
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Anion exchange membrane water electrolysis over superparamagnetic ferrites†
Tiago
Fernandes
 a,
Ramsundar Rani
Mohan
a,
Laura
Donk
b,
Wei
Chen
b,
Chiara
Biz
c,
Mauro
Fianchini
cd,
Saeed
Kamali
ef,
Siavash Mohammad
Alizadeh
a,
Anna
Kitayev
g,
Aviv
Ashdot
g,
Miles
Page
g,
Laura M.
Salonen
ai,
Sebastian
Kopp
h,
Ervin Tal
Gutelmacher
g,
José
Gracia
c,
Marta Costa
Figueiredo
b and
Yury V.
Kolen’ko
*a
a,
Ramsundar Rani
Mohan
a,
Laura
Donk
b,
Wei
Chen
b,
Chiara
Biz
c,
Mauro
Fianchini
cd,
Saeed
Kamali
ef,
Siavash Mohammad
Alizadeh
a,
Anna
Kitayev
g,
Aviv
Ashdot
g,
Miles
Page
g,
Laura M.
Salonen
ai,
Sebastian
Kopp
h,
Ervin Tal
Gutelmacher
g,
José
Gracia
c,
Marta Costa
Figueiredo
b and
Yury V.
Kolen’ko
*a
aInternational Iberian Nanotechnology Laboratory, Braga 4715-330, Portugal. E-mail: yury.kolenko@inl.int
bDepartment of Chemical Engineering and Chemistry, Eindhoven University of Technology, Eindhoven 5600 MB, the Netherlands
cMagnetoCat SL, Calle General Polavieja 9, 3Izq, Alicante 03012, Spain
dDepartamento de Química Física, Universidad de Alicante, Carretera San Vicente del Raspeig s/n, 03690 San Vicente del Raspeig, Alicante E-03080, Spain
eDepartment of Physics and Astronomy, Middle Tennessee State University, Murfreesboro, TN, USA
fDepartment of Mechanical, Aerospace and Biomedical Engineering, University of Tennessee Space Institute, Tullahoma, TN, USA
gHYDROLITE, Hatohen 2, Caesarea 3088900, Israel
hHydrogen Technologies, Fraunhofer Institute for Solar Energy Systems ISE, Freiburg 79110, Germany
iCINBIO, Universidade de Vigo, Department of Organic Chemistry, Vigo 36310, Spain
First published on 30th August 2024
Abstract
The oxygen evolution reaction (OER) is usually the bottleneck in water electrolysis due to its sluggish kinetics, resulting in increased costs in the production of green hydrogen. Therefore, there is a need for more efficient, stable, and ideally, critical-raw-material-free catalysts. To this end, we have synthesized nanosized spinel ferrites CoFe2O4, NiFe2O4, and ZnFe2O4, and a high-entropy spinel ferrite Zn0.2Mn0.2Ni0.2Co0.2Fe2.2O4 through a simple coprecipitation reaction in an automated reactor on a gram scale. The powder X-ray diffraction and transmission electron microscopy studies revealed crystallite sizes of 20–35 nm. Insight into the oxidation states and cation distribution in the mixed spinel systems was gained through X-ray photoelectron and Mössbauer spectroscopy studies. The activity of all spinel ferrites was tested for the OER through half-cell laboratory measurements and full-cell anion exchange membrane electrolysis (AEMEL), where Zn0.2Mn0.2Ni0.2Co0.2Fe2.2O4 showed the lowest overpotential of 432 mV at a current density of 10 mA cm−2. All the synthesized ferrites demonstrated good stability up to 20 h, with NiFe2O4 being the most active in high current density experiments up to 2 A cm−2. In addition, studies on the magnetic properties at room temperature revealed a largely superparamagnetic response of the prepared materials, indicating that quantum spin-exchange interactions facilitate oxygen electrochemistry. Computational calculations shed light on the superior catalytic activities of NiFe2O4 and Zn0.2Mn0.2Ni0.2Co0.2Fe2.2O4, the two strongly correlated oxides that exhibit the highest magnetization and the smallest band gaps, corroborating the recent principles determining the activity of magnetic oxides in electron transfer reactions.
Introduction
Hydrogen is an important energy vector with regard to achieving the targeted net-zero CO2 emissions by 2050, allowing for the elimination of our dependence on fossil fuels.1 When powered by renewable electricity, water electrolysis is of profound and long-standing interest for green H2 production.2,3 In the last few decades, the industry has focused on two electrolysis systems that operate at low temperatures. Traditional alkaline water electrolysis (ALK) typically operates in the temperature range of 60–80 °C with potassium hydroxide as the electrolyte, Ni-based electrodes, and a diaphragm (e.g., Zirfon).4,5 The usual maximum operating geometric current density is less than 0.4 A cm−2, with an energy efficiency of about 60%.6 ALK is recognized as a mature technology, which is commercially available due to its low cost. However, its major limitation is related to its poor dynamic operation capability, e.g., the ability of the electrolysis system to rapidly adjust its operation to match the fluctuating power output of renewable energy sources.7 Proton exchange membrane water electrolysis (PEMEL) is the other key electrolysis technology in the near-term. PEMEL is advantageous in that it uses a semi-solid electrolyte in the form of proton exchange membranes (PEMs) with high proton conductivity (e.g., Nafion),8 pressurized set-up, and good operation under dynamic load.4,9 Despite its inherent advantages, PEMEL is defined by high CAPEX, due to the need for rare and expensive platinum-group-metal (PGM) electrodes,10 along with costly titanium gas-diffusion layers11 and bipolar plates.12The relatively recent anion exchange membrane electrolysis (AEMEL) offers a great opportunity to generate green H2 in a cost-efficient and environmentally benign fashion. The use of anion exchange membranes (AEMs) may mitigate some of the limitations of ALK. Similar to PEMEL, AEMEL is characterized by pressurized operation and high dynamic responsiveness, but it allows for the use of cheap, non-PGM catalysts and low-cost cell/stack materials.13,14 For instance, various hydroxide/oxyhydroxide/oxide nanoparticles (NPs) based on abundant non-PGM 3d transition metals, such as Mn, Fe, Co, Ni, Cu, Zn, and Mo, are commonly investigated and employed as catalysts for the alkaline oxygen evolution reaction (OER) over AEMEL anodes. The fact that these types of non-PGM catalysts are more abundant and less costly than PGM-containing ones, while also featuring good chemical stability under alkaline conditions, shows the potential for the large-scale implementation of AEMEL.15–18 Despite great efforts in the recent years to develop non-PGM catalysts, a systematic analysis regarding their physicochemical properties such as the effect of composition, particle size, surface area, and physical properties is of high relevance for the future implementation of AEMEL technology at the industrial level.4
The efficiency of AEMEL relies highly on that of the OER due to its slower reaction kinetics and associated high overpotentials as compared to the hydrogen evolution reaction (HER), the other half-reaction of water electrolysis.19–21 Notably, the most active catalysts for the OER have strongly correlated electrons,22 associated with itinerant spin conduction.23,24 For example, ferrites AFe2O4 with a spinel structure are strongly correlated oxides with favorable spin-orbital orderings, and thus these have been postulated as promising catalysts for the OER.25 High activity and durability of ferrites are commonly associated with their open-shell electronic configurations. Moreover, the structural and chemical stability of ferrites under alkaline conditions combined with their multiple cation valences (i.e., A3+/A2+ and Fe3+/Fe2+) renders them interesting catalysts for AEMEL anodes.14,26–30 While several ferrites with various compositions, structures, and nanostructures have been synthesized, characterized, and investigated in the laboratory in a half-cell alkaline OER experimental setting,31 their performance in industrially relevant full-cell AEMEL has not been evaluated in great detail to date.
Our laboratories have been exploring the chemical synthesis of ferrite NPs to modulate their structural and magnetic properties.32,33 Herein, we report the preparation of a series of ferrite AFe2O4 (A = Fe2+, Ni2+, Co2+, Zn2+, and Mn2+) NPs on a gram scale and their performance in alkaline OER, and importantly, in full-cell AEMEL. The spin-dependent theory in catalysis22 and computational calculations provided comprehensive insight into the structural, magnetic, and electronic properties of the prepared ferrites, leading to structure–property relationships that will assist in the future design of active catalysts.
Experimental section
Materials
Iron(III) chloride hexahydrate (FeCl3·6H2O, >98%, Sigma-Aldrich), nickel(II) chloride hexahydrate (NiCl2·6H2O, >98%, Thermo Scientific), cobalt(II) chloride (CoCl2·6H2O, >98%, Thermo Scientific), zinc(II) chloride anhydrous (ZnCl2, >98%, Thermo Scientific), manganese(II) chloride tetrahydrate (MnCl2·4H2O, 99%, Thermo Scientific), sodium hydroxide (NaOH, pellets, EMSURE®, Supelco), potassium hydroxide (KOH, flakes, 90%, Sigma-Aldrich), 30% hydrogen peroxide solution (H2O2, Sigma-Aldrich), potassium permanganate (KMnO4, ≥99.0%, Sigma-Aldrich), isopropanol (≥99.5%, Sigma-Aldrich), carbon black Vulcan XC 72 (Fuel Cell Store) and ethanol (≥99.5%, Honeywell) were used as received. Ultrapure water (18.2 MΩ cm) was produced using a Milli-Q Advantage A10 system (Millipore).Synthesis of ferrites
All ferrites, namely, CoFe2O4, NiFe2O4, and ZnFe2O4, as well as high-entropy Zn0.2Mn0.2Ni0.2Co0.2Fe2.2O4 ferrite [4A1/4Fe2.2O4] were prepared using the robust coprecipitation method adapted from elsewhere12 using NaOH as a precipitating agent. Typically, the molar ratio of base to metal precursors was fixed as 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. The reproducible gram-scale synthesis of the ferrites (≫5 g) was achieved using a 2 L automated Atlas potassium synthesis system (Syrris) equipped with two syringe pumps (Syrris), a temperature control system LH85 PLUS (Julabo), an overhead stirrer, and a temperature probe. In a typical reaction, 11.0 g of NaOH (0.225 mol) was dissolved in 400 mL of ultrapure water while heating at 80 °C under mechanical stirring of 300 rpm for 30 min. This step was followed by a controlled addition of the metal precursors previously dissolved in 200 mL of ultrapure water at a feeding rate of 5 mL min−1. Typically, for two-metal ferrites, 42 mmol of Fe(III) salt and 21 mmol of the corresponding A(II) salt were mixed and used as the precursor solution. For the synthesis of the high-entropy ferrite containing five metals, an aqueous solution was prepared with 42 mmol of Fe(III) salt and 4.2 mmol of each M(II) salt. After complete addition of the precursor solution to the NaOH solution in a 2 L reactor, the reaction mixture was allowed to gradually cool to room temperature. The resulting solid products were either collected magnetically with the aid of a permanent NdFeB magnet for magnetically active products or through centrifugation at 12000 rpm for 10 min for weakly magnetic products. The resulting ferrites were extensively washed with water and ethanol and dried under vacuum. The obtained powders were calcined at 400 °C in air with a heating rate of 3 °C min−1 and holding time of 5 h. The calcination products were converted into fine powders through ball milling in an 8000 M high-energy mixer/mill (SPEX) using a zirconia vial and spheres at a sample-to-sphere w/w ratio of 9:1 (1 cycle at 1425 rpm of 30 min).
