
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Assessing the purity of model glycoconjugate vaccines by low field NMR†
Victoria
Leadley
 a,
David
Egan
b,
Kelly
Sackett
*c and
Simon B.
Duckett
*a
a,
David
Egan
b,
Kelly
Sackett
*c and
Simon B.
Duckett
*a
aCentre for Hyperpolarisation in Magnetic Resonance, Department of Chemistry, University of York, UK. E-mail: simon.duckett@york.ac.uk
bGlobal Technology and Engineering (GT&E), Manufacturing Science and Technology Process Development (MSAT-PD), Grange Castle, Clondalkin, Co., Dublin, Ireland
cAnalytical Research and Development, Pfizer Research and Development, 1 Burtt Road, Andover, MA, 01810, USA. E-mail: Kelly.Sackett@pfizer.com
First published on 12th March 2025
Abstract
Glycoconjugate vaccines are of growing importance to modern healthcare where they provide an opportunity for high efficacy prophylactic treatment against a growing number of infectious bacterial diseases. Unfortunately, their preparation is highly complex and involves multiple stages of analysis prior to product release. Such analyses must quantify the degree of successful conjugation and the amount of relevant co-expressed and co-purified process impurities (i.e. cell-wall polysaccharide). Whilst nuclear magnetic resonance (NMR) spectroscopy can be used for these assessments, the cost of high field systems is significant and hence there is a need to evaluate the performance of low-cost benchtop apparatus. Here, we set a goal of achieving a satisfactory analysis within 20 min on a series of model glycoconjugates and sought to use hyperpolarization methods based on signal amplification by reversible exchange (SABRE) to enable higher sample throughput. Our analyses demonstrate that a 1 Tesla (T) benchtop NMR can achieve satisfactory dextran-conjugation analysis results without the need for hyperpolarization, although SABRE hyperpolarization offers a route to improvement. The assessment of the common impurity cell-wall polysaccharide proved more challenging, and its hyperpolarization failed due to the necessary solvent system. At high field satisfactory analyses were possible at 10 wt%, 5 wt%, 1 wt% and 0.5 wt% loadings where the resulting signals are distinguishable. However, at 1 T signal overlap precluded simple signal integration and a T1 filter was implemented. This allowed the overlapping signal contributions to be differentiated and made quantification possible for the 10 wt% sample where signal to noise ratios remained high.
Introduction
Glycoconjugate vaccines have been proven to produce a stronger immune response than polysaccharide vaccines alone1 whilst evoking a much-improved immune response in infants and the elderly.2 They also have an excellent safety record.3 Their formation involves linking a polysaccharide antigen, typically produced by bacterial fermentation, to a carrier protein, which collectively provides multiple triggers for the immune system.4 To date, fully licenced glycoconjugate vaccines exist for humans that target Haemophilus influenzae (Haemophilus influenzae), Meningococcus (Neisseria meningitidis) serogroups ACWY, alongside 20 serotypes of bacterial pneumonia(Streptococcus pneumoniae), and typhoid fever (Salmonella typhi),5 although many others are in development.6 The importance of this clinical pathway is reflected in the predicted market worth of these vaccines, which is greater than $10 billion.4Unfortunately, current production methods for these vaccines involve long, and complicated multi-step processes where polysaccharide purification from the pathogenic organism and purification of the carrier protein, reflect the most challenging steps.7,8 These steps are followed by chemically activating the polysaccharide, and then conjugating it to the protein through covalent bonds, each stage needing further purification. Most modern glycoconjugate vaccines utilize either reductive amination or cyanylation chemistry to chemically conjugate polysaccharides to carrier proteins. It is necessary to use these chemical platforms as most polysaccharide structures do not contain a convenient chemical handle for conjugation. The first reported use of cyanylation for the production of conjugate vaccines used cyanogen bromide (CNBr), which requires a relatively high pH and is difficult to work with owing to its inherent toxicity.9 A report for the use of 1-cyano-4-dimethylaminopyridinium tetrafluoroborate (CDAP) as an alternative cyanylating reagent was published by Lees et al. in 1996. It offered significant advantages in terms of handling and processing conditions. Furthermore, unlike CNBr, CDAP activated polysaccharides can be directly bound to proteins, dramatically simplifying processing.10 Emphasizing the importance of this synthetic pathway, there have been several follow-up publications from Lees and other authors, as recently as 2024, that have offered significant improvements on their first publication. These included the use of alternative bases, which limits the possibility of pH fluctuations, and optimized temperature conditions.11 Although there have been multiple new novel designs for chemical conjugation, including N-hydroxysuccinimide (NHS) activation and lysine activated iso-thiocyanates, among many others, the use of CDAP activation chemistry is still highly prevalent in commercial glycoconjugate vaccines. It is currently used in GSK's pneumococcal conjugate vaccine (Synflorix®) and Pfizer's meningococcal conjugate vaccine (Nimenrix®). Furthermore, a modified version has even been incorporated into one of the most novel conjugation platforms currently being evaluated in industry; the Multiple Antigen Presenting System (MAPS).12 Affinivax labs (GSK) are currently developing a novel 24-valent pneumococcal vaccine using a CDAP-constructed MAPS platform that has shown promising results in Phase 2 clinical trials.13 This novel utilization, in tandem with the longevity and incremental improvements that have been published since CDAP chemistry was first described is the basis for our current investigation into the application of low-field NMR to better understand this chemical pathway.
Consequently, the protracted manufacturing process produces batch to batch variations which leads to time-consuming and costly analysis.4 Analytical techniques are also required to test for a diverse range of potential impurities including host cell proteins, charged protein species and residual process components, such as unconjugated polysaccharide.14 An example manufacturing process for Pfizer's Prevnar 13 pneumococcal vaccine requires 700 distinct quality control tests.4 Cell-wall polysaccharide (C-poly), a common, cell-wall-associated polysaccharide antigen, is an impurity that continues to be present alongside pneumococcal polysaccharide during such preparations even after many years of manufacturing development.15 Because C-poly can consume the reagent used to activate the target polysaccharide, and can potentially conjugate to carrier proteins, it's level must be controlled and quantified. New cheaper and faster ways of quantifying this impurity in glycoconjugate vaccine preparations would reflect a significant benefit for manufacturing companies. However, this task is not straightforward due to the similar nature of the C-poly to the product of interest.
