
Development and validation of a one-step SMN assay for genetic testing in spinal muscular atrophy via MALDI-TOF MS†
Xiaodong
Xing‡
 ab,
Xing
Ji‡
c,
Xinzhu
Liu‡
a,
Xiaohui
Jin
d,
Zhenglei
He
d,
Ajing
Xu
a,
Wengao
Jiang
b,
Wenbo
Ji
e,
Yan
Liu§
a,
Jian
Zhang§
a and
Xiaohui
Huang§
*a
ab,
Xing
Ji‡
c,
Xinzhu
Liu‡
a,
Xiaohui
Jin
d,
Zhenglei
He
d,
Ajing
Xu
a,
Wengao
Jiang
b,
Wenbo
Ji
e,
Yan
Liu§
a,
Jian
Zhang§
a and
Xiaohui
Huang§
*a
aDepartment of Clinical Pharmacy, Xinhua Hospital Affiliated to Shanghai Jiaotong University School of Medicine, Shanghai 200092, China. E-mail: huangxiaohui@xinhuamed.com.cn
bCollege of Pharmacy, Chongqing Medical University, Chongqing 400016, China
cDevelopmental and Behavioral Pediatric Department and Child Primary Care Department, Xinhua Hospital Affiliated to Shanghai Jiaotong University School of Medicine, Shanghai 200092, China
dWuXi Diagnostic Medical Testing Institute (Shanghai), Shanghai 200137, China
eDepartment of Neurology, Second Affiliated Hospital (Shanghai Changzheng Hospital) of Naval Medical University, Shanghai 201209, China
First published on 25th November 2024
Abstract
Spinal muscular atrophy (SMA) is a fatal neuromuscular disorder primarily attributed to the homozygous deletion of the survival motor neuron 1 (SMN1) gene, with disease severity closely correlated to the copy number variations (CNV) of SMN2. Conventional methodologies, however, fail to provide a comprehensive gene overview of SMN and are often both time-intensive and costly. In this study, we present a novel one-step MALDI-TOF MS assay for SMA gene testing. To accurately quantify CNV, we incorporated RPPH1 as an internal control alongside normal samples and competing templates targeting SMN1, SMN2, and RPPH1 for multiple corrections. The CNV assay enables precise quantification of exons 7/8 in both SMN1 and SMN2 genes, achieving a kappa value of 0.935 (P < 0.001) when compared with multiple ligation-dependent probe amplification (MLPA) during its development phase. This accuracy was further corroborated in a cohort comprising 78 individuals. To identify patients harboring compound heterozygous mutations or silent carriers, prevalent pathogenic variants along with sequence variants of SMN1 were integrated into our analysis framework; plasmids were constructed for methodological validation purposes. Utilizing these combinatorial assays for SMN detection, we identified one patient exhibiting a compound heterozygous mutation characterized by genotype [0 + 1d] and another subject presenting genotype [2 + 1], who harbored simultaneous variants of g.27134T > G and g.27706_27707delAT. The CNV assessment combined with pathogenic variants analysis developed through MALDI-TOF MS provides a comprehensive gene profile of SMN within a single analytical run. Given its unparalleled cost-effectiveness and time efficiency, this approach holds significant promise for further application in clinical diagnosis as well as newborn screening for SMA.
Introduction
Spinal muscular atrophy (SMA) is a rare autosomal recessive neuromuscular disorder primarily resulting from the homozygous deletion of the survival motor neuron gene (SMN1; OMIM 600354), which is located proximal to the telomere on the long arm of chromosome 5.1 Inadequate levels of SMN protein lead to the degeneration of alpha motor neurons in the anterior horn of the spinal cord,2 resulting in muscle atrophy and movement disorders. In severe cases, SMA can compromise respiratory and swallowing functions.1,2 SMA exhibits a markedly high incidence of disability and mortality, rendering it one of the leading genetic causes of untimely demise among infants aged less than 2 years.3Biallelic variants in SMN1 were found to be inherited from both parents in 98% of SMA patients, while de novo variants affecting one allele were identified in the remaining 2%.4 There are two primary categories of SMA genotypes: approximately 95% are attributed to the homozygous deletion of SMN1, denoted as genotype [0 + 0]; while the remaining 5% stem from compound heterozygous mutation involving SMN1, characterized by the absence of one allele and an intragenic pathogenic variant in the other, referred to as genotype [0 + 1d].2 Individuals harboring minor pathogenic variants within biallelic SMN1 alleles, specifically the genotype [1d + 1d], are exceedingly rare.2 The estimated prevalence of carriers with SMA is approximately between 1 in 100 and 1 in 45 globally, with a frequency of about 1 in 48 observed within Asian population.5 The SMN1 genotypes detected among carriers predominantly fall into four distinct categories: [1 + 0], [2 + 0], [1 + 1d], or [2 + 1d], with population frequencies recorded as 2.58%, 0.11%, 0.05%, and 0.002%, respectively.6SMN2 (OMIM 601627), the orthologous gene to SMN1, is located on the centromere side and exhibits only 5 base disparities compared to its counterpart. Among these variations, c.840C > T results in selective splicing that leads to the expression of only full-length viable SMN protein from approximately ten percent of exon7-containing transcripts derived from SMN2 mRNA, thus resulting in a predominance of truncated forms.1 The copy number variation (CNV) associated with SMN2 has been recognized as a significant modifier for SMA; it has been noted that patients possessing higher copy numbers tend to exhibit milder phenotypic manifestations.7 In pharmaceutical clinical trials, CNV analysis for SMN2 is frequently employed as a critical reference point for therapeutic intervention strategies.7 Although correlations between copies of SMN2 and clinical phenotype may not always be consistent,1,2,7 it remains an essential component within diagnostic frameworks according to current management consensus.8
One of the hallmark characteristics of SMA is its manifestation across multiple clinical types, each exhibiting significant variability in disease progression and prognosis among affected individuals.1 This considerable heterogeneity underscores the complex nature of SMA and poses challenges for clinicians in tailoring treatment strategies to meet the needs of individual patients. As a rare genetic disorder, those afflicted with SMA have long grappled with the formidable challenge posed by the absence of any definitive cure. Until 2016, when the U.S. Food and Drug Administration approved nusinersen, the first SMN-dependent therapy designed to target SMN2 and restore viable SMN protein expression.9,10 This breakthrough marks a substantial advancement in addressing SMA and has brought renewed optimism to those impacted by this condition. Subsequently, onasemnogene abeparvovec and risdiplam, both gene-targeting therapeutics, were added to the list, prompting SMA research into the forefront of research interest.11,12 Clinical evidence indicates that disease-modifying therapies yield improved outcomes in pediatric patients when administered prior to the symptom onset;13,14 nevertheless, their efficacy remains limited for individuals with a protracted disease course or older age cohorts.14,15 Given the exorbitant costs associated with gene-targeting therapeutics alongside the benefits of early intervention, it is imperative to shift treatment paradigms for SMA towards a “pre-symptomatic” stage. Newborn screening (NBS) emerges as an optimal strategy for identifying pre-symptomatic patients effectively. A nonrandomized controlled trial (DRKS00012699) has demonstrated significantly better functional outcomes and treatment responses within NBS cohort compared to the unscreened groups experiencing clinical symptom onset.16 Therefore, implementing NBS for SMA has become a focal point of global interest.
