
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA UOx@HMnO2 biozyme–nanozyme driven electrochemical platform for specific uric acid bioassays†
Chenlong
He‡
a,
Huawei
Liu‡
a,
Ming
Yin
a,
Jing
Chen
a,
Wensi
Huang
b,
Han
Zhou
a,
Shengming
Wu
a and
Yilong
Wang
 *a
*a
aState Key Laboratory of Cardiology and Medical Innovation Center, Shanghai East Hospital, The Institute for Biomedical Engineering & Nano Science, School of Medicine, Tongji University, Shanghai, 200092, P.R. China. E-mail: yilongwang@tongji.edu.cn
bSchool of Biomedical Engineering, Shanghai Jiao Tong University, Shanghai, 200030, P.R. China
First published on 21st February 2025
Abstract
Uric acid (UA) is a key end product of purine metabolism in the human body, and its abnormal levels are associated with many diseases, so accurate monitoring is essential. Existing detection methods have many limitations. For example, chromatography is cumbersome, time-consuming, and not cost-effective, while serum uric acid analysis requires specialized equipment and venous blood collection. In the field of uric acid sensors, electrochemical detection is commonly used but prone to interference, and nanomaterials offer improvements but are complicated to modify. To better block interference via an easily-made nanocomposite-based system, in this study, MnO2 with peroxidase-mimicking activity was used as a protective shell to encapsulate natural uric acid oxidase (UOx), realizing a good combination of nanozymes and biocatalysts. UOx can selectively catalyze UA and generate H2O2, and the MnO2 nanozymes can make up for the insufficiency of UOx, and the two main components synergistically enhance the activity of UOx@HMnO2, resulting in ultra-high performance. This provides a simple and general method for the preparation of efficient hybridized biocatalysts in the fields of biosensors and biocatalysis. The detection limit of the fabricated uric acid sensor is as low as 0.74 μM, and the concentration of the actual sample is consistent with that of mass spectrometry, which provides a means of non-invasive detection of uric acid with high sensitivity, high specificity and good accuracy.
Introduction
Uric acid (UA, C5H4N4O3) is a key end product of purine metabolism in the human body and is excreted primarily in urine to maintain homeostasis of uric acid levels in the body. In healthy individuals, serum uric acid concentrations are usually maintained between 120.0 and 520.0 μM, while urinary uric acid concentrations range from 1.4 to 4.4 mM.1,2 Abnormal uric acid levels are closely associated with a variety of diseases, including but not limited to gout, kidney disease, Lesch–Nyhan syndrome, cardiovascular disease and metabolic syndrome.3 Therefore, accurate monitoring of uric acid levels is essential for early diagnosis, effective treatment and long-term follow-up of related diseases.4 However, existing uric acid testing methods, such as high-performance liquid chromatography (HPLC) and gas chromatography (GC), are not only cumbersome and time-consuming, but also rely on specialized reagents and techniques, and are relatively less cost-effective.5–8 In addition, serum uric acid analysis relies on specialized medical equipment and involves the invasive procedure of venous blood collection.9 In contrast, urinalysis, as a non-invasive test, has the potential for rapid diagnosis and prediction of diseases in clinical settings and is therefore expected to become a new means of uric acid testing.10,11In the field of uric acid sensors, commercially available products commonly utilize electrochemical detection methods, attributed to their high sensitivity.12,13 However, conventional electrochemical methods are susceptible to interference from other molecules in complex biological fluids, which can affect detection accuracy.14 To solve this problem, the introduction of nanomaterials has significantly improved the stability of the sensors, but they still require complex modification procedures.15,16 In contrast, uricase (UOx) exhibits significant advantages in enzymatic sensing schemes due to its high specificity for uric acid and resistance to interference. However, UOx lacks metal cofactors, leading to inefficient electron transfer and consequently influencing electrochemical signal generation.17,18 To overcome this challenge, researchers typically employ a two-enzyme system strategy, whereby concomitant enzymes capable of decomposing hydrogen peroxide (H2O2), such as horseradish peroxidase (HRP), a common byproduct of the oxidoreductase reaction, are introduced to effectively eliminate their interference with uric acid detection.19,20 This approach is also applicable to UOx-based uric acid sensors. In recent years, numerous studies have reported the results of uric acid detection using the UOx–HRP dual enzyme system on different detection platforms. However, there are still some problems with this method, such as the high cost of HRP and the lack of stability of free UOx and HRP.21,22
Nanozymes, such as iron oxide nanoparticles (Fe3O4 NPs), have attracted much attention in recent years as a class of innovative nanomaterials with intrinsic enzymatic activity.23–25 Among them, MnO2 nanozymes are particularly prominent due to their excellent H2O2 decomposition mimetic enzyme activity, which can effectively catalyze the decomposition of H2O2 into water and oxygen.26,27 Compared with natural enzymes, MnO2 nanozymes not only have lower cost, but also have the advantages of tunable catalytic activity, stable structure, and unique surface chemistry as well as easy recyclability and large-scale preparation.28,29 In addition, MnO2 nanozymes are able to maintain higher catalytic activity for a longer period of time and are less prone to agglomeration or deactivation compared to gold nanoparticles and Fe3O4 NPs.30–33 Specifically structured MnO2 nanozymes, such as MnO2 nanoflowers and hollow MnO2, also exhibit highly efficient drug-carrying capacity, making them ideal carriers for biological enzymes.34 In particular, hollow MnO2 nanozymes are able to protect biomolecules from harsh environments and reduce the leakage of loadings due to their stable porous and hollow structures, which allow the transfer of substances such as electrons.35,36 This property not only improves the stability of the biological enzyme, but also promotes efficient diffusion, transfer and reaction of substrates or intermediates, which in turn improves the mass transfer efficiency and assay accuracy.37
In this study, we innovatively utilized MnO2, which has peroxidase-mimicking activity, as a protective shell to encapsulate natural uric acid oxidase (UOx), realizing an organic combination of nanozymes and biological enzymes (Fig. 1). The results showed that UOx was able to selectively catalyze the substrate uric acid (UA) and produce H2O2, while effectively reducing interference from common biomolecules such as ascorbic acid, dopamine, L-norepinephrine, glucose, lactate, etc. In addition, the MnO2 nanozymes were able to compensate for the absence of metal cofactors or concomitant enzymes in UOx to efficiently reduce H2O2. Due to the cascade effect, UOx@MnO2 exhibited an ultra-high activity, which was 4.39 times higher than that of the equivalent MnO2. Ultimately, the prepared UOx@HMnO2/Glassy Carbon Electrode (GCE)-based uric acid sensor exhibited high sensitivity and good stability with a detection limit as low as 0.74 μM. Therefore, this study may provide a simple and versatile method for fabricating efficient hybrid biocatalysts in the fields of biosensors and biocatalysis.