One method of analysis that stands out in the identification and assessment of polysaccharide molecules is high-field NMR spectroscopy which benefits from good resolution and sensitivity. However, this technique is expensive, largely due the equipment cost and requires on-going cryogenic cooling. Consequently, a rapid lower cost low-field NMR analysis pathway using cost-effective stable benchtop magnets that don't require cryogenic cooling is highly desirable. Low intrinsic sensitivity and the lower spectral resolution of low-field NMR instruments would suggest this route to be unfeasible without some form of signal enhancement. Several techniques are available to overcome this sensitivity problem. One such technique is Dynamic Nuclear Polarisation (DNP), which uses unpaired electron spins at liquid helium temperatures to boost NMR signals after polarisation transfer and dissolution.16 An alternative and more rapid room temperature method based on parahydrogen (pH2), called SABRE (Signal Amplification by Reversible Exchange) is finding growing utility in chemical analysis.17 SABRE works by catalytically improving NMR signal strengths through interactions with protons derived from parahydrogen in seconds according to the pathway outlined in Scheme 1.
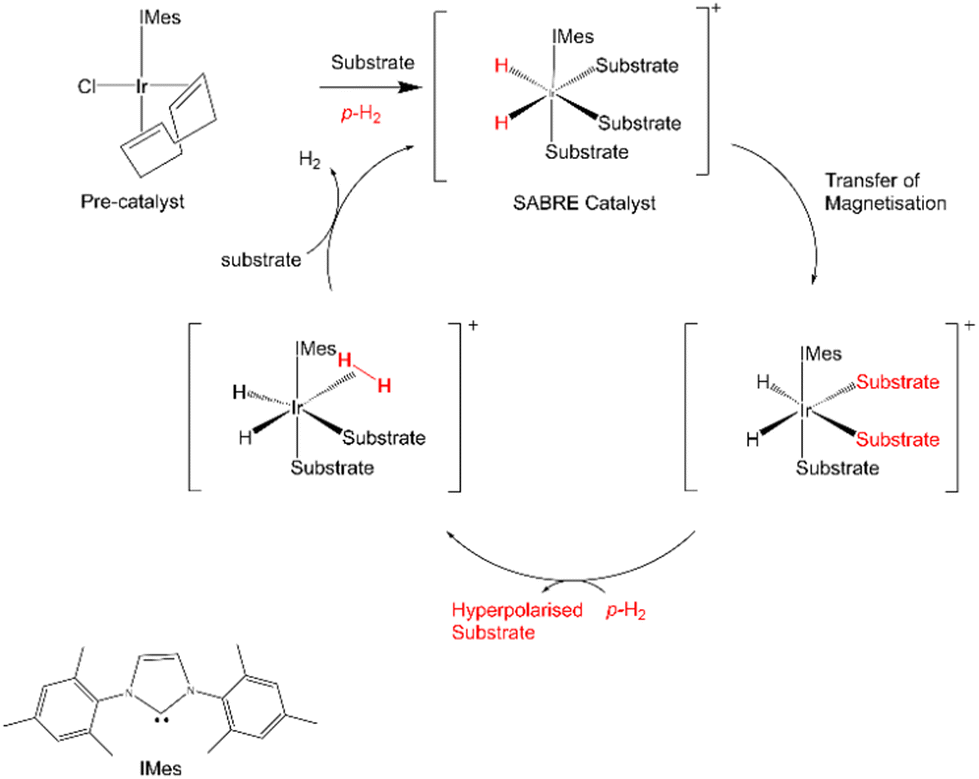 | ||
| Scheme 1 Schematic details of the SABRE process where in molecular catalysis amplifies the NMR response of a target substrate by magnetisation transfer. | ||
NMR has been used to assess protein–ligand binding during the drug discovery process for many years.18 It can be used for screening ligand binding and the determination of binding epitope structures.19 However, due to insensitivity of the technique, large concentrations of protein and ligand are needed, which are greater than physiological concentrations.20 Hyperpolarization has been proven to offer improved insight where for example it has demonstrated the protein–ligand interactions in trypsin,20 quantified Fentalogues,21 and been used to probe amino acid reactions.22
We aim here to show that SABRE can be used to improve our ability to analyse glycoconjugates by NMR spectroscopy at both high (400 MHz, 9.4 Tesla; 500 MHz, 11.7 Tesla) and low field (43 MHz, 1 Tesla). Our studies target dextrans as model polysaccharides due to the fact they are readily available at 6 kDa, 40 kDa and 250 kDa sizes, which reflect those seen in the bacterial polysaccharides used in glycoconjugate vaccines. The dextrans are activated for conjugation by the addition of 1-cyano-4-dimethylaminopyridine (CDAP), which acts as a source of CN+ and converts a hydroxyl group into a reactive cyanoester.10 Subsequent trapping of the resulting cyanoester by isonicotinic acid hydrazide (INAH) is then used to model glycoconjugate formation (Scheme 2). The resulting products were analysed to determine the extent of derivatization by standard NMR techniques and SABRE hyperpolarization, with their INAH aromatic proton resonances proving to be well resolved from the aliphatic dextran resonances and therefore excellent handles for tracking this process.
