gibbs-Davis, Chem. Commun., 2015, 51, 9101–9104 RSC.
- B. S. Alladin-Mustan, Y. Liu, Y. Li, D. R. Q. De Almeida, J. Yuzik, C. F. Mendes and J. M. Gibbs, Anal. Chim. Acta, 2021, 1149, 238130 CrossRef CAS PubMed.
- A. Kausar, E. A. Osman, T. Gadzikwa and J. M. Gibbs-Davis, Analyst, 2016, 141, 4272–4277 RSC.
- D. S. Mouliou and K. I. Gourgoulianis, Expert Rev. Respir. Med., 2021, 15, 993–1002 CrossRef CAS PubMed.
- J. Pugh, D. Wilkinson and J. Savulescu, J. Med. Ethics, 2022, 48, 329–333 Search PubMed.
- H. Park, S. Parshotam, S. C. Hales, A. K. Mittermaier and J. M. Gibbs, Chem. – Eur. J., 2023, 29, e202300080 CrossRef CAS PubMed.
- L. Ferretti and V. Sgaramella, Nucleic Acids Res., 1981, 9, 3695–3705 CrossRef CAS PubMed.
- K. Hayashi, M. Nakazawa, Y. Ishizaki and A. Obayashi, Nucleic Acids Res., 1985, 13, 3261–3271 CrossRef CAS PubMed.
- I. R. Lehman, Science, 1974, 186, 790–797 CrossRef CAS PubMed.
- A. V. Cherepanov and S. De Vries, Eur. J. Biochem., 2003, 270, 4315–4325 CrossRef CAS PubMed.
- A. V. Cherepanov and S. De Vries, Eur. J. Biochem., 2002, 269, 5993–5999 CrossRef CAS PubMed.
- R. Rossi, A. Montecucco, G. Ciarrocchi and G. Biamonti, Nucleic Acids Res., 1997, 25, 2106–2113 CrossRef CAS PubMed.
- S. El-Ashram, I. Al Nasr and X. Suo, Biotechnol. Rep., 2016, 12, 33–39 CrossRef PubMed.
- A. J. Raae, R. K. Kleppe and K. Kleppe, Eur. J. Biochem., 1975, 60, 437–443 CrossRef CAS PubMed.
- D. Y. Wu and R. B. Wallace, Gene, 1989, 76, 245–254 CrossRef CAS PubMed.
- M. R. Taylor, J. A. Conrad, D. Wahl and P. J. O'Brien, J. Biol. Chem., 2011, 286, 23054–23062 CrossRef CAS PubMed.
- B. Tian, G. A. S. Minero, J. Fock, M. Dufva and M. F. Hansen, Nucleic Acids Res., 2020, 48, e30 CrossRef CAS PubMed.
- E. J. H. Wee and M. Trau, ACS Sens., 2016, 1, 670–675 CrossRef CAS.
- J. C. Rolando, E. Jue, N. G. Schoepp and R. F. Ismagilov, Anal. Chem., 2019, 91, 1034–1042 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: DNA sequences, experimental details, graphical presentation and gel images. See DOI: https://doi.org/10.1039/d5an00047e |
| This journal is © The Royal Society of Chemistry 2025 |