:1. The reproducible gram-scale synthesis of the ferrites (≫5 g) was achieved using a 2 L automated Atlas potassium synthesis system (Syrris) equipped with two syringe pumps (Syrris), a temperature control system LH85 PLUS (Julabo), an overhead stirrer, and a temperature probe. In a typical reaction, 11.0 g of NaOH (0.225 mol) was dissolved in 400 mL of ultrapure water while heating at 80 °C under mechanical stirring of 300 rpm for 30 min. This step was followed by a controlled addition of the metal precursors previously dissolved in 200 mL of ultrapure water at a feeding rate of 5 mL min−1. Typically, for two-metal ferrites, 42 mmol of Fe(III) salt and 21 mmol of the corresponding A(II) salt were mixed and used as the precursor solution. For the synthesis of the high-entropy ferrite containing five metals, an aqueous solution was prepared with 42 mmol of Fe(III) salt and 4.2 mmol of each M(II) salt. After complete addition of the precursor solution to the NaOH solution in a 2 L reactor, the reaction mixture was allowed to gradually cool to room temperature. The resulting solid products were either collected magnetically with the aid of a permanent NdFeB magnet for magnetically active products or through centrifugation at 12000 rpm for 10 min for weakly magnetic products. The resulting ferrites were extensively washed with water and ethanol and dried under vacuum. The obtained powders were calcined at 400 °C in air with a heating rate of 3 °C min−1 and holding time of 5 h. The calcination products were converted into fine powders through ball milling in an 8000 M high-energy mixer/mill (SPEX) using a zirconia vial and spheres at a sample-to-sphere w/w ratio of 9:1 (1 cycle at 1425 rpm of 30 min).
Characterization
The materials were characterized by powder X-ray diffraction (XRD, X’Pert PRO diffractometer, PANalytical), Raman spectroscopy (alpha300 R confocal microscope, WITec), Mössbauer spectroscopy (MS4 spectrometer, SEE Co.), transmission electron microscopy (TEM, JEM-2100 microscope, Jeol), high-angle annular dark field scanning TEM (HAADF–STEM), energy-dispersive X-ray spectroscopy (EDX) in STEM mode (STEM–EDX, Titan ChemiSTEM microscope at 200 kV; FEI, 0.08 nm point resolution, Super-X EDX System), N2 physisorption at 77 K (Autosorb IQ2 apparatus, Quantachrome), scanning electron microscopy (SEM, Quanta 650 FEG microscope, FEI), energy dispersive X-ray spectroscopy (EDX, INCA 350 spectrometer, Oxford Instruments), vibrating sample magnetometry (VSM, EV9 instrument, Lot-Oriel), UV/vis diffuse reflectance spectroscopy (LAMBDA 950 UV/Vis/NIR spectrophotometer, PerkinElmer), X-ray photoelectron spectroscopy (XPS, ESCALAB 250 Xi, Thermo Scientific), and inductively coupled plasma–optical emission spectroscopy (ICP–OES, ICPE–9000 spectrometer, Shimadzu); all powders were digested using a concentrated HCl 37% solution. The NiFe2O4 nanoparticles were further digested under heat treatment at 170 °C using an autoclave reactor.Half-cell alkaline OER measurements
The electrochemical OER activity of the synthesized ferrites was studied by cyclic voltammetry (CV) and linear sweep voltammetry (LSV) in a conventional three-electrode glass cell. Before use, the glassware was cleaned following a standard cleaning procedure: first, the glassware was submerged overnight in a saturated KMnO4 solution. Next, the glassware was rinsed with a dilute H2O2 solution and then boiled three times in ultrapure water (5 minutes each time). The working electrode consisted of a catalyst ink drop-casted on a glassy carbon (GC) rotating disk electrode (RDE) (Metrohm, ∅ = 3 mm, S = 0.0071 cm2). The counter electrode and reference electrode were a GC rod and a Hg|HgO electrode, respectively. All potentials E in the half-cell study are quoted with respect to the reversible hydrogen electrode (RHE), converted via ERHE = E(RHE) = E(Hg/HgO) + 0.059 × pH + E0(Hg/HgO) = E(Hg/HgO) + 0.924 V.34 The applied potential was controlled using an Autolab PGSTAT302N potentiostat (Metrohm) while the RDE rotation rate was controlled using an Autolab RRDE rotator (Metrohm).The catalyst ink was prepared by mixing ultrapure water, isopropanol (volume ratio of water/isopropanol = 3:1), Sustainion ionomer solution (5% in solvent, Dioxide Materials) at a 5% volume ratio to the overall volume, a physical mixture of the synthesized ferrite powder (8 mg mL−1), and conductive carbon black (16 mg mL−1). Before drop-casting the catalyst ink, the GC RDE was polished with alumina powder for 5 min, and then rinsed with water and ethanol. This procedure was repeated three times. After ultrasonication treatment for 15 min, the as-formulated catalyst ink was drop-casted on the GC RDE, and then left in a solvent–vapor–saturated water + isopropanol atmosphere overnight for drying. The mass loading (0.3 mg cm−2 ferrite + 0.6 mg cm−2 conductive carbon black) was precisely controlled by the volume of the drop-casted ink.
Aqueous 1 M NaOH electrolyte solution was used, and all experiments were performed under N2-saturated (5.0, LindeGas) conditions at room temperature. Before electrochemical characterization, the working electrode containing the catalyst was carefully rinsed with ultrapure water, followed by electrochemical cleaning for 15 min by performing CV from 0 to 1.2 VRHE at a scan rate of 200 mV s−1. All data shown are iR-corrected for a voltage drop of 80%. The electrochemical surface area (ECSA) of the working electrode was determined from the double-layer capacitance (Cdl) measured at 1 VRHE, assuming a uniform specific double layer of 40 μF cm−2.
OER properties of the synthesized ferrites in KOH electrolyte were also investigated in non-RDE mode at a higher catalyst mass loading of 1 mg cm−2 on a 1 cm2 glassy carbon plate. The catalyst ink was prepared by dispersing a mixture of 4 mg of catalyst and 1 mg of conductive carbon black in a 3:2 ethanol/water solution while adding 60 μL of Nafion ionomer solution (5% in aliphatic alcohols and water, Sigma-Aldrich) as a binder, to a total volume of 1 mL.
Full-cell AEMEL testing
Full-cell testing was conducted in an in-house-built AEMEL electrolyzer using a cell with a 4 cm2 active area (Fuel Cell Store). The catalyst was coated on the gas diffusion/porous transport layers (GDL/PTL) using the catalyst coated-on-substrate method, thus affording gas diffusion/porous transport electrodes (GDE/PTE). The cathode GDE was carbon paper GDL SGL 39BC (Sigracet) loaded with 0.3 mg cm−2 of 40%Pt/C catalyst (Premetek), while the anode PTE was nickel PTL 2Ni 18–0.5 (Bekaert) loaded with 3 mg cm−2 of the synthesized ferrite catalysts. Proprietary Membrane B 70 μm (HYDROLITE) was employed as the AEM, and PTFE gaskets of 200 μm and 400 μm were used for sealing the cathode and the anode, respectively. After the assembly, the AEMEL cell was connected to the electrolyte inlets and outlets, thermocouple, as well as voltage and current sensors in the testing workstation. The 1 M KOH electrolyte was supplied on the OER side at a flow rate of 170 mL h−1. Membrane electrode assembly (MEA) activation was performed by gradually increasing the voltage from 1.4 to 2 V to achieve the maximum current density for the cell at a specific temperature. All catalysts were tested at both 60 °C and 80 °C. After activation, the current–voltage (I–V) curve was measured from 1.4 to 2 V and backwards. The short-term durability of the AEMEL cells was evaluated through chronoamperometric measurements under a constant current density of 0.5 A cm−2 for 20 h.Results
CoFe2O4, NiFe2O4, ZnFe2O4, and high-entropy Zn0.2Mn0.2Ni0.2Co0.2Fe2.2O4 were prepared following a straightforward coprecipitation method using NaOH as the precipitating agent. We avoided the use of aqueous ammonia solution for precipitation because of the possible formation of soluble complexes with several transition metals (e.g., Ni and Zn). An automated benchtop synthesis system was employed, which allows for the synthesis of NPs on a gram scale in a highly controlled and reproducible fashion.32,35 The as-synthesized products were calcined at a moderate temperature to clean the surface of the catalysts and remove any residuals from the synthesis. Finally, the catalysts were subjected to ball milling to decrease their degree of agglomeration to facilitate the formulation of catalytic inks.Powder XRD patterns of the catalysts (Fig. S1, ESI†) evidenced all final products to be single-phase nanocrystalline ferrite powders exhibiting a cubic spinel structure (space group Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m, no. 227). No diffraction peaks corresponding to secondary phases were detected. The average size of the crystallites and reliable unit cell parameters were estimated from the collected XRD data and are summarized in Table S1 (ESI†). Next, Raman spectra were recorded for the prepared catalysts (Fig. S2, ESI†), which exhibited broad peaks with a low signal-to-noise ratio, which is common for nanocrystalline powders. The catalysts displayed typical Raman band profiles associated with the spinel structure of the O7h (Fdm) space group while featuring characteristic Raman-active bands arising from A1g (≫690 cm−1), T2g (≫470 cm−1), and Eg (≫320 cm−1) modes.36–39 As expected, the different elemental compositions of the prepared ferrites were mostly reflected in the small shifts and splitting of the observed Raman bands.40 Based on the XRD and the Raman spectroscopy results, the synthesized catalysts were obtained as phase pure ferrites.
m, no. 227). No diffraction peaks corresponding to secondary phases were detected. The average size of the crystallites and reliable unit cell parameters were estimated from the collected XRD data and are summarized in Table S1 (ESI†). Next, Raman spectra were recorded for the prepared catalysts (Fig. S2, ESI†), which exhibited broad peaks with a low signal-to-noise ratio, which is common for nanocrystalline powders. The catalysts displayed typical Raman band profiles associated with the spinel structure of the O7h (Fdm) space group while featuring characteristic Raman-active bands arising from A1g (≫690 cm−1), T2g (≫470 cm−1), and Eg (≫320 cm−1) modes.36–39 As expected, the different elemental compositions of the prepared ferrites were mostly reflected in the small shifts and splitting of the observed Raman bands.40 Based on the XRD and the Raman spectroscopy results, the synthesized catalysts were obtained as phase pure ferrites.