 | ||
| Scheme 2 Dextran polysaccharide derivatization using 1-cyano-4-dimethylaminopyridine (CDAP) and subsequent reaction with an INAH hydrazide linker is used to create a mimic for the carrier protein that is used in glycoconjugate vaccine preparations. CDAP derivatization involves substrate dextran hydroxyl groups, and while representative position 4 dextran derivatization is shown, site specificity and selectivity is not elucidated. | ||
Experimental
The NMR experiments described in this work were completed on Magritek 1 T Spinsolve Carbon, Bruker Avance III 400 MHz (9.4 T, with a 5 mm BBI probe) and Bruker 500 MHz (11.7 T) NMR spectrometers (5 mm BBI probe). Dextrans of molecular weight 6 kDa (sourced from Sigma), 40 kDa and 250 kDa (sourced from Alfa Aesar), Isonicotinic acid hydrazide (INAH, Acros Organics), and the deuterated solvents D2O (Sigma), DMSO-d6 (Sigma) and DMF-d7 (Apollo Scientific) were used without further purification. For the NMR measurements an internal standard, the sodium salt of 3-(trimethylsilyl)propionic-2,2,3,3,-d4 acid (TSP, Merck) at a 0.0278 M concentration was employed for quantification. Purification of the model glycoconjugates was achieved by using Slide-a-lyzer dialysis cassettes with a 2000 Da molecular weight cut-off sourced from Thermo-Scientific. A Labconco FreeZone 2.5 L −50 °C freeze dryer was used for lyophilisation.Synthesis of the glycoconjugate models
The three dextrans and INAH were linked using a 1-cyano-4-dimethylaminopyridine (CDAP, Fluorochem) derived reductive amination according to a variation on the published method.23 This necessitated the use of a solution of 50 mg of the dextran in 5 ml of deionized water. The solution was stirred vigorously at 0 °C and a further solution containing 50 mg of CDAP in 0.5 ml of CH3CN was added slowly. Once this addition was complete, stirring was halted and the solutions pH measured (pH 7). An aliquot of 0.1 M NaOH was then added to increase the pH to 9. The resulting solution was then stirred for 3 minutes before 1 ml of 0.5 M aqueous INAH was added. As the measured pH was found to lie between 8 and 9 no further NaOH was added. This solution was then stirred for 15 min before being placed in a fridge overnight.At this stage, the solution was removed from the fridge and placed in a pre-hydrated Slide-a-Lyzer dialysis cassette. The loaded cassette was then placed in phosphate-buffered saline (Fisher bioreagents) and left for 24 h; the buffer solution was changed 4 times during this period. In the last dialysis step, deionized water replaced the buffer. The resulting solutions were lyophilized for ∼24 hours at 0.133 mbar and 223 K. Typically ∼55 mg of product resulted.
SABRE detection
The hyperpolarisation method SABRE involves the transfer of signal enhancing spin order from parahydrogen into a substrate whilst they are both bound to an iridium metal centre.24 The process of spin order transfer is mediated by the spin–spin coupling network of the complex and occurs optimally when their interaction takes place in a specific magnetic field.25 For transfer between protons this magnetic field is ∼60 G.26 In practice, this process is achieved by taking a solution of a SABRE precatalyst, like IrCl(COD)(IMes), and the analyte to be sensitised and exposing it to parahydrogen gas. A rapid reaction takes place to form the active catalyst. The resulting NMR tube is then shaken in an ∼60 G field for 10 seconds under fresh parahydrogen, and sensitisation of the nuclear spins of the analyte is expected. The enhanced NMR signals are then readout in an appropriate spectrometer to aid analysis.27Results and discussion
Demonstration of dextran limits of quantification (LOQ)
In order to benchmark the subsequent hyperpolarization data, we first set out to determine the limits of quantification (the lowest concentration of an analyte that can be reliably measured by a particular measurement procedure) using standard NMR procedures at both high and low field. This process involved a series of control measurements at 298 K on samples of the free 6 kDa dextran. These samples were prepared by taking 100 mg of dextran in 5 ml of D2O, and then using this stock solution to prepare five standard solutions each containing the internal standard TSP at 0.0278 M, and dextran concentrations ranging from 55.6 mM to 11.1 mM. These solutions were then analysed at both 9.4 T and 1 T by 1H NMR spectroscopy. Quantification relative to the internal standard was achieved at high field by reference to the area of signals for the 1 dextran CHO glucose anomeric proton at 4.9 ppm (T1 1.065 s, interscan delay 20 s), whilst the six broad CH signals clustered around 3.6 ppm (T1 0.132 s, interscan delay 20 s), and representing positions 2–6 (glucose methine and methylene resonances) were used at 1 T. As expected, the response from both spectrometers proved to vary linearly with concentration, with both providing similar accuracy as the R2 fitting values were 0.9995 and 0.9999 respectively.Following this step, the preparation methods accuracy was assessed. This involved taking three separate solutions, prepared in triplicate by stock solution dilution, aiming for absolute loadings of 11.11 mM, 33.3 mM and 55.6 mM respectively. These solutions were analysed by four scan high field NMR measurements and the results compared with the predicted values. The 9.4 T analysis results displayed a % Recovery (i.e. the result obtained compared to the predicted value of the sample) of 96.2–109%, whilst for the corresponding 16 scan 1 T measurements the value was 97.9–115.8%. Furthermore, the concentration repeatability measures for these triplicates were 2.8%, 0.7% and 0.8% for the 11.11 mM, 33.34 mM and 55.6 mM samples respectively at 9.4 T. This testing strategy is aligned with the recommended ICH qualification regimen for repeatability of a minimum of 9 determinations across three concentrations (ICH Q2(R2) & Q14 Guidelines (page 15)).28 The results obtained all lie within the chosen acceptance criterion for repeatability of <5%. The corresponding %RSD values for the corresponding 16-scan low field measurements were 3.3%, 2.0% and 1.8% respectively and hence, the resulting data is of comparable precision to the high field measurements.
A series of further dilution measurements were conducted to determine the LOQ, with a threshold of less than 15%RSD between concentration measurements being used to define this value alongside a %Recovery value of between 80 and 120. In order for the resulting control signal to have comparable intensity, a lower concentration of TSP (2.78 mM) was used. The corresponding sample with 5.56 mM loading yielded an %RSD of 1.8 and a %Recovery of 106.1 at low field to define this limit; a 2.778 mM measurement produced an %Recovery of 120.04. The low field results were obtained by averaging data across four 16 scan measurements on the same sample; these were collected using a 90° excitation pulse, a relaxation delay of 20 s, a receiver gain of 28 and a spectral width of 5000 Hz. Each of these measurements took ∼5 min.
The corresponding LOQ measurements at high field were selected to employ 4 scans, a receiver gain of 144 and a spectral width of 8012 Hz with identical relaxation delays and pulse angle; measurements now took ∼1 min. The same sample concentrations were analysed and the new LOQ defined as 0.56 mM, with a 11.5%RSD and 104%Recovery.
Determination of the substrates LOQ is relevant for understanding the feasibility of quantitative NMR analysis, particularly when moving from higher sensitivity and field to lower sensitivity and field. Importantly, purified polysaccharide drug substances used for downstream activation and conjugation in conjugate vaccine development can be found at concentrations of ∼3 to 7 mM based on the repeat unit. These data confirm therefore that polysaccharide concentrations typical of those found during conjugate drug manufacture lie within the accessible quantification range available to 1 T NMR analysis, albeit under thermal conditions through 4 × 5 minutes of triplicate measurement. Accessing lower LOQ by increasing the measurement time to ranges typical for NMR release assays for polysaccharide drug substances (60 to 90 minutes), would also be possible and generate a corresponding decrease in LOQ by ∼3.5–4.25-fold at low field and ∼7.25–9.5-fold at high field from these values but at a further cost in instrument time.
