
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGlucose-based highly-porous activated carbon nanospheres (g-ACNSs) for high capacity hydrogen storage†
Fan-Gang
Tseng
 abcd,
Dinesh
Bhalothia
a,
Kuan-Hou
Lo
a,
Cheng-Huei
Syu
a,
Ying-Cheng
Chen
a,
Amita
Sihag
a,
Che-Wen
Wang
a,
Hsin-Yi Tiffany
Chen
*ae and
Tsan-Yao
Chen
*a
abcd,
Dinesh
Bhalothia
a,
Kuan-Hou
Lo
a,
Cheng-Huei
Syu
a,
Ying-Cheng
Chen
a,
Amita
Sihag
a,
Che-Wen
Wang
a,
Hsin-Yi Tiffany
Chen
*ae and
Tsan-Yao
Chen
*a
aDepartment of Engineering and System Science, National Tsing Hua University, Hsinchu 30013, Taiwan. E-mail: chencaeser@gmail.com; Tel: +886-3-5715131#34271
bFrontier Research Center on Fundamental and Applied Sciences of Matters, National Tsing Hua University, Hsinchu 300044, Taiwan
cDepartment of Chemistry, National Tsing Hua University, Hsinchu 300044, Taiwan
dResearch Center for Applied Sciences, Academia Sinica, Taiwan
eCollege of Semiconductor Research, National Tsing Hua University, Hsinchu 300044, Taiwan
First published on 29th April 2024
Abstract
Nanoporous glucose-based active carbon nanospheres (g-ACNSs) with high efficiency and stability in hydrogen (H2) storage are synthesized by a hydrothermal method followed by multiple KOH activation processes. For the optimized conditions (g-ACNS24), they exhibit a high specific surface area of 2291 m2 g−1 and a large defect ratio (ID/IG = 1.77) in the carbon structure. With these structure characteristics, the g-ACNS24 demonstrates an H2 storage capacity of 5.04 wt% and a high hydrogen uptake capacity (>80%) in the durability test for more than 100 storage cycles at 77 K and 100 bar. DFT calculation results show that the chemisorption hydrogen adsorption enhances in an amorphous model with mixed coordinated carbon atoms compared to a perfect six-membered graphene surface. This once again proves that the superior hydrogen storage performance of g-ACNSs can be attributed not only to their high specific surface area and large pore volume, but also to the distribution ratio of micropores and associated defects. Overall, the findings suggest that g-ACNS materials hold promise as efficient and cyclically stable materials for hydrogen storage, with potential applications in the field of hydrogen energy.
Introduction
Hydrogen is considered a clean energy source due to its zero greenhouse gas emissions.1 However, to build a sustainable hydrogen energy ecosystem, efficient and economic methods for hydrogen production, transportation, and storage are crucial.1 While there have been promising solutions proposed for hydrogen production, storage methods are still not very satisfactory.2 Common methods for hydrogen storage can be categorized into compressed gas, cryogenic storage, chemical hydrides, and sorbent materials.3 Compressed gas involves high-pressure storage of hydrogen, which requires thick and sturdy containers, resulting in low-weight storage density.3 Polymer composite hydrogen storage tanks have been proposed to reduce weight and increase storage density.4 Cryogenic storage requires bringing hydrogen to very low temperatures (20.3 K) to maintain it in a liquid state, which requires high energy consumption and special cryogenic containers.5 Chemical hydrides can store hydrogen at high volume density but low weight density, but their hydrogen release at higher temperatures requires high energy input.6 Alkali metals such as LiNH2, LiBH4, and NaBH4 can provide better hydrogen release conditions at lower temperatures (<77 K),7 but the hydrogenation process usually requires high-pressure environments (>10 MPa).8As for non-metal hydrides, two types are considered, including carbon-containing and non-carbon hydrides. Carbon-containing hydrides, such as natural gas and low-carbon alkane molecules, are greenhouse gases; while the non-carbon-containing ones, mainly borane ammonia and its derivatives, form irreversible chemicals.9 Non-metal hydrides can be either catalyzed for hydrolysis or thermal cracking to produce controllable hydrogen.10 However, it is troublesome that either vast carbon dioxide can be generated from carbon-containing hydrides or the waste from borane ammonia dehydrogenation is hard to handle, and both of them are difficult to regenerate.11