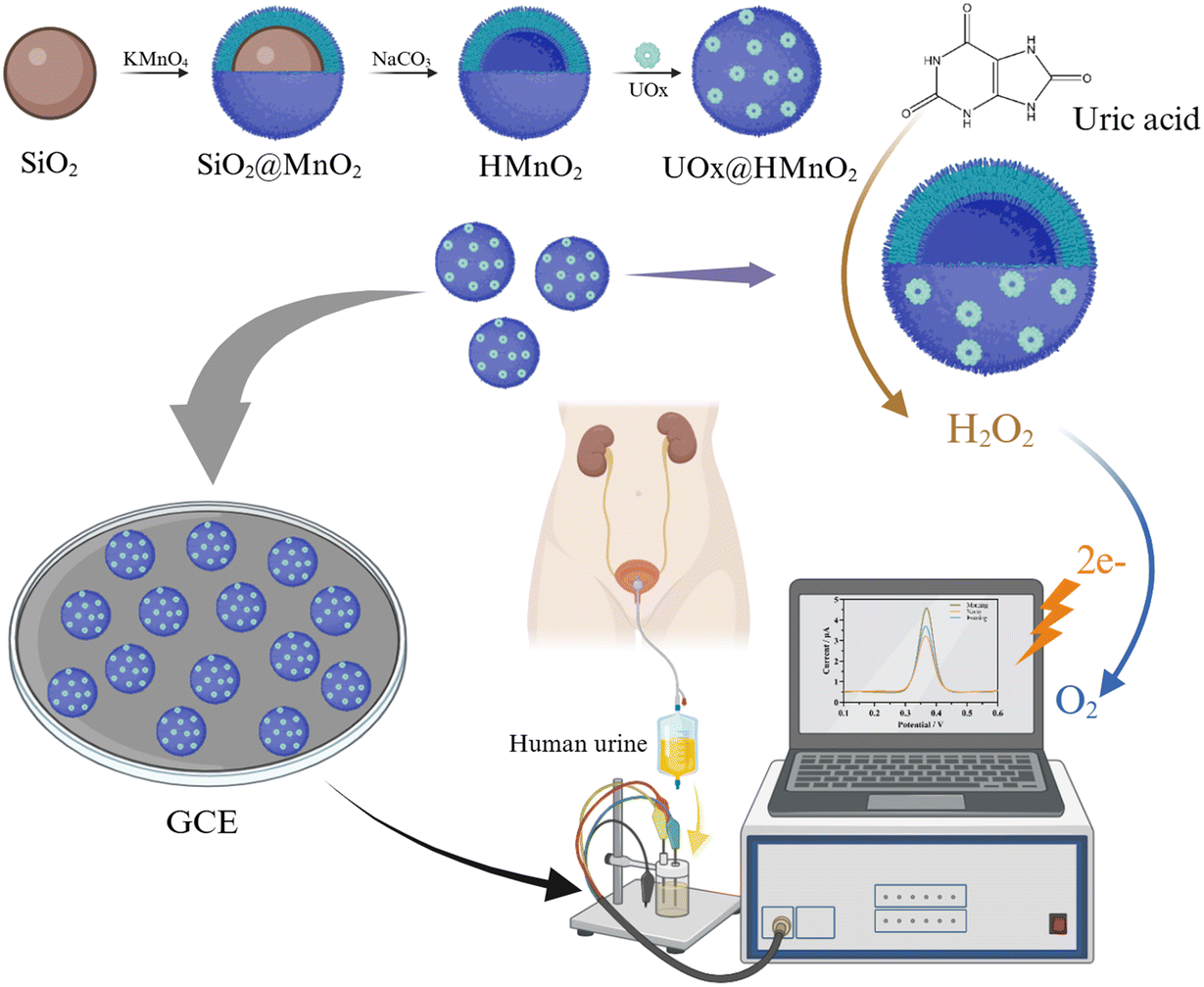 | ||
| Fig. 1 Schematic representation of the preparation and catalytic enhancement of uric acid redox signaling by the bio-enzyme–nanozyme biosensor UOx@HMnO2/GCE. | ||
Experimental
Materials
All chemicals and reagents were of the highest purity grade commercially available and used as received. Tetraethyl orthosilicate (TEOS), ethanol, hydrogen peroxide (H2O2, 30% aqueous solution), potassium permanganate (KMnO4) and sodium carbonate (Na2CO3) were purchased from Shanghai Reagent Co. (China). Uric acid, dopamine, L-ascorbic acid, potassium ferrocyanide (K4Fe(CN)6), potassium ferricyanide (K3Fe(CN)6), potassium chloride (KCl), (3-aminopropyl) triethoxysilane (APTES) and uricase were purchased from Sigma-Aldrich (USA). Uricase was purchased from Aladdin (China).Apparatus
All electrochemical measurements were performed on a CHI660E (Shanghai Chenhua Instrument Co., Ltd).Synthesis of SiO2 nanoparticles
SiO2 nanoparticles were prepared by Stöber's method: 50 mL of ethanol was added to a 100 mL flask, followed by 2 mL of deionized water and 2 mL of ammonia under magnetic stirring, and after mixing the solution, 3 mL of TEOS was added, and the reaction was continued with stirring for 6 h. Finally, SiO2 nanoparticles were obtained by centrifugation with ethanol (12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm) three times. For the amination of SiO2 nanoparticles, 500 mg of SiO2 nanoparticles were dispersed in a mixture of 45 mL of ionized water and 100 mL of ethanol, stirred for 5 min and then 0.5 mL of APTES was added to the reaction solution. After stirring for 1 h at room temperature, the reaction solution was heated to 80 °C for 2 h, and after the reaction solution was cooled down to room temperature, the SiO2–NH2 nanoparticles were obtained by centrifugal washing with ethanol 3 times.