To date, the majority of European and American nations, along with certain Asian countries such as Japan and South Korea, have implemented comprehensive nationwide NBS programs for SMA.17–19 Meanwhile, a variety of clinical techniques are employed for SMA gene testing. Among these methods, multiple ligation-dependent probe amplification (MLPA) directly quantifies the CNV of SMN1 and SMN2, effectively distinguishing between patients, carriers, and unaffected individuals. This positions MLPA as the gold standard for diagnosing SMA according to management consensus guidelines.8 However, MLPA is unable to detect pathogenic variants within SMN1 or identify carriers with the [2 + 0] genotype. Similarly, real-time PCR can be utilized to quantify SMN genes; nevertheless, it exhibits marginally inferior specificity compared to MLPA and requires distinct probes engineered specifically for detecting SMN2 while also failing to identify [2 + 0] genotype or point mutations. Consequently, reliance solely on traditional sequencing techniques alone such as Sanger sequencing or next-generation sequencing in genetic testing for SMA proves exceedingly limited.20 Certain long-read sequencing techniques may be better suited for these scenarios;21,22 nevertheless, their prohibitive costs and stringent sample quality requirements hinder large-scale implementation—such as in NBS—beyond the scientific inquiry.
In the global promotion of NBS for SMA, there is an urgent need for rapid and cost-effective detection modalities that can facilitate broader-scale screening and diagnosis of SMA. The detecting techniques should ideally possess the capability to (1) discern CNV of SMN1 and SMN2, (2) identify pathogenic variants beyond c.840C deletions, and (3) aid in identifying carriers with a [2 + 0] genotype. Due to the cost-effectiveness and moderate throughput capabilities, previous studies have sought to utilize matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) to develop genetic tests for SMA.23–25 However, the MALDI-TOF MS methodologies established in these investigations either failed to accurately quantify CNV of SMN genes or were unable to simultaneously detect pathogenic variants, hindering comprehensive information retrieval regarding the SMN gene. This limitation poses a risk of misdiagnosis or missed diagnosis in patients with [0 + 1d] and [1d + 1d] genotypes as well as carriers with [2 + 0], [1 + 1d], and [2 + 1d] genotypes, significantly constraining SMA diagnosis and large-scale NBS efforts. To address these issues, integrating MALDI-TOF MS with other genetic testing techniques such as capillary electrophoresis, MLPA, or sequencing methods is necessary; however, this increases labor costs, reagent expenses, and processing times—rendering it less suitable for NBS applications. In this study, we employed MALDI-TOF MS to develop a novel one-step SMN assay that accurately quantifies SMN copies while simultaneously identifying variants within SMN1, facilitating clear identification of individual SMN genes within a single analytical run.26 Upon the development phase of the CNV assay, internal reference controls and competing templates were introduced. The obtained data revealed a high degree of concordance when compared with MLPA. Based on prevalent pathogenic and sequence variants of SMN1, we accurately identified one patient exhibiting compound heterozygous mutations characterized by the [0 + 1d] genotype as well as an asymptomatic individual with a [2 + 1] genotype via MALDI-TOF MS. The SMN assay developed through MALDI-TOF MS is well-suited for diagnosing SMA, assessing disease severity, and supporting large-scale screening initiatives.
Materials and methods
Sample collection and DNA extraction
All procedures were conducted in accordance with the approval of the Ethics Committee of Xinhua Hospital, Shanghai Jiaotong University School of Medicine (No. XHEC-C-2023-072-1) and adhered to principles of the Helsinki Declaration and International Standards on Good Clinical Practice standards.27 Whole peripheral blood samples from all participants were collected in EDTA tubes between 2023 and 2024, with 200 μl subsequently processed using a commercial magnetic beads-based extraction kit (Xi'an TianLong Science and Technology Co., Ltd, China). Genomic DNA (gDNA) was then extracted utilizing an automated nucleic acid extractor (GeneRotex 96). The concentration and quality of the extracted nucleic acids were assessed spectrophotometrically using a NanoDrop™ 2000 spectrophotometer (NanoDrop Technologies Inc., DE, USA), with samples exhibiting an A260/A280 ratio between 1.6 and 2.0 deemed acceptable. Coriell DNAs (IDs: NA03813, NA03814, NA12548) were obtained from Coriell Institute (https://www.coriell.org). DNA concentration was further quantified employing the Qubit dsDNA BR Assay Kit on Qubit® 2.0 Fluorometer (Life Technologies, Grand Island, NY, USA). All samples were stored at −80 °C until experimental procedures commenced. Blood samples were collected following informed consent provided by a parent or legal guardian.Panels and primer design
Two distinct assays (CNV and pathogenic variants) were designed for the genetic analysis of SMA. The CNV panel was designed with amplification primers targeting exon 7 (E7) and exon 8(E8) of SMN1 and SMN2, utilizing RPPH1 as an internal reference control. To ensure precise copy number quantification, competitive templates that correspond to SMN1/SMN2 and RPPH1 were synthesized based on previously established principles.28 For the variants panel, seven prevalent pathogenic variants of SMN1 in the Chinese population (detection frequency > 2), as identified by the American College for Medical Genetics and Genomics,29,30 along with two polymorphic variations associated with the [2 + 0] genotype were incorporated.31 Primers were designed using Agena Bioscience's online software Assay Design Suite (https://support.agenabio.com/s/login/) and subsequently synthesized by Sangon Biotech (Shanghai) Co., Ltd. In instances where software design proved inadequate, primers were manually crafted for optimal assay performance. Detailed primer sequences are provided in Tables S1–S4.†MALDI-TOF MS systems
Unless otherwise specified, all reagents utilized in the MALDI-TOF MS system were sourced from Agena Biosciences. The experimental procedure can be roughly divided into PCR amplification of the target DNA fragments, Shrimp Alkaline Phosphatase (SAP) reaction (treating PCR products with SAP to remove excess ddNTPs), single nucleotide extension, and subsequent mass spectrometry analysis.PCR amplification
The PCR reagent set comprised 0.8 μl of ddH2O, 0.5 μl of buffer, 0.4 μl of MgCl2, 0.1 μl of dNTP mix, 1 μl of Primer Mix, 0.2 μl of PCR Enzyme, and 20 ng of gDNA (2 μl) to form a total volume of 5 μl PCR system. In the overall volume of the Primer Mix (200 μl), all amplified primers were included at a concentration of 1 μl each, except for SMN Exon7_F and SMN Exon7_R which were incorporated at a concentration of 2 μl each. For the CNV panel, to maintain the integrity of 5 μl PCR system, ddH2O was substituted with an equivalent volume competitive template mix containing approximately 62![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 160 copies for exon 7_C, 92000 copies for exon 8_C, and 424000 copies for RPPH1_C respectively. The PCR reaction procedure consisted of the following stages: Stage 1 involved an initial denaturation step at 95 °C for 2 minutes. Stage 2 comprised 45 cycles of denaturation at 95 °C for 30 seconds, annealing at 56 °C for 30 seconds, and extension at 72 °C for 60 seconds. Stage 3 involved a final extension step at 72 °C for 5 minutes before holding the reaction mixture at 4 °C for storage.
160 copies for exon 7_C, 92000 copies for exon 8_C, and 424000 copies for RPPH1_C respectively. The PCR reaction procedure consisted of the following stages: Stage 1 involved an initial denaturation step at 95 °C for 2 minutes. Stage 2 comprised 45 cycles of denaturation at 95 °C for 30 seconds, annealing at 56 °C for 30 seconds, and extension at 72 °C for 60 seconds. Stage 3 involved a final extension step at 72 °C for 5 minutes before holding the reaction mixture at 4 °C for storage.