After confirming the phase composition of the catalysts, the cation distribution within the ferrite catalysts was studied. Notably, the spinel structure AB2O4 has tetrahedral (A) and octahedral (B) sites, and for ferrites with normal and inverse spinel crystal structures, the compositions [M1]A{Fe2}BO4 and [Fe1]A{M1Fe1}BO4 are expected, respectively. To estimate the cation distribution in the synthesized catalysts, the products were analyzed by Mössbauer spectroscopy at 6 K (Fig. 1). The collected Mössbauer spectra for all four catalysts can be fitted with three magnetically split sextets, Q1, Q2, and Q3, and the extracted Mössbauer parameters, namely, the centroid shift, δ, quadrupole shift, ε, magnetic hyperfine field, Bhf, magnetic hyperfine field distribution, σ, and intensity, I, for all measurements are summarized in Table S2 (ESI†). Analysis of the Mössbauer data revealed that the cation distribution for CoFe2O4, NiFe2O4, ZnFe2O4, and Zn0.2Mn0.2Ni0.2Co0.2Fe2.2O4, within the experimental errors, are [Co0.36Fe0.64]A{Co0.64Fe1.36}BO4, [Fe]A{NiFe}BO4, [Zn0.36Fe0.64]A{Zn0.64Fe1.36}BO4, and [M0.01Fe0.99]A{M0.79Fe1.21}BO4, respectively (Table S3, ESI†). These distributions confirm that while CoFe2O4, ZnFe2O4, and Zn0.2Mn0.2Ni0.2Co0.2Fe2.2O4 catalysts are partially inverse ferrites, the NiFe2O4 catalyst is an ideal inverse ferrite.
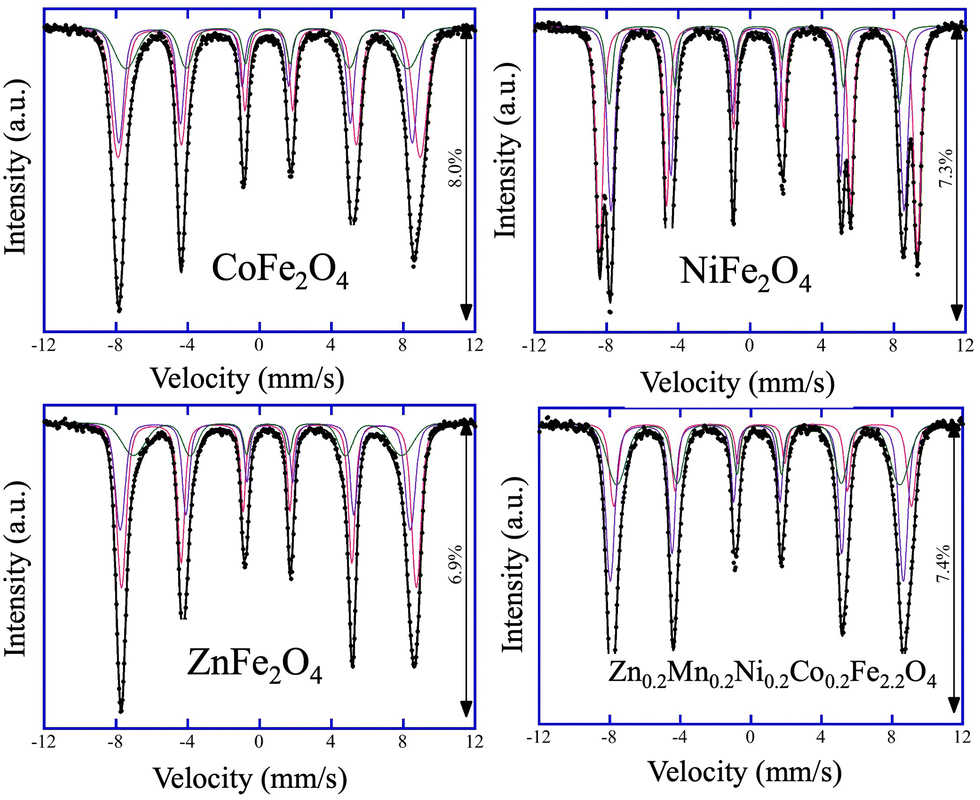 | ||
| Fig. 1 Mössbauer spectra of CoFe2O4, NiFe2O4, ZnFe2O4, and high-entropy Zn0.2Mn0.2Ni0.2Co0.2Fe2.2O4 catalysts collected at 6 K. Experimental data: black circles. Calculated spectrum: black line. Q1, Q2, and Q3 components: red, violet, and green, respectively. | ||
TEM was next employed to gather information about the size of the obtained catalysts. Fig. 2 shows the representative low-magnification TEM images of the ferrites. The NPs were found to be moderately agglomerated and reasonably polydisperse with average sizes of 20 ± 7 nm, 26 ± 6 nm, 18 ± 4 nm, and 21 ± 5 nm for CoFe2O4, NiFe2O4, ZnFe2O4, and Zn0.2Mn0.2Ni0.2Co0.2Fe2.2O4, respectively (Table S1 and Fig. S3, ESI†). Notably, the observed nanocrystalline nature of the catalysts gave rise to relatively high specific surface areas, SBET, which were estimated to be 65, 42, 54, and 84 m2 g−1 for CoFe2O4, NiFe2O4, ZnFe2O4, and Zn0.2Mn0.2Ni0.2Co0.2Fe2.2O4, respectively (Table S1, ESI†). Furthermore, SEM–EDX/STEM–EDX was used for the elemental analysis of the catalysts. The obtained data confirmed the chemical composition of the samples (Fig. S4 to S6, ESI†), and indicated that the elements were uniformly distributed, even within the high-entropy Zn0.2Mn0.2Ni0.2Co0.2Fe2.2O4 ferrite (Fig. S6, ESI†).
 | ||
| Fig. 2 Representative low-magnification TEM images of CoFe2O4 (A), NiFe2O4 (B), ZnFe2O4 (C), and high-entropy Zn0.2Mn0.2Ni0.2Co0.2Fe2.2O4 (D) catalysts. | ||
Since the catalysis takes place at the surface of the materials, X-ray photoelectron spectroscopy (XPS) was used to probe the surface of the catalysts. Fig. 3 and Fig. S7 (ESI†) show the survey and the high-resolution XPS data for the synthesized CoFe2O4, NiFe2O4, ZnFe2O4, and Zn0.2Mn0.2Ni0.2Co0.2Fe2.2O4 materials. The deconvolution of the XPS data revealed that the as-synthesized ferrites display the expected elemental composition at the surface. In the ferrites containing Co, the detailed XPS peaks (Co 2p3/2) located at 780.5 eV, 782.1 eV, and 787.5 eV can be attributed to Co2+ and Co3+, together with the corresponding shakeup satellites,41,42 which is consistent with the Mössbauer data above. Here the peak located at around 782 eV is attributed to the Augur line of Fe LMM (its contribution is much more pronounced in the case of Zn0.2Mn0.2Ni0.2Co0.2Fe2.2O4 due to the higher Fe:Co ratio). The same applies to the other synthesized ferrite catalysts (Fig. 3 and Fig. S7, ESI†), where the corresponding ionic species were observed, namely, for Ni2+/Ni3+,43,44 Zn2+,45 and Mn2+.46 In the case of Fe 2p XPS spectra, binding energies (BEs) at 710.5 eV, 712.0 eV, and 716.0 eV can be attributed to Fe2+ and Fe3+, together with the corresponding shakeup satellites.47,48 Moreover, the asymmetric nature of the Fe 2p3/2 bands for all the ferrites further suggests the existence of Fe3+ ions in the octahedral and tetrahedral sites of the structures.49 In all the catalysts, in the O 1s XPS data, the bands located at around 529.2 eV, 530.4 eV, and 531.5 eV are attributed to O2− (metal oxide), OH− (metal hydroxide) and O− (deficiencies, or adsorbed oxygen), confirming the formation of M–O bonds.