Therefore, sorbent materials, consisting of large surface area, stable material properties, and easy hydrogen-releasing process, have been becoming attractive and drawn intensive attention in the past few decades.12 There are many types of sorbent materials, which usually have several important characteristics from three aspects: structure, stability, and production cost.12 A good adsorbent material should have good pore channels and surface structure so that it has enough pore volume and surface contact area to facilitate adsorption.13 Porous materials become good adsorbent materials because of their high specific surface area, suitable pore volume, and size.14 The stability aspect mainly considers whether the material will chemically react with the adsorbate and the medium to change the mechanical strength.15 On the other hand, low cost, convenient manufacturing, and easy regeneration are also important considerations for sorbent materials.15 Among many sorbent materials, carbon materials possess all the above-mentioned properties required for good hydrogen storage and thus have been considerably investigated in the past few decades.16 The influencing factors of hydrogen storage on the surface of carbon materials include specific surface area, pores, and defects.13 High specific surface area means that hydrogen has more adsorption sites, the size distribution of pores affects the rate of hydrogen adsorption and desorption, and the type and density of defects determines the reversibility of adsorption and desorption.17 There are several common types of carbon materials for hydrogen storage, including NanoFibers, A-NFs (Activated NanoFiber), SWCNTs, MWNTs, graphene, and activated carbon (AC).18–21 The physical properties and hydrogen storage capacity of the above carbon materials together with the materials proposed by this study (g-ACNSs) are listed in Table S1 (ESI†) at 77 K or 300 K and 2–10 MPa for comparison. Besides hydrogen storage, carbonaceous materials are also extensively employed in other fields, such as energy storage.22–24
The activated carbon nanospheress (CNSs) proposed in this article are synthesized using a simple hydrothermal method from glucose combined with KOH activation. They have an average particle size of 50 nm, a specific surface area of more than 2500 m2 g−1, a pore distribution between 1 and 6 nm, and a defect ID/IG ratio of 1.7. These characteristics suggest a decent hydrogen storage capacity, with hydrogen storage exceeding 5% at 77 K, and good reversibility, with cycling for more than 10 times and irreversibility of less than 5%. The hydrogen storage performance and cycle life of the CNSs were evaluated using a high-pressure volumetric analyzer (HPVA, Micromeritics, USA). The structure and physical properties of the CNSs were characterized using TEM (transmission electron microscopy), SEM (scanning electron microscopy), BET (Brunauer–Emmett–Teller) analysis for specific surface area, Raman spectroscopy and XRD (X-ray diffraction), and modelled by DFT (density functional theory) calculations to understand the underlying mechanisms. The obtained results indicate the superiority of the as-prepared CNSs over previously reported carbonaceous materials owing to several distinct features including: (1) high surface area: the CNSs possess a high surface area, providing abundant active sites for hydrogen adsorption. The porous structure allows for efficient utilization of the carbon material, enhancing the overall storage capacity; (2) tunable pore size and volume: the synthesis methodology of the CNSs allows for precise control over pore size and volume distribution. This tunability enables optimization of the material's hydrogen storage properties, tailoring it to specific storage requirements and conditions; (3) chemical stability: these CNSs exhibit excellent chemical stability, minimizing the risk of degradation or decomposition during hydrogen storage cycles. This ensures the long-term stability and durability of the porous carbon spheres as hydrogen storage media, and (4) low cost and abundant availability: the as-prepared CNSs are relatively inexpensive and can be derived from a variety of renewable carbon sources, making them a cost-effective and environmentally friendly option for hydrogen storage compared to other materials.