As commented on earlier, the hydrazide functionality can be used to introduce linker groups between polysaccharides and carrier proteins for the manufacture of conjugates with specific physico-chemical properties, the feasibility for SABRE polarization enhancement of hydrazides and CDAP activated dextran with subsequent INAH hydrazide derivatization (refer to Scheme 2) was therefore investigated in order to test whether any improvement in the accuracy and speed of data collection was possible.
SABRE analysis of uncoupled INAH
This aspect of the project started out by considering the signals for free INAH itself and demonstrating that it was amenable to SABRE, whose efficiency is sensitive to the lifetime of the catalyst and hence the duration of the spin–spin interactions between the hydride ligands and bound substrate.Consequently, the precatalyst IrCl(COD)(IMes) was dissolved with INAH in methanol-d4, (MeOD) and examined under parahydrogen by 1H NMR spectroscopy at 11.7 T after transfer from a 60 G field.29 As expected hydride ligand signals were seen for the SABRE active catalyst between −21 and −27 ppm, alongside an aromatic proton signal for INAH at δ 8.7 (referenced to MeOD at 3.3 ppm) which was 300 times larger than that observed when the corresponding thermally polarised NMR spectrum was recorded.
Hence, INAH was confirmed to be SABRE active and potentially suitable for probing dihydrazide linkers located on activated polysaccharide drug-substance intermediates. However, as the dextran itself is insoluble in methanol, validation of any SABRE activity would need to take place in a solvent suitable for dextran solubilization. Unfortunately, due to low H2 solubility in water, SABRE is not normally conducted in this solvent.30 Solubility screening resulted in the identification of a solvent mixture comprising of 50 μl of D2O, 10 μl of DMSO-d6 and 600 μl of DMF-d7 as potentially suitable. When SABRE was repeated in this medium on a fresh INAH sample, the resulting NMR signal gain for the analogous δ 8.9 signal rose to 500-fold, a better result than that achieved in methanol alone. The role of DMSO-d6 in these solutions is two-fold, it promotes reagent solubility and acts as a co-ligand to change the SABRE catalysts identity (to Ir(H)2Cl(DMSO)(INAH)(IMes)) and this acts to boost SABRE efficiency.31,32
The efficiency of SABRE is also known to depend on the substituents located on the N-heterocyclic carbene (NHC) ligand of the catalyst.33 We therefore evaluated the level of signal gain seen in the signals for free INAH as a function of the five precatalysts detailed in Fig. 1 and 2 in the presence of DMSO-d6. The results are shown in Table 1, and reveal that all five catalysts were effective in generating at least 2-orders of magnitude signal enhancement in the INAH ortho-proton signals at 11.7 T and 1 T. In this instance, the largest signal gains resulted from use of the IMes-d22 derived catalyst (610-fold) at 11.7 T, with the IMes-d22 and NMe-IMes catalysts showing similar performance at 1 T.
 | ||
| Fig. 1 Structures, and compound labels, for the five pre-catalysts used here to evaluate the level of 1H NMR signal gain seen with free INAH under SABRE. | ||
 | ||
| Fig. 2 Typical single scan 1H NMR spectra, recorded under SABRE conditions, showing the relative signal strengths and hence gains delivered by the indicated catalyst precursors (see Fig. 1) for INAH detection at 11.7 T after transfer from a 60 Gauss field using 3 bar of parahydrogen and a polarization transfer time of 10 s. | ||
| SABRE precatalyst identity | 11.7 T signal intensity increase (fold) | T 1 value 11.7 T (s) | 1 T signal intensity increase (fold) | T 1 value 1 T (s) |
|---|---|---|---|---|
| IMes (no DMSO) | 310 | 4.29 ± 0.06 | 2230 | 4.12 ± 0.05 |
| IMes | 500 | 4.09 ± 0.04 | 2170 | 2.76 ± 0.05 |
| ClIMes | 310 | 4.25 ± 0.04 | 1850 | 4.99 ± 0.06 |
| NMeIMes | 470 | 4.26 ± 0.05 | 2470 | 3.51 ± 0.02 |
| IMes-d22 | 610 | 4.33 ± 0.09 | 2440 | 4.72 ± 0.09 |
| IPrCl | 370 | 4.76 ± 0.03 | 1300 | 2.93 ± 0.03 |
Fig. 3 shows a 16 scan thermally polarized 1H NMR spectrum of INAH at 1 T for comparison, alongside a single scan hyperpolarized trace whose vertical expansion is ×1/16th of the thermal spectrum. It is clear from this data that the ortho protons of INAH receive the strongest uplift in signal intensity through SABRE and that the effect of SABRE is substantial.
 | ||
| Fig. 3 1H NMR spectra of a 11 mM INAH sample in DMF-d7 at 1 T and 298 K. (a) Thermally polarized NMR spectrum recorded with 16 scans. (b) Single scan, hyperpolarized NMR spectrum, recorded using IMes-d22 as the precatalyst with 3 bar of parahydrogen, and a hyperpolarization transfer time of 10 s in a polarization transfer field of 60 Gauss; vertical expansion is 1/16 relative to (a). | ||
Table 1 also details the corresponding T1 values for the ortho INAH proton under SABRE conditions at 11.7 T and 1 T. Whilst there is a small T1 variation with catalyst identity at 11.7 T, the four second T1 values will not significantly degrade the SABRE data due to the delay that exists between the polarization transfer step and measurement with all catalysts. In contrast, the corresponding T1 values at 1 T show a larger variation and fall to 2.76 ± 0.05 s with IMes. As one T1 period results in a 63% polarization loss, the corresponding enhancement values at low field will be more affected by the delay between the polarisation step and measurement. This suggests that the IMes-d22 catalyst should be preferred for both high and low field studies. Furthermore, as free INAH itself provides a strong SABRE response, it will be important to either remove any residual unreacted INAH from the final analysis or identify specific spectroscopic differences between derivatized and underivatized INAH signals to achieve satisfactory analysis.