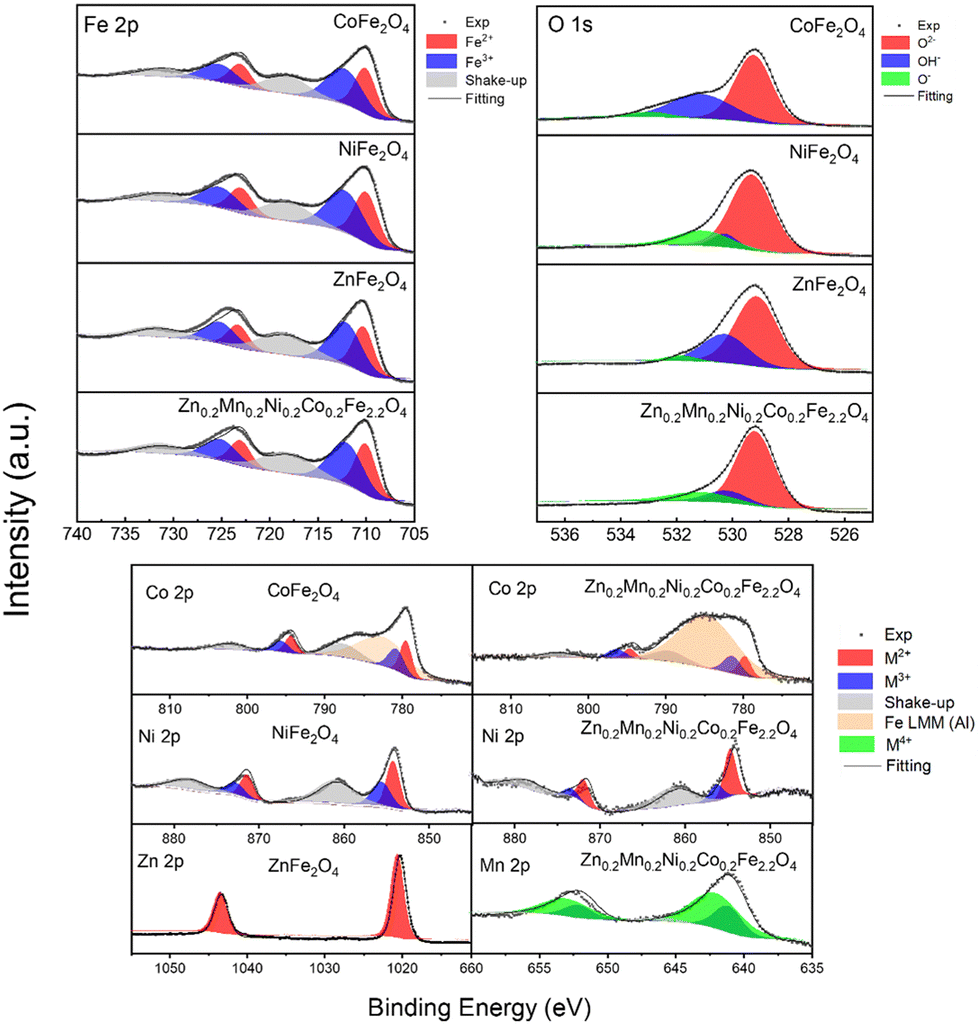 | ||
| Fig. 3 XPS spectra collected from the CoFe2O4, NiFe2O4, ZnFe2O4, and Zn0.2Mn0.2Ni0.2Co0.2Fe2.2O4 catalysts synthesized by the coprecipitation method followed by calcination and ball milling. High-resolution XPS data for the Fe 2p and O 1s region collected from all studied catalysts (top panel). High-resolution XPS data for the remaining metallic elements Co 2p, Ni 2p, Zn 2p, and Mn 2p for the catalysts (bottom panel). | ||
Next, the physical properties of the synthesized materials were studied. The magnetization versus magnetic field M(H) dependence at 300 K is shown in Fig. 4. While NiFe2O4, ZnFe2O4, and high-entropy Zn0.2Mn0.2Ni0.2Co0.2Fe2.2O4 samples demonstrate superparamagnetic-like behavior, CoFe2O4 features ferromagnetic-like behavior but with rather small coercivity and remanence in the M(H) curve (Table S4, ESI†). The lowest saturation magnetization Ms = 15 emu g−1 was expectedly observed for ZnFe2O4. At the same time, CoFe2O4, NiFe2O4, and Zn0.2Mn0.2Ni0.2Co0.2Fe2.2O4 exhibited reasonably high Ms values of 39, 44, and 50 emu g−1. Furthermore, the optical bandgap Eg of the synthesized catalysts was experimentally estimated by means of UV/vis diffuse reflectance spectroscopy. From the Tauc plots, optical bandgap values of 1.48 eV, 1.58 eV, 1.70 eV, and 1.51 eV were obtained for CoFe2O4, NiFe2O4, ZnFe2O4, and Zn0.2Mn0.2Ni0.2Co0.2Fe2.2O4, respectively (Table S1 and Fig. S8, ESI†). Despite the limitation of this method for band gap determination,50 the observed results are in line with reported data, wherein the optical band gap of the ferrites is strongly influenced by the preparation method, heat treatment procedures, mean particle size, and the degree of structural disorder in the lattice.51–55
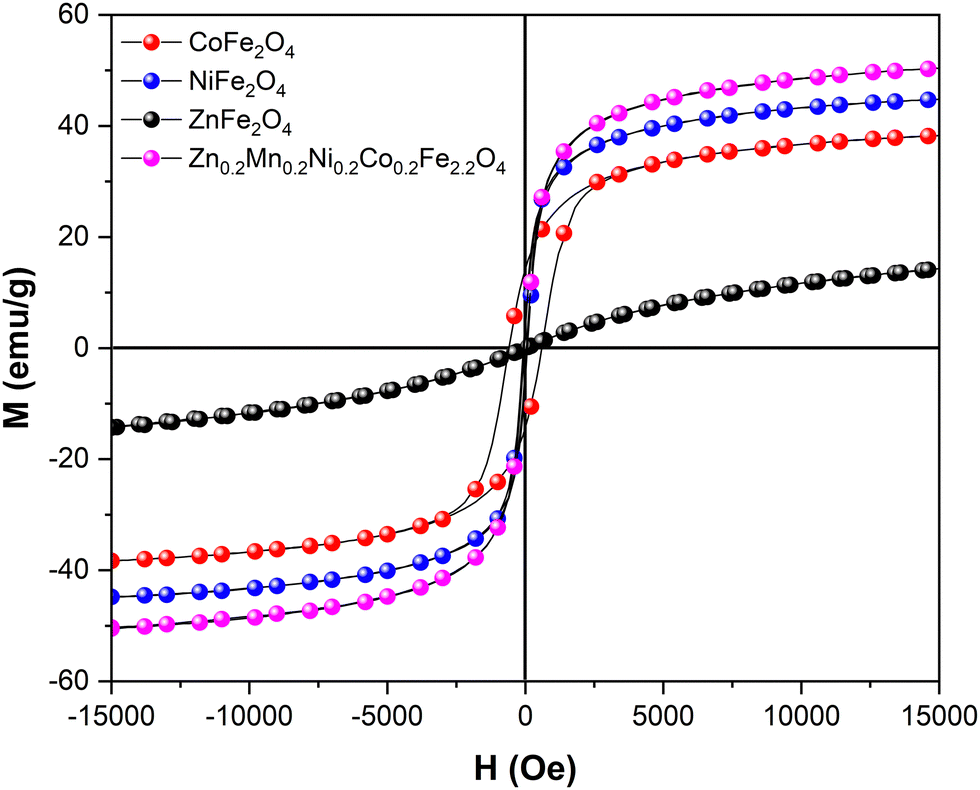 | ||
| Fig. 4 Room temperature M(H) dependence data for the synthesized catalysts. | ||
The magnetic and electronic properties of CoFe2O4, NiFe2O4, ZnFe2O4, and Zn0.2Mn0.2Ni0.2Co0.2Fe2.2O4 were also investigated by the DFT+U+J method (see the Computational study section on spinel ferrites in the ESI† for further information). The cation distributions provided by Mössbauer spectroscopy were used as plausible input to build the computational bulk models for all the ferrites under study. Two possible models were constructed in the case of Zn0.2Mn0.2Ni0.2Co0.2Fe2.2O4 (Fig. 5a) and NiFe2O4 (Fig. 5b). The Zn0.2Mn0.2Ni0.2Co0.2Fe2.2O4 bulk models were built based on both the cation distribution provided by Mössbauer spectroscopy (T = 6 K) and the high-resolution XPS data shown in Fig. 3, in which both Zn2+ and Co2+ can occupy tetrahedral positions. Thus, [Zn0.125Fe0.875]A{M0.750Fe1.250}BO4 (M = Co, Ni, and Mn) and [Co0.125Fe0.875]A{M0.750Fe1.250}BO4 (M = Co, Ni, Mn, and Zn) are model a and model b, respectively, for Zn0.2Mn0.2Ni0.2Co0.2Fe2.2O4. These models align with the available experimental data on Zn0.2Mn0.2Ni0.2Co0.2Fe2.2O4, which report challenges in including Zn2+ and Ni2+ species into the spinel structure.56 Regarding NiFe2O4, Mössbauer spectroscopy showed an intensity of 46% at the B-sites (see the first sub-spectrum Q1 in Table S2, ESI†). This value is slightly smaller than the expected 50%. A possible explanation for the observed small reduction in Q1 could be the presence of some Fe vacancies (≈12%) at the octahedral sites of NiFe2O4. This has also been suggested experimentally in the available literature data, since the cation distribution and the cation occupancy for nickel ferrite depends on several factors, such as the preparation method, calcination temperature, and particle size.57 Thus, two computational bulk models for the inverse NiFe2O4 ferrite were built, one with no Fe vacancies at the octahedral sites and one with 15% of Fe vacancies at the B-sites (Fig. 5b). The calculated lattice parameters at 0 K for all the investigated ferrites are in good agreement with the experimental ones presented in Table S1 (ESI†). Regarding the magnetic properties, all possible electronic ground states (i.e., ferromagnetic, antiferromagnetic, ferrimagnetic, and non-magnetic) were computationally explored for each ferrite (Tables S8–S13 and Fig. S9–S20, ESI†). Calculation data show that all the bulk structures possess ferrimagnetic ordering as the most stable electronic ground state at 0 K. Indeed, all the samples have non-zero magnetization at 300 K (Fig. 3 and Table S4, ESI†), suggesting that the blocking temperature falls between 1–300 K in the case of superparamagnetic samples. Moreover, the calculated minimum band gaps (Fig. 5a and b) show that all the ferrites under study are semiconductors, in good agreement with the experimental optical band gaps estimated by UV/vis diffuse reflectance spectroscopy, which are known to depend on the particle size.58,59
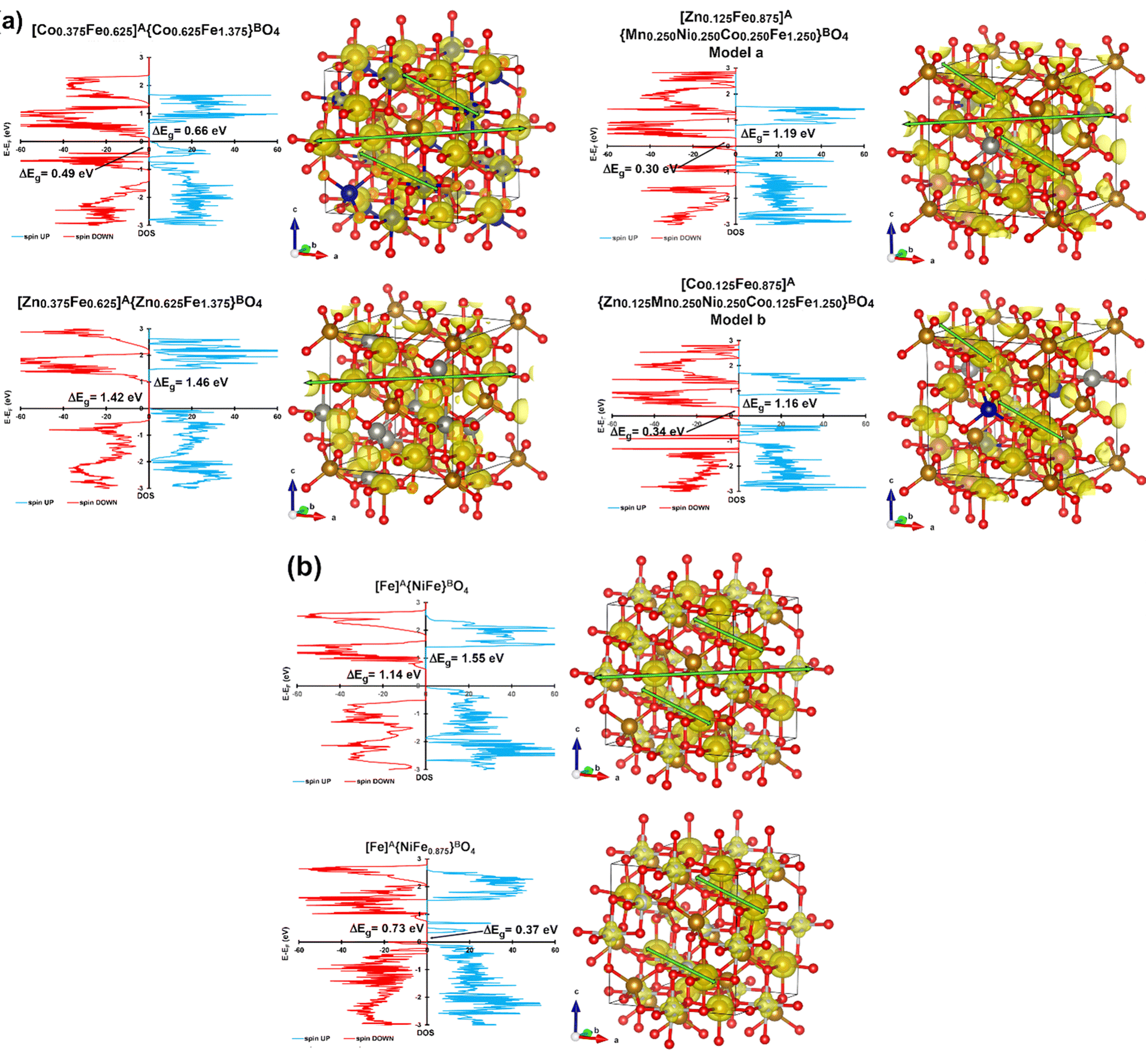 | ||
| Fig. 5 (a) Density of states (DOS, left) and α–β > 0 spin density (spin up, 0.04 a0−3, right) for the most stable ground states of partially inverse spinel model a and model b of Zn0.2Mn0.2Ni0.2Co0.2Fe2.2O4, and (b) DOS (left) and α–β > 0 spin density (spin up, 0.04 a0−3, right) for the most stable ground states of perfect NiFe2O4 and defective NiFe2–xO4. Possible spin channels are marked with green arrows. | ||
Regarding conductivity properties (Table S14, ESI†), defective NiFe1.875O4 is the best among the investigated bulk models, exhibiting the smallest minimum band gaps (0.37 and 0.73 eV as spin ↑ and spin ↓ band gaps, respectively). Partially inverse Zn0.2Mn0.2Ni0.2Co0.2Fe2.2O4 (model a and model b) and CoFe2O4 are the two ferrites displaying the smallest minimum band gaps within the stoichiometric ferrites, demonstrating 1.19–1.16 eV (spin ↑)/0.30–0.34 eV (spin ↓) and 0.66 eV (spin ↑)/0.49 eV (spin ↓), respectively.