Experimental
Chemicals and characterization
D-(+)-Glucose (Sigma-Aldrich, USA) was used as the precursor for the synthesis of carbon nano spheres (CNSs) without any purification. The graphene powder for the hydrogen adsorption test was 60 nm flakes (AO-4, UR-Graphene 60, Graphene Supermarket, USA). The commercial activated carbon was Cabot XC-72 and acquired from CABOT (USA). The high-resolution confocal Raman microscope employed for material characterization was made by HORIBA (USA) with a 532 nm laser for excitation. For morphology observation and material characterization, a scanning electron microscope (JEOL JSM-5600, Japan), a transmission electron microscope (JEOL JEM-F200, Japan), and X-ray diffraction (BL01C2, NSRRC, Taiwan) were also employed. For the measurement of surface area and pore sizes/distributions, a Brunauer–Emmett–Teller surface area analyzer (autoSorb iQ-TPX, Quantachrome Instruments, USA) was utilized. The hydrogen storage and cycle life were tested by a hydrogen absorption and desorption test system (HPVA-II, Micromeritics, USA).Synthesis of CNSs
The CNSs were synthesized from D-(+)-glucose by a hydrothermal process.15 First, 5 g of D-(+)-glucose as the precursor and 1 g of polystyrene sulfonate (or kayexalate) as a structure-directing agent were dissolved in 25 ml DI water. After becoming transparent, the mixture was placed into an autoclave (DH-400N) for a hydrothermal process. During the process, it went through a heat-up stage for 8 hours under 220 °C, and then cooled down naturally.Activation of highly porous CNSs
Before the chemical activation process, the fabricated CNSs went through several times of cleaning and drying-out processes. Two repeating activations with the same thermal history are applied to the CNSs. During heat treatment, the CNS powder was mixed with KOH with a weight ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, 2:4, and 4:4, respectively, for CNS22, CNS24, and CNS44. After mixing, the mixtures were placed in a furnace under 500 c.c. min−1 flow of nitrogen. Two stages of the heating process were then conducted, the furnace was heated up to 350 °C for the first half an hour and then to 800 °C for 2 hours.25–27 The heating rate is 5 °C min−1 for both stages. In the 2nd activation, the weight ratio is 2 for CNS22, 4 for CNS24 and 4 for CNS44. After the activation process, 1 M of HCl was utilized to neutralize the product. Before the hydrogen adsorption testing, two times of surface activation process were employed by using the solid-state method to ensure a proper hydrogen uptake ability of the newly fabricated porous CNSs. The inlet carbon loading is 8.0 g per batch. The resulting powder is 7.2 g for the 1st and 6.4 g for the 2nd activation. After the acid treatment and washing, the final product (CNSs) is 5.5 g.
:2, 2:4, and 4:4, respectively, for CNS22, CNS24, and CNS44. After mixing, the mixtures were placed in a furnace under 500 c.c. min−1 flow of nitrogen. Two stages of the heating process were then conducted, the furnace was heated up to 350 °C for the first half an hour and then to 800 °C for 2 hours.25–27 The heating rate is 5 °C min−1 for both stages. In the 2nd activation, the weight ratio is 2 for CNS22, 4 for CNS24 and 4 for CNS44. After the activation process, 1 M of HCl was utilized to neutralize the product. Before the hydrogen adsorption testing, two times of surface activation process were employed by using the solid-state method to ensure a proper hydrogen uptake ability of the newly fabricated porous CNSs. The inlet carbon loading is 8.0 g per batch. The resulting powder is 7.2 g for the 1st and 6.4 g for the 2nd activation. After the acid treatment and washing, the final product (CNSs) is 5.5 g.