000 rpm) three times. For the amination of SiO2 nanoparticles, 500 mg of SiO2 nanoparticles were dispersed in a mixture of 45 mL of ionized water and 100 mL of ethanol, stirred for 5 min and then 0.5 mL of APTES was added to the reaction solution. After stirring for 1 h at room temperature, the reaction solution was heated to 80 °C for 2 h, and after the reaction solution was cooled down to room temperature, the SiO2–NH2 nanoparticles were obtained by centrifugal washing with ethanol 3 times.
Synthesis of hollow manganese dioxide nanoparticles (HMnO2)
For SiO2@MnO2 nanoparticle synthesis, 200 mg of SiO2–NH2 nanoparticles were dispersed in 100 mL of deionized water and sonicated in a water bath for 10 min. Subsequently, KMnO4 (50 mL, 20 mg mL−1) solution was added dropwise to the reaction solution under ultrasonic stirring, and the reaction was continued with stirring for 12 h. SiO2@MnO2 nanoparticles were obtained by washing with deionized water 3–6 times. The prepared SiO2@MnO2 was dissolved in aqueous Na2CO3 solution (2 M) and allowed to stand at 60 °C for 6 h. Hollow manganese dioxide (HMnO2) nanoparticles were obtained after centrifugal washing with deionized water 3 times.Synthesis of UOx@HMnO2
For UOx loading, UOx (2 U) was loaded onto HMnO2 (1 mg) to assess the loading efficiency and catalytic activity.17,18,38 1 mg of HMnO2 was dispersed in 1 mL of deionized water, followed by the addition of 100 μg of UOx, and then the reaction was allowed to proceed overnight. The nanoparticles were subsequently centrifuged and washed three times to obtain UOx@HMnO2. The supernatant from each wash was collected to determine the concentration of eluted UOx, and the loading rate was determined by differential subtraction.Characterization
Dynamic light scattering (DLS) measurements were carried out using a Zetasizer Nano-ZS (Malvern, UK) with a standard 633 nm laser at 298.0 K. A JEM-2010 transmission electron microscope (TEM) was used to characterize the morphology of the nanoparticles. High-angle annular dark field scanning TEM (HAADF-STEM) images and elemental maps were obtained using a Titan Themis 60-300 G2. UV-Vis spectra of different samples were recorded using a UV-Vis spectrophotometer (Lambda 35, PerkinElmer).Preparation of electrodes
1 mg of prepared UOx@HMnO2 was uniformly dispersed in 1 mL of deionized water. 5 μL of the above solution was added dropwise onto the surface of the working electrode and dried under infrared light.Electrochemical measurements
All electrochemical measurements were performed on a CHI660E with an electrolyte of 0.1 M KCl, 5 mM [Fe(CN)6]3−/4− for Cyclic Voltammetry (CV), Electrochemical Impedance Spectroscopy (EIS) and Differential Pulse Voltammetry (DPV). A glassy carbon electrode was used as the working electrode, and a silver/silver chloride reference electrode was set as the reference electrode. The counter electrode was a platinum wire.Results and discussion
Synthesis and characterization of UOx@HMnO2
The synthesis pathway of hollow manganese dioxide nanoparticles (HMnO2) is detailed in Fig. 1.35 First, SiO2@HMnO2 composites with a shell–core structure were constructed through a well-designed synthesis strategy using SiO2 spheres with a diameter of 100 nm as a template (Fig. S1†). Subsequently, the chemical etching effect of sodium bicarbonate was utilized to precisely remove the SiO2 cores, resulting in hollow manganese dioxide nanoparticles (HMnO2) with good monodispersity. Transmission electron microscopy (TEM) observations clearly revealed a unique hollow structure (Fig. 2a). The hollow nanoparticles have a hydrodynamic diameter of about 140 nm and a zeta potential value of −30 mV, indicating excellent dispersion stability (Fig. S2a†). Then, the UOx@HMnO2 composites were successfully prepared by mixing uric acid oxidase (UOx) with HMnO2. The TEM images visualized the typical morphological features of UOx@HMnO2, and the results showed that the original homogeneous hollow structure of HMnO2 was preserved without any significant changes in the UOx loading process (Fig. 2b). In addition, the hydrodynamic diameter of the composites increased to 190 nm and the zeta potential was tuned to −20 mV (Fig. S2b†), reflecting that the successful introduction of UOx had a slight effect on the material properties. To further investigate the morphological features and elemental composition of UOx@HMnO2, high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) combined with elemental mapping was used for in-depth analysis. The results show that in the prepared UOx@HMnO2, the elements Mn and O, as well as the N element derived from UOx, present clear elemental signals (Fig. 2c). These elemental signals are also further confirmed by energy dispersive spectroscopy (EDS) recordings (Fig. S3†), demonstrating the successful preparation of hollow manganese dioxide nanoparticles (UOx@HMnO2) loaded with UOx and their structural features as expected. | ||
| Fig. 2 Fabrication and characterization of UOx@HMnO2. (a) Representative TEM images of HMnO2 and (b) UOx@HMnO2, with insets showing higher magnification images of UOx@HMnO2. (c) STEM image of UOx@HMnO2 showing the elemental distribution of Mn, O, Si and N. | ||
To further clarify the structure and chemical composition of UOx@HMnO2, we characterized UOx@HMnO2 using X-ray diffraction (XRD) and X-ray photoelectron spectroscopy (XPS). From the XRD patterns, we clearly observed that HMnO2 exhibits sharp and distinct characteristic diffraction peaks (Fig. 3a). This indicates that we successfully prepared HMnO2 with the characteristics of the crystal structure of MnO2. The XRD peaks of HMnO2 did not change significantly before and after the loading of UOx. This phenomenon indicates that the loading process of UOx did not significantly affect the crystal structure of HMnO2, i.e., the lattice structure of HMnO2 remained relatively stable. Through XPS analysis, we obtained high-resolution spectra of each element in UOx@HMnO2 and determined the presence of Mn, O, C, and N and their chemical states (Fig. 3b). Among them, the Mn 2p spectra showed typical MnO2 characteristic peaks (Fig. 3c), and the N 1s spectra helped to determine the presence of UOx in the composite (Fig. 3d).