With phase-pure, nanocrystalline, and chemically uniform ferrite materials in hand, we investigated the anodic OER half-reaction of electrochemical water splitting in alkaline electrolyte at room temperature through half-cell laboratory measurements. First, we studied the OER properties of the synthesized materials in the kinetic regime with low catalyst mass loading (0.3 mg cm−2) while using the RDE testing mode so that the effects of mass transport limitations are minimized.60 As shown in the inset of Fig. 6a, the basic CV characteristics of all the ferrite catalysts were quite similar: (i) capacitor-like behavior over a wide potential range, which may be ascribed to the capacitance current of both the ferrite catalyst and the conductive carbon additive; (ii) a pair of redox peaks located at ≈1.3 VRHE, which may be ascribed to the redox behavior of metal hydroxide/oxyhydroxide (A2+–to–A3+, e.g., Ni(OH)2 to NiOOH in the case of NiFe2O4) at potentials prior to the OER onset potential.
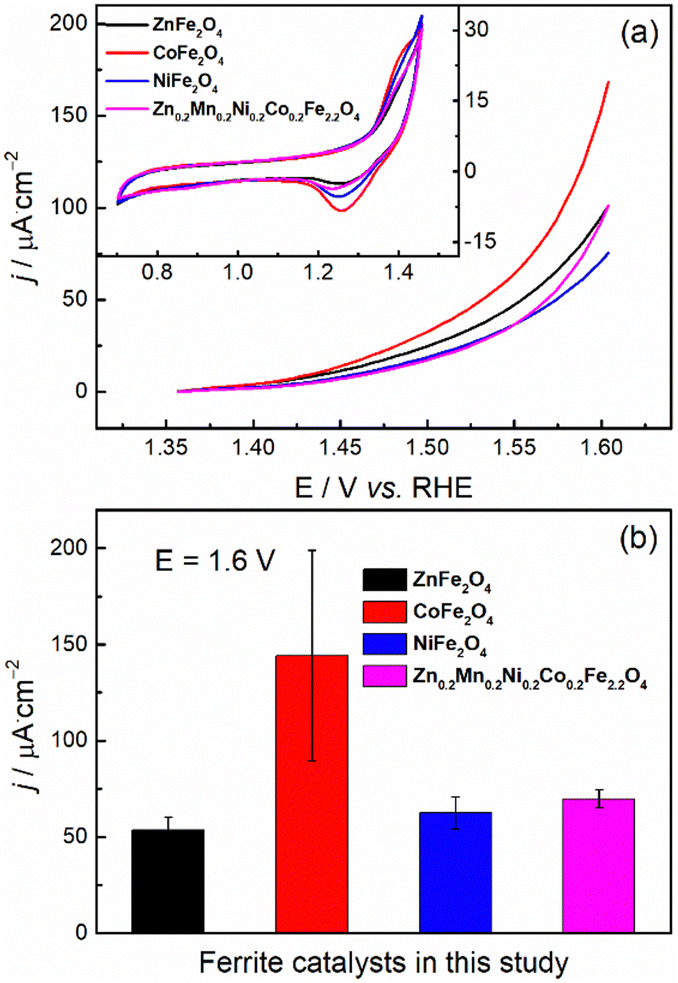 | ||
| Fig. 6 (a) Representative positive going linear sweep voltammograms at OER potentials and (b) the current densities (normalized by ECSA) at 1.6 VRHE towards the OER for the synthesized catalysts. The corresponding basic cyclic voltammograms are given as the inset in (a). | ||
In Fig. 6a, the OER current may overlap with the oxidation current of the ferrites, as shown by the redox peaks at ≈1.3 VRHE (inset of Fig. 6a). According to the literature,31,61,62 the onset potential for the OER over ferrite catalysts (e.g., NiFe2O4 and CoFe2O4) is expected to be ≈1.5 VRHE. This is also the case with most of our ferrites, with CoFe2O4 and ZnFe2O4 featuring a slightly earlier onset. Nevertheless, for the precise determination of the OER onset potential (i.e., to track the onset potential for the production of O2 rather than the anodic current), the application of in situ techniques, such as differential electrochemical mass spectrometry63,64 is necessary.
The electrochemical activity towards OER was evaluated by comparing the current densities (normalized by electrochemically active surface area) at 1.6 VRHE (Fig. 6b). Since the measured overall current is ≈0.1 mA, the Ohmic drop corrections (viz, we applied 80%) will not make a large difference.65 All the synthesized ferrite catalysts have similar OER activities in alkaline media except for CoFe2O4, the activity of which was measured to be about twice that of the others, albeit with significant uncertainty. The observed higher OER activity of CoFe2O4 in the kinetic regime could originate from the suitable binding strength of the reactive intermediates during the OER.66
Second, to be closer to realistic AEMEL conditions, we examined the OER properties of the synthesized ferrites with a higher catalyst mass loading of 1 mg cm−2 on a glassy carbon plate to drive higher current densities.67 For this purpose, we employed commercially available Ni foam. From the recorded LSV voltammograms (Fig. 7a), the OER activity decreases in the order of Zn0.2Mn0.2Ni0.2Co0.2Fe2.2O4 > NiFe2O4 > CoFe2O4 > ZnFe2O4, with overpotential values of 432, 455, 484, and 500 mV, respectively, at a current density of 10 mA cm−2. Similarly, the Tafel slopes increase in the same trend (Fig. 7b). Also, Zn0.2Mn0.2Ni0.2Co0.2Fe2.2O4 shows a very low charge transfer resistance (0.66 Ohm) in the Nyquist plot (Fig. 7c). Fig. 7d shows the capacitive current at different scan rates recorded in the non-faradaic potential window, where Zn0.2Mn0.2Ni0.2Co0.2Fe2.2O4 again shows the highest positive slope, implying the highest roughness factor. From the slope, the calculated roughness values are 61, 47, 37, and 21 for Zn0.2Mn0.2Ni0.2Co0.2Fe2.2O4, NiFe2O4, CoFe2O4, and ZnFe2O4, respectively.
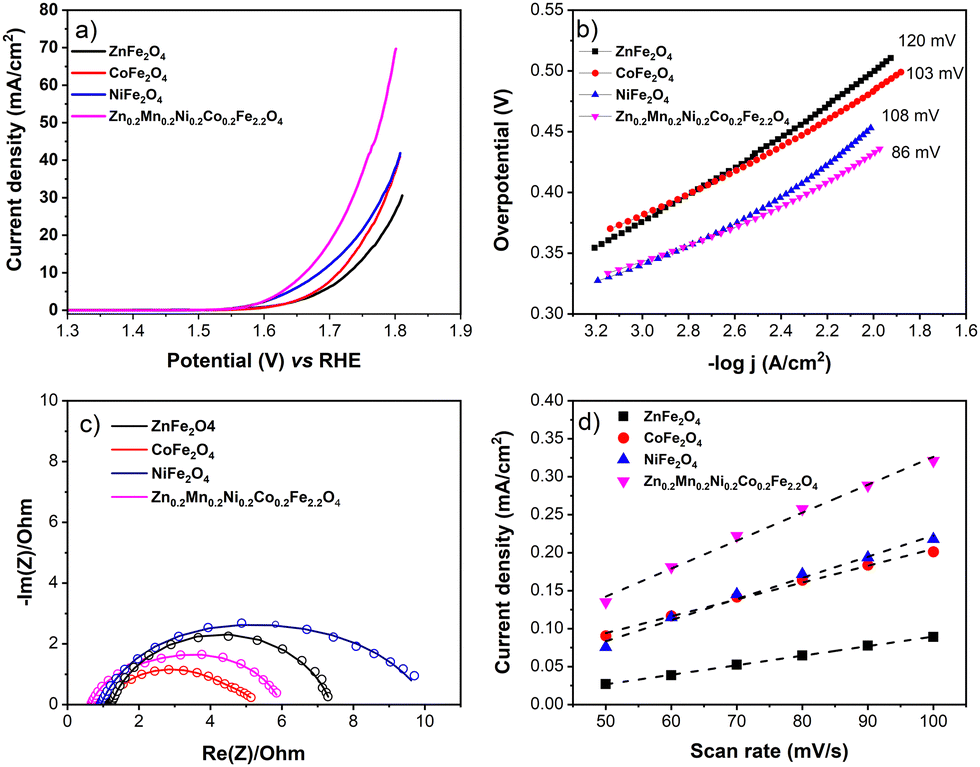 | ||
| Fig. 7 (a) Representative LSV voltammograms recorded at the 5 mV s−1 scan rate and (b) Tafel plots extracted from the respective LSV curves; (c) Nyquist plot; (d) capacitive current density as a function of scan rate for the synthesized ZnFe2O4, CoFe2O4, NiFe2O4, and Zn0.2Mn0.2Ni0.2Co0.2Fe2.2O4 catalysts. | ||
With the promising OER data in hand and to gain insight into structure–property relationships of the synthesized ferrite catalysts, we next implemented the materials in the AEMEL cell to study their system-level performance. Fig. 8 shows the polarization curves of AEMEL cells with the four different ferrite anode catalysts, operated at 60 °C (Fig. 8a) and 80 °C (Fig. 8b). At a cell potential of 2.0 V, the AEMEL cells with ZnFe2O4 and CoFe2O4 anode catalysts were found to deliver the lowest current densities of ≈0.7 and ≈1.2 A cm−2 at 60 and 80 °C, respectively. The AEMEL cells with high-entropy Zn0.2Mn0.2Ni0.2Co0.2Fe2.2O4 as the anode showed a significantly higher performance, delivering current densities of ≈1.0 A cm−2 (60 °C) and ≈1.5 A cm−2 (80 °C) at a cell potential of 2.0 V. Finally, at a cell potential of 2.0 V, AEMEL cells with the NiFe2O4 anode catalyst were found to deliver ≈1.3 and ≈1.9 A cm−2 at 60 and 80 °C, respectively.