Gas adsorption measurement
Hydrogen adsorption and desorption tests were performed under 77 k from 1 to 100 bars by an HPVA-II system (Micromeritics, USA). The degas condition was kept at 523 k for 14 hours to ensure the vacuum value was lower than 0.013 mbar. For pore analysis and specific surface area measurement, BET measurements were conducted. From the BET results, the specific surface area was calculated by the Brunauer–Emmett–Teller (BET) equation.28 The total pore volume was then evaluated by converting the amount of nitrogen adsorbed to the equivalent liquid volume of the adsorbate at a relative pressure of 0.99 and using the t-plot method to obtain the micropore volume.28Computational details
To understand the correlation between the number of defects and the hydrogen storage ability of CNSs with a high defect ratio of ID/IG = 1.77, a computational model of amorphous carbon (42% 2-fold, 52% 3-fold, and 6% 4-fold coordinated carbon atoms) was built to mimic the experimental CNS samples. The correlation between the ID/IG ratios and 2-fold/3-fold coordinated carbon atom ratios was reported.29 Spin-polarized density functional theory (DFT)30 and ab initio molecular dynamics31 (AIMD) calculations were performed using the Vienna ab initio Simulation Package32–34 (VASP 5.4). The projector augmented wave (PAW) method with a plane wave basis set was used for the calculations. The Perdew–Burke–Ernzerhof (PBE)35 was used to describe the exchange–correlation function. The amorphous carbon structure was built using the liquid-quench method, used in the previous studies to build amorphous structures.36–38 The initial structure contains 100 carbon atoms placed randomly in the unit cell of the dimensions a = b = c = 11 Å with constant volume and periodic boundaries; the density of this amorphous carbon is 1.5 g cm−3 in alignment with our experimental samples. The amorphous carbon surface was created using a vacuum 24 Å along the z direction to avoid the interaction between atoms due to the periodic repetition. The energy cut-off for the plane wave basis was set to be 500 eV. All the structures were optimized using a conjugate gradient algorithm until the forces and energies converged up to 0.01 eV Å−1 and 1.00 × 10−5 eV, respectively. Grimme's D3-dispersion39,40 corrections were included in all calculations to account for the role of long-range dispersion forces. The k-points meshes of 2 × 2 × 2 and 2 × 2 × 1 were used for the bulk and slab models, respectively. The hydrogen adsorption energy per hydrogen atom on amorphous carbon was calculated bywhere a-C represents the amorphous carbon surface and n is the number of hydrogen atoms. The gravimetric capacity (wt%) was calculated by the following equation:
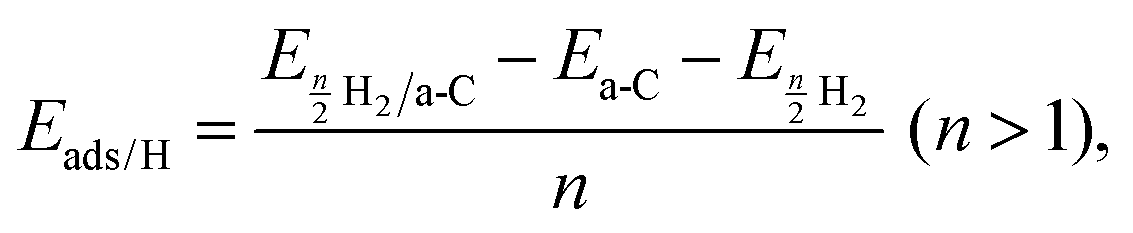
where nH is the number of H atoms, nC is the number of C atoms in the amorphous carbon and MH and MC are the molar mass of hydrogen and carbon.