 | ||
| Fig. 3 Structure and chemical composition of UOx@HMnO2. (a) XRD plots of HMnO2 and UOx@HMnO2. (b) XPS spectra of UOx@HMnO2. (c) Mn 2p spectrum of UOx@HMnO2. (d) N 1s spectrum of UOx@HMnO2. | ||
Catalytic properties of UOx@HMnO2
To check the payload of nanocarriers on UOx, the UV-vis absorption spectra of pure UOx, HMnO2 and UOx@HMnO2 are shown in Fig. 4a. The UV absorption peak exhibited by UOx@HMnO2 at 280 nm was identical to that of pure UOx, indicating that the UOx loading was successful. In addition, the loading of HMnO2 on UOx was calculated by determining the elution amount of UOx using the BCA method, and its payload was 1.24 w/w% (Table 1). HMnO2 loaded with UOx exhibits efficient H2 O2 conversion. In order to investigate its catalytic activity, we investigated the ability of UOx@HMnO2 to generate ·OH in 10 μM UA solution, specifically by observing the oxidation of TMB to oxTMB (Fig. S4†). This process was confirmed by the UV-Vis absorption spectra, especially the change in the absorption peak at 652 nm (Fig. 4b). These experimental results fully demonstrated that the material possesses not only excellent oxidase activity but also excellent peroxidase activity. | ||
| Fig. 4 Catalytic properties of UOx@HMnO2. (a) UV-Vis absorption spectra of free UOx, HMnO2 and UOx@HMnO2. (b) UV-Vis absorption spectra of the catalytic oxidation of TMB (oxTMB) catalyzed by 50 μg mL−1 UOx@HMnO2 and 10 μL H2 O2 in reaction buffer (pH 6.5). | ||
| Add amount (μg) | Found amount (μg) | Loading amount (μg) |
|---|---|---|
| 2.45 | 1.21 ± 0.079 | 1.24 ± 0.084 |
Although we have demonstrated that UOx@HMnO2 is capable of converting UA to –OH, the difference between its catalytic activity and that of free UOx has not been clarified. It is worth noting that UOx converts UA to H2O2,39 while UOx@HMnO2 directly generates –OH through a cascade reaction. To compare the difference in catalytic activity between the two, we used an equivalent amount of HMnO2 to convert the H2O2 produced by free UOx to –OH. We also compared the difference in the UV absorption of UOx + HMnO2 with that of UOx@HMnO2 through a TMB experiment. We found that compared to free uric acid oxidase, UOx@HMnO2 catalyzed the generation of H2O2 from uric acid with only a slight reduction in activity, but still maintained a high catalytic activity (Fig. S5†).
Electrochemical study of the prepared UOx@HMnO2/GCE electrodes
CV and EIS methods were used to analyze the electrochemical behavior of the different modified biosensors. Fig. 5a shows the CV curves with distinct redox separation peaks obtained at the GCE, HMnO2/GCE and UOx@HMnO2/GCE in the presence of 5 mM [Fe(CN)6]3−/4− containing 0.1 M KCl with a fixed scan rate of 50 mV s−1. The modification of the GCE surface with HMnO2 and UOx@HMnO2 resulted in a slight change in the peak cathodic and anodic currents compared to the GCE, which could be attributed to the “blocking” of the electrode due to the multiple modifications. However, this repulsive effect was very limited and did not significantly change the difficulty of charge transfer between the electrode surface and the [Fe(CN)6]3−/4− solution. Fig. 5b shows the Nyquist plots of the GCE, HMnO2/GCE, and UOx@HMnO2/GCE. The calculated Rct values for the three electrodes are 241.30, 706.60, and 712.53 Ω. This is attributed to the conformational change of the electrode surface due to the increase in the thickness of the modified layer. This indicates that HMnO2 and UOx@HMnO2 have been successfully immobilized on the GCE. Therefore, the fabrication of the UOx@HMnO2 electrode was completely successful. | ||
| Fig. 5 Electrochemical study of the prepared UOx@HMnO2/GCE electrodes. (a) CV plots of GCE, HMnO2 and UOx@HMnO2 electrodes in a solution containing 5 mM [Fe(CN)6]3−/4− and 0.1 M KCl. (b) EIS plots of GCE, HMnO2 and UOx@HMnO2 electrodes in 5 mM [Fe(CN)6]3−/4− indicator solution containing 0.1 M KCl. (c) CV analysis of UOx@HMnO2/GCE electrodes at different scan rates and (d) corresponding calibration plots. | ||
In 0.1 M KCl solution (pH = 7.0) containing 5 mM concentration of [Fe(CN)6]3−/4− solution at different scan rates ranging from 50 to 300 mV s−1, cyclic voltammetry (CV) was performed to deeply investigate the electron transfer properties of this redox pair at the UOx@HMnO2/GCE interface. The results showed that the peak currents (Ip) of the redox reaction, including the anodic peak current (Ipa) and cathodic peak current (Ipc),40 showed a corresponding increasing trend with the gradual increase of the scanning rate on the UOx@HMnO2/GCE electrode (Fig. 5c). In particular, the ratio of anodic (Ipa) to cathodic (Ipc) peak currents was close to 1:1 at a scanning rate of 100 mV s−1, a result that demonstrated the remarkable quasi-reversibility feature of the redox reaction within the UOx@HMnO2/GCE electrode system. In addition, as the scanning rate increased, the potential corresponding to Ipa shifted in the positive direction, while the potential corresponding to Ipc shifted in the negative direction, which led to an increase in the peak separation potential (ΔE), and further verified the quasi-reversibility of the redox process occurring on the surface of the electrodes from an experimental point of view (Fig. 5d).