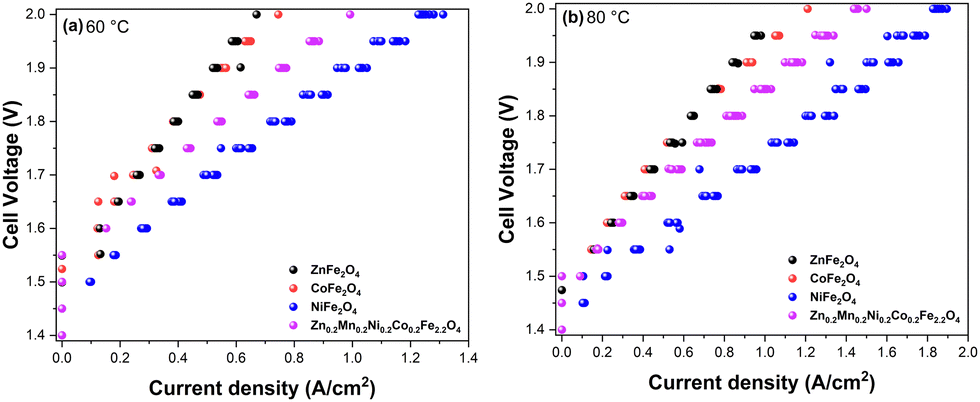 | ||
| Fig. 8 Polarization curves of AEMEL cells with the synthesized ZnFe2O4, CoFe2O4, NiFe2O4, and Zn0.2Mn0.2Ni0.2Co0.2Fe2.2O4 anode catalysts recorded at 60 °C (a) and 80 °C (b) under atmospheric pressure in 1 M KOH electrolyte. | ||
After determining the viability of the synthesized ferrites as anode catalysts on a single-cell AEMEL level, we then investigated the short-term durability of the AEMEL cells with different ferrite anodes. Fig. 9 shows the cell–voltage changes under a constant current density as a function of time. Notably, the recorded cell voltages demonstrate merely small fluctuations over 20 h of chronoamperometric measurements, thus evidencing good short-term durability of the AEMEL cells with ferrite anode catalysts. At a constant current density of 0.5 A cm−2, the AEMEL cell performance decreased in the order of NiFe2O4 > Zn0.2Mn0.2Ni0.2Co0.2Fe2.2O4 > CoFe2O4 > ZnFe2O4, reinforcing the I–V measurement results presented in Fig. 8. Overall, from all the prepared ferrites with different chemical compositions and material properties, NiFe2O4 showed the best performance at the single-cell AEMEL level, affording the highest current density of ≈1.9 A cm−2 with a cell voltage of 2.0 V at 80 °C while demonstrating high stability.
 | ||
| Fig. 9 Cell–voltage changes at a constant current density of 0.5 A cm−2 as a function of testing time for the AEMEL cells with the synthesized ZnFe2O4, CoFe2O4, NiFe2O4, and Zn0.2Mn0.2Ni0.2Co0.2Fe2.2O4 anode catalysts, recorded at 80 °C under atmospheric pressure in 1 M KOH electrolyte. | ||
Discussion
The spinel ferrites, such as NiFe2O4 or CoFe2O4, are among the most explored earth-abundant OER catalysts because of their high OER activity.31,43,68–71 For example, NixCu1–xFe2O4 ferrites anchored onto S-doped graphitic carbon nitride have been reported to exhibit low overpotential η10 = 250 mV at current density j = 10 mA cm−2 and good stability.72 In another study, doped CoFe2O4-based hollow nanospheres displayed high OER activity due to heteroatom substitution and vacancy engineering through Cr3+ doping and S2− exchange.73 Doping with Cr3+ at the octahedral Fe sites induces a Co vacancy that activates the adjacent Fe3+ present at the tetrahedral sites. The S2− exchange resulted in a structural distortion of tetrahedral Fe, leading to enhanced adsorption of intermediate *OOH species in the OER. Another work described the preparation of mesoporous first-row transition metal ferrites based on Mn2+, Fe2+, Co2+, Ni2+, Cu2+, and Zn2+,74 and found NiFe2O4 to show the highest OER activity with low η10 = 278 mV. The best OER activity was also found for NiFe2O4 in a study of tubular ferrite microstructures (AFe2O4, A = Fe2+, Co2+, and Ni2+),75 where the high activity was attributed to the large specific surface area, high active surface area, and low charge-transfer resistance. In this context, MnFe2O4 has also been reported as an alternative OER catalyst by fine-tuning the Mn occupancy at the octahedral sites along with the conversion of Mn2+–to–Mn3+ through heat treatment.76 The mixed valence of Mn resulted in improved alkaline OER activity, wherein Mn3+ was suggested to be more catalytically active than Mn2+.In this work, we show that high-quality ferrite nanoparticle catalysts can be prepared at large-scale (5 g) using an automated synthesis system. The magnetic property measurements confirmed that the synthesized materials largely exhibit a superparamagnetic state at room temperature as a consequence of the small size of the particles, allowing the spontaneous flipping of their magnetization at room temperature. We further found that the OER properties of the synthesized ferrites are quite similar in the kinetic regime at low current densities of ≫0.1 mA (Fig. 6). At the same time, the alkaline OER measurements with higher catalyst mass loading over glassy carbon plate, i.e., at higher current densities ≫0.1 mA, clearly show that the catalysts demonstrate different performance compared to that observed in the kinetic regime. Specifically, NiFe2O4 and CoFe2O4 ferrites feature OER activities that align with the literature data (Table S5, ESI†), with overpotentials of 455 and 484 mV at 10 mA cm−2, respectively. In addition, high-entropy Zn0.2Mn0.2Ni0.2Co0.2Fe2.2O4 ferrite outperforms the two-metal ferrites showing an overpotential of η10 = 432 mV at j = 10 mA cm−2. Interestingly, the number of reports regarding the OER activity of high-entropy spinels is still limited as compared to one- or two-metal spinel oxides (Table S5, ESI†), and the results in Fig. 7 highlight the potential of such materials as highly active OER catalysts.77,78
Previously, many descriptors have been identified for the OER,79 such as eg electrons, M–O–M bond angle, and M–O bond length. The magnetic moment is also considered as one of the descriptors.80 In the case of spinel ferrites, therefore, the saturation magnetization can be identified as an important macroscopic experimental descriptor, as anticipated from theory,23,81,82 as the increase in the saturation magnetization results in an increase in the OER activity in the case of ferrites AFe2O4 (A = Mg, Ni, Mn, and Co).83 This trend is clearly observed in the current study (Fig. 10), with Zn0.2Mn0.2Ni0.2Co0.2Fe2.2O4 exhibiting the highest saturation magnetization and showing the highest OER activity at moderated current densities (Fig. 6). On the other hand, the ZnFe2O4 catalyst with the lowest saturation magnetization showed the lowest OER activity among the spinel ferrites (Fig. 10). Also, the surface state of the high-entropy ferrite is unique compared to the ferrites with fewer elements. Namely, the presence of a higher number of elements leads to highly dispersed active sites, and may provide various active sites for the OER.84 In the present case, Zn0.2Mn0.2Ni0.2Co0.2Fe2.2O4 has very low charge transfer resistance, a low overpotential, a low Tafel slope, a low bandgap, high saturation magnetization, and a high roughness factor, leading to the observed highest OER activity in the moderate current density range (Fig. 7). Interestingly, in full-cell AEMEL testing at high current densities, NiFe2O4 outperforms Zn0.2Mn0.2Ni0.2Co0.2Fe2.2O4 (Fig. 8 and 9), and the observed AEMEL performance for these two anodes compares favorably with the literature (Table S6, ESI†). Two explanations can be proposed for the superior activity of NiFe2O4 and Zn0.2Mn0.2Ni0.2Co0.2Fe2.2O4 at high current densities. First, the computational calculations revealed that nickel and high-entropy ferrites show the lowest band gaps, i.e., exhibit high conductivity (Fig. 5a and b and Table S13, ESI†), due to the presence of Fe vacancies that help to reduce the band gap and enhance the electron transfer. Second, after careful examination of the spin polarization of the oxygen atoms along the spin channels (Fig. 5a and b), we can suggest that nickel ferrite should advantageously possess more catalytically active centers along the spin channels (i.e., more metal sites with high spin-polarized oxygen atoms) than Zn0.2Mn0.2Ni0.2Co0.2Fe2.2O4 ferrite, which showed spin polarization on the oxygen atoms only at very few metal sites.