SAP reaction
The 2 μl SAP system comprises 1.53 μl of ddH2O, 0.17 μl of SAP buffer, and 0.3 μl of SAP Enzyme. The reaction protocol is as follows: incubation at 37 °C for 40 minutes, followed by a 5-minute incubation at 85 °C, and then maintained at 4 °C.Extension reaction
The 2 μl reagent for the extension reaction consists of 0.62 μl of ddH2O, 0.2 μl of Buffer Plus, 0.2 μl of Termination Mix, 0.04 μl of Extend Enzyme, and 0.94 μl of Extend Primer Mix. For the CNV panel, all primers in the 100 μl Extend Primer Mix were prepared at a concentration that is 4.5 times higher than the initial concentration (500 μM). The Extend Primer Mix system for pathogenic panel had a total volume of 200 μl, with all primers maintained at a consistent concentration of 500 μM. The reaction protocol comprises three sequential stages: (1) an initial heat denaturation step at 95 °C for 30 seconds; (2) a subsequent stage divided into three parts: (i) initial denaturation at 95 °C for 5 seconds; (ii) 5 cycles consisting of 5 seconds at 52 °C followed by 5 seconds at 80 °C; (iii) implementation of 40 cycles combining steps (i) and (ii); (3) the final stage entails incubation at 72 °C for 3 minutes, followed by a cooling step to 4 °C.Mass spectrometry analysis
Extension products were subjected to desalting and subsequently dispensed onto a 384-format SpectroCHIP® (Agena Bioscience). MALDI-TOF mass spectra were obtained using a linear time-of-flight mass spectrometer (Agena Bioscience) equipped with a 337 nm nitrogen laser source operating at a frequency of 20 Hz. The extraction voltage was set at 3 kV, the acceleration voltage maintained at 20 kV, the delay time configured to 4000 ns, and the grid voltage established at 5 kV. The obtained data were analyzed utilizing Typer Analyzer (Agena Biosciences, version 4.1). For copy number quantification, the target gene was initially normalized against the peak area of the corresponding competing template, followed by normalization with an internal reference corrected for variations (RPPH1/RPPH1_C), and ultimately calibrated using samples containing two copies each of SMN1 and SMN2 E7/8 (2N). The calculation formula is as follows:
MLPA assay
MLPA analysis in this study was performed at Shanghai Medicore Technology Co., Ltd (China). The SALSA MLPA kit MRC – P060-B2 SMA Carrier, Applied Biosystems capillary sequencer, and the publicly available Coffalyser.Net software (MRC-Holland, Amsterdam, The Netherlands) were utilized in accordance with the general protocol (https://www.mrcholland.com). Specifically, copy number classification was based on the final ratio (FR) as follows: 0.8 < FR < 1.20 indicates a normal copy number, FR = 0 denotes homozygous deletion, 0.4 < FR < 0.65 signifies heterozygous deletion, 1.30 < FR < 1.65 represents heterozygous duplication, and 1.75 < FR < 2.15 indicates either heterozygous triplication or homozygous duplication.Construction of standard mutation plasmids
Due to the insufficient availability of mutation samples, we constructed mutant plasmids for all identified mutation sites, encompassing both pathogenic and sequence variants, to assess the specificity of the MALDI-TOF MS system in distinguishing mutants from their wild-type counterparts. All mutant plasmids were generated by Sangon Biotech (Shanghai) Co., Ltd. In brief, the synthesized target gene was transferred into the pUC57 vector, followed by plasmid extraction and Sanger sequencing. Subsequently, plasmid samples demonstrating successful mutagenesis were selected for further analysis via MALDI-TOF MS. The sequence of the synthesized plasmids is detailed in the Sanger sequencing results (see ESI†).Statistical analysis
In this study, data were analyzed and plotted by IBM SPSS Statistics (version 29) and GraphPad Prism (version 10). All quantitative data were shown as the mean ± standard deviation. P < 0.05 indicated statistically significant.Results and discussion
The workflow and schematic illustration of CNV assay via MALDI-TOF MS
Drawing upon the genetic attributes of SMA and leveraging the capabilities of the MALDI-TOF MS platform, we developed a quantitative panel to quantify the copy number of the pathogenic gene SMN1 and the prognostic gene SMN2 (Fig. 1). The exogenous oligonucleotide, referred to as a competitive template, shares an identical sequence with the amplified target region except for a single base variation. For example, the competitive template targeting SMN E7 contains a single base extension of G, differing from C in SMN1 E7 and the T in SMN2 E7 (Fig. 1A); therefore, the mass spectrum would reveal peaks corresponding to C, G, T or C and G (indicating zero copies of SMN2) in non-SMA subjects (regardless of carriers status) (Fig. 1B), while most patients with SMA exhibit peaks for G and T (Fig. 1E). Similarly, peak patterns for SMN E8 and RPPH1 are observed in MALDI-TOF MS analysis. The quantity of copies can be determined after rectifying the peak area for the intended region as described in the Mass spectrometry analysis section. In 384-well plate format, the entire experimental process takes about 8 hours. | ||
| Fig. 1 Workflow and schematic illustration of the CNV assay utilizing MALDI-TOF MS. (A) The experimental procedure consists of four steps: a 2.5-hour PCR amplification, a 45-minute SAP reaction, approximately 3.5 hours for the extension reaction, and a 1-hour mass spectrometry analysis. Schematic diagram of mass spectrometry results for SMN E7 (B and E) and SMN E8 (C and F) in non-SMA individuals and patients with SMA, respectively. Internal calibration is performed with (D) or without (G) competing templates. | ||
Notably, the original design scheme aimed to compare the target gene with internal references (RPPH1 or TERT) and 2N samples (Fig. S1†). We have tried to employ instrument parameters of MALDI-TOF MS, such as peak area, height, and signal-to-noise (SNR), for copy quantification. Periodic findings demonstrated the protocol's capability to accurately distinguish between SMA patients exhibiting homozygous deletion and 2N individuals; meanwhile, quantitative analysis revealed a high level of consistency in peak area, height, and SNR (data not shown). However, quantification for non-homozygous deletion or non-2N samples—specifically among certain SMA carriers and individuals with copy number duplications—remained unsatisfactory despite iterative adjustments to primer ratios and refinements in the reaction procedure. Additionally, significant inter-batch disparities were observed, rendering it challenging to extrapolate findings for large-scale applications. To achieve accurate quantitation, we introduced competitive templates for further correction with CNV assay.