UA detection using UOx@HMnO2/GCE electrodes
In order to investigate the effect of the amount of electrically modified materials on electrochemical measurements, working electrodes were assembled on GCEs with 3 μL, 5 μL and 7 μL of UOx@HMnO2, respectively. The results show that when the input is 5 μL, a more satisfactory current value and specific surface area are obtained under different CV conditions to achieve the best catalytic effect (Fig. 6a). The possible reason for this result is due to the balance between the impedance and the catalytic performance of the material immobilized on the surface of the working electrode. That is, the more material accumulated on the electrode surface, the greater the loss of the active surface of the material. Conversely, when insufficient material was deposited on the electrode surface, the catalytic capacity failed to reach its peak. Therefore, the input of 5 μL was chosen for modifying the GCE. | ||
| Fig. 6 CV plots of UOx@HMnO2/GCE in PBS solution, sweep speed: 100 mV s−1, (a) different volumes, and (b) different pH levels. | ||
Additionally, the pH of the electrolyte solution has a great influence on the performance of enzyme biosensors.41Fig. 6b shows the influence of different pH values at a fixed concentration of UA (50 μM). As the pH value decreased from 8.5 to 5.7, the peak current increased significantly, which was attributed to the fact that MnO2 had better catalytic performance under acidic conditions,42 although uricase remained stable in the wide pH range of 5.5 to 10.0 (kept at 25 °C for 16 h). Therefore, pH 5.7 was chosen as the optimal pH condition.43,44
DPV was used to study the responses of UOx@HMnO2/GCE and HMnO2/GCE electrodes to the increase in the concentration of UA (10 to 1000 μM) in a PBS solution with a pH of 5.7 at a scan rate of 100 mV s−1. The results are shown in Fig. 7a and b. The DPV curves show the increasing current response of the UOx@HMnO2/GCE and HMnO2/GCE biosensors over the entire concentration range. That is, the current values increased with increasing uric acid concentration. The HMnO2/GCE electrode also exhibited a linearly increasing oxidation peak at a +0.3 V potential, but did not respond at lower concentrations of UA (Fig. 7c). The UOx@HMnO2/GCE electrode (Fig. 7d), on the other hand, shows a clear and well-defined UA oxidation peak at a potential of +0.35 V, which tends to increase consistently with increasing UA analyte concentration, and demonstrates excellent UA detection performance. It has a stronger current response signal. The peak current (μA) versus uric acid concentration (μM) is also plotted in Fig. S6.† From this plot, it can be seen that the UOx@HMnO2/GCE electrode has two regions (linear and nonlinear), while the HMnO2/GCE electrode has a continuous linear region (Fig. 7e). Typically, the nonlinear region represents the saturation of the current response of the UOx@HMnO2/GCE sensor to high uric acid concentrations. In addition, the linear region of the sensor response was extracted and plotted on a calibration graph (peak current (μA) versus uric acid concentration (μM)) as shown in Fig. 7f. The sensor responded linearly (R2 = 0.9947) to uric acid concentrations up to 100 μM. In addition, the limit of detection (LOD) was 0.75 μM according to the 3σ rule.45,46 The good sensing performance was attributed to the strong oxidase and peroxidase catalytic activity of UOx@HMnO2/GCE.
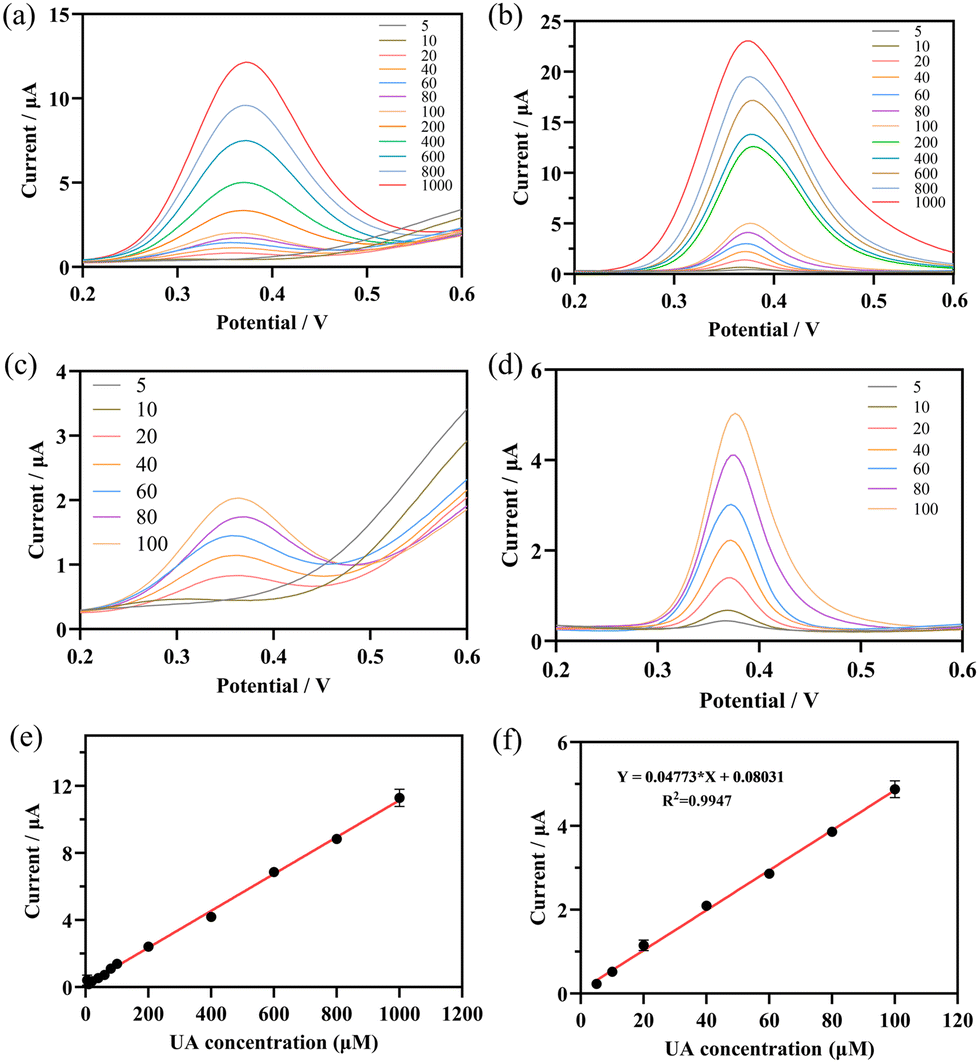 | ||
| Fig. 7 Current response of DPV to (a) HMnO2 and (b) UOx@HMnO2 modified electrodes at different concentrations of UA (10–1000 μM). Current response of DPV to (c) HMnO2 and (d) UOx@HMnO2 modified electrodes at low UA concentrations (10–100 μM) and (e and f) linear relationship between UA concentration and current. | ||
Stability and anti-interference testing
Meanwhile, to determine the stability and reproducibility of the UOx@HMnO2/GCE electrode, independent experiments were repeated on 100 μM UA. The results are shown in Fig. 8a. The RSD was 3.56%, respectively, which indicates that the electrode has good reproducibility. In the long-term storage stability test, we placed the prepared sensor at 4 °C at different time points and recorded the response current signal of the sensor. As shown in Fig. S7†, the response signal of the sensor showed only 6.43% fluctuation during the 30 day storage period, which remained within the acceptable error range, indicating that our sensor has good long-term storage stability. The specificity or