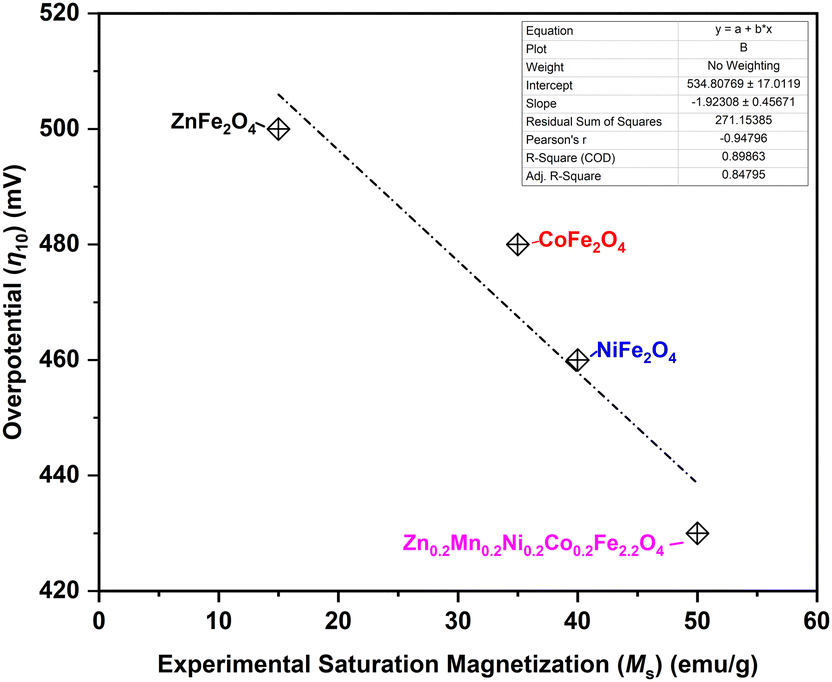 | ||
| Fig. 10 Alkaline OER overpotential η10 needed to drive current density j = 10 mA cm−2 as a function of saturation magnetization of the synthesized ferrite catalysts. | ||
Conclusions
The spinel ferrites, CoFe2O4, NiFe2O4, and ZnFe2O4, and high entropy Zn0.2Mn0.2Ni0.2Co0.2Fe2.2O4 were successfully synthesized on a large scale by the coprecipitation method. XRD, Mössbauer spectroscopy, and XPS confirmed the mixed spinel compositions of the samples. The magnetic measurements evidenced that the synthesized nanocrystalline materials predominantly exhibit a superparamagnetic ground state at room temperature. In the kinetic regime at very low current densities, the ferrites demonstrate similar alkaline OER properties, while high entropy Zn0.2Mn0.2Ni0.2Co0.2Fe2.2O4 ferrite was found to be the best-performing OER catalyst in the moderate current density range. An increase in the OER activity was observed with increasing saturation magnetization, indicating that saturation magnetization can be considered a macroscopic experimental descriptor for magnetic spinels. Finally, the AEMEL measurements at high current densities revealed NiFe2O4 and Zn0.2Mn0.2Ni0.2Co0.2Fe2.2O4 as the most promising anode catalysts among the studied spinel ferrites, most likely due to their favorable electronic and magnetic structures, as corroborated theoretically. Future efforts should focus on (i) the electrode level optimization of AEMEL, since this hydrogen generation technology is in the developing stage at present; and (ii) investigation of the OER properties of ferrimagnetic/ferromagnetic counterparts of the ferrites presented in the current study.Author contributions
Tiago Fernandes: conceptualization, data curation, formal analysis, investigation, methodology, validation, visualization, and writing – original draft. Ramsundar Mohan: data curation, investigation, methodology, and writing – original draft. Laura Donk: investigation and methodology. Wei Chen: investigation, methodology, and writing – original draft. Chiara Biz: investigation, methodology, and writing – original draft. Mauro Fianchini: investigation, methodology, and writing – original draft. Anna Kitayev: investigation and writing – review & editing. Saeed Kamali: investigation, data curation, and writing – review & editing. Siavash Mohammad Alizadeh: data curation. Aviv Ashdot: investigation and writing – review & editing. Miles Page: conceptualization, funding acquisition, methodology, and writing – review & editing. Laura M. Salonen: writing – review & editing. Sebastian Kopp: writing – review & editing. Ervin Tal Gutelmacher: funding acquisition, resources, supervision, validation, and writing – review & editing. José Gracia: funding acquisition, resources, supervision, validation, and writing – review & editing. Marta Costa Figueiredo: funding acquisition, resources, supervision, validation, and writing – review & editing. Yury V. Kolen’ko: funding acquisition, resources, supervision, validation, and writing – review & editing.Data availability
The data supporting this article have been included as part of the ESI.† Data related to materials synthesis and characterization are openly available in a public repository that issues datasets with DOIs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the European Union's Horizon 2020 research and innovation program through the SpinCat Project under grant agreement no. 964972. The authors also thank Dr. Oleksandr Bondarchuk for providing feedback during the XPS analysis of the samples.References
- G. Singh, K. Ramadass, V. D. B. C. DasiReddy, X. Yuan, Y. Sik Ok, N. Bolan, X. Xiao, T. Ma, A. Karakoti, J. Yi and A. Vinu, Prog. Mater. Sci., 2023, 135, 101104 CrossRef CAS.
- K. Veeramani, G. Janani, J. Kim, S. Surendran, J. Lim, S. C. Jesudass, S. Mahadik, H. lee, T.-H. Kim, J. K. Kim and U. Sim, Renewable Sustainable Energy Rev., 2023, 177, 113227 CrossRef CAS.
- A. Vazhayil, J. Thomas, A. Kumar PP and N. Thomas, in Transition Metal-Based Electrocatalysts: Applications in Green Hydrogen Production and Storage, American Chemical Society, 2023, vol. 1435, ch. 4, pp. 73–119 Search PubMed.
- D. Hua, J. Huang, E. Fabbri, M. Rafique and B. Song, ChemElectroChem, 2023, 10, e202200999 CrossRef CAS.
- J. Brauns and T. Turek, Processes, 2020, 8, 248 CrossRef CAS.
- Hydrogen from renewable power: Technology outlook for the energy transition, IRENA, 2018.
- S. A. Grigoriev, V. N. Fateev, D. G. Bessarabov and P. Millet, Int. J. Hydrogen Energy, 2020, 45, 26036–26058 CrossRef CAS.
- Nafion, https://www.nafion.com/en, (accessed March 5th, 2024).
- T. Smolinka and J. Garche, Electrochemical power sources: fundamentals, systems, and applications: hydrogen production by water electrolysis, Elsevier, 2021 Search PubMed.
- S.-C. Sun, H. Jiang, Z.-Y. Chen, Q. Chen, M.-Y. Ma, L. Zhen, B. Song and C.-Y. Xu, Angew. Chem., Int. Ed., 2022, 61, e202202519 CrossRef CAS PubMed.
- M. Maier, J. Dodwell, R. Ziesche, C. Tan, T. Heenan, J. Majasan, N. Kardjilov, H. Markötter, I. Manke, L. Castanheira, G. Hinds, P. R. Shearing and D. J. L. Brett, J. Power Sources, 2020, 455, 227968 CrossRef CAS.
- A. Kellenberger, N. Vaszilcsin, D. Duca, M. L. Dan, N. Duteanu, S. Stiber, T. Morawietz, I. Biswas, S. A. Ansar, P. Gazdzicki, F. J. Wirkert, J. Roth, U. Rost, M. Brodmann, A. S. Gago and K. A. Friedrich, Mater., 2022, 15, 1628 CrossRef CAS PubMed.
- H. A. Miller, K. Bouzek, J. Hnat, S. Loos, C. I. Bernäcker, T. Weißgärber, L. Röntzsch and J. Meier-Haack, Sustainable Energy Fuels, 2020, 4, 2114–2133 RSC.
- N. Du, C. Roy, R. Peach, M. Turnbull, S. Thiele and C. Bock, Chem. Rev., 2022, 122, 11830–11895 CrossRef CAS PubMed.
- D. Li, A. R. Motz, C. Bae, C. Fujimoto, G. Yang, F.-Y. Zhang, K. E. Ayers and Y. S. Kim, Energy Environ. Sci., 2021, 14, 3393–3419 RSC.
- L. Royer, J. Guehl, M. Zilbermann, T. Dintzer, C. Leuvrey, B. P. Pichon, E. Savinova and A. Bonnefont, Electrochim. Acta, 2023, 446, 141981 CrossRef CAS.
- M. Klingenhof, P. Hauke, M. Kroschel, X. Wang, T. Merzdorf, C. Binninger, T. Ngo Thanh, B. Paul, D. Teschner, R. Schlögl and P. Strasser, ACS Energy Lett., 2022, 7, 3415–3422 CrossRef CAS.
- H. Wan, L. Hu, X. Liu, Y. Zhang, G. Chen, N. Zhang and R. Ma, Chem. Sci., 2023, 14, 2776–2798 RSC.
- F. Zeng, C. Mebrahtu, L. Liao, A. K. Beine and R. Palkovits, J. Energy Chem., 2022, 69, 301–329 CrossRef CAS.
- S. Han, J. Park and J. Yoon, Adv. Funct. Mater., 2024, 2314573, DOI:10.1002/adfm.202314573.
- M. K. Adak, L. Mallick, K. Samanta and B. Chakraborty, J. Phys. Chem. C, 2023, 127, 154–168 CrossRef CAS.
- C. Biz, M. Fianchini and J. Gracia, ACS Catal., 2021, 11, 14249–14261 CrossRef CAS.
- T. Lim, J. W. Niemantsverdriet and J. Gracia, ChemCatChem, 2016, 8, 2968–2974 CrossRef CAS.
- Y. Sun, S. Sun, H. Yang, S. Xi, J. Gracia and Z. J. Xu, Adv. Mater., 2020, 32, 2003297 CrossRef CAS PubMed.
- L. Li, S. Lu, Y. Dai, H. Li, X. Wang and Y. Zhang, ACS Appl. Nano Mater., 2023, 6, 2184–2192 CrossRef CAS.
- Ö. N. Avcı, L. Sementa and A. Fortunelli, ACS Catal., 2022, 12, 9058–9073 CrossRef PubMed.
- L. He, H. Kang, G. Hou, X. Qiao, X. Jia, W. Qin and X. Wu, Chem. Eng. J., 2023, 460, 141675 CrossRef CAS.
- J. O. Olowoyo and R. J. Kriek, Small, 2022, 18, 2203125 CrossRef CAS PubMed.
- S. Campagna Zignani, M. L. Faro, A. Carbone, C. Italiano, S. Trocino, G. Monforte and A. S. Aricò, Electrochim. Acta, 2022, 413, 140078 CrossRef CAS.
- A. Martinez-Lazaro, A. Caprì, I. Gatto, J. Ledesma-García, N. Rey-Raap, A. Arenillas, F. I. Espinosa-Lagunes, V. Baglio and L. G. Arriaga, J. Power Sources, 2023, 556, 232417 CrossRef CAS.
- Q. Zhao, Z. Yan, C. Chen and J. Chen, Chem. Rev., 2017, 117, 10121–10211 CrossRef CAS.
- Y. V. Kolen’ko, M. Bañobre-López, C. Rodríguez-Abreu, E. Carbó-Argibay, A. Sailsman, Y. Piñeiro-Redondo, M. F. Cerqueira, D. Y. Petrovykh, K. Kovnir, O. I. Lebedev and J. Rivas, J. Phys. Chem. C, 2014, 118, 8691–8701 CrossRef.