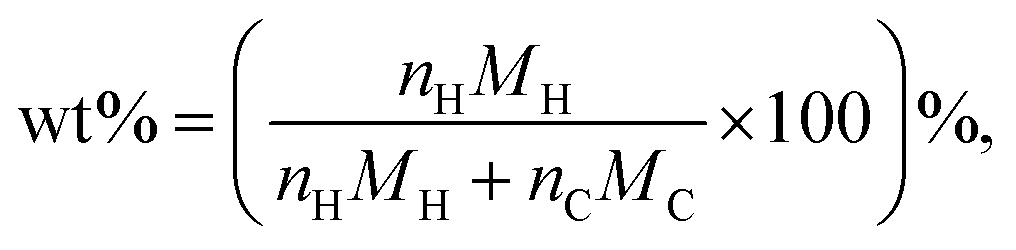
Results and discussion
Nanoscale structures of the CNSs, XC-72, and graphene
The SEM and TEM images in Fig. 1 illustrate the structures of three different carbon materials: CNSs, XC-72, and graphene. Fig. 1(a) shows that the CNS carbon nanospheres have an average size of about 50 nm with a uniform size distribution. The center part of each particle appears darker and shows a gradient in color, while the edge is brighter. This suggests that there might be defects or pore structures inside the CNS particles, which create a three-dimensional contrast difference in the SEM and TEM images. This is further supported by the relatively uneven atomic structure arrangement observed in the TEM image in Fig. 1(d), indicating the presence of defects or pores in the carbon spheres. | ||
| Fig. 1 (a)–(c) SEM images, (d)–(f) TEM images, and (g)–(i) schematic structures of the CNSs, XC-72, and graphene, respectively. The scale bar is 500 nm for the SEM images and 10 nm for the TEM images. The enlarged region is ∼50 × 50 nm2 for the SEM images. | ||
In comparison, Fig. 1(b) shows the SEM image of XC-72, a commercially available activated carbon material, which appears as local long flakes stacked layer by layer. The flakes have irregular shapes and vary in size between 100 and 250 nm, with some possibly larger. There is no obvious contrast of light and dark colors on the flake surfaces, even at high SEM magnification, suggesting relatively flat and defect-free surfaces. This is further supported by the relatively uniform and complete atomic structure arrangement observed in the TEM image in Fig. 1(e). Fig. 1(c) shows the SEM image of graphene, which consists of a complete sheet structure with a smooth surface. The TEM image in Fig. 1(f) also shows a similar result, with a dark part exhibiting an orderly stripe shape, indicating a multi-layered structure. Based on the comparison of the SEM and TEM images, the schematic structures of the three carbon materials can be redrawn in Fig. 1(g), (h), and (i), respectively. It can be observed that the CNSs possess distinct characteristics compared to XC-72 and graphene, including more uniform size distribution and abundant holes and defects inside the particles. These characteristics are likely to contribute to the high surface area and proper pore size, which could be advantageous for hydrogen storage in the CNS material.
XRD analysis for the CNSs, XC-72, and graphene
