
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA bis-pyrene polyamine receptor for fast optical detection of ketoprofen: synthesis, characterization and application in all-solid-state fluorescent sensors†
Giammarco Maria
Romano
 *a,
Pierangela
Di Menna
b,
Andrea
Bencini
a,
Yschtar Tecla
Simonini Steiner
a,
Massimo
Innocenti
a,
Corrado
Di Natale
c,
Roberto
Paolesse
b and
Larisa
Lvova
*b
*a,
Pierangela
Di Menna
b,
Andrea
Bencini
a,
Yschtar Tecla
Simonini Steiner
a,
Massimo
Innocenti
a,
Corrado
Di Natale
c,
Roberto
Paolesse
b and
Larisa
Lvova
*b
aDepartment of Chemistry “Ugo Schiff”, University of Florence, Via della Lastruccia 3, 50019 Sesto Fiorentino, Italy. E-mail: giammarcomaria.romano@unifi.it
bDepartment of Chemical Sciences and Technologies, University “Tor Vergata”, Via della Ricerca Scientifica 1, 00133 Rome, Italy. E-mail: larisa.lvova@uniroma2.it
cDepartment of Electronic Engineering, University “Tor Vergata”, Via del Politecnico 1, 00133 Roma, Italy
First published on 23rd January 2025
Abstract
Herein, we report on a polyamine receptor L1 bearing pyrene fluorogenic groups for the optical assessment of the non-opioid analgesic drug ketoprofen (KP). L1, composed of a diethylenetriamine moiety linked at its extremities to the 1 position of two pyrene units via methylene linkers, produced an emission at 460 nm in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v) water/ethanol mixture at pH 7, which can be attributed to the excimer formation between the two aromatic groups. In the presence of KP, a salt-bridging interaction between the carboxylate group of the analyte and the central ammonium group of L1 induced a redistribution of acidic protons in the polyamine chain, causing a marked increase in the emission. This optical signal was used to detect KP in aqueous media. Based on this observation, the properties of all-solid-state optodes with plasticized PVC membranes doped with L1 and deposited on an appropriate solid support material were further investigated. The best membrane contained 1 wt% of fully protonated L1, plasticized with DOS and doped with 3 equiv. of the TDMACl anion-exchanger, which detected KP in the 2 μM–0.1 mM range with a low influence of interfering ions. Furthermore, this membrane was applied for the assessment of the amount of KP in OkiTask, achieving an RSD of 2.1% and recovery of 102%. Moreover, the possibility to decrease the LOD of KP to 0.84 μM (0.21 mg L−1) through the application of L1-based fluorescent sensor arrays and chemometrics was studied.
:1 (v/v) water/ethanol mixture at pH 7, which can be attributed to the excimer formation between the two aromatic groups. In the presence of KP, a salt-bridging interaction between the carboxylate group of the analyte and the central ammonium group of L1 induced a redistribution of acidic protons in the polyamine chain, causing a marked increase in the emission. This optical signal was used to detect KP in aqueous media. Based on this observation, the properties of all-solid-state optodes with plasticized PVC membranes doped with L1 and deposited on an appropriate solid support material were further investigated. The best membrane contained 1 wt% of fully protonated L1, plasticized with DOS and doped with 3 equiv. of the TDMACl anion-exchanger, which detected KP in the 2 μM–0.1 mM range with a low influence of interfering ions. Furthermore, this membrane was applied for the assessment of the amount of KP in OkiTask, achieving an RSD of 2.1% and recovery of 102%. Moreover, the possibility to decrease the LOD of KP to 0.84 μM (0.21 mg L−1) through the application of L1-based fluorescent sensor arrays and chemometrics was studied.
1. Introduction
Among the non-steroidal anti-inflammatory drugs (NSAIDs), ketoprofen (KP) is one of the most widely used to reduce various types of inflammations, muscle pain and headache; to lower fever as well as counter cold and flu symptoms.1 The main mechanism of action of KP is via the inhibition of prostaglandin synthesis with the cyclooxygenase enzyme, making it effective against acute and chronic pain in arthrosis and arthritis. In medicine, either its acidic form (KPH) or more soluble salt form (ketoprofen lysine salt, KP-Lys) is the main component of many pharmacological compositions for oral submission for adults and children and in skin-treatment pastes and gels, wound treat dressings, etc.2,3Although KP is a readily degradable compound characterized by low environmental stability, its inflow to ground and surface waters is constant owing to its excessive use and production.4,5 Existing wastewater treatment plants (WWTPs) are not specifically designed to eliminate NSAIDs, allowing only partial reduction of their concentration. Thus, more hydrophilic drugs may pass through treatment processes, resulting in significant environmental concentrations and exposure to living organisms.6 Besides, several side-effects, such as poor tolerance of the gastrointestinal tract, allergy induction, liver and kidney dysfunctions, and light sensitization, can result upon high dose consumption or long-term medication. Several studies have proved that KP toxicity is potentially associated with many chronic medical conditions such as cardiovascular diseases, renal failure and endocrine disruption.7 KP has recently been acknowledged as an emerging contaminant with considerable environmental implications, attributable to its long-term toxic effects, absence of effective removal methods from waste, and marked increase in consumption during the COVID-19 pandemic and the subsequent period.8,9
The detection of anti-inflammatory KP drug in pharmaceutical compositions and environmental samples is an important analytical task, and several analytical techniques have been previously reported for the assessment of NSAIDs, in particular KP, including chromatography,10 spectrophotometry,11,12 electrochemical,13–15 optical sensors,16–20 and multisensory arrays.21,22 Among different transduction-type sensors, optical sensors have the advantages of fast response time, simplicity of preparation and signal acquisition, as well as the possibility to achieve low detection limits and high sensitivity.23–28 Moreover, these devices do not require sophisticated hardware, are wireless and their output may be estimated with common optoelectronic devices or even through visual observation in a “naked-eye” mode.29–31 In the last few years, several nanostructured assemblies have been used for the optical signaling of KP, among them are differently modified quantum dots (QDs),16,17 metallic nanoparticles,18 and composite materials such as molecularly imprinted polymers (MIPs),19 hydrogels,20 or specific polymer-based sensors arrays.22 Delgado-Pérez et al. developed CdSe/ZnS quantum dots bearing a chiral organic ligand for the enantioselective recognition of KP; however, no selective recognition among the NSAIDs KP, flurbiprofen, and naproxen was achieved.17 Bhogal et al. reported a hybrid fluorescent material composed of carbon dots–molecularly imprinted polymer (CDs–MIPs), which was selectively quenched by KP in a water solution, with applications in the determination of KP in human serum and urine samples.19 A simultaneous detection system in a water/ethanol mixture for naproxen and ketoprofen was also proposed by Wang et al. exploiting two different FMIPs. The system showed high specificity towards each NSAID and LODs at the nanomolar level.32
Significantly less focus has been directed towards fluorescent small molecules for the detection of ketoprofen. Examples of supramolecular arrays exploiting hydrogen-bonding chemosensors, such as calix[4]pyrroles as receptor units,33 BINOL derivatives for the enantiomeric recognition of NSAIDs,34 and calixpyrrole-type macrocycles for KP sensing,35 have been previously explored. In this regard, polyamine receptors bearing aromatic signaling units36 are potentially appealing receptors for NSAIDs as they present common structural features, being normally composed of a carboxylic group, which are usually deprotonated at neutral pH, linked to an aromatic portion. Polyamines can protonate in aqueous solution even at pH 7, and the resulting polyammonium cations can give H-bonding and charge–charge contacts between NSAID carboxylate groups. In polyamine-based fluorescent receptors, this interaction is expected to result in an overall fluorescence intensity emission change, which may be correlated to the pain-killer compound concentration in the analyzed sample.37,38 A recent work by Romano et al. reported on open-chain polyamine receptors equipped with two anthracene fluorogenic fragments at their extremities for KP sensing in water/ethanol solutions upon excitation at 340 nm.36 However, chemosensor response to KP was registered in quite a narrow concentration range from 0 to 5.0 equiv. with respect to the ligand. 1H NMR titrations demonstrated no direct involvement of the aromatic anthracene units in KP binding via π-stacking interactions, which may be the reason for such a small KP concentration working range.
To increase the linear working range, the introduction of fluorescent signaling units other than anthracene on polyamine scaffolds can be useful. To this purpose, we linked two pyrene units at the terminal amine groups of a diethylenetriamine chain to explore the ability of the two large aromatic moieties to stack together and give excimer emission in aqueous solution, which can be modulated upon substrate coordination. In the present study, we investigated the properties of this receptor for selective and rapid assessment of KP in aqueous media. Since the construction of all-solid-state optical sensors is more practical and easier to handle in comparison to the common optical sensing methodology when an analyte is added to the solution containing an optically active receptor, we have focused on the development of all-solid-state fluorescent sensors for KP detection. To this purpose, we tested PVC polymeric membranes containing either the three-hydrochloride form of the receptor, L1·3HCl, and its unprotonated form, L1 (Scheme 1), for KP sensing. Our purpose was the improvement of sensor analytical characteristics through the accurate tuning of sensing membrane composition to select an appropriate solid support material, together with the interpretation of the signal transduction mechanism. Moreover, a multisensory approach was employed to lower the KP detection limit of the developed optodes.
 | ||
| Scheme 1 Structures of KP and the L1 receptor. | ||
2. Experimental section
2.1 Reagents and L1·3HCl, L1 syntheses
The detailed list of reagents used is provided in section S1 of ESI,† while the synthetic details for L1 and its hydrochloride salt are reported in section S2.† L1 was synthesized, as summarized in Scheme S1,† using a slightly modified procedure commonly used for the terminal alkylation of linear polyamines.39,40 The characterization data for L1·3HCl and L1 are given in Fig. S2–S6.†2.2 Electronic absorption and fluorescence measurements
Absorption spectra were recorded on a Jasco V-670 spectrophotometer using ligand concentrations of 5 × 10−7 M. Emission spectra were recorded on a Horiba Scientific Fluoromax Plus fluorimeter using an excitation wavelength of 360 nm and ligand concentrations of 5 × 10−7 M. All measurements were performed in a water/ethanol 1:1 (v/v) mixture at pH 7 using a 0.005 M Tris/HCl buffer at 298.0 ± 0.1 K. Measurements at different pH values were performed in a 1:water/ethanol 1 (v/v) mixture with 0.1 M NMe4Cl to maintain the ionic strength (pKw = 14.52(1) in the above stated condition).41 The binding tests were performed by spectrophotometric and spectrofluorimetric titrations at the pH of an L1 solution in the presence of increasing amounts of different analytes. Luminescence quantum yields were determined using quinine sulfate in a 0.05 M H2SO4 aqueous solution (Φ = 0.53) as a standard reference.42
2.3 NMR Measurements
NMR spectra were recorded on a Bruker 400 MHz (Bruker Corp., Billerica, MA, USA) instrument. NMR titrations were carried out in a D2O/CD3OD 40:60 (v/v) mixture at pH 7 by the addition of KP to 0.001 M solutions of L1. The pH was adjusted to 7 by adding small amounts of NaOD or DCl after each addition of KP to the solutions of L1.
2.4 Membranes preparation and sensing materials testing
Polymeric membranes were prepared by the incorporation of approximately 1 wt% of L1·3HCl or L1 and 1–3 equiv. of anion-exchanger (TDACl or TDMACl) into a polymeric matrix containing PVC and plasticizer (TOP or DOS) in 1:2 ratio by weight. The overall membrane weight was 100 mg; all the membrane components were dissolved in 1 mL of THF. Planar optodes were prepared through drop-casting method, for which a series of sensing spots was produced by deposition of 2 μL of each membrane cocktail on different solid supports: Whatman 1003–110 (390 mm, 185 g m−2) filter paper (FP), color catcher cellulose tissue (CC), and glass slides (GS). After THF evaporation, optodes were tested for the optical assessment of primary KP and KP-Lys and interfering ions in 2 × 10−6–1 × 10−4 M concentration range upon illumination with UV light at 365 nm and direct spiking of 5 μL of calibration solutions of different concentration every time on the new sensing spot.
All the solutions for optodes testing were prepared with distilled water at 0.005 M Tris/HCl background, pH 7.00. The pH was adjusted by adding small aliquots of 1 M HCl and simultaneously controlled with an AMEL pH-meter (Italy), preliminarily calibrated with pH 7.00 and pH 10.00 standard buffer solutions (Sigma-Aldrich, Germany).
2.5 Optical signal acquisition and data processing
For the quantification of optical response and its correlation to the KP content in the calibration and test solutions, optode images were acquired with a smartphone from a fixed distance of 10 cm upon excitation at 365 nm (see section S3 of ESI† for details). The whole sensing spot surface was considered as a region of interest (ROI); ROI luminescence intensity variations were then transformed into analytically useful digital signals by application of Colorpicker (https://imagecolorpicker.com/) and ImageJ (https://ij.imjoy.io/) freeware. Optode luminescent response was converted into three main colors of the visible spectrum–red (630 nm), green (530 nm), and blue (480 nm)–according to the RGB scale. The RGB values were extracted for each pixel of ROI, the color components were averaged, and these mean values were used for data analysis after subtraction of background without analyte and background without sensing spot. Measurements were performed for 3 solid supports and repeated twice for 3 consecutive measurement days (n = 6).Standard addition method was used to estimate the KP amount in OkiTask by Dompé pharmaceutical composition. Details on sample preparation and analytical procedure are reported in section S4 of ESI.†
The data obtained from fluorescent multisensor array were treated with Unscrambler (CAMO PROCESS AS, Norway). PLS1 regression model was used to fit multisensor array optical response with KP concentrations in the calibration solutions; leave-one-out validation was applied due to the restricted number of measurements composing the dataset. RMSEC, RMSEV (Root Mean Square Error of Calibration and Validation respectively) and correlation coefficient, R2, of predicted vs. measured correlation lines were used to evaluate the efficiency of the constructed regression models. The low detection limit of KP detection was estimated by 3σ method (LOD = 3σ/S, where σ is the RMSEC recalculated in mg L−1 and S is the slope of the regression line at calibration stage) (for details on LOD calculations, see section S5 of ESI†).
3. Results and discussion
3.1 Acid–base properties of L1
The determination of protonation characteristics of a polyamine-based fluorescent host is the preliminary step to analyze the effect of pH on its emission properties and binding/detection ability for anionic substrates. To this purpose, UV-Vis and fluorescence emission spectroscopy were used to study the photophysical characteristics of the present receptor at different pH values. Measurements were performed in a H2O/EtOH 1:1 (v/v) mixture to ensure the complete solubility of the ligand in a wide pH range (3–11.5). The absorption spectrum of L1 shows the classic pyrene absorption band with a maximum at 345 nm and a red-shifted tail (ca. 360 nm), which increases with increasing pH (Fig. 1a). This suggests the formation of an intramolecular ground-state dimer (GSD) between the two pyrene units, which is more favored at alkaline pH values.43 In fact, the triamine chain is likely to be protonated at acidic pH values. Its progressive deprotonation with increasing pH enhances its flexibility and allows the whole receptor to assume a conformation in which the two pyrene units are close to one another, favoring their interaction via hydrophobic and/or π-stacking. Direct photoexcitation of this GSD at 360 nm gives rise to a broad emission band centered at 463 nm typical of the excimer form of pyrene.44–46 As shown in Fig. 1b, at slightly acidic, neutral, and alkaline conditions, L1 exhibits both a typical structured emission band at 370–430 nm (λexc = 360 nm) corresponding to monomer emission, and a second red-shifted band at 460 nm, attributed to the formation of intramolecular excimer species. The progressive deprotonation of the ammonium groups with increasing pH results in a decrease in intensity of the monomer emission at 395 nm and, to a lesser extent, in an increase of the excimer emission at 460 nm. Most likely, deprotonation of the triamine chain induces an increase in the electron density at the nitrogen atoms, favoring a photo-induced electron transfer (PET) effect from an amine group to the excited fluorophore that leads to quenching of the monomer emission at 395 nm and justifies the slight increase in dimer emission at 460 nm. The emission intensity at 395 nm remains almost constant between pH 4 and 5 (Fig. 1a, inset). At higher pH values, the emission decreases again to achieve a constant value in the pH range of 6.5–7.5. Above pH 7.5, the emission increases again and finally assumes a nearly constant value above pH 9. Treatment of the emission intensity data at 395 nm using the HypSpec program47 allows for the calculation of protonation constants of 8.1(1) and 5.9(1) log units, corresponding to the formation of a mono- and di-protonated receptor L1, respectively.
 | ||
| Fig. 1 Absorption spectra (a) and selected fluorescence emission spectra (b) of L1 at different pH values in H2O:EtOH (1:1 v/v) mixture and 0.1 M NMe4Cl. Inset of (b) shows a plot of the fluorescence emission intensity at 395 nm and 463 nm ([L1] = 1 × 10−6 M, λexc = 360 nm). | ||
These values are very similar to those found for the corresponding bis-anthracenyl derivative of 1,4,7-diethylenetriamine,36 for which a third protonation step was also observed at acidic pH values (pKa = 3.3), as normally found in diethylenetriamine and its derivatives.48 In the present case, precipitation in H2O/EtOH below pH 3.5 prevents spectrum collection and determination of the constant relative to the third protonation step of the receptor. The calculated constants for the first and second protonation equilibrium allow to conclude that the receptor is mainly in its mono-protonated form at neutral pH. Considering that benzylic amine groups adjacent to aromatic moieties are normally less basic,49 we can reasonably conclude that in the monoprotonated species, the acidic proton is localized on the central amine groups of the triamine chain at a larger distance from the pyrene units.
3.2 Binding properties of L1 toward KP in water/ethanol solution
:1 v/v) buffered at pH 7 with Tris. Preliminary tests did not demonstrate evident changes in the UV-vis spectra upon increase in the KP concentration, suggesting that no L1 structural changes occur upon binding with KP in the ground state (Fig. S7†). On the contrary, a significant increase of the emission band of the excimer species, centered at ca. 460 nm, was recorded upon excitation of L1 at 360 nm upon the addition of KP (Fig. 2).
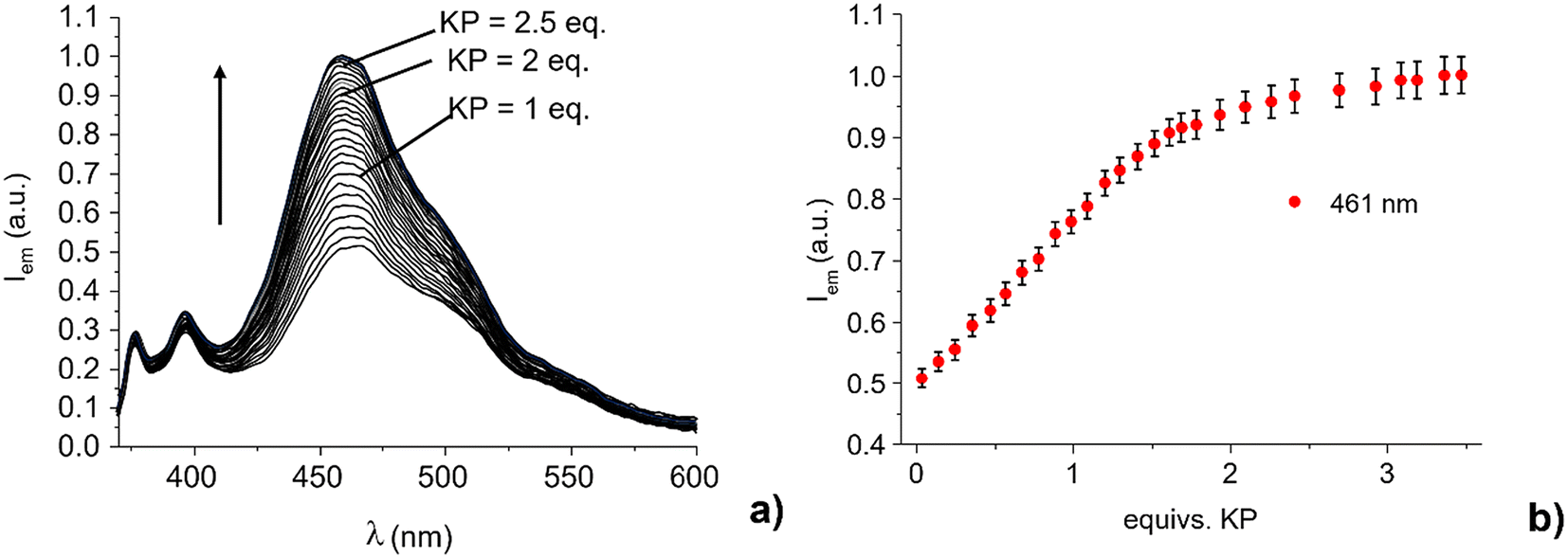 | ||
| Fig. 2 (a) Fluorescence spectra of L1 and (b) plot of the emission intensity at 396 nm and 461 nm in the presence of increasing amount of KP (H2O:EtOH 1:1 v/v, pH = 7, Tris 0.005 M, [L1] = 5 × 10−7 M, λexc = 360 nm). Error bars represent the standard deviation for triplicate measurements. | ||
The intensity of the monomer band at about 375–400 nm remains almost constant. Notably, the emission of the 460 nm band increases almost linearly up to a 1.8:1 KP:L1 molar ratio (R). At higher R values, the emission achieves an almost constant value for molar ratio greater than 2.5, with a quantum yield Φ of 0.37 (Φ = 0.25 for L1 in the absence of KP). This suggests the formation of adducts with both 1:1 and 1:2 receptor to substrate stoichiometry. Indeed, the analysis of the spectrofluorimetric data with HypSpec program48 shows the formation of complexes with receptor to substrate stoichiometry of 1:1 and 1:2 with estimated apparent binding constants at pH 7.0 (ligand speciation not analytically considered) for the successive addition of one (HL1+ + KP− = [HL1(KP)]) and two ([HL1(KP)] + KP− = [HL1(KP)2]−) KP anions to L1 of 4.1(1) and 3.8(1) log units, respectively. The receptor is in the protonated (positively charged) form at neutral pH (the main form present in solution at neutral pH is the monoprotonated one). Therefore, it can interact via non-directional charge–charge interactions with the anionic carboxylate groups of ketoprofen. At the same time, the ammonium and amine groups can act as hydrogen bonding donors toward the carboxylate moiety. Finally, the pyrene units can give hydrophobic interactions with the aromatic portion of KP. The presence of multiple interaction modes, which likely act cooperatively in anion binding, may allow the formation of adducts containing more than one anionic host. This result is in good agreement with that previously found for the adduct of KP with the corresponding bis-anthracenyl derivatives of both diethylene- and triethylenetriamine, for which the formation of adducts with both 1:1 and 1:2 stoichiometry was observed.36 Under these experimental conditions, a detection limit (LOD) of 17 μM (4.3 mg L−1) was estimated for KP sensing by L1 (Fig. S9†). As reported above, L1 is in its monoprotonated form at neutral pH value, the acidic proton being localized on the central amine group. Hydrogen bonding and electrostatic interactions may occur between the carboxylate group of KP (the pKa value of KP is 5.31 in 1:1 (v/v) H2O/EtOH mixture) and the protonated central –NH2+ group of L1, as shown in Scheme 2. However, as already observed for the bis-anthracenyl derivative adducts with KP,36 the presence of H-bonding interactions between the carbonyl function of KP and a benzyl amino-group, which contribute to the stabilization of the complex, cannot be excluded.
 | ||
| Scheme 2 Proposed interaction mode between KP− and L1 in its monoprotonated form. | ||
In polyamine compounds bearing aromatic fluorogenic units, the benzylic amine groups generally exert the most effective PET effect. Their protonation inhibits the electron transfer process due to the involvement of their lone pair in proton binding, leading to a renewal of the emission of the adjacent fluorogenic unit. This would suggest that interaction of the carboxylate group of KP induces a proton transfer process from the central amine group to a benzylic amine, with a consequently reduced PET effect and increased excimer emission.
To corroborate this suggestive hypothesis, we performed 1H NMR titration by adding KP to an aqueous solution of L1.
:4 (v/v) mixture at neutral pH. This solvent mixture was chosen to avoid precipitation of L1 adducts with KP observed in 1:1 D2O/EtOH mixture at the concentration used in 1H NMR experiments (1 × 10−2 M). As shown in Fig. 3, the increase in KP concentration in an L1 solution induces a progressive downfield shift of the resonance of both the methylene bridge 1AL (see Fig. 3 for atom numbering) and the methylene groups 2AL adjacent to the lateral amine groups, while the chemical shift of the signal relative to the 3AL methylene group remains unchanged.
 | ||
| Fig. 3 Aliphatic signals of the 1H NMR spectra of L1 at pH 7 in the presence of increasing amounts of KP (D2O/DMSO-d6 1:4 v/v, [L1] = 1 × 10−3 M). | ||
In aliphatic polyamines, protonation and deprotonation processes are accompanied by a downfield and upfield shift, respectively, of the signal of the adjacent methylene units.49 Therefore, the downfield shift of the resonance of methylene groups 1AL and 2AL can be ascribed to a proton transfer process from the central protonated amine group to the benzylic one upon KP binding. The increased positive charge on the nitrogen atom adjacent to the fluorophore can induce an inhibition of the PET process, justifying the partial restoration of the fluorescence emission. The aromatic signals of pyrene are not affected by the interaction with the substrate, as shown in Fig. S10,† suggesting that the aromatic units of receptor and substrate are not involved in substrate coordination via π-stacking interactions, at least in the ground state. The proposed interaction mode in the ground state is confirmed by the analysis of the shift of the signals of KP, in which both the signals relative to the CH and CH3 groups of KP are not significantly shifted, as displayed in Fig. S11 and S12.†
3.3 Optical properties of L1-based optodes
To transfer the optical signal of bis-pyrene-polyamine receptors in the presence of KP from solution into all-solid-state fluorimetric optical sensors, we investigated the properties of PVC plasticized membranes doped with L1·3HCl and L1 as ‘free’ amine, both deposited on three different solid supports (FP, CC and GS, see Experimental section for details). In total, 7 membrane compositions were prepared, as summarized in Table 1. Due to the low solubility of L1·3HCl in the solvent (THF) used for membrane Mb1.1 cocktail preparation, the optical properties of membranes Mb1.2–Mb5 doped with the non-protonated L1 (present in oil form) ligand were further studied.| Membrane | Ligand | Anion-exchanger | Ligand:anion exchanger, molar ratio |
Plasticizer | TFA |
|---|---|---|---|---|---|
| Mb1.1 | L1·3HCl | 1:0.5 |
TOP | — | |
| Mb1.2 | 1:0.7 |
TOP | — | ||
| Mb1.3 | 1:0.7 |
TOP | 3 equiv. | ||
| Mb2 | L1 | TDACl | 1:0.7 |
DOS | 3 equiv. |
| Mb3 | 1:0.7 |
DOS | — | ||
| Mb4 | 1:1.5 |
DOS | 3 equiv. | ||
| Mb5 | TDMACl | 1:2.8 |
DOS | 3 equiv. |
TDACl and TDMACl (in membrane Mb5) anion-exchangers were added to membrane cocktails in 1:1, 1:2 or 1:3 ratio with respect to L1 receptor to stabilize membrane neutrality and ensure the analyte anions’ membrane permselectivity. The influence of L1 protonation state in the membrane phase as well as the plasticizer nature on the optical properties of tested membranes and their sensitivity and selectivity toward ketoprofen have been also investigated.
As expected, preliminary tests on the optical response of membrane Mb1.2 deposited on CC support have shown the preferable binding of KP compared to ibuprofen (IBU) and benzoate anions, which were tested as potential interfering ions. However, an evident luminescence signal quenching was registered upon KP concentration increase, in contrast to the L1 behavior in solution (Fig. S13†). A similar response was registered for Mb1.2 spots deposited on glass and filter paper support, thus evidencing the necessity of careful tuning of the fluorophore protonation state and competitive salt-bridging effects among the fluorophore and target KP− and/or Cl− ions coming from the anion-exchanger or other interfering ions. To clarify these phenomena, a new series of membranes was prepared, in which 3 equivalents of protons (H+), in the form of trifluoroacetic acid (TFA), were introduced into the freshly prepared membranes (Mb1.3, Mb2, Mb4, and Mb5) to protonate the L1 ligand directly in the membrane phase.
The DOS plasticizer with lower dielectric constant (εDOS = 4.0 vs. εTOP = 7.9) was used instead of highly anion-solvating and basic TOP plasticizer for membranes Mb2–Mb5 preparation. To study the influence of the anion-exchanger, the ratio of L1/anion-exchanger was changed from approximately 1:1 to 1:1.5 and 1:3 equiv. in membranes Mb3, Mb4 and Mb5, respectively. Moreover, to keep pH at a constant value, all the measurements were done in 0.01 M Tris/HCl pH 7.00 buffer. Under these conditions, KP is completely ionized, considering that its pKa is 4.45.
 | ||
| Fig. 4 Photogram of L1-based all-solid-state optodes deposited on the glass support GS to increasing concentrations of KP (1–10 equiv.) on 0.005 M Tris pH 7.0 background solution. In membrane Mb3, the L1 ligand is in its not-protonated form, and in membranes Mb4 and Mb5, 1.5 and 2.8 equiv. of TFA, respectively, were added to protonate L1 directly in the membrane phase. The image was acquired from 10 cm distance with a common smartphone, λexc = 365 nm. | ||
Moreover, considering the acid–base properties of L1 in solution (see section 3.1), the receptor is most likely present in the membrane as a monoprotonated species, with the acidic proton localized on the central amine group of the triamine chain, making it form 1:1 or 1:2 receptor to analyte adducts.
:1 ligand to KP complex is prevalent and the 1:1 molar ratio of lipophilic TDA+ cations with respect to the carrier L1 would be sufficient to stabilize the ion-exchange process. However, the trifluoroacetate anions from TFA will remain in the membrane phase due to their higher lipophilicity, while smaller and more mobile Cl− anions from TDACl will be quickly leached into the solution. Hence, additional TDA+ sites are essential for maintaining the membrane neutrality and ion-exchange efficacy. The overall equilibrium, which ensures the mass balance of neutral and charged species in the membrane, can be represented as follows.where L1org, KP−aq corresponds to the neutral ligand L1 form in the membrane phase and ketoprofen anion (deprotonated form) in solution, respectively. Additionally, the asymmetric anion-exchanger, for instance, TDMACl, may better stabilize the asymmetric complex between KP and L1 in its monoprotonated form given in Scheme 2 and enhance the sensor's optical response to ketoprofen.

The tests on membranes Mb1.3–Mb5 response to KP were run in the concentration range of 2.0 μM–0.1 mM (two orders of magnitude of KP− ions concentration) and corresponding to 1–20 equiv. ratio of L1 in the membrane and KP in the analyzed solution.
The increase in the fluorescence intensity of sensing spots upon the excitation at 365 nm as a function of the growing concentration of KP was recorded, and this change was strongly correlated to the L1/anion-exchange ratio. Thus, as shown in Fig. 5, in comparison to the membranes Mb1.3–Mb3 doped with 1 equiv. of TDACl, Mb4 and especially Mb5 containing 2 and 3 equiv. of anion-exchanger, respectively, have shown the best response in a wider linear range of KP concentration. The best optical response in all the tested concentration range with R2 = 0.984 was registered for membrane Mb5, containing 1 wt% of L1, 3 equiv. of TFA, and 3 equiv. of asymmetric TDMACl anion-exchanger.
 | ||
| Fig. 5 (a) Luminescence intensity variation of L1-doped membranes Mb1.3–Mb5 deposited on the GS substrate upon KP concentration variation; (b) details of optical response the optimum membrane Mb5 to KP, n = 3. λexc = 365 nm. | ||
To acquire the 2D images of all-solid-state optodes prepared by the drop-casting of a small amount (2 μL) of L1-doped PVC membranes on a solid support, a conventional smartphone was used. Two cellulose-based materials, a filter paper (FP) and color catcher tissue (CC) treated/non-treated with 0.005 M Tris pH 7 buffer solution, and glass slides (GC) were employed to test the KP response in the fluorescence readout mode. The laboratory UV-lamp operating at a wavelength of 365 nm served as an excitation light source. While no significative differences were registered for optodes on CC, a clear luminescence intensity variation for previously selected best membrane composition, Mb5, containing 1 wt% of L1, 3 equiv. of TFA, and 3 equiv. of TDMACl anion-exchanger, was observed on GS and non-treated FP. The obtained calibration curves and corresponding photos are shown in Fig. S14.† Notably, the faster optical response of PVC membranes deposited on FP in comparison to the GC support (fully luminescent at the 5th day after the exposure of KP solutions) indicates FP as the most adapted solid support material for optodes development. While the diffusion of KP solutions inside the membrane deposited on GS support is limited and could occur only from the external top side of the membrane, the faster response of FP-based optodes is due to high permeability of the FP support and possible diffusion of the sample solution inside the membrane phase not only top down but also laterally and from the bottom side. For this reason, further tests on selectivity and KP detection in pharmaceutical compositions were run for optodes on FP support. Moreover, GS and FP are eco-friendly and cost-effective materials, providing “green solutions” in the development of all-solid-state optical sensors.
 | ||
| Fig. 6 Emission intensity at 495 nm of L1 in the presence of increasing amounts of (a) KP, acetate, benzoate, ibuprofen (IBU), naproxen (NPX) and (b) Na+, K+, Ca2+, Mg2+. H2O:EtOH 1:1 v/v, pH = 7, Tris 0.005 M, [L1] = 5 × 10−7 M, λexc = 360 nm. | ||
The selectivity evaluation of all-solid-state optodes indicated the highest influence of KP through the analysis of luminescence intensity in the blue channel (Fig. 7 and Fig. S15†). The selectivity coefficients of all-solid-state optodes with Mb5 were calculated by a modified separate solution method (SSM) as previously reported51 and are listed in Table 2. Details on the optodes’ selectivity evaluation are provided in section S5 of ESI.† No significant influence from the parent compounds such as naproxen, ibuprofen, and benzoate, which can all be present simultaneously in pharmaceutical formulations, were observed. Likewise, the optical response of the L1-based membrane (Mb5) towards KP was not affected either by other highly lipophilic anions tested, such as perchlorate (ClO4−) and thiocyanate (SCN−), which do not contain carboxyl groups (–COOH) in their structures, or by all the tested interfering cations (Table 2 and Fig. S16†), were in good accordance with selectivity tests run in the solution.
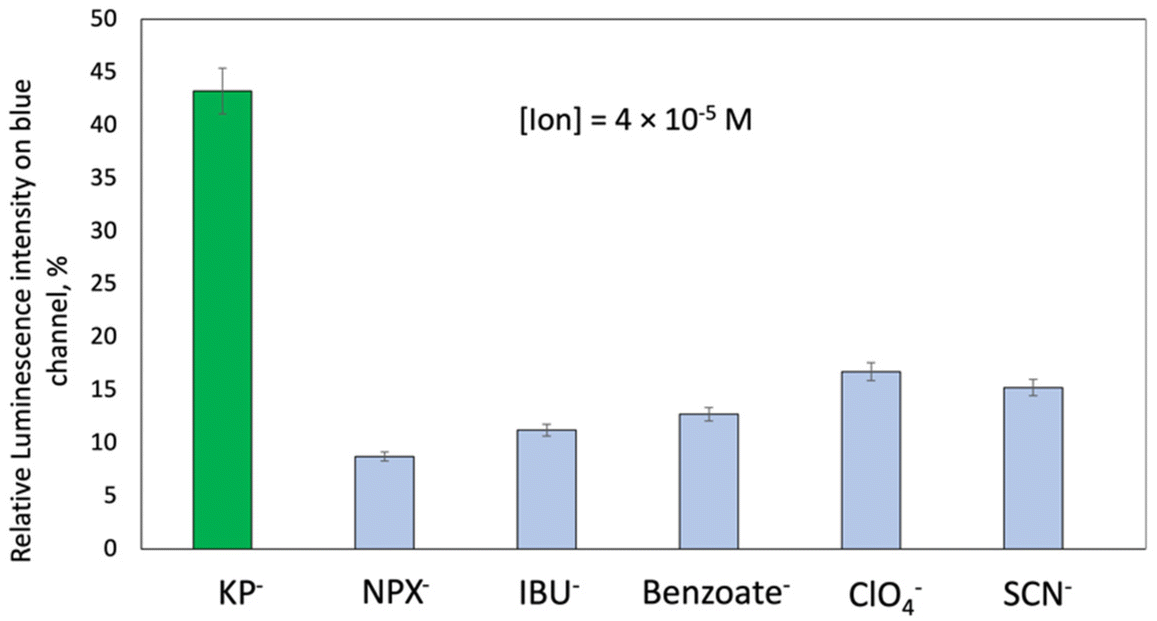 | ||
| Fig. 7 Relative luminescence intensity of membrane Mb5 sensing spots on the blue channel in the presence of equal amounts of KP−, naproxen (NPX−), ibuprofen (IBU−), benzoate (Benzoate−), ClO4− and SCN− interfering anions. [Ion] = 4 × 10−5 M, pH = 7.0, Tris 0.005 M, and λexc = 365 nm. | ||
| Interfering iona | logKKP/ION |
|---|---|
|
a KP was considered as a primary ion; the slope of the semilogarithmic calibration curve toward KP ions was used to calculate logKKP/ION; see ESI† for calculation details.
|
|
| NPX− | −2.21 |
| IBU− | −2.66 |
| Benzoate− | 2.70 |
| ClO4− | −2.74 |
| SCN− | −2.94 |
| Na+ | −2.50 |
| K+ | −2.22 |
| Mg2+ | −4.78 |
| Ca2+ | −4.98 |
| Zn2+ | −5.79 |
| Cu2+ | −6.31 |
In Table 3, the comparison of the main characteristics of NSAIDs optical sensors previously reported in the literature16–19,32,36,52–57 and the characteristics of the developed all-solid-state KP optode in this work based on protonated bis-pyrene polyamine receptor L1 is reported. The wide linear range of response, low detection limit, together with easy application procedure and low operational costs of the developed all-solid-state optical sensor, demonstrate its potential for KP assessment.
| Ligand | Optical output, working media | Sensing range | Limit of detection | Ref. |
|---|---|---|---|---|
| Polyethyleneimine passivated copper-doped CdS quantum dots and zinc oxide nanorods | Fluorescence quenching | 0.05–35.5 μM | 1.36 nM | 16 |
| Tap water, lake water, waste water and human urine samples | ||||
| CdSe/ZnS core–shell nanoparticles (chiral quantum dots) (QDs) bearing a chiral organic ligand | Fluorescence quenching; enantioselective recognition of KP | 50.0 × 10−8 to 250.0 × 10−4 M | — | 17 |
| One-pot water-dispersed chitosan stabilized silver nanoparticles (AgNPs) | Fluorescence quenching | 0.05 to 0.4 μM | — | 18 |
| Aqueous solutions pH 4.00, Ketorolac ampules, spiked river water | ||||
| Carbon Dot core Molecularly Imprinted Polymer | Fluorescence quenching | 0.039 to 3.9 μM | 0.01 μM | 19 |
| Aqueous solutions, human serum and urine samples | ||||
| MIPs with rhodamine B fluorescein-functionalized | Fluorescence quenching water/ethanol 2:1 (v/v) mixture |
1–20 μM (NPX) | 0.27 μM (NPX); 0.011 μM (KP) | 32 |
| 1–10 nM (KP) | ||||
| Bis-anthracene substituted polyamines | Fluorescence enhancement | 0–3 equiv. of KP | 0.21 μM (L1); 0.28 μM (L2) | 36 |
| Water/EtOH 50:50 (v/v) solutions, pH 7.00 |
||||
| 3,3′-bis[N,N-bis(pyridine-2-ylmethyl)aminomethyl]-2,2′-dihydroxybiphenyl (L) complex with Zn2+, [Zn2(H − 2L)]2+ | Fluorescence quenching | 0–10 equiv. of KP and IBU | — | 52 |
| Water/CH3CN 80:20 (v/v) solutions, pH 10.00 |
||||
| Carbon quantum dots in silica molecularly imprinted polymer (CQDs-SiO2@MIP) | Fluorescence quenching, distinction between S- and R-enantiomers. | 0.5–35.0 μM for S-NPX; | 0.20 μM (S-NPX); 0.37 μM (R-NPX). | 53 |
| Solutions on 0.05 M acetate buffer, pH = 4.00 background | 0.5–30.0 μM for R-NPX | |||
| Bis-squaramide receptors | Specific fluorescence quenching of L1 and L2 to KP | 0–5 equiv. of KP, NPX, Benz- | — | 54 |
| CH3CN/DMSO solution 9:1(v/v); paper strip-test able to work in water |
||||
| Hollow core microstructure optical fibers modified with deep eutectic solvent based on natural monoterpenoid and fatty acid. | HC-MOF transmission spectra were recorded over the wavelength range of 400–800 nm | 0.1–3 mg L−1 of NSAIDs | 3 μg L−1 for KP | 55 |
| Human urine samples spiked with KP, mefenamic acid, diclofenac, flurbiprofen, indomethacin | ||||
| ITO-LMR probe ITO electrode | Lossy-mode resonance (LMR) effect for KP detection | 0.25–250 μg mL−1 | 0.5 mM | 56 |
| Aqueous solutions upon electropolymerisation of KP on ITO probe | ||||
| AgNPs in solution | Fluorescence quenching | 0.5–5.0 mg mL−1 | — | 57 |
| Pharmaceutical and prepared dosage forms, spiked human plasma | ||||
| Protonated L1 (N1,N3-bis(1-pyrenylmethyl)diethylenetriamine) ligand inside PVC-based membrane and sensor array | Fluorescence enhancement | 2 μM–0.1 mM | 0.84 μM (0.21 mg L−1) | This work |
| In water/ethanol 1:1 (v:v) solution; on-paper and on-glass all-solid-state optode for aqueous KP solutions |
0.5–20 equiv. KP |
3.4 KP assessment in pharmaceutical compositions
The KP amount in pharmaceutical composition OkiTask by Dompé was estimated with planar all-solid-state optodes based on Mb5 membrane deposited on FP support since this membrane has shown the best analytical characteristics and wider dynamic optical response range. The application of the double addition method has shown the possibility to determine the KP amount in unknown samples with a relative error, RSD of 2.1% and recovery of 102%, as shown in Table 4, which indicates the suitability of L1 receptor for the fluorimetric detection of ketoprofen in real samples.| [KP] real | [KP] found | RSD, % | Recovery, % |
|---|---|---|---|
| 2.00 × 10−5 M | 2.04 × 10−5 M | 2.1 | 102 |
3.5 Multisensor approach for KP low detection limit lowering
Finally, membranes Mb1.3-Mb5 deposited on GS were used in multisensory arrangement to lower the KP detection limit. The membranes’ luminescence intensities were measured simultaneously in individual aqueous solutions of KP in 2 × 10−6–1 × 10−4 M concentration range. The obtained data matrix was used to construct the PLS1 model to correlate optical sensor array output to the KP concentration in calibration solutions (Fig. 8). | ||
| Fig. 8 PLS1 regression model based on the luminescence response of the all-solid-state planar optical sensor array in KP aqueous solutions of different concentrations. | ||
The PLS1 regression model (R2 = 0.944, RMSEV = 0.17 log[KP], 3 PCs, 96% of total variance, 30 samples, 4 variables – luminescence intensity, RGB optical intensities) permitted to improve the KP detection limit to 0.84 μM (0.21 mg L−1).
4. Conclusions
This study investigates a tri-amine receptor with two pyrene fluorogenic groups, in its protonated L1·3HCl and non-protonated L1 forms, for detecting the non-steroidal painkiller ketoprofen (KP). Results highlight that coupling a polar, hydrophilic structure with two large hydrophobic units can create a molecular receptor capable of forming stable complexes with KP. Optical studies of L1 in a 1:1 (v/v) water/ethanol solution at neutral pH revealed salt-bridging interactions between the carboxylate group of KP (in its anionic form at pH 7) and the protonated polyamine chain of the receptor. This interaction induces a significant enhancement of the 396 nm excimer emission band. Coordination of KP also triggered a translocation of the acidic proton within the triamine chain from the central nitrogen to the benzylic nitrogen, inhibiting the PET process from the triamine chain to the excited fluorophores.
The receptor's optical signal was used to develop an all-solid-state sensor based on L1 incorporated into PVC membranes for the optical sensing of KP in aqueous media. The sensor composition was optimized by considering the impact of different membrane components on the optical response. The best response was achieved with membrane Mb5, containing 1 wt% L1, 3 equivalents of TFA, and 3 equivalents of asymmetric TDMACl anion-exchanger. Various solid supports were tested. Paper and glass were selected due to their cost-effectiveness and environmental friendliness. Selectivity tests showed minimal interference from ions and other active pharmaceutical ingredients, including naproxen, ibuprofen, and benzoate. The developed sensor was successfully applied to analyze the OkiTask pharmaceutical formulation, with an RSD of 2.1% and a recovery of 102%. This work demonstrates that polyamine chains coupled with fluorescent hydrophobic moieties provide an effective strategy for developing optical KP chemosensors with high stability, fluorescence response, and selectivity. Additionally, the optical sensor array based on bis-pyrene-substituted receptors (Mb1.3–Mb5) offers a fast and indirect method for screening and discriminating between common non-steroidal painkillers, reducing the detection limit of KP from 4.3 mg L−1 to 0.21 mg L−1.
Data availability
The data supporting this article have been included as part of the ESI.†The data analysis scripts of this article are freeware and available online: Colorpicker (https://imagecolorpicker.com/) ImageJ (https://ij.imjoy.io/).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors acknowledge financial support from MIUR (PRIN 2017 project 2017EKCS35). L. Lvova acknowledges financial support from Tor Vergata University (project ORIENTATE). A. Bencini, M. Innocenti and G. M. Romano thank Regione Toscana for financial support of the MetalRec Project (CUP B97G23000210009) within the program “Progetti di alta formazione attraverso l'attivazione di assegni di ricerca nell'ambito della transizione verde”.References
- P. M. Brooks and R. O. Day, N. Engl. Med., 1991, 324, 1716–1725 CrossRef CAS PubMed.
- https://www.nhs.uk/conditions/nsaids/ .
- N. Halfter, E. Espinosa-Cano, G. M. Pontes-Quer, R. A. Ramírez-Jiménez, C. Heinemann, S. Möller, M. Schnabelrauch, H. P. Wiesmann, V. Hintze and M. R. Aguilar, J. Funct. Biomater., 2003, 14, 160 CrossRef PubMed.
- E. Koumaki, D. Mamais and C. Noutsopoulos, J. Hazard. Mater., 2017, 323, 233–241 CrossRef CAS PubMed.
- K. Świacka, A. Michnowska, J. Maculewicz, M. Caban and K. Smolarz, Environ. Pollut., 2021, 273, 115891 CrossRef PubMed.
- P. Izadi, P. Izadi, R. Salem, S. A. Papry, S. Magdouli, R. Pulicharla and S. K. Brar, Environ. Pollut., 2020, 267, 115370 CrossRef CAS PubMed.
- E. Tyumina, M. Subbotina, M. Polygalov, S Tyan and I. Ivshina, Front. Microbiol., 2023, 14, 1200108 CrossRef PubMed.
- J. Wilkinson, P. S. Hooda, J. Barker, S. Barton and J. Swinden, Environ. Pollut., 2017, 231, 954–970 CrossRef CAS PubMed.
- L. M. Gomez-Olivan, Non-Steroidal Anti-Inflammatory Drugs in Water, Springer International Publishing, 2020 Search PubMed.
- Y. Fan, Y. Feng, S. Da and Z. Wang, Talanta, 2005, 65, 111–117 Search PubMed.
- M. J. Martin, F. Pablos and A. G. Gonzalez, Talanta, 1999, 49, 453–459 CrossRef CAS PubMed.
- M. El-Sadek, S. El-Adl, M. Abou-Kull and S. M. Sakr, Talanta, 1993, 40, 585–588 Search PubMed.
- J. Lenik, J. Anal. Chem., 2012, 67, 543–549 Search PubMed.
- M. Y. Fares, N. S. Abdelwahab, M. A. Hegazy, M. M. Abdelrahman, A. M. Mahmoud and G. M. El-Sayed, Anal. Sci., 2022, 38, 23–37 CrossRef PubMed.
- G. Picci, S. Farotto, J. Milia, C. Caltagirone, V. Lippolis, M. C. Aragoni, C. Di Natale, R. Paolesse and L. Lvova, ACS Sens., 2023, 8, 3225–3239 CrossRef CAS PubMed.
- S. Chen, S. Zhou, J. Fu, S. Tang, X. Wu, P. Zhao and Z. Zhang, Anal. Methods, 2021, 13, 2836–2846 RSC.
- T. Delgado-Pérez, L. M. Bouchet, M. de la Guardia, R. E. Galian and J. Pérez-Prieto, Chem. – Eur. J., 2013, 19, 11068–11076 CrossRef PubMed.
- M. Mabrouk, S. F. Hammad, A. A. Abdella and F. R. Mansour, Colloids Surf., A, 2021, 614, 126182 CrossRef CAS.
- S. Bhogal, K. Kaur, S. Maheshwari and A. K. Malik, J. Fluoresc., 2019, 29, 145–154 CrossRef CAS PubMed.
- Y. Liu, T. Minami, R. Nishiyabu, Z. Wang and P. Anzenbacher Jr., J. Am. Chem. Soc., 2013, 135, 7705–7712 Search PubMed.
- T. V. Shishkanova, G. Broncová, A. Skálová, V. Prokopec, M. Člupek and V. Král, Electroanalysis, 2019, 31, 2024–2031 CrossRef CAS.
- J. Han, B. Wang, M. Bender, S. Kushida, K. Seehafer and U. H. F. Bunz, ACS Appl. Mater. Interfaces, 2017, 9, 790–797 Search PubMed.
- X. Liu, N. Zhang, J. Zhou, T. Chang, C. Fang and D. Shangguan, Analyst, 2013, 138, 901–906 Search PubMed.
- D. Zhang, S. Li, R. Lu, G. Liu and S. Pu, Dyes Pigm., 2017, 146, 305 Search PubMed.
- J. Li, Y. Chen, T. Chen, J. Qiang, Z. Zhang, T. Wei, W. Zhang, F. Wang and X. Chen, Sens. Actuators, B, 2018, 268, 446–455 CrossRef CAS.
- A. Garau, M. C. Aragoni, M. Arca, A. Bencini, A. J. Blake, C. Caltagirone, C. Giorgi, V. Lippolis and M. A. Scorciapino, ChemPlusChem, 2020, 85, 1789–1799 CrossRef CAS PubMed.
- M. C. Aragoni, M. Arca, A. Bencini, C. Caltagirone, A. Garau, F. Isaia, M. E. Light, V. Lippolis, C. Lodeiro, M. Mameli, R. Montis, M. C. Mostallino, A. Pintus and S. Puccioni, Dalton Trans., 2013, 42, 14516–14530 Search PubMed.
- M. Formica, G. Ambrosi, V. Fusi, L. Giorgi, M. Arca, A. Garau, A. Pintus and V. Lippolis, New J. Chem., 2018, 42, 7869–7883 Search PubMed.
- D. Paderni, E. Macedi, L. Lvova, G. Ambrosi, M. Formica, L. Giorgi, R. Paolesse and V. Fusi, Chem. – Eur. J., 2022, 28, e202201062 Search PubMed.
- A. Garau, L. Lvova, E. Macedi, G. Ambrosi, M. C. Aragoni, M. Arca, C. Caltagirone, S. J. Coles, M. Formica, V. Fusi, L. Giorgi, F. Isaia, V. Lippolis, J. B. Orton and R. Paolesse, New J. Chem., 2020, 44, 20834–20852 RSC.
- L. Lvova, C. Di Natale and R. Paolesse, in Electronic Tongues Fundamentals and recent advances, ed. F. M. Shimizu, M. L. Braunger and A. Riul Jr, IOP Publishing, Bristol, UK, 2021, pp 9–1–9–51 Search PubMed.
- T. Wang, Q. Li, M. Wang, J. Xu, J. Li and F. Wang, Food Chem., 2023, 410, 135419 CrossRef CAS PubMed.
- Y. Liu, T. i Minami, R. Nishiyabu, Z. Wang and P. Anzenbacher Jr., J. Am. Chem. Soc., 2013, 135, 7705–7712 CrossRef CAS PubMed.
- A. Akdeniz, T. Minami, S. Watanabe, M. Yokoyam, T. Ema and P. Anzenbacher Jr., Chem. Sci., 2016, 7, 2016–2022 RSC.
- M. Pushina, P. Koutnik, R. Nishiyabu, T. Minami, P. Savechenkov and P. Anzenbacher Jr., Chem. – Eur. J., 2018, 24, 4879–4884 CrossRef CAS PubMed.
- G. M. Romano, L. Mummolo, M. Savastano, P. Paoli, P. Rossi, L. Prodi and A. Bencini, Chem. Commun., 2022, 58, 7022–7025 RSC.
- (a) N. Busschaert, C. Caltagirone, W. Van Rossom and P. A. Gale, Chem. Rev., 2015, 115, 8038–8155 CrossRef CAS PubMed; (b) P. A. Gale, Chem. Commun., 2011, 47, 82–86 RSC; (c) C. Bazzicalupi, A. Bencini and V. Lippolis, Chem. Soc. Rev., 2010, 39, 3709–3728 RSC; (d) C. Caltagirone and P. A. Gale, Chem. Soc. Rev., 2009, 38, 520–563 RSC; (e) V. Amendola, M. Bonizzoni, D. Esteban-Gómez, L. Fabbrizzi, M. Licchelli, F. Sancenón and A. Taglietti, Coord. Chem. Rev., 2006, 2590, 1451–1470 CrossRef.
- (a) E. García-España, P. Diaz, J. M. Llinares and A. Bianchi, Coord. Chem. Rev., 2006, 250, 2952–2986 CrossRef; (b) A. E. Hargrove, S. Nieto, T. Zhang, J. L. Sessler and E. V. Anslyn, Chem. Rev., 2011, 111, 6603–6782 CrossRef CAS PubMed; (c) E. Arturoni, C. Bazzicalupi, A. Bencini, C. Caltagirone, A. Danesi, A. Garau, C. Giorgi, V. Lippolis and B. Valtancoli, Inorg. Chem., 2008, 47, 6551–6563 CrossRef CAS PubMed; (d) P. Mateus, R. Delgado, P. Brandão and V. Félix, J. Org. Chem., 2009, 74, 8638–8646 CrossRef CAS PubMed; (e) A. Bianchi, K. Bowman-James and E. García-España, Supramolecular Chemistry of Anions, Wiley-VCH, New York, 1997 Search PubMed.
- J. A. Sclafani, M. T. Maranto, T. M. Sisk and S. A. Van Arman, J. Org. Chem., 1996, 61, 3221–3222 CrossRef CAS PubMed.
- Y. Shiraishi, Y. Tokitoh and T. Hirai, Org. Lett., 2006, 8, 3841–3844 CrossRef CAS PubMed.
- Y. T. Simonini Steiner, G. M. Romano, L. Massai, M. Lippi, P. Paoli, P. Rossi, M. Savastano and A. Bencini, Molecules, 2023, 28, 4552 CrossRef CAS PubMed.
- M. J. Adams, J. G. Highfield and G. F. Kirkbright, Anal. Chem., 1977, 49, 1850–1852 CrossRef CAS.
- (a) A. Ueno, I. Suzuki and T. Osa, J. Am. Chem. Soc., 1990, 62, 2461–2466 CAS; (b) J. Strauss and J. Daub, Org. Lett., 2002, 4, 683–686 CrossRef CAS PubMed.
- S. Reiter, M. K. Roos and R. de Vivie-Riedle, ChemPhotoChem, 2019, 3, 881–888 CrossRef CAS.
- J. Hoche, H. Schmitt, A. Humeniuk, I. Fischer, R. Mitric and M. I. S. Rohr, Phys. Chem. Chem. Phys., 2017, 19, 25002–250015 RSC.
- G. Jones and V. I. Vullev, J. Phys. Chem. A, 2001, 105, 6402–6406 CrossRef CAS.
- P. Gans, A. Sabatini and A. Vacca, Talanta, 1996, 43, 1739–1753 CrossRef CAS PubMed.
- (a) A. Bencini, A. Bianchi, E. García-España, M. Micheloni and J. A. Ramirez, Coord. Chem. Rev., 1999, 188, 97–156 CrossRef CAS; (b) A. Bianchi, B. Escuder, E. García-España, S. V. Luis, V. Marcelino, J. F. Miravet and J. A. Ramirez, J. Chem. Soc., Perkin Trans. 2, 1994, 1253–1259 RSC.
- (a) C. Bazzicalupi, A. Bencini, A. Bianchi, C. Giorgi, V. Fusi, B. Valtancoli, M. A. Bernardo and F. Pina, Inorg. Chem., 1999, 38, 3806–3813 CrossRef CAS; (b) J. Seixas de Melo, M. T. Albelda, P. Daíz, E. García-España, C. Lodeiro, S. Alves, J. C. Lima, F. Pina and C. Soriano, J. Chem. Soc., Perkin Trans., 2002, 2, 991–998 RSC.
- F. A. Cotton and G. Wilkinson, Advanced Inorganic Chemistry, Wiley-Interscience, 5th edn, 1988 Search PubMed.
- L. Lvova, G. Pomarico, F. Mandoj, F. Caroleo, C. Di Natale, K. M. Kadish and S. Nardis, J. Porphyrins Phthalocyanines, 2020, 24, 964–972 CrossRef CAS.
- D. Paderni, E. Macedi, G. E. Giacomazzo, M. Formica, L. Giorgi, B. Valtancoli, P. Rossi, P. Paoli, L. Conti, V. Fusi and C. Giorgi, Dalton Trans., 2024, 53, 9495–9509 RSC.
- Z. Dehghani, M. Akhond and G. Absalan, Microchem. J., 2021, 160, 105723 CrossRef CAS.
- G. Picci, M. C. Aragoni, M. Arca, C. Caltagirone, M. Formica, V. Fusi, L. Giorgi, F. Ingargiola, V. Lippolis, E. Macedi, L. Mancini, L. Mummolo and L. Prodi, Org. Biomol. Chem., 2023, 21, 2968 RSC.
- P. Pidenko, C. Vakh, A. Shishov, J. Skibina, cN. Burmistrova and A. Bulatov, Talanta, 2021, 232, 122305 CrossRef CAS PubMed.
- R. Bogdanowicz, P. Niedziałkowski, M. Sobaszek, D. Burnat, W. Białobrzeska, Z. Cebula, P. Sezemsky, M. Koba, V. Stranak, T. Ossowski and M. Smietana, Sensors, 2018, 18, 1361 CrossRef PubMed.
- N. A. Abdallah, M. E. Fathy, M. M. Tolba, A. M. El-Brashy and F. A. Ibrahim, RSC Adv., 2022, 12, 33540 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4an01469c |
| This journal is © The Royal Society of Chemistry 2025 |