- F. L. Deepak, M. Bañobre-López, E. Carbó-Argibay, M. F. Cerqueira, Y. Piñeiro-Redondo, J. Rivas, C. M. Thompson, S. Kamali, C. Rodríguez-Abreu, K. Kovnir and Y. V. Kolen’ko, J. Phys. Chem. C, 2015, 119, 11947–11957 CrossRef CAS.
- K. Kawashima, R. A. Márquez, Y. J. Son, C. Guo, R. R. Vaidyula, L. A. Smith, C. E. Chukwuneke and C. B. Mullins, ACS Catal., 2023, 13, 1893–1898 CrossRef CAS.
- E. Carbó-Argibay, X.-Q. Bao, C. Rodríguez-Abreu, M. Fátima Cerqueira, D. Y. Petrovykh, L. Liu and Y. V. Kolen’ko, J. Colloid Interface Sci., 2015, 456, 219–227 CrossRef PubMed.
- M. H. Sousa, F. A. Tourinho and J. C. Rubim, J. Raman Spectrosc., 2000, 31, 185–191 CrossRef CAS.
- A. Ahlawat, V. G. Sathe, V. R. Reddy and A. Gupta, J. Magn. Magn. Mater., 2011, 323, 2049–2054 CrossRef CAS.
- Ž. Cvejić, E. Đurđić, G. Ivković Ivandekić, B. Bajac, P. Postolache, L. Mitoseriu, V. V. Srdić and S. Rakić, J. Alloys Compd., 2015, 649, 1231–1238 CrossRef.
- Z. Tong, Q. Yao, J. Deng, L. Cheng, T. Chuang, J. Wang, G. Rao, H. Zhou and Z. Wang, Mater. Sci. Eng. B, 2021, 268, 115092 CrossRef CAS.
- S. W. d Silva, F. Nakagomi, M. S. Silva, A. F. Jr., V. K. Garg, A. C. Oliveira and P. C. Morais, J. Appl. Phys., 2010, 107, 09B503 CrossRef.
- G. Lee, M. Jeong, H. R. Kim, M. Kwon, S. Baek, S. Oh, M. Lee, D. Lee and J. H. Joo, ACS Appl. Mater. Interfaces, 2022, 14, 48598–48608 CrossRef CAS.
- Y. Zhang, T. Lu, Y. Ye, W. Dai, Y. a Zhu and Y. Pan, ACS Appl. Mater. Interfaces, 2020, 12, 32548–32555 CrossRef CAS PubMed.
- D. Lim, H. Kong, N. Kim, C. Lim, W.-S. Ahn and S.-H. Baeck, ChemNanoMat, 2019, 5, 1296–1302 CrossRef CAS.
- L. Yao, H. Zhang, M. Humayun, Y. Fu, X. Xu, C. Feng and C. Wang, J. Alloys Compd., 2023, 936, 168206 CrossRef CAS.
- N. Son, J. Lee, T. Yoon and M. Kang, Ceram. Int., 2021, 47, 20317–20334 CrossRef CAS.
- H. Etemadi and P. G. Plieger, J. Mater. Sci., 2021, 56, 17568–17583 CrossRef CAS.
- L. Gao, C. Tang, J. Liu, L. He, H. Wang, Z. Ke, W. Li, C. Jiang, D. He, L. Cheng and X. Xiao, Energy Environ. Mater., 2021, 4, 392–398 CrossRef CAS.
- L. Jia, F. Xian, Y. Sugahara, N. Sakai, E. Picheau, H. Xue, Y. Yamauchi, T. Sasaki and R. Ma, Chem. Mater., 2023, 35, 1769–1779 CrossRef CAS.
- K. K. Jani, P. Y. Raval, N. H. Vasoya, M. Nehra, M. Singh, N. Jakhar, S. Kumar, K. B. Modi, D.-K. Lim and R. K. Singhal, Ceram. Int., 2022, 48, 31843–31849 CrossRef CAS.
- M. Meinert and G. Reiss, J. Phys.: Condens. Matter, 2014, 26, 115503 CrossRef.
- W. E. Pottker, R. Ono, M. A. Cobos, A. Hernando, J. F. D. F. Araujo, A. C. O. Bruno, S. A. Lourenço, E. Longo and F. A. La Porta, Ceram. Int., 2018, 44, 17290–17297 CrossRef CAS.
- S. Asiri, M. Sertkol, S. Guner, H. Gungunes, K. M. Batoo, T. A. Saleh, H. Sozeri, M. A. Almessiere, A. Manikandan and A. Baykal, Ceram. Int., 2018, 44, 5751–5759 CrossRef CAS.
- J. P. Singh, J. Y. Park, V. Singh, S. H. Kim, W. C. Lim, H. Kumar, Y. H. Kim, S. Lee and K. H. Chae, RSC Adv., 2020, 10, 21259–21269 RSC.
- M. H. Habibi and H. J. Parhizkar, Spectrochim. Acta, Part A, 2014, 127, 102–106 CrossRef CAS PubMed.
- N. Kislov, S. S. Srinivasan, Y. Emirov and E. K. Stefanakos, Mater. Sci. Eng. B, 2008, 153, 70–77 CrossRef CAS.
- F. Arteaga-Cardona, U. Pal, J. María Alonso, P. de la Presa, M.-E. Mendoza-Álvarez, U. Salazar-Kuri and M. Á. Méndez-Rojas, J. Magn. Magn. Mater., 2019, 490, 165496 CrossRef CAS.
- C. Simon, M. B. Zakaria, H. Kurz, D. Tetzlaff, A. Blösser, M. Weiss, J. Timm, B. Weber, U.-P. Apfel and R. Marschall, Chem. – Eur. J., 2021, 27, 16990–17001 CrossRef CAS PubMed.
- H. Salazar-Tamayo, K. E. G. Tellez and C. A. B. Meneses, Mater. Res., 2019, 22, 1–11 Search PubMed.
- J. Klein, L. Kampermann, B. Mockenhaupt, M. Behrens, J. Strunk and G. Bacher, Adv. Funct. Mater., 2023, 33, 2304523 CrossRef CAS.
- S. Anantharaj and S. Kundu, ACS Energy Lett., 2019, 4, 1260–1264 CrossRef CAS.
- M. Plevová, J. Hnát and K. Bouzek, J. Power Sources, 2021, 507, 230072 CrossRef.
- M. Gong and H. Dai, Nano Res., 2015, 8, 23–39 CrossRef CAS.
- H. Baltruschat, J. Am. Soc. Mass Spectrom., 2004, 15, 1693–1706 CrossRef CAS PubMed.
- H. M. A. Amin, P. Königshoven, M. Hegemann and H. Baltruschat, Anal. Chem., 2019, 91, 12653–12660 CrossRef CAS PubMed.
- W. Zheng, ACS Energy Lett., 2023, 8, 1952–1958 CrossRef CAS.
- K. Chakrapani, G. Bendt, H. Hajiyani, I. Schwarzrock, T. Lunkenbein, S. Salamon, J. Landers, H. Wende, R. Schlögl, R. Pentcheva, M. Behrens and S. Schulz, ChemCatChem, 2017, 9, 2988–2995 CrossRef CAS.
- W. Zheng, M. Liu and L. Y. S. Lee, ACS Energy Lett., 2020, 5, 3260–3264 CrossRef CAS.
- W. Xiang, N. Yang, X. Li, J. Linnemann, U. Hagemann, O. Ruediger, M. Heidelmann, T. Falk, M. Aramini, S. DeBeer, M. Muhler, K. Tschulik and T. Li, Nat. Commun., 2022, 13, 179 CrossRef CAS PubMed.
- C. Mahala, M. D. Sharma and M. Basu, Electrochim. Acta, 2018, 273, 462–473 CrossRef CAS.
- C. Simon, J. Timm, D. Tetzlaff, J. Jungmann, U.-P. Apfel and R. Marschall, ChemElectroChem, 2021, 8, 227–239 CrossRef CAS.
- L. S. Ferreira, T. R. Silva, V. D. Silva, R. A. Raimundo, T. A. Simões, F. J. A. Loureiro, D. P. Fagg, M. A. Morales and D. A. Macedo, Adv. Powder Technol., 2022, 33, 103391 CrossRef CAS.
- S. B. Sundararaj and S. Thangavelu, J. Phys. Chem. C, 2023, 127, 4953–4966 CrossRef CAS.
- J. Sun, H. Xue, Y. Zhang, X.-L. Zhang, N. Guo, T. Song, H. Dong, Y. Kong, J. Zhang and Q. Wang, Nano Lett., 2022, 22, 3503–3511 CrossRef CAS PubMed.
- L. A. Achola, S. Shubhashish, Z. Tobin, Y. Su, L. F. Posada, Y. Dang, J. Shi, A. G. Meguerdichian, M. Jain and S. L. Suib, Chem. Mater., 2022, 34, 7692–7704 CrossRef CAS.
- F. Yuan, X. Cheng, M. Wang and Y. Ni, Electrochim. Acta, 2019, 324, 134883 CrossRef CAS.
- Y. Zhou, Y. Du, S. Xi and Z. J. Xu, Electrocatalysis, 2018, 9, 287–292 CrossRef CAS.
- X. Wang, H. Singh, M. Nath, K. Lagemann and K. Page, ACS Mater. Au, 2024, 4(3), 274–285 CrossRef CAS PubMed.
- X. Wang, P. Metz, E. Calì, P. R. Jothi, E. A. Lass and K. Page, Chem. Mater., 2023, 35, 7283–7291 CrossRef CAS.
- W. T. Hong, R. E. Welsch and Y. Shao-Horn, J. Phys. Chem. C, 2016, 120, 78–86 CrossRef CAS.
- D. Deng and L.-M. Yang, ACS Appl. Mater. Interfaces, 2023, 15, 22012–22024 CrossRef CAS PubMed.
- J. Gracia, Phys. Chem. Chem. Phys., 2017, 19, 20451–20456 RSC.
- L. Zhang, A. Cheruvathur, C. Biz, M. Fianchini and J. Gracia, Phys. Chem. Chem. Phys., 2019, 21, 2977–2983 RSC.
- C. Iwakura, M. Nishioka and H. Tamura, Nippon Kagaku Kaishi, 1982, 1982, 1136–1140 CrossRef.
- D. Stenzel, B. Zhou, C. Okafor, M. V. Kante, L. Lin, G. Melinte, T. Bergfeldt, M. Botros, H. Hahn and B. Breitung, Front. Energy Res., 2022, 10, 942314 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ya00170b |
| This journal is © The Royal Society of Chemistry 2024 |