X-Ray diffraction (XRD) analysis was used to analyze the lattice planes of three different carbon materials, including CNS22, CNS24, and CNS44, to investigate the effects of KOH activation on the lattice structure. The XRD patterns in Fig. 2(a) showed that the CNSs have three main composite diffraction peaks, with the 20°–28° angle range corresponding to the [002] crystal plane. The [002] main peak of the CNSs appeared wider and less well-defined compared to that of graphene, indicating that KOH activation caused more damage to the [002] plane along the C-axis (vertical direction), as OH− ions can easily destroy the van der Waals bonds along the C-axis.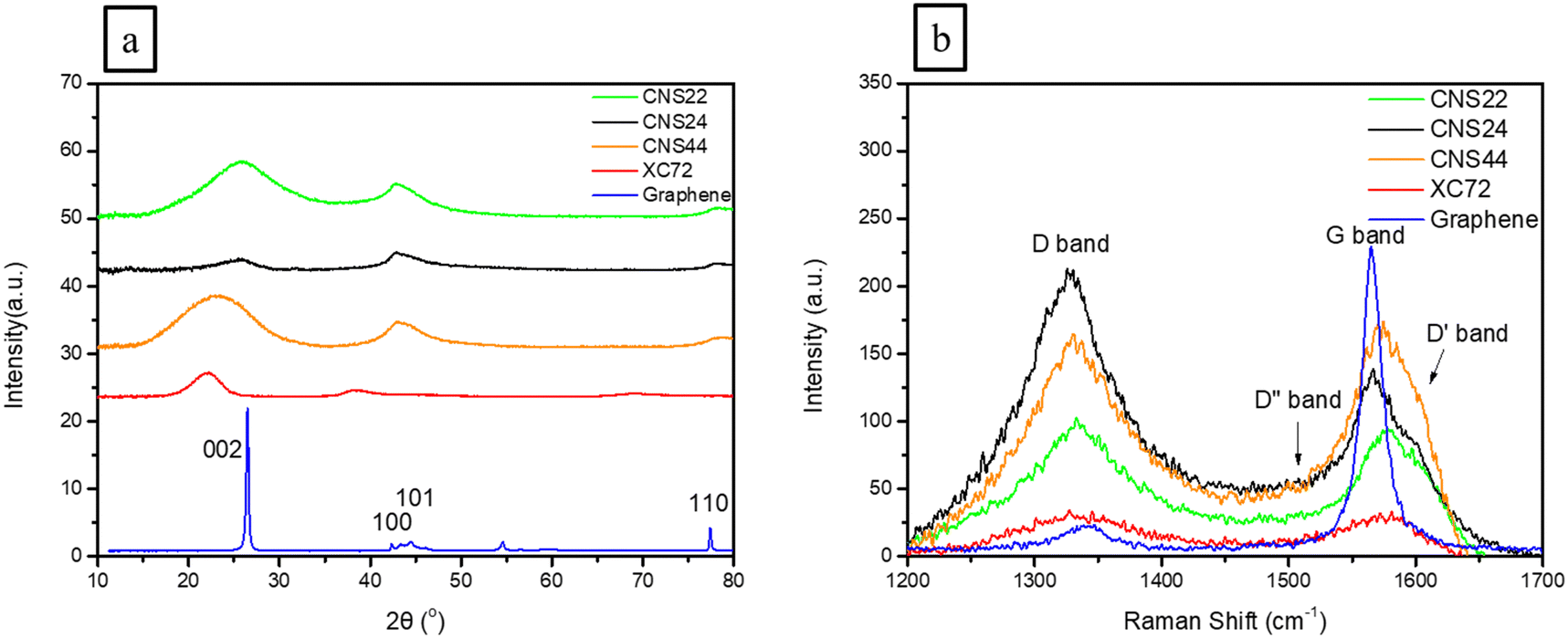 | ||
| Fig. 2 (a) XRD and (b) Raman analysis for CNS, XC-72, and graphene. | ||
Comparing CNS22, CNS24, and CNS44, it was observed that CNS24 showed more lattice damage, suggesting that the second addition of a higher concentration of KOH selectively destroyed more lattice planes. Tables 1 and 2 provide information on the lattice size and lattice plane spacing, revealing that OH− ions had the most significant impact on the C-axis along the [002] plane, as these could easily enter the C-axis and expand the plane spacing. In contrast, other directions showed less influence on the sp2 bonding in the horizontal direction.
| Lattice | [002] | [100] | [101] | [110] | [110] |
|---|---|---|---|---|---|
| Sample | Coherence lengtha (Å) | Coherence lengtha (Å) | Coherence lengtha (Å) | Coherence lengtha (Å) | Coherence lengtha (Å) |
a Grain size is derived from 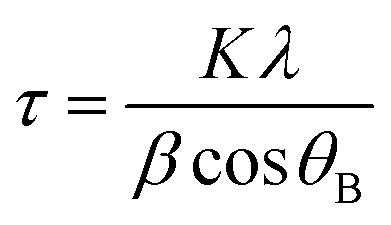 (Scherrer equation, where K: grain constant, λ: X-ray wavelength, β: half-width of characteristic peak, and θB: Bragg angle). (Scherrer equation, where K: grain constant, λ: X-ray wavelength, β: half-width of characteristic peak, and θB: Bragg angle).
|
|||||
| CNS22 | 4.98 | 8.93 | 7.72 | 19.46 | 13.53 |
| CNS24 | 8.96 | 9.93 | 3.67 | 31.65 | 10.96 |
| CNS44 | 4.79 | 9.91 | 3.91 | 13.92 | 11.35 |
| XC-72 | 12.96 | 14.27 | 5.56 | 10.74 | 4.62 |
| Graphene | 113.37 | 258.43 | 159.62 | 174.97 | 174.97 |
| Lattice | [002] | [100] | [101] | [110] | [110] | I D/IGb |
|---|---|---|---|---|---|---|
| Sample | Lattice spacinga (Å) | Lattice spacinga (Å) | Lattice spacinga (Å) | Lattice spacinga (Å) | Lattice spacinga (Å) | |
a Lattice spacing is derived from 2d![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) sinθ = nλ (Bragg's law, where n: integer, λ: X-ray wavelength, d: plane spacing in the atomic lattice, and θ: angle between the incident wave and the scattering plane).