anti-interference properties of the PUM were also evaluated. First, 1 mL of buffer solution was added to UOx@HMnO2/GCE, and after the electrode current was stabilized, 10 μL of 1 mM of various interfering substances were added gradually. The stabilized currents before and after the addition of the interfering substances were compared. The results in Fig. 8b show that a significant signal response can only be induced by UA, while the other control samples could not induce any significant signal response. This excellent specificity can be attributed to the UOx-assisted catalytic electrochemical sensing mechanism employed, which also ensures better immunity to interference in clinical urine samples that can be directly detected without any pre-treatment. | ||
| Fig. 8 Stability and anti-interference testing. (a) Current response values from 4 independent replicate experiments for 100 μM UA. (b) IT curve after addition of 6 interfering substances (10 μM). | ||
Detection of UA from real urine samples with UOx@HMnO2/GCE biosensors
Urine is an essential biofluid and contains many important metabolites that can convey physiological information relevant to the organism. Therefore, we evaluated the ability of the developed sensors for practical applications in urine detection. As shown in Fig. 9a, we collected real urine samples from a healthy volunteer at three time points (morning, noon, and evening). The detection of UA was carried out using the UOx@HMnO2/GCE biosensor in PBS solution at pH 5.7. The results show that DPV responds to UA in urine with clearly visible signal peaks at three different time points (Fig. 9b). The calculated UA levels detected in urine at the three time points were 3.998 ± 0.127, 2.802 ± 0.136 and 3.227 ± 0.086 μM, respectively. This trend is consistent with the results of a study by Xing et al., in which the participants in serial uric acid tests had the lowest levels of uric acid at noon (11:30 a.m.), regardless of whether they consumed high-purine and high-protein foods or followed a normal daily diet.17 The above results indicate that our prepared sensor can be used to detect uric acid in human urine under different physiological conditions. | ||
| Fig. 9 Detection of real samples with UOx@HMnO2/GCE biosensors. (a) Schematic diagram of human urine sample collection and testing. (b) Current response of DPV to the UOx@HMnO2/GCE electrode in urine collected at different time points. (c) Uric acid concentration in human urine samples was determined using high-performance liquid chromatography-tandem mass spectrometry (UHPLC-MS/MS). | ||
To further confirm the accuracy of the developed UOx@HMnO2/GCE biosensor for the detection of uric acid in urine, we compared the results with those obtained using mass spectrometry. This is due to the fact that mass spectrometry is the gold standard technology in the field of detection due to its high sensitivity and accuracy. Specifically, we determined the concentration of uric acid in real urine samples using high performance liquid chromatography-tandem mass spectrometry (UHPLC-MS/MS) (Fig. 9c). Using toluenesulfonylurea as an internal standard, chromatographic peaks from uric acid were detected (Fig. S8†), and their true concentrations were calculated from the standard curves to be 2.839 ± 0.126, 4.169 ± 0.160, and 3.185 ± 0.075 mM, respectively (Fig. 10a). Compared with our prepared UOx@HMnO2/GCE biosensor, no significant difference was found using a paired t-test (Fig. 10b). These results indicate that the method possesses comparable detection capability to mass spectrometry and can be used to accurately detect uric acid content in actual urine samples.
 | ||
| Fig. 10 (a) Calibration curve and (b) T-test analysis of uric acid concentration in real samples at three time points detected using the UOx@HMnO2/GCE electrode and UHPLC-MS/MS. | ||
Spiking recovery experiments are essential for assessing the accuracy and reliability of the assay in the analysis of real samples and can be used as a complementary validation method for the UOx@HMnO2–GCE and UHPLC-MS/MS comparison. We selected three actual samples. Known concentrations of uric acid standard (10 μM, 20 μM, and 40 μM) were added to each sample, followed by analysis using our established assay method. The results showed that the recoveries ranged from 96.00% to 103.27%, which indicated that our assay had good accuracy and reliability in the analysis of real samples and could effectively detect the real content of uric acid in the samples (Table S1†).