Development and validation of CNV assay
During the development stage, samples from ten randomly selected subjects and three Coriell DNA samples (IDs: NA03813, patient with SMA; NA03814, SMA carrier; NA12548, [2 + 1] genotype) were utilized for methodological refinement. For these 13 samples, the SMN copy numbers detected by MALDI-TOF MS exhibited a high degree of concordance with MLPA results (Fig. 2 and Table S5†), yielding a kappa value of 0.935 (P < 0.001; Table S6†). These samples can be reliably detected at a concentration of 10 ng μl−1 (equivalent to 20 ng gDNA) per well, exhibiting an in-batch precision of 96.15%. Of them, five samples exhibited duplication of SMN1 and SMN2 E7/8 as determined by MLPA analysis. The quantifications obtained via MALDI-TOF MS were recorded as follows: 2.00 ± 0.18, 2.03 ± 0.27, 2.03 ± 0.08, and 2.01 ± 0.13, respectively. Additionally, sample NA03814 exhibited an atypical result with five copies for E7/8 of the SMN2 gene (refer to MLPA product brochure). The quantitative measurement for E7 of SMN2 in the MALDI-TOF MS system was found to be 4.4 ± 0.90, which falls within the ambiguous range (Fig. 2B).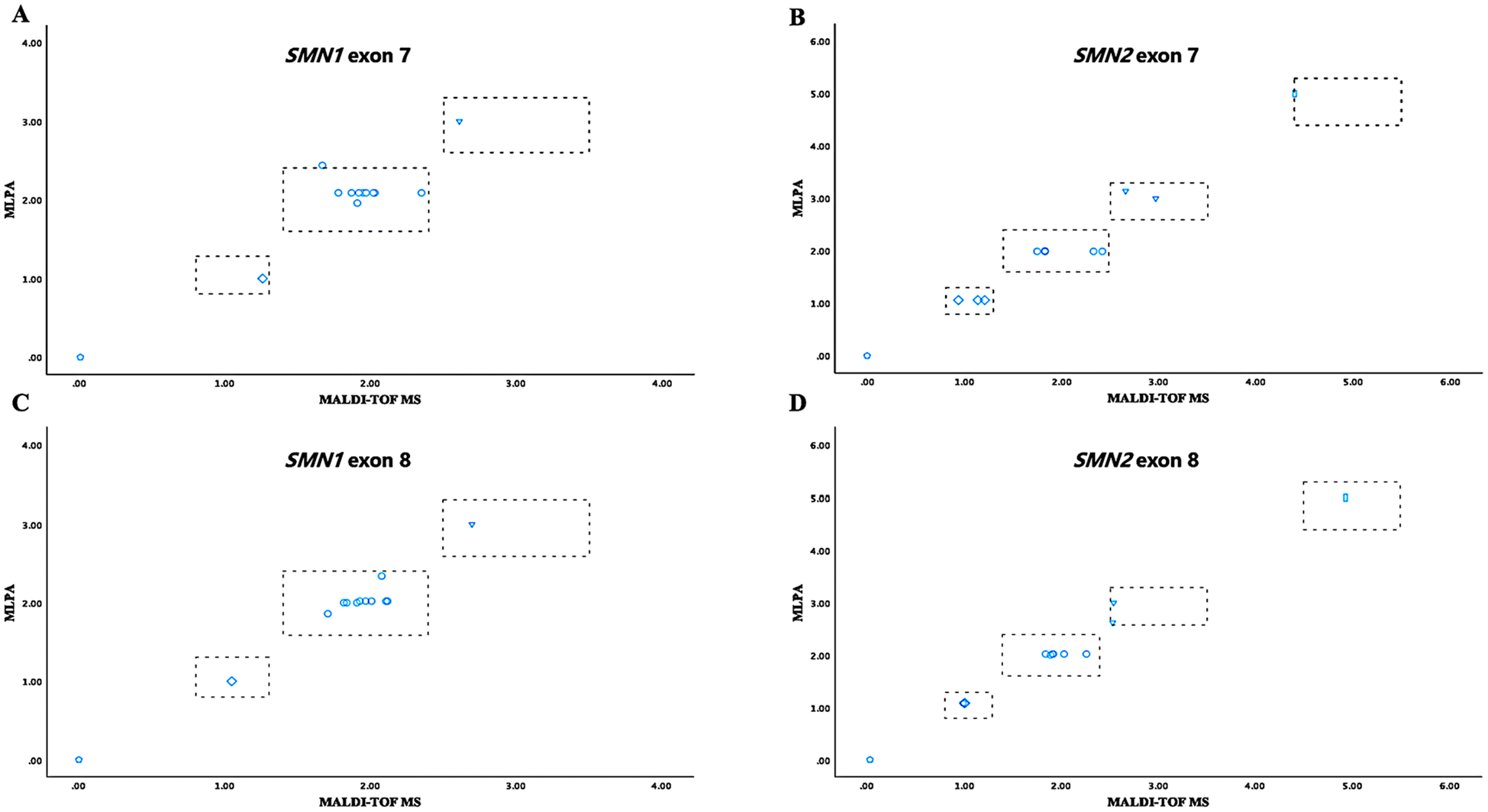 | ||
| Fig. 2 Comparison of SMN copy numbers between MLPA and MALDI-TOF MS across 13 DNA samples. Using two distinct detection methodologies, the copy numbers for SMN1 E7 (A), SMN2 E7 (B), SMN1 E8 (C), and SMN2 E8 (D) were obtained. The gray dotted lines symbolize the cut-off values for both MLPA and MALDI-TOF MS. | ||
To further validate the performance of the CNV assay, we applied it to a clinical cohort consisting of 78 individuals. In this phase, DNA samples were assigned blind numbers by personnel not involved in this study. The quantification findings from MLADI-TOF MS exhibited a robust concordance with MLPA (Fig. 3 and Table S7†). Among these cases, 16 exhibited deletion of both exons 7 and 8, while three instances showed exclusive deletion of exon 7. Additionally, there were also five carriers and 38 normal individuals; the remaining subjects presented varying copy number alterations. Most samples (77 out of 78) displayed three or fewer SMN copies. Using a gDNA volume of 20 ng per well, we conducted additional repetitions of the 2N samples across multiple batches, confirming the inter-batch stability of the CNV panel (Fig. S2†). Moreover, quantification results of 20 ng gDNA per well were consistent with those of 40 ng (Fig. S3†).
 | ||
| Fig. 3 Comparison of SMN copy numbers between MLPA and MALDI-TOF MS in a clinical cohort. By employing two distinct detection methodologies, the copy numbers for SMN1 E7 (A), SMN2 E7 (B), SMN1 E8 (C), and SMN2 E8 (D) were acquired. The gray dotted lines represent the cut-off values for MLPA and MALDI-TOF MS, respectively. Samples outside the box (red) exhibit ambiguous copy numbers. | ||
Our findings indicate that the copies identified through the CNV panel exhibit a high concordance with MLPA (Fig. 2 and 3). Significantly, two researchers from our group (X. X. and Z. H.) in separate laboratories successfully replicated the quantitative findings on a limited set of samples using the CNV assay (Fig. S4†), achieving 100% specificity and accuracy. It is worth noting that while MLPA is regarded as the gold standard for detecting SMN copies, variations may occur across different laboratory settings.32 Thus, certain samples detected by MLPA within an ambiguous range can be juxtaposed with the measurements of MALDI-TOF MS to obtain more precise information (Table S8†). This alternative provides clarity and accuracy where ambiguous data may introduce uncertainty, offering a more reliable option for analysis and decision-making. Additionally, the amount of gDNA required for MALDI-TOF MS is minimal, thereby not imposing an additional burden on newborn blood collection efforts. Studies have shown that DNA extracted from dried blood spots can also be effectively utilized for MALDI-TOF MS testing.24
Detection of intragenic SMN pathogenic variants and sequence variants
Approximately 5% of patients with SMA harbor a point mutation in one SMN1 allele and a deletion in the other, or, exceptionally rarely, biallelic mutations within any of the SMN1 exons.2De novo mutations occur at a relatively elevated frequency due to the inherent instability of the 5q region.1,2 Thus, in addition to quantifying copy numbers, we have also developed a panel of pathogenic variants (Fig. 4 and Fig. S5†) based on common SMN1 point mutations identified in the Chinese population.29,30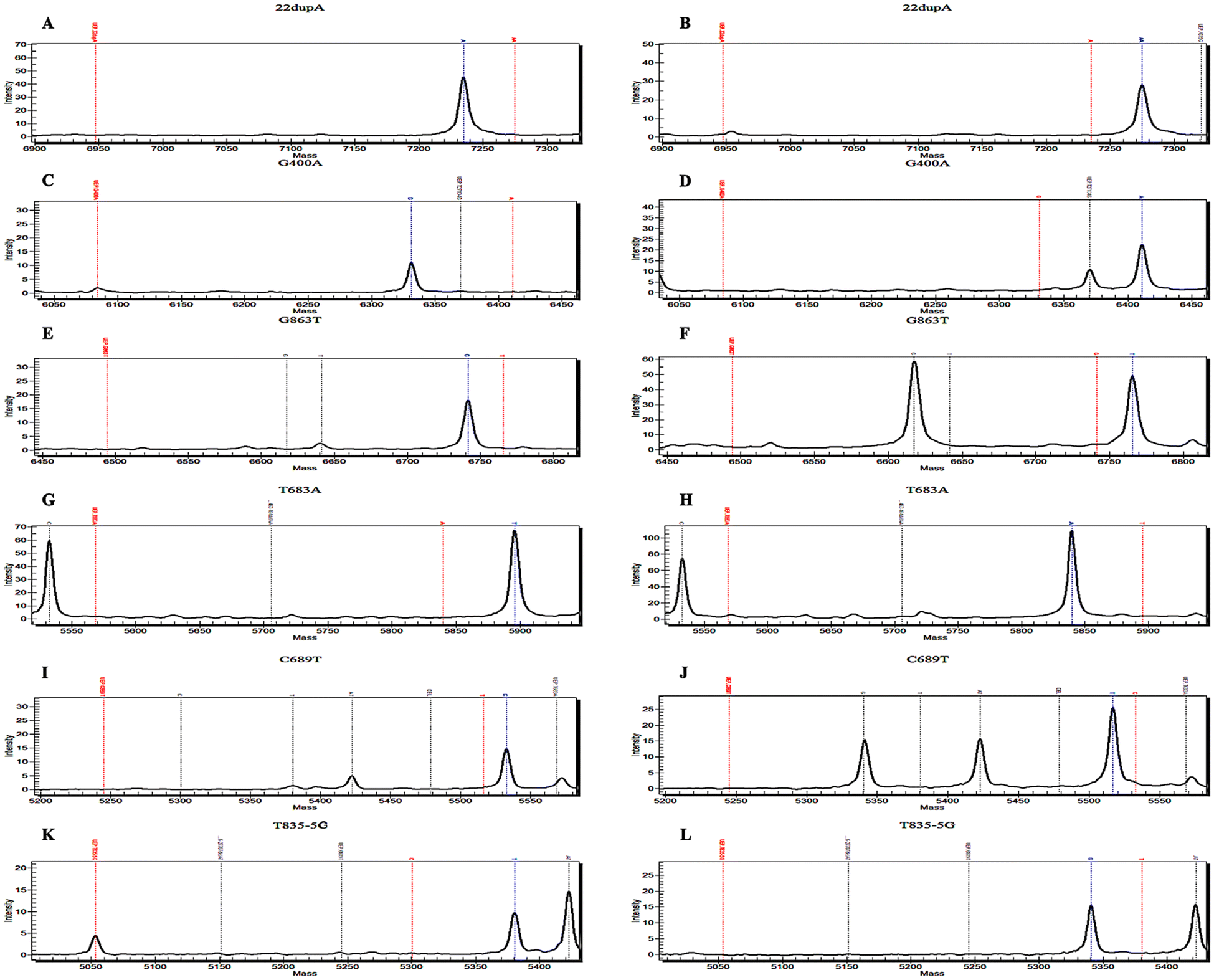 | ||
| Fig. 4 Schematic representation of the detection of common SMN1 pathogenic variants using MALDI-TOF MS. (A and B) 22dupA represents c.22dupA (p. Ser8Lysfs*23), located on exon 1 of SMN1. (C and D) G400A represents c.400G > A (p. Glu134Lys) on exon 3 of SMN1. (E and F) G863T represents c.863G > T (p. Gly279Glufs*5) on exon 7 of SMN1. (G and H) T683A represents c.683T > A (p. Leu228*), located on exon 5 of SMN1. (I and J) C689T represents c.689C > T (p. Ser230Leu) on exon 5 of SMN1. (K and L) T835-5G represents c.835-5T > G (p. Gly279Glufs*5) on intron 6 of SMN1. The figure on the left (A, C, E, G, I and K) represents the wild type of pathogenic variants, while the figure on the right (B, D, F, H, J and L) represents the mutant type. | ||
The pathogenic variant panel demonstrated high sensitivity across the entire cohort, with stable and accurate detections at 20 ng gDNA. Notably, c.463_464delAA, initially part of this panel, is a common mutation site within SMN1.29,30 However, an additional SNP (most likely rs4915) proximal to its mass spectrum peak has impeded accurate interpretation, leading to its ultimate exclusion. Besides, a unique [2 + 0] genotype exists among SMA carriers, characterized by two functional copies of SMN1 on one chromosome and either an SMN1 deletion or conversion on the other. The frequency of the [2 + 0] genotype in SMA carriers is approximately 4%.33 Studies have demonstrated that polymorphic site variation g.27134T > G and g.27706_27707delAT exert a suggestive influence on individuals with the [2 + 0] genotype across several ethnicities.31 Thus, we incorporated these two polymorphic sites to identify potential type [2 + 0] carriers of SMA (Fig. S5A–D†). Upon validation of these polymorphisms, we observed concurrent mutations of g.27134T > G and g.27706_27707delAT in a subject possessing three copies of SMN1 (Fig. S6E and F†); nevertheless, no mutations were identified in two other samples also possessing three SMN1 copies (one being NA12548), indicating that this suggestive effect may only be evident in certain individuals (Table 1).31
Overall, individuals who possess two copies of SMN1 and test positive for both (either) g.27134T > G and (or) g.27706_27707delAT can be considered potential silent SMA carriers. When one parent harbors the [1 + 0] genotype and the other carries three or more copies of SMN1, it strongly indicates that the individual under suspicion possesses a [2 + 0] genotype. The suggestive impact of these two polymorphic sites on individuals with either a [2 + 0] or [2 + 1] genotype still requires further validation through extensive population studies.
Clinical application of SMN detection utilizing MALDI-TOF MS
Remarkably, one patient in the cohort presented with a compound heterozygous deletion of SMN1, specifically characterized by a deletion of one SMN1 gene combined with an intragenic allele pathogenic variant. By employing integrated assays for CNV and pathogenic variants, we successfully elucidated the pathogenic mechanism underlying this compound heterozygous deletion (Fig. 5). The MALDI-TOF MS assays accurately identified the c.A815G mutation in this patient (Fig. 5D), a finding that was validated through DNA-level sequencing encompassing eight exons within the coding region of SMN and RNA reverse transcription monoclonal sequencing. The c.815A > G mutation was confirmed to be located on exon 6 of the patient's sole remaining copy of SMN1. Further, sequencing of parental genomes revealed that the father harbored two copies of SMN1, one of which bore the same c.815A > G mutation as observed in the patient, while the mother was identified as a carrier for SMA (Fig. 5E). Beyond that, no additional pathogenic variants were detected within the cohort. | ||
| Fig. 5 Genetic profiling of one patient with SMA characterized by heterozygous deletion and pathogenic variant of SMN1. (A) Results from the patient's MLPA revealed one copy for exon 7/8 of SMN1 and three copies for exon 7/8 of SMN2. (B) CNV results obtained via MALDI-TOF MS indicated quantifications for E7 of SMN1/2 and E8 of SMN1/2 as 0.98 ± 0.14, 2.64 ± 0.16, 0.87 ± 0.03, and 2.79 ± 0.13, respectively (n = 3). (C) Schematic plot of A815G detection (wild type) from DNA mass spectrometry. (D) Schematic diagram of the patient's pathogenic variant panel featuring A815G as detected by DNA mass spectrometry. A815G variant represents c.815A > G (p. Tyr272Cys) located on exon 6 of SMN1. (E) Genetic map depicting the SMA patient with compound heterozygous mutations. | ||
Currently, approximately 90 documented minor pathogenic variants exist (https://www.hgmd.cf.ac.uk), with nearly 30 identified in Chinese patients.34–40 Adding additional variants can enhance screening for potential [1 + 1d] carriers. Interestingly, the combination of CNV and pathogenic variants assays effectively identified a patient with a compound heterozygous mutation (Fig. 5). This 13-year-old girl with type II SMA displayed typical proximal muscle weakness, particularly in the lower extremities—achieving a maximum motor milestone of standing but unable to walk independently and exhibiting an inability to lift heavy objects with both upper limbs, as indicated by Revised Upper Limb Module rating of 28/37. However, due to possessing only one copy of SMN1, this patient was long misclassified as an SMA carrier rather than as an affected individual. The c.815A > G in SMN1 is a known pathogenic variant reported in previous studies.41,42 This case illustrates the significant potential of MALDI-TOF MS for genetic detection of SMA.
In the domain of SMA genetic testing, our MALDI-TOF MS methodology has exhibited substantial methodological and clinical advantages. From the methodological standpoint, the quantitative variability of prior methods arising from peak intensity fluctuations of SMN genes was effectively mitigated through multiple calibrations. Moreover, the incorporation of constructed positive plasmids addresses the challenges associated with mutation samples scarcity and enhances both the accuracy and specificity of mutation detection via MALDI-TOF MS. Compared to prior MALDI-TOF MS methods, our approach enables absolute quantification of exons 7 and 8 of SMN genes while simultaneously detecting pathogenic variants. In contrast to combined detection methods (such as MALDI-TOF MS in conjunction with other techniques), our method necessitates lower sample volumes and quality requirements. Furthermore, employing MALDI-TOF MS alone offers comprehensive coverage of SMN genes within a single assay, which is time-efficient, user-friendly, and cost-effective—rendering it highly suitable for SMA diagnosis and NBS.
From the clinical application perspective, our method effectively integrates molecular diagnosis and NBS for SMA, significantly reducing the risk of missed screenings. Through timely diagnosis, patients with genotypes such as [0 + 1d] or [1d + 1d] can access gene-targeting therapeutics at the earliest opportunity. Meanwhile, carriers exhibiting genotypes like [2 + 0], [1 + 1d], or [2 + 1d], who are often challenging to identify using conventional methods, can be informed of the possibility of having children with genetic disorders through timely screening. Thus, they can make better preparations for reproduction. To our knowledge, this study represents the most comprehensive investigation of the SMN gene utilizing MALDI-TOF MS thus far and is also the first report suggesting that MALDI-TOF MS can be employed for detecting [0 + 1d] genotype while identifying potential [2 + 0] or [2 + 1] genotypes.25
Conclusions
In general, utilizing MALDI-TOF MS, we have developed a CNV and pathogenic variants assay for SMA. This approach demonstrates the capability of MALDI-TOF MS not only in delivering highly consistent copy number measurements with MLPA in a clinical cohort but also in accurately identifying patients with the [0 + 1d] genotype. Additionally, it offers valuable insights for identifying [2 + 0] silent carriers. Given its capacity for one-step detection of SMN, this assay is expected to be utilized in the clinical diagnosis and NBS of SMA in the foreseeable future.Author contributions
X. X., X. J. and X. L.: writing – original draft, review & editing, methodology, data curation; Xh. J., Z. H.: investigation, visualization, software, validation; A. X., Wg. J. and Wb. J.: investigation, visualization; Y. L., J. Z. and X. H.: conceptualization, project administration, writing – review & editing. All authors read and approved the final manuscript.Ethics approval and consent to participate
The ethics committee of the local hospital approved this study. All procedures followed complied with the ethical standards of the responsible committee on human experimentation (institutional and national) and the Helsinki Declaration of 1975, as revised in 2000(5). Informed consent was obtained from all individual participants included in the study.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors of this study declare that there are no conflicts of interest.Acknowledgements
This work was funded by the Collaborative Innovation Center for Clinical and Translational Science by Chinese Ministry of Education & Shanghai, No. CCTS-2022205, the “Double World-Class Project” of Shanghai Jiaotong University School of Medicine (both to JZ) and the Talent Project established by the Chinese Pharmaceutical Association Hospital Pharmacy department, No. CPA-Z05-ZC-2022-03 (to Xh H).References
- E. Mercuri, C. J. Sumner, F. Muntoni, B. T. Darras and R. S. Finkel, Spinal muscular atrophy, Nat. Rev. Dis. Primers, 2022, 8, 52, DOI:10.1038/s41572-022-00380-8.
- B. Wirth, Spinal Muscular Atrophy: In the Challenge Lies a Solution, Trends Neurosci., 2021, 44, 306–322, DOI:10.1016/j.tins.2020.11.009.
- H. Chaytow, K. M. E. Faller, Y. T. Huang and T. H. Gillingwater, Spinal muscular atrophy: From approved therapies to future therapeutic targets for personalized medicine, Cell Rep. Med., 2021, 2, 100346, DOI:10.1016/j.xcrm.2021.100346.
- B. Wirth, T. Schmidt, E. Hahnen, S. Rudnik-Schöneborn, M. Krawczak, B. Müller-Myhsok, J. Schönling and K. Zerres, De novo rearrangements found in 2% of index patients with spinal muscular atrophy: mutational mechanisms, parental origin, mutation rate, and implications for genetic counseling, Am. J. Hum. Genet., 1997, 61, 1102–1111, DOI:10.1086/301608.
- I. E. C. Verhaart, A. Robertson, I. J. Wilson, A. Aartsma-Rus, S. Cameron, C. C. Jones, S. F. Cook and H. Lochmüller, Prevalence, incidence and carrier frequency of 5q-linked spinal muscular atrophy - a literature review, Orphanet J. Rare Dis., 2017, 12, 124, DOI:10.1186/s13023-017-0671-8.
- M. Smith, V. Calabro, B. Chong, N. Gardiner, S. Cowie and D. du Sart, Population screening and cascade testing for carriers of SMA, Eur. J. Hum. Genet., 2007, 15, 759–766, DOI:10.1038/sj.ejhg.5201821.
- B. Wirth, M. Karakaya, M. J. Kye and N. Mendoza-Ferreira, Twenty-Five Years of Spinal Muscular Atrophy Research: From Phenotype to Genotype to Therapy, and What Comes Next, Annu. Rev. Genomics Hum. Genet., 2020, 21, 231–261, DOI:10.1146/annurev-genom-102319-103602.
- E. Mercuri, R. S. Finkel, F. Muntoni, B. Wirth, J. Montes, M. Main, E. S. Mazzone, M. Vitale, B. Snyder, S. Quijano-Roy, E. Bertini, R. H. Davis, O. H. Meyer, A. K. Simonds, M. K. Schroth, R. J. Graham, J. Kirschner, S. T. Iannaccone, T. O. Crawford, S. Woods, Y. Qian and T. Sejersen, Diagnosis and management of spinal muscular atrophy: Part 1: Recommendations for diagnosis, rehabilitation, orthopedic and nutritional care, Neuromuscular Disord., 2018, 28, 103–115, DOI:10.1016/j.nmd.2017.11.005.
- R. S. Finkel, C. A. Chiriboga, J. Vajsar, J. W. Day, J. Montes, D. C. De Vivo, M. Yamashita, F. Rigo, G. Hung, E. Schneider, D. A. Norris, S. Xia, C. F. Bennett and K. M. Bishop, Treatment of infantile-onset spinal muscular atrophy with nusinersen: a phase 2, open-label, dose-escalation study, Lancet, 2016, 388, 3017–3026, DOI:10.1016/s0140-6736(16)31408-8.
- S. M. Hoy, Nusinersen: First Global Approval, Drugs, 2017, 77, 473–479, DOI:10.1007/s40265-017-0711-7.
- S. M. Hoy, Onasemnogene Abeparvovec: First Global Approval, Drugs, 2019, 79, 1255–1262, DOI:10.1007/s40265-019-01162-5.
- G. Baranello, B. T. Darras, J. W. Day, N. Deconinck, A. Klein, R. Masson, E. Mercuri, K. Rose, M. El-Khairi, M. Gerber, K. Gorni, O. Khwaja, H. Kletzl, R. S. Scalco, T. Seabrook, P. Fontoura and L. Servais, Risdiplam in Type 1 Spinal Muscular Atrophy, N. Engl. J. Med., 2021, 384, 915–923, DOI:10.1056/NEJMoa2009965.
- C. R. R. Alves, M. Petrillo, R. Spellman, R. Garner, R. Zhang, M. Kiefer, S. Simeone, J. Sohn, E. J. Eichelberger, E. Rodrigues, E. A. Arruda, E. L. Townsend, W. Farwell and K. J. Swoboda, Implications of circulating neurofilaments for spinal muscular atrophy treatment early in life: A case series, Mol. Ther. – Methods Clin. Dev., 2021, 23, 524–538, DOI:10.1016/j.omtm.2021.10.011.
- A. Reilly, L. Chehade and R. Kothary, Curing SMA: Are we there yet?, Gene Ther., 2023, 30, 8–17, DOI:10.1038/s41434-022-00349-y.
- X. Xing, X. Liu, X. Li, M. Li, X. Wu, X. Huang, A. Xu, Y. Liu and J. Zhang, Insights into spinal muscular atrophy from molecular biomarkers, Neural Regener. Res., 2025, 20, 1849–1863, DOI:10.4103/nrr.Nrr-d-24-00067.
- O. Schwartz, K. Vill, M. Pfaffenlehner, M. Behrens, C. Weiß, J. Johannsen, J. Friese, A. Hahn, A. Ziegler, S. Illsinger, M. Smitka, A. von Moers, H. Kölbel, G. Schreiber, N. Kaiser, E. Wilichowski, M. Flotats-Bastardas, R. A. Husain, M. Baumann, C. Köhler, R. Trollmann, A. Schwerin-Nagel, A. Eisenkölbl, M. Schimmel, M. Fleger, B. Kauffmann, G. Wiegand, M. Baumgartner, C. Rauscher, S. Cirak, D. Gläser, G. Bernert, T. Hagenacker, S. Goldbach, K. Probst-Schendzielorz, H. Lochmüller, W. Müller-Felber, U. Schara-Schmidt, M. C. Walter, J. Kirschner and A. Pechmann, Clinical Effectiveness of Newborn Screening for Spinal Muscular Atrophy: A Nonrandomized Controlled Trial, JAMA Pediatr., 2024, 178, 540–547, DOI:10.1001/jamapediatrics.2024.0492.
- T. Dangouloff, A. Burghes, E. F. Tizzano and L. Servais, 244th ENMC international workshop: Newborn screening in spinal muscular atrophy May 10-12, 2019, Hoofdorp, The Netherlands, Neuromuscular Disord., 2020, 30, 93–103, DOI:10.1016/j.nmd.2019.11.002.
- D. S. T. Kariyawasam, J. S. Russell, V. Wiley, I. E. Alexander and M. A. Farrar, The implementation of newborn screening for spinal muscular atrophy: the Australian experience, Genet. Med., 2020, 22, 557–565, DOI:10.1038/s41436-019-0673-0.
- T. Dangouloff, E. Vrščaj, L. Servais and D. Osredkar, Newborn screening programs for spinal muscular atrophy worldwide: Where we stand and where to go, Neuromuscular Disord., 2021, 31, 574–582, DOI:10.1016/j.nmd.2021.03.007.
- Y. Feng, X. Ge, L. Meng, J. Scull, J. Li, X. Tian, T. Zhang, W. Jin, H. Cheng, X. Wang, M. Tokita, P. Liu, H. Mei, Y. Wang, F. Li, E. S. Schmitt, W. V. Zhang, D. Muzny, S. Wen, Z. Chen, Y. Yang, A. L. Beaudet, X. Liu, C. M. Eng, F. Xia, L. J. Wong and J. Zhang, The next generation of population-based spinal muscular atrophy carrier screening: comprehensive pan-ethnic SMN1 copy-number and sequence variant analysis by massively parallel sequencing, Genet. Med., 2017, 19, 936–944, DOI:10.1038/gim.2016.215.
- X. Chen, J. Harting, E. Farrow, I. Thiffault, D. Kasperaviciute, A. Hoischen, C. Gilissen, T. Pastinen and M. A. Eberle, Comprehensive SMN1 and SMN2 profiling for spinal muscular atrophy analysis using long-read PacBio HiFi sequencing, Am. J. Hum. Genet., 2023, 110, 240–250, DOI:10.1016/j.ajhg.2023.01.001.
- J. Bai, Y. Qu, W. Huang, W. Meng, J. Zhan, H. Wang, W. Hou, Y. Jin, A. Mao and F. Song, A high-fidelity long-read sequencing-based approach enables accurate and effective genetic diagnosis of spinal muscular atrophy, Clin. Chim. Acta, 2024, 553, 117743, DOI:10.1016/j.cca.2023.117743.
- W. Jin, Z. Yang, X. Tang, X. Wang, Y. Huang, C. Hui, J. Yao, J. Luan, S. Tang, S. Wu, S. Jin and C. Ding, Simultaneous quantification of SMN1 and SMN2 copy numbers by MALDI-TOF mass spectrometry for spinal muscular atrophy genetic testing, Clin. Chim. Acta, 2022, 532, 45–52, DOI:10.1016/j.cca.2022.05.017.
- Y. Lin, C. H. Lin, X. Yin, L. Zhu, J. Yang, Y. Shen, C. Yang, X. Chen, H. Hu, Q. Ma, X. Shi, Y. Shen, Z. Hu, C. Huang and X. Huang, Newborn Screening for Spinal Muscular Atrophy in China Using DNA Mass Spectrometry, Front. Genet., 2019, 10, 1255, DOI:10.3389/fgene.2019.01255.