b Defect ratio: using the intensity ratio of the D peak and G peak to quantitatively express the number of defects in atoms. sinθ = nλ (Bragg's law, where n: integer, λ: X-ray wavelength, d: plane spacing in the atomic lattice, and θ: angle between the incident wave and the scattering plane).
b Defect ratio: using the intensity ratio of the D peak and G peak to quantitatively express the number of defects in atoms.
|
||||||
| CNS22 | 1.74 | 1.05 | 0.96 | 0.96 | 0.96 | 1.14 |
| CNS24 | 1.78 | 1.04 | 0.94 | 0.94 | 0.94 | 1.77 |
| CNS44 | 1.93 | 1.04 | 1.01 | 1.01 | 1.01 | 1.12 |
| XC-72 | 2.04 | 1.17 | 1.03 | 1.03 | 1.03 | 1.09 |
| Graphene | 1.51 | 0.95 | 0.93 | 0.93 | 0.93 | 0.08 |
The high angle diffraction peaks, corresponding to the non-C-axis directions, showed less change, as the interatomic distance on these planes was less than the size of the hydrogen atom (1.1 Å), making them unsuitable for hydrogen storage. However, some sp3 structures were still present along the C-axis, indicating bond breakage and the formation of defects and pore structures on the material's surface. These defects and pore structures could potentially improve hydrogen storage by providing additional surface area and pore volume for hydrogen adsorption.
Raman analysis for the CNSs, XC-72, and graphene
Raman analysis was used in this study to confirm the existence of defect structures in the carbon materials, which can enhance hydrogen adsorption. Raman spectra of highly porous activated carbon nanospheres (CNSs), XC-72, and graphene were obtained, as shown in Fig. 2(b), showing two major peaks: the G band at 1580 cm−1, which is the characteristic peak of graphitization (sp2) and the D band located at 1270–1450 cm−1, which is an amorphous peak (sp3) resonated by the disordered structure in the sample and represents the corresponding defects in the sample. The D′ band at around 1600–1620 cm−1 and the D′′ band are also peaks that appear in high-defect structures, connecting the G band and the D band.From the Raman spectra in Fig. 2(b) and the curving fitting in the ESI† (Fig. S1), it can be observed that compared to graphene, the CNSs have both the D band of the amorphous material and the G band of the graphitized carbon material. The ratio (ID/IG) of the D band and G band of the CNSs, XC-72, and graphene was calculated and tabulated in Table 2, which represents the proportion of carbon material defects. It was found that the ID/IG value of the CNSs is significantly higher (ranging from 1.123 to 1.770) compared to graphene's value of 0.077, indicating that the CNSs have a similar content of graphite and amorphous carbon, suggesting that the structure of the CNSs consists of activated carbon nanospheres with a high volume of pores and defects, which can be beneficial for hydrogen adsorption.
On the other hand, XC-72 also exhibits a decent amount of defects similar to the CNSs, as shown in Table 2. This suggests that XC-72 may also have the ability to adsorb hydrogen. However, as revealed in the results of BET analysis in the next section, it is evident that high defects are not the only factor contributing to good hydrogen adsorption capacity, but also the pore sizes and structures of the carbon materials.
BET analysis for the CNSs, graphene, and XC-72
Fig. 3 (left) shows the nitrogen adsorption capacity of various materials at 77 K from 0 to 1 bar, and it is evident that the CNSs have a much higher nitrogen adsorption capacity compared to XC-72 and graphene. This is attributed to the fine pore structure of the CNSs, with most of the pores being micropores and mesopores in the size range of 2 to 6 nm. In contrast, XC-72 and graphene primarily consist of large mesopores between 40 and 60 nm, with fewer micropores.