To make a comparison between our sensor and previously reported sensors for UA detection, as shown in Table 2, it is clear that our sensor exhibited better performance than conventional sensors in terms of the limit of detection and is able to achieve accurate detection of much smaller amounts of uric acid. However, the linear range is relatively narrow, which may be due to the fact that UOx@HMnO2–GCE has two linear regions. The concentration and trend of UA in the actual samples after being diluted were proved consistent with those measured by UHPLC-MS/MS, demonstrating the method's ability to detect a wider range of concentrations. Moreover, in terms of selectivity, UOx@HMnO2–GCE can tolerate interfering substances and maintain a stable detection performance over a longer period of time.
| Methods | Detection range | LOD | R 2 | Ref. |
|---|---|---|---|---|
| Electrochemical | 50–1000 μM | 18.7 μM | 0.998 | 18 |
| Electrochemical | 0–4 mM | 8.87 μM | 0.9933 | 17 |
| Colorimetric | 0.5–6 mM | 0.25 mM | 0.9922 | 25 |
| Electrochemical | 0–1.6 mM | 11.4 μM | 0.9898 | 15 |
| Electrochemical | 2–500 μM | 0.411 μM | 0.998 | 14 |
| Electrochemical | 100–1000 μM | 1 μM | 0.9956 | 47 |
| Electrochemical | 0.2–50 μM | 0.067 μM | 0.994 | 48 |
| Electrochemical | 0–1000 μM | — | 0.9978 | 16 |
| Colorimetric | 1–50 μg mL−1 | — | 0.9995 | 49 |
| LC-MS/MS | 0.06–30 μM | 0.15 μM | 0.9993 | 7 |
| This study | 5–100 μM | 0.74 μM | 0.9947 | — |
Conclusions
Focusing on the important field of uric acid detection, this study explores the many problems faced by existing detection methods and uric acid sensors and achieves remarkable results. The core innovation of the study is the successful construction of the UOx@HMnO2 composite system. By wrapping natural uric acid oxidase (UOx) with MnO2 nanozymes, which has peroxidase mimetic activity, as a protective shell, the organic combination of nanozymes and biological enzymes was realized. In this system, UOx can specifically catalyze the production of H2O2 from uric acid and effectively reduce the interference of common biomolecules such as ascorbic acid, dopamine, L-norepinephrine, glucose, and lactic acid, while MnO2 nanozymes make up for the lack of metal cofactors in UOx, and efficiently catalyze the decomposition of H2O2. The cascade effect produced by the synergy of the two resulted in the sensitivity of the UOx@HMnO2 uric acid sensor as high as 0.74 μM with good stability. In the future, the results of this research are expected to be further expanded and deepened in exploring the structure–property relationship of the MnO2 composite system, the expansion of material applications, and translation into clinical applications.Author contributions
Chenlong He: conceptualization, investigation, validation, and writing – original draft. Huawei Liu: investigation, validation, and writing – review & editing. Ming Yin: conceptualization, methodology, investigation, and writing – original draft. Jing Chen: conceptualization, methodology and writing – original draft. Wensi Huang: investigation, validation. Han Zhou: conceptualization and writing – original draft. Shengming Wu: writing – original draft and writing – review & editing. Yilong Wang: writing – review & editing, supervision, project administration and funding acquisition.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 32271439 and 32071395).References
- S. Wen, H. Arakawa and I. Tamai, Pharmacol. Ther., 2024, 256, 108615 Search PubMed.
- L. Zheng, H. Ma, Y. Ma, Q. Meng, J. Yang, B. Wang, Y. Yang, W. Gong and G. Gao, Int. J. Electrochem. Sci., 2019, 14, 9573–9583 CrossRef CAS.
- L. Du, Y. Zong, H. Li, Q. Wang, L. Xie, B. Yang, Y. Pang, C. Zhang, Z. Zhong and J. Gao, Signal Transduction Targeted Ther., 2024, 9, 212 Search PubMed.
- Y. Zhao, T. Ye, H. Liu, Y. Kou, M. Li, Y. Shao, Z. Zhu and Q. Zhuang, Front. Biosci.-Landmark, 2006, 11, 2976–2982 Search PubMed.
- X. Luo, N. Cai and Z. Cheng, Anal. Sci., 2013, 29, 709–713 CrossRef CAS PubMed.
- W. Huang, J. Yu and X. Diao, J. Pharm. Biomed. Anal., 2023, 224, 115188 Search PubMed.
- X.-Y. Liu, Y. Luo, C.-Y. Zhou, A. Peng and J.-Y. Liu, Bioanalysis, 2017, 9, 1751–1760 CrossRef CAS PubMed.
- W. Huang, W. Li, X. Yu, M. Xue, Y. Yuan, C. Chen, Y. Wu, J. Yu and X. Diao, J. Chromatogr. B:Anal. Technol. Biomed. Life Sci., 2024, 1234, 123991 Search PubMed.
- Y. Zhang, H. Yu, S. Chai, X. Chai, L. Wang, W.-C. Geng, J.-J. Li, Y.-X. Yue, D.-S. Guo and Y. Wang, Adv. Sci., 2022, 9, 2104463 CrossRef CAS PubMed.
- S. Yeasmin, G. Ammanath, A. Onder, E. Yan, U. H. Yildiz, A. Palaniappan and B. Liedberg, TrAC, Trends Anal. Chem., 2022, 157, 116786 CrossRef CAS.
- N. Balhara, M. Devi, A. Balda, M. Phour and A. Giri, Urine, 2023, 5, 40–52 CrossRef.
- L. Chelmea, M. Badea, I. Scarneciu, M. A. Moga, L. Dima, P. Restani, C. Murdaca, D. Ciurescu and L. E. Gaman, Chemosensors, 2023, 11, 341 Search PubMed.
- L. Hao, X. Li, H. Liang, W. Lei, W. Yang and B. Zhang, Microchim. Acta, 2024, 38, 191, DOI:10.1007/s00604-023-06113-2.
- Y. Liu, Y. Zhang, C. Wang, X. Zeng, J. Lei, J. Hou, D. Huo and C. Hou, ACS Appl. Nano Mater., 2024, 7, 6273–6283 CrossRef CAS.
- R. Stephanie, D. Y. Lee, C. Y. Park and T. J. Park, Analyst, 2023, 148, 1442–1450 Search PubMed.
- S. Masrat, V. Nagal, M. Khan, I. Moid, S. Alam, K. S. Bhat, A. Khosla and R. Ahmad, Biosensors, 2022, 12, 1140 CrossRef CAS PubMed.