SABRE analysis of derivatized dextran
In the first instance, the 6 kDa derived sample, without dialysis to remove underivatized free INAH, was assessed at high field by routine 1H NMR spectroscopy in D2O.† The resulting 1 scan 1H NMR spectra, collected on a 2 mg sample (Fig. S1† top trace), showed sharp, strong and well resolved signals for free INAH at δ 8.6 and 7.6, DMAP at δ 7.9 and 6.8, and much weaker broader signals at δ 8.6 and 7.7 for conjugated INAH. Integration suggested, the mole percent of free INAH and DMAP leaving groups were ∼50% and 100% respectively, relative to that of the dextran glucose repeat unit (integrals are 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:2.5; protons 1:2:2). In contrast, while the corresponding 1 T NMR spectrum yielded the expected free INAH and DMAP signals, the expected broad signals for the conjugated dextran were now unresolved in the corresponding 512 scan measurement (Fig. S18†).
:1:2.5; protons 1:2:2). In contrast, while the corresponding 1 T NMR spectrum yielded the expected free INAH and DMAP signals, the expected broad signals for the conjugated dextran were now unresolved in the corresponding 512 scan measurement (Fig. S18†).
This sample was then analysed by hyperpolarized 1H NMR SABRE at high field (Fig. S19†). This involved diluting 10 μl of the above NMR solution 67-fold into a preformed solution containing 2 mg IMes, 10 μl DMSO-d6, 50 μl of D2O and 600 μl of DMF-d7-followed by SABRE 1H NMR analysis. A 260-fold signal gain was determined for the free INAH ortho C–H proton at 11.7 T consistent with strong hyperpolarization observed for INAH alone in solution (Table 1). However, the broad, potentially conjugated INAH signals were masked by this strong free INAH hyperpolarized response.
Dialysis of the CDAP activated and INAH derivatized dextran samples was then carried out to remove the free INAH and DMAP (Fig. S1, S4 and S6†). The efficiency of dialysis was monitored by high field 1H NMR analysis of the corresponding lyophilized samples that were re-dissolved in D2O. It was found that five cycles of dialysis were needed to clear both INAH and DMAP to levels below routine 1H NMR detection. With effective dialysis, the broad purported INAH conjugated ortho and meta aromatic signals became fully resolved, and proved to exhibit an approximate 1:1 integral ratio (Fig. S8–S10†).
Confirmation that these broad signals arise from conjugated INAH came from high field 2D Nuclear Overhauser spectroscopy (NOESY) and diffusion ordered spectroscopy (DOSY) spectra. NOESY spectroscopy identified correlations between the aromatic protons of the derivatized INAH and glucose repeat unit CHOH signals at δ 3.87–3.48 (Fig. S2, S5 and S7†). Importantly, DOSY spectroscopy confirmed that the derivatized INAH aromatic resonances and backbone glucose repeat unit resonances exhibit similar diffusion coefficients (Fig. S3†). A significant increase in line width for the dextran conjugated INAH signals at δ 8.6 and 7.7, relative to those of free INAH was also observed (Fig. S1, S4 and S6†). Hence, we confirm CDAP activation and stable INAH derivatisation of the dextran. It should be noted that the hyperpolarized responses for the derivatized samples can be observed for several hours without any change in their relative intensities. Hence measurement promoted leaching or sample degradation does not influence these results.
In order to quantify the amount of derivatisation, the INAH derivatized aromatic ortho signal at ∼δ 8.6 was integrated and compared to the dextran glucose anomeric at δ 5.2 in a 298 K NMR spectrum. Signal overlap was reduced by warming the sample to 323 K, which moved the residual water signal away from the δ 5.2 resonance. The extent of CDAP activation and INAH derivatization of the 6 kDa, 40 kDa and 250 kDa dextrans were 27%, 35.5% and 21.5% respectively (Fig. S8–S10†) and hence there is no apparent correlation between average polymer size and efficiency of combined CDAP activation and INAH derivatisation.
The 5-cycle dialysed 6 kDa INAH derivatized dextran was then screened for SABRE activity at high field with the five catalysts used previously. These results are tabulated in Table 2 and reveal that successful SABRE polarization takes place in all cases provided DMSO is present. However, the SABRE enhancements proved to generate both positive and negative lineshapes (Fig. S20†) as a consequence of the creation of longitudinal one, two, three and four spin order terms whose efficiency of creation varies with the value of the polarisation transfer field.34 In these experiments, we are seeking to create a hyperpolarisation response that is readily integrable in order to compare data. Hence the creation of single spin order hyperpolarisation alone is desired. The results at high field suggest that the two catalyst precursors NMeIMes and IPrCl are most suitable in this regard as they yield no antiphase character in their NMR response, with the latter catalyst proving to provide a slightly improved product hyperpolarisation level (see Table 2).
| SABRE precatalyst (with DMSO-d6 unless otherwise stated) | ortho-Proton signal enhancement, ∼δ 8.9 (relative to signal in thermal spectrum) | Signal character |
|---|---|---|
| IMes – no DMSO | None | No catalytic complex formed |
| IMes | Impossible to quantify | Very complex signal with antiphase character |
| ClIMes | 9-Fold | 2 signals seen to develop over time – the free signal and catalyst-bound signal |
| NMeIMes | 3.1-Fold | No antiphase character or second signal |
| IMes-d22 | Impossible to quantify | Complex signal with antiphase character |
| IPrCl | 5-Fold | No antiphase character or second signal |
The SABRE process might be expected to create the same level of polarization at both high and low field. However, when these polarization levels are assessed the apparent signal gains will be higher at low field (fold) due to the weaker inherent response. Such an effect was observed as the corresponding signal enhancements delivered with the IPrCl catalyst were significantly higher (41-fold) at 1 T relative to 11.5 T indicating that detection of moderate levels of INAH derivatized (∼20–30%) dextran is feasible at low field (See Fig. 4). In addition, these conjugated hydrazide signals now appear like well-defined doublets (Fig. S21†), in contrast to their broad appearance at high field. This is reflective of the fact that the frequency separation (chemical shift difference) between resonances reduces at low field and hence the differing sites in the derivatized conjugate can no longer be distinguished from one another.33
 | ||
| Fig. 4 1H NMR spectra of 5 mM INAH derivatized 6 kDa dextran in DMF at 1 T and 298 K. (a) Thermally polarized NMR spectrum recorded with 16 scans and a vertical expansion relative to (b) of ×10. (b) Single scan hyperpolarized NMR spectrum recorded using IPrCl as the precatalyst with 3 bar of parahydrogen, and a hyperpolarization transfer time of 10 s in a polarization transfer field of 60 Gauss. | ||
Next, the extensively dialysed 40 kDa and 250 kDa dextran conjugates were analysed for hyperpolarization at high field. The NMeIMes precatalyst resulted in SABRE enhancements for the ortho INAH conjugate protons of 3.8-fold and 6.4-fold respectively. The IPrCl precatalyst giving enhancements of 6-fold and 5-fold respectively.
This is consistent with the enhancement observed using the 6 kDa INAH derivatized dextran. Importantly, we find that there is no steric barrier to interaction between the large polysaccharides and the SABRE catalysts, supporting the feasibility for SABRE transfer using polysaccharides from low to high average molecular weight.
In order to eliminate any concern regarding the hyperpolarization of free INAH in the low field NMR spectra of the derivatized samples, the contributing signal enhancements need to be separated. This proved to be possible by examination of the weaker enhanced meta-proton signal for the derivatized INAH which are now distinct from those in the free material. This aspect is particularly important as the relative hyperpolarization of free INAH is approximately two orders of magnitude greater than that of derivatized INAH.
This differentiation was achieved by comparing the ortho-signal in the routine NMR spectrum and the meta-signal in the hyperpolarized spectrum. Consequently, we are able to distinguish between the basal residual free INAH (δ 8.0) and derivatized INAH populations (δ 8.3) based on the chemical shift of the meta-aromatic INAH proton once the sample is hyperpolarized. The derivatized INAH signal is not observed in the routine thermal 1H NMR spectrum due to overlap with the DMF-d6 solvent signal. Furthermore, the solvent signal is not hyperpolarized and the enhanced INAH signal is of much larger intensity than that of the solvent under SABRE.
These experiments were then repeated for the 40 kDa and 250 kDa INAH derivatized dextrans, at 1 T (Fig. S22 and S23†). Similar signals are seen in the 40 kDa spectrum and an enhancement factor of 31-fold has been calculated.16 However, while a doublet is observed in the hyperpolarized spectrum for the para-proton, no signal is seen above the noise for the meta-proton and therefore an enhancement factor cannot be calculated for the 250 kDa sample.
Analysis of C-polysaccharide
As indicated earlier, the manufacturing process is further complicated by the presence of cell wall polysaccharide (C-poly) which contains multiple functional groups with exchangeable protons (OH, NH and POH). The quantification of this impurity is also essential for sample validation. Despite the presence of these exchangeable protons, assessments using SABRE or SABRE-relay protocols failed to result in any successful sensitisation using IMES-d22 in the D2O, DMSO-d6, DMF-d7 matrices. In the latter protocol, it will be the presence of too high a concentration of exchangeable protons in the solvent that precludes signal enhancement.35However, we hypothesised that as the C-poly quaternary –N+Me3 signal, which is both relatively strong and a singlet, is readily detectable it could be used as a diagnostic marker without hyperpolarization. Measurements revealed its chemical shift to be 3.15 ppm in our solvent matrix, just on the edge of the dextrans carbohydrate signals.
A spiking experiment was then conducted to determine whether the quantification of C-poly within a dextran matrix using 1H NMR was possible at high or low field. This involved taking four 6 kDa dextran samples, and spiking them with C-poly at 10, 5, 1 and 0.5 wt% concentrations. The use of spiking in this way is a common protocol used in analysis laboratories where a known amount of a known substance is introduced into a sample to evaluate the performance of an analytical method. The resulting high field analysis, obtained via a series of 2 scan measurements produced predicted ratios of added dextran to C-poly that differed from the expected ratios by 1.05, 0.89, 1.05 and 1.12 (Table S3†); one would indicate agreement. As these values lie within the allowed tolerance limits referred to previously in the Limits of Quantification section, they confirm that direct high field quantification by simple integral analysis is possible. Unfortunately, when the same samples were examined at 1 T, the corresponding values proved to be 0.53, 0.84, 1.97 and 2.59. Hence signal overlap (refer to Fig. 5) means that accurate quantification via simple integral analysis is not feasible in the absence of analysis or processing modifications to compensate for partial dextran-C-poly N+Me3 resolution at low field.
 | ||
| Fig. 5 1H NMR spectra recorded at 1 T on a 6 kDa dextran sample doped with 10 mol% C-poly in D2O. The NMR trace in red, involved 512 scans and reveals overlap between signals from the dextran and the CH3 choline peak of C-poly. The NMR trace in green reflects a T1 filtered measurement, recorded with 512 scans and a 125 ms relaxation delay to address peak overlap. | ||
As a consequence, a protocol involving a T1 filter to remove the overlapping signal contribution from the C-poly N+Me3 response was implemented. This approach relies on the fact that the overlapping resonances will have different T1 values. These were determined for individual samples in D2O at 1.4 s for the dextran and 0.65 s for the pure N+Me3 C-poly signal at 11.7 T, and 0.18 s and 0.37 s for the corresponding signals at 1 T. T1 inversion spectra were then collected on a 6 kDa dextran sample doped with C-poly at 10 wt% concentration. The signal intensity for the peak at 3.2 ppm of Fig. 5 was then measured through 20 separate NMR spectra that encode relaxation after the inversion pulse. The resulting intensity data were then fit to a bi-exponential function, representing overlapping signals with differing initial populations and specified relaxation times. The difference between the real and calculated intensities were then minimised to determine the relative C-poly and overlapping signal contributions, which were calculated to be 1:0.19. The corresponding TSP signal has an intensity of 1.034 under these conditions, and hence this analysis predicts the C-poly concentration to be 9.97 wt%, which is the same as the chosen experimental concentration. A plot associated with this analysis is presented in the ESI.†
A second set of measurements were then completed where only a partial T1 curve made up of six points was collected between 0.2 and 1 second time points. Fitting this more limited data to a growing C-poly signal with constant offset, resulted in the same proportions being predicted for the two species. However, as the collection of 6 data points rather than the initial 20 is reflective of a significant measurement time saving such an analysis will be more cost effective.
In support of this, the high-field NMR trace was then reconsidered in order to predict the ratio of C-poly signal to overlapping signals that would move to lie under it at low field. A value of 1:0.20 was obtained which further validates this process.
Low-field analysis was then attempted on the 1 mol% sample using three 2k measurements and total acquisition times of 5 h per measurement. Even after line broadening and polynomial baseline correction low integral accuracy due to spectral overlap precluded satisfactory analysis with the predicted ratio being 1:0.6 C-poly to dextran rather than 1:1 as determined from the high-field data. A more rigorous approach using resolution enhancing data manipulations that would reduce the effect of C-poly peak overestimation in conjunction with peak deconvolution was deemed inappropriate due to the low signal to noise of the data and the lack of such facilities in a more general QC-environment.
This suggested that 1 T is insufficient to produce the signal to noise values needed for analysis and that moving to 1.88 T (80 MHz), with high temperatures to enable sharper peak profiles would be necessary if this approach were to be used in practice.
Conclusions
This study has demonstrated that the high and low field assessment of a dextran conjugate (CDAP linker with INAH model) by 1H NMR is possible without hyperpolarization. The resonances for the ortho and meta sites of free INAH show visible fine structure associated with the JHH splitting that connects them. In contrast, the derivatized materials show much broader signals for these groups at high field, due to the variety of environments they experience in the oligomeric model glycoconjugate. Remarkably these subtle chemical shift variations are attenuated on moving to low field such that well resolved multiplets are again observed. Hence, despite low sensitivity there is a discernible advantage associated with the informative shape of these resonances. Furthermore, the limits of quantification were assessed at both high and low field for the dextrans as 0.56 mM and 5.56 mM respectively, given a measurement time limit of no longer than 5 min. These values lie within the ranges expected for glycoconjugate vaccines and suggest their analysis via thermally equilibrated measurements is possible.C-poly, an impurity carried through the manufacturing process, also needs to be assessed if a batch is to meet quality control standards. This has again been demonstrated at high and low field by reference to a dextran/C-poly sample containing a 10 wt% loading. Here a T1 filter was used to remove the effects of overlapping signals on the diagnostic choline derived N+Me3CH2– response. Unfortunately, low signal to noise ratios precluded the meaningful analysis of a 1 wt% sample at 1 T. This situation could be addressed by moving to 1.86 T or 2.3 T, available on more recent instrumentation, and possibly working at 60 °C where sharper peak profiles would result.
Further results were presented to explore the possibility of using a SABRE hyperpolarized approach by reference to the model INAH conjugate. The SABRE hyperpolarization of free INAH was successful with signal gains of 610 and 2440-fold being observed for high and low field respectively. These would reflect compressions in data acquisition times of factors of hours and confirm very significant improvements could be made through the application of SABRE. The generation of a model conjugate involved the activation of dextran hydroxyls with CDAP and derivatization with INAH. These chemistries were chosen because they are described in the literature, have been applied to real-world glycoconjugates, and utilize functional groups (hydrazide, amine, cyanate) that may interact preferentially with SABRE catalysts. The expected INAH derivatization of the 6, 40, and 250 kDa dextran samples was confirmed by NMR diffusion, through-space correlation, and lineshape analysis. However, when the resulting model conjugate system was examined, a solvent mixture of D2O/DMF was needed for dissolution. This medium uses water, which has been avoided due to low H2 solubility and poor catalyst solubility in many SABRE studies. Nonetheless, when this solvent medium was used here, remarkably better enhancements were achieved than with the more usual methanol solvent for INAH. Unfortunately, when the model glycoconjugate was examined the resulting SABRE enhancements were significantly lower than those of the free INAH. The low relative SABRE signal from derivatized INAH is most likely related to the steric bulk of the glycoconjugate preventing efficient binding to the SABRE catalyst. Hence, further time-consuming catalyst development would be needed if these values were to be improved.
One of the major challenges in dealing with these SABRE assessments was associated with the remarkably strong free INAH signal gains, which can swamp those of the derivatized sample. Even after 5 dialysis runs, a weak SABRE response for free INAH was evident in the resulting 1H NMR spectra. Based on spectral examination, we estimated the ratio of derivatized INAH to free INAH to be greater than 200:1. Given the degree of SABRE amplification of the free material, very careful removal of free INAH is necessary for accurate analysis.
It is clear therefore, that the high and low field SABRE responses provided useful signal amplification resulting in a compression of measurement time. At low field though, the signal overlap proved challenging to resolve and very careful analysis was needed. It is likely that the latest generation of 80 or 100 MHz (1.88 and 2.35 T respectively) benchtop spectrometers will provide the necessary chemical shift dispersion to overcome both of these issues. Hence, in the future we predict that SABRE hyperpolarized validation methods will be appropriate for the analysis of glycoconjugates.
Author contributions
The manuscript was written through contributions from all authors. VL made the measurements. KS, DE and SBD directed the research. All authors have given approval to the final version of the manuscript.Data availability
NMR data used in the preparation of this article, are available at University of York repository – https://doi.org/10.15124/ef1b69d0-f200-488d-a66f-dacb949fd99f.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We are extremely grateful to Dr Victoria Annis (University of York, UK) for synthesis of the [IrCl(η2-η2-COD)(IMes)] precatalyst and Dr Peter J. Rayner (University of York, UK) for synthesis of the other precatalysts deployed here. Dr Peter J. Rayner and Callum A. Gater (University of York, UK) are thanked for helpful discussions. We are grateful to Pfizer (R23324) and UK Research and Innovation (UKRI) under the UK government's Horizon Europe funding guarantee [grant number EP/X023672/1]. NMR data for this work can be found at https://doi.org/10.15124/ef1b69d0-f200-488d-a66f-dacb949fd99f.References
- O. T. Avery and W. F. Goebel, J. Exp. Med., 1929, 50, 533–550 CrossRef CAS PubMed.
- R. Rappuoli, Sci. Transl. Med., 2018, 10, eaat4615 CrossRef PubMed.
- G. Stefanetti, F. Borriello, B. Richichi, I. Zanoni and L. Lay, Front Cell Infect Microbiol, 2021, 11, 808005 CrossRef CAS PubMed.
- E. Kay, J. Cuccui and B. W. Wren, npj Vaccines, 2019, 4, 16 CrossRef PubMed.
- R. Adamo, Glycoconjugate J., 2021, 38, 397–398 CrossRef CAS PubMed.
- F. Micoli, P. Costantino and R. Adamo, FEMS Microbiol. Rev., 2018, 42, 388–423 CrossRef CAS PubMed.
- P. Gronemeyer, R. Ditz and J. Strube, Bioengineering, 2014, 1, 188–212 CrossRef PubMed.
- S. J. Wu, Z. Y. Jin, J. M. Kim, Q. Y. Tong and H. Q. Chen, Carbohydr. Polym., 2009, 77, 750–753 CrossRef CAS.
- R. Schneerson, O. Barrera, A. Sutton and J. B. Robbins, J. Exp. Med., 1980, 152, 361–376 CrossRef CAS PubMed.
- A. Lees, B. L. Nelson and J. J. Mond, Vaccine, 1996, 14, 190–198 CrossRef CAS PubMed.
- R. Nappini, R. Alfini, S. Durante, L. Salvini, M. M. Raso, E. Palmieri, R. Di Benedetto, M. Carducci, O. Rossi, P. Cescutti, F. Micoli and C. Giannelli, Vaccines, 2024, 12, 1272 CrossRef PubMed.
- V. Morais and N. Suarez, Bioengineering, 2022, 9, 774 CrossRef CAS PubMed.
- G. R. Chichili, R. Smulders, V. Santos, B. Cywin, L. Kovanda, C. Van Sant, F. Malinoski, S. Sebastian, G. Siber and R. Malley, Vaccine, 2022, 40, 4190–4198 CrossRef CAS PubMed.
- WHO, Recommendations to Assure the Quality, Safety and Efficacy of Pneumococcal Conjugate Vaccines, Expert Committee on Biological Standardization, WHO, Geneva, 2009 Search PubMed.
- Q. W. Xu, C. Abeygunawardana, A. S. Ng, A. W. Annie, B. J. Harmon and J. P. Hennessey, Anal. Biochem., 2005, 342, 358–358 CrossRef CAS.
- J. H. Ardenkjaer-Larsen, B. Fridlund, A. Gram, G. Hansson, L. Hansson, M. H. Lerche, R. Servin, M. Thaning and K. Golman, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 10158–10163 CrossRef CAS PubMed.
- P. J. Rayner and S. B. Duckett, Angew. Chem., Int. Ed., 2018, 57, 6742–6753 CrossRef CAS PubMed.
- M. Pellecchia, D. S. Sem and K. Wuthrich, Nat. Rev. Drug Discovery, 2002, 1, 211–219 CrossRef CAS PubMed.
- Y. Wang and C. Hilty, Anal. Chem., 2020, 92, 13718–13723 CrossRef CAS PubMed.
- R. Mandal, P. Pham and C. Hilty, Chem. Sci., 2021, 12, 12950–12958 RSC.
- T. B. R. Robertson, L. H. Antonides, N. Gilbert, S. L. Benjamin, S. K. Langley, L. J. Munro, O. B. Sutcliffe and R. E. Mewis, ChemistryOpen, 2019, 8, 1375–1382 CrossRef CAS PubMed.
- F. Fernandez Diaz-Rullo, F. Zamberlan, R. E. Mewis, M. Fekete, L. Broche, L. A. Cheyne, S. Dall'Angelo, S. B. Duckett, D. Dawson and M. Zanda, Bioorg. Med. Chem., 2017, 25, 2730–2742 CrossRef CAS PubMed.
- A. Lees, J. F. Barr and S. Gebretnsae, Vaccines, 2020, 8, 777 CrossRef CAS PubMed.
- R. W. Adams, J. A. Aguilar, K. D. Atkinson, M. J. Cowley, P. I. Elliott, S. B. Duckett, G. G. Green, I. G. Khazal, J. Lopez-Serrano and D. C. Williamson, Science, 2009, 323, 1708–1711 CrossRef CAS PubMed.
- D. A. Barskiy, S. Knecht, A. V. Yurkovskaya and K. L. Ivanov, Prog. Nucl. Magn. Reson. Spectrosc., 2019, 114–115, 33–70 CrossRef CAS PubMed.
- P. M. Richardson, S. Jackson, A. J. Parrott, A. Nordon, S. B. Duckett and M. E. Halse, Magn. Reson. Chem., 2018, 56, 641–650 CrossRef CAS PubMed.
- K. D. Atkinson, M. J. Cowley, S. B. Duckett, P. I. Elliott, G. G. Green, J. Lopez-Serrano, I. G. Khazal and A. C. Whitwood, Inorg. Chem., 2009, 48, 663–670 CrossRef CAS PubMed.
- E. M. Agency, ICH Q2(R2) Guideline on validation of analytical procedureshttps://www.ema.europa.eu/, 2023.
- M. J. Cowley, R. W. Adams, K. D. Atkinson, M. C. R. Cockett, S. B. Duckett, G. G. R. Green, J. A. B. Lohman, R. Kerssebaum, D. Kilgour and R. E. Mewis, J. Am. Chem. Soc., 2011, 133, 6134–6137 CrossRef CAS PubMed.
- P. Purwanto, R. M. Deshpande, R. V. Chaudhari and H. Delmas, J. Chem. Eng. Data, 1996, 41, 1414–1417 CrossRef CAS.
- W. Iali, S. S. Roy, B. Tickner, F. Ahwal, A. J. Kennerley and S. B. Duckett, Angew. Chem., Int. Ed., 2019, 58, 10271–10275 CrossRef CAS PubMed.
- B. J. Tickner, M. Dennington, B. G. Collins, C. A. Gater, T. F. N. Tanner, A. C. Whitwood, P. J. Rayner, D. P. Watts and S. B. Duckett, ACS Catal., 2024, 14, 994–1004 CrossRef CAS PubMed.
- P. J. Rayner, P. Norcott, K. M. Appleby, W. Iali, R. O. John, S. J. Hart, A. C. Whitwood and S. B. Duckett, Nat. Commun., 2018, 9, 4251 CrossRef PubMed.
- R. E. Mewis, K. D. Atkinson, M. J. Cowley, S. B. Duckett, G. G. R. Green, R. A. Green, L. A. R. Highton, D. Kilgour, L. S. Lloyd, J. A. B. Lohman and D. C. Williamson, Magn. Reson. Chem., 2014, 52, 358–369 CrossRef CAS PubMed.
- M. Fekete, C. Gibard, G. J. Dear, G. G. Green, A. J. Hooper, A. D. Roberts, F. Cisnetti and S. B. Duckett, Dalton Trans., 2015, 44, 7870–7880 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic and NMR methods. See DOI: https://doi.org/10.1039/d4an01503g |
| This journal is © The Royal Society of Chemistry 2025 |