- H. Y. Kao, Y. N. Su, H. K. Liao, M. S. Liu and Y. J. Chen, Determination of SMN1/SMN2 gene dosage by a quantitative genotyping platform combining capillary electrophoresis and MALDI-TOF mass spectrometry, Clin. Chem., 2006, 52, 361–369, DOI:10.1373/clinchem.2005.061192.
- M. W. Duncan, D. Nedelkov, R. Walsh and S. J. Hattan, Applications of MALDI Mass Spectrometry in Clinical Chemistry, Clin. Chem., 2016, 62, 134–143, DOI:10.1373/clinchem.2015.239491.
- W. M. Association, World Medical Association Declaration of Helsinki: ethical principles for medical research involving human subjects, J. Am. Med. Assoc., 2013, 310, 2191–2194, DOI:10.1001/jama.2013.281053.
- C. Ding and C. R. Cantor, A high-throughput gene expression analysis technique using competitive PCR and matrix-assisted laser desorption ionization time-of-flight MS, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3059–3064, DOI:10.1073/pnas.0630494100.
- A. N. Abou Tayoun, T. Pesaran, M. T. DiStefano, A. Oza, H. L. Rehm, L. G. Biesecker and S. M. Harrison, Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion, Hum. Mutat., 2018, 39, 1517–1524, DOI:10.1002/humu.23626.
- S. Richards, N. Aziz, S. Bale, D. Bick, S. Das, J. Gastier-Foster, W. W. Grody, M. Hegde, E. Lyon, E. Spector, K. Voelkerding and H. L. Rehm, Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology, Genet. Med., 2015, 17, 405–424, DOI:10.1038/gim.2015.30.
- M. Luo, L. Liu, I. Peter, J. Zhu, S. A. Scott, G. Zhao, C. Eversley, R. Kornreich, R. J. Desnick and L. Edelmann, An Ashkenazi Jewish SMN1 haplotype specific to duplication alleles improves pan-ethnic carrier screening for spinal muscular atrophy, Genet. Med., 2014, 16, 149–156, DOI:10.1038/gim.2013.84.
- T. W. Prior, P. Bayrak-Toydemir, T. C. Lynnes, R. Mao, J. D. Metcalf, K. Muralidharan, A. Iwata-Otsubo, H. T. Pham, V. M. Pratt, S. Qureshi, D. Requesens, J. Shen, F. Vetrini and L. Kalman, Characterization of Reference Materials for Spinal Muscular Atrophy Genetic Testing: A Genetic Testing Reference Materials Coordination Program Collaborative Project, J. Mol. Diagn., 2021, 23, 103–110, DOI:10.1016/j.jmoldx.2020.10.011.
- M. D. Mailman, T. Hemingway, R. L. Darsey, C. E. Glasure, Y. Huang, R. B. Chadwick, J. W. Heinz, A. C. Papp, P. J. Snyder, M. S. Sedra, R. W. Schafer, D. N. Abuelo, E. W. Reich, K. S. Theil, A. H. Burghes, A. de la Chapelle and T. W. Prior, Hybrids monosomal for human chromosome 5 reveal the presence of a spinal muscular atrophy (SMA) carrier with two SMN1 copies on one chromosome, Hum. Genet., 2001, 108, 109–115, DOI:10.1007/s004390000446.
- Z. Sheng-Yuan, F. Xiong, Y. J. Chen, T. Z. Yan, J. Zeng, L. Li, Y. N. Zhang, W. Q. Chen, X. H. Bao, C. Zhang and X. M. Xu, Molecular characterization of SMN copy number derived from carrier screening and from core families with SMA in a Chinese population, Eur. J. Hum. Genet., 2010, 18, 978–984, DOI:10.1038/ejhg.2010.54.
- C. C. Wang, J. G. Chang, Y. L. Chen, Y. J. Jong and S. M. Wu, Multi-exon genotyping of SMN gene in spinal muscular atrophy by universal fluorescent PCR and capillary electrophoresis, Electrophoresis, 2010, 31, 2396–2404, DOI:10.1002/elps.201000124.
- J. Zeng, Y. Lin, A. Yan, L. Ke, Z. Zhu and F. Lan, Establishment of a molecular diagnostic system for spinal muscular atrophy experience from a clinical laboratory in china, J. Mol. Diagn., 2011, 13, 41–47, DOI:10.1016/j.jmoldx.2010.11.009.
- W. L. Liu, F. Li, Z. X. He, R. Ai and H. W. Ma, Molecular analysis of the SMN gene mutations in spinal muscular atrophy patients in China, Genet. Mol. Res., 2013, 12, 3598–3604, DOI:10.4238/2013.September.13.4.
- J. L. Bai, Y. J. Qu, Y. Y. Cao, E. Z. Li, L. W. Wang, Y. Li, Y. L. Zhu, W. H. Zhang, Y. W. Jin, H. Wang and F. Song, Subtle mutation detection of SMN1 gene in Chinese spinal muscular atrophy patients: implication of molecular diagnostic procedure for SMN1 gene mutations, Genet. Test. Mol. Biomarkers, 2014, 18, 546–551, DOI:10.1089/gtmb.2014.0002.
- Y. J. Qu, J. L. Bai, Y. Y. Cao, H. Wang, Y. W. Jin, J. Du, X. S. Ge, W. H. Zhang, Y. Li, S. X. He and F. Song, Mutation Spectrum of the Survival of Motor Neuron 1 and Functional Analysis of Variants in Chinese Spinal Muscular Atrophy, J. Mol. Diagn., 2016, 18, 741–752, DOI:10.1016/j.jmoldx.2016.05.004.
- Y. Y. Cao, W. H. Zhang, Y. J. Qu, J. L. Bai, Y. W. Jin, H. Wang and F. Song, Diagnosis of Spinal Muscular Atrophy: A Simple Method for Quantifying the Relative Amount of Survival Motor Neuron Gene 1/2 Using Sanger DNA Sequencing, Chin. Med. J., 2018, 131, 2921–2929, DOI:10.4103/0366-6999.247198.
- L. Alías, S. Bernal, P. Fuentes-Prior, M. J. Barceló, E. Also, R. Martínez-Hernández, F. J. Rodríguez-Alvarez, Y. Martín, E. Aller, E. Grau, A. Peciña, G. Antiñolo, E. Galán, A. L. Rosa, M. Fernández-Burriel, S. Borrego, J. M. Millán, C. Hernández-Chico, M. Baiget and E. F. Tizzano, Mutation update of spinal muscular atrophy in Spain: molecular characterization of 745 unrelated patients and identification of four novel mutations in the SMN1 gene, Hum. Genet., 2009, 125, 29–39, DOI:10.1007/s00439-008-0598-1.
- Y. Xu, B. Xiao, Y. Liu, X. X. Qu, M. Y. Dai, X. M. Ying, W. T. Jiang, J. M. Zhang, X. Q. Liu, Y. W. Chen and X. Ji, Identification of novel SMN1 subtle mutations using an allelic-specific RT-PCR, Neuromuscular Disord., 2020, 30, 219–226, DOI:10.1016/j.nmd.2019.11.010.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4an01225a |
| ‡ These authors contributed equally to this work. |
| § Yan Liu, Jian Zhang and Xiaohui Huang share senior authorship. |
| This journal is © The Royal Society of Chemistry 2025 |