- X. Xing, B. Yao, Q. Wu, R. Zhang, L. Yao, J. Xu, G. Gao and W. Chen, Biosens. Bioelectron., 2022, 198, 113804 Search PubMed.
- S. H. Han, Y.-J. Ha, E. H. Kang, K. Shin, Y. J. Lee and G.-J. Lee, Sci. Rep., 2022, 12, 12033 CrossRef CAS PubMed.
- Y. Zhang, S. Tsitkov and H. Hess, Nat. Catal., 2018, 1, 276–281 CrossRef.
- A. Chellachamy Anbalagan and S. N. Sawant, Microchim. Acta, 2021, 11, 188, DOI:10.1007/s00604-021-05059-7.
- M. Dai, T. Huang, L. Chao, Q. Xie, Y. Tan, C. Chen and W. Meng, Talanta, 2016, 149, 117–123 CrossRef CAS PubMed.
- Y. Jiao, Y. Xing, K. Li, Z. Li and G. Zhao, Luminescence, 2021, 36, 1110–1116 CrossRef CAS PubMed.
- W. Yang, X. Yang, L. Zhu, H. Chu, X. Li and W. Xu, Coord. Chem. Rev., 2021, 448, 214170 CrossRef CAS.
- M. Yin, L. Zhang, Y. Chen, S. Qi, X. Wei, X. Tian, J. Qiu and D. Xu, Mater. Chem. Phys., 2022, 282, 125992 CrossRef CAS.
- T. Leelasattarathkul, T. Trakoolwilaiwan and K. Khachornsakkul, Analyst, 2024, 149, 5518–5526 RSC.
- S. Sisakhtnezhad, M. Rahimi and S. Mohammadi, Biomed. Pharmacother., 2023, 163, 114833 CrossRef CAS PubMed.
- B. Song, Z. Wang, H. Yan, X. Zhang, Q. Liu, J. Luo and J. Yuan, Analyst, 2024, 149, 3547–3554 Search PubMed.
- A. M. Ashrafi, Z. Bytesnikova, J. Barek, L. Richtera and V. Adam, Biosens. Bioelectron., 2021, 192, 113494 Search PubMed.
- M. Li, T. Zhang and Y. Zhang, Analyst, 2023, 148, 2180–2188 RSC.
- H. Yuan, J. Chen, Y. Guo, H. Wang and P. Hu, J. Phys. Chem. C, 2018, 122, 25365–25373 CrossRef CAS.
- S. Xiong, R. Tang, D. Gong, Y. Deng, C. Zhang, J. Zheng, M. Zhong, L. Su, L. Yang and C. Liao, Appl. Mater. Today, 2020, 21, 100798 CrossRef.
- Y. Wang, F. Wang, B. Chen, H. Xu and D. Shi, Chem. Commun., 2011, 47, 10350–10352 RSC.
- H.-Y. Xia, B.-Y. Li, Y. Zhao, Y.-H. Han, S.-B. Wang, A.-Z. Chen and R. K. Kankala, Coord. Chem. Rev., 2022, 464, 214540 Search PubMed.
- Q. Yang, Y.-Y. Mao, Q. Liu and W.-W. He, Rare Met., 2023, 42, 2928–2948 CrossRef CAS.
- H. Ji, C. Zhang, F. Xu, Q. Mao, R. Xia, M. Chen, W. Wang, S. Lv, W. Li and X. Shi, Adv. Sci., 2022, 9, 2201696 Search PubMed.
- C. Lin, H. Zhang, X. Zhang, Y. Liu and Y. Zhang, ACS Appl. Mater. Interfaces, 2023, 15, 14388–14398 CAS.
- G. Yang, L. Xu, Y. Chao, J. Xu, X. Sun, Y. Wu, R. Peng and Z. Liu, Nat. Commun., 2017, 1, 8, DOI:10.1038/s41467-017-01050-0.
- S. A. A. Noma, Process Biochem., 2023, 131, 52–58 CrossRef CAS.
- Y.-C. Chiu, T.-S. Hsu, C.-Y. Huang and C.-H. Hsu, Int. J. Biol. Macromol., 2021, 188, 914–923 Search PubMed.
- Q. Yan, N. Zhi, L. Yang, G. Xu, Q. Feng, Q. Zhang and S. Sun, Sci. Rep., 2020, 10, 10607 Search PubMed.
- W. Liu, D. Zhang, F. Zhang, Z. Hao, Y. Li, M. Shao, R. Zhang, X. Li and L. Zhang, Analyst, 2023, 148, 1579–1586 Search PubMed.
- M. Wekesa and Y. H. Ni, Tappi J., 2003, 2, 23–26 Search PubMed.
- D. Iswantini, N. Nurhidayat, T. Trivadila and O. Widiyatmoko, Pak. J. Biol. Sci., 2014, 17, 277–281 Search PubMed.
- D. Iswantini, N. Nurhidayat, T. Trivadila and E. Mardiah, J. Ilmu Pertanian Indones., 2009, 14, 163–169 Search PubMed.
- J. Chen, L. Hao, J. Hu, K. Zhu, Y. Li, S. Xiong, X. Huang, Y. Xiong and B. Z. Tang, Angew. Chem., 2022, 7, 134, DOI:10.1002/ange.202112031.
- J. Chen, L. Hao, Y. Wu, T. Lin, X. Li, Y. Leng, X. Huang and Y. Xiong, Chem. Commun., 2019, 55, 10312–10315 RSC.
- M. Yang, H. Wang, P. Liu and J. Cheng, Biosens. Bioelectron., 2021, 179, 113082 CrossRef CAS PubMed.
- G. Ren, F. Dong, Z. Zhao, K. Li and Y. Lin, ACS Appl. Mater. Interfaces, 2021, 13, 52987–52997 CrossRef CAS PubMed.
- Y. Bai, X. Fan, G. Chen and Z. Zhao, Anal. Methods, 2024, 16, 5896–5901 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Other TEM, DLS, EDS, TMB, optimization and mass spectrometry data. See DOI: https://doi.org/10.1039/d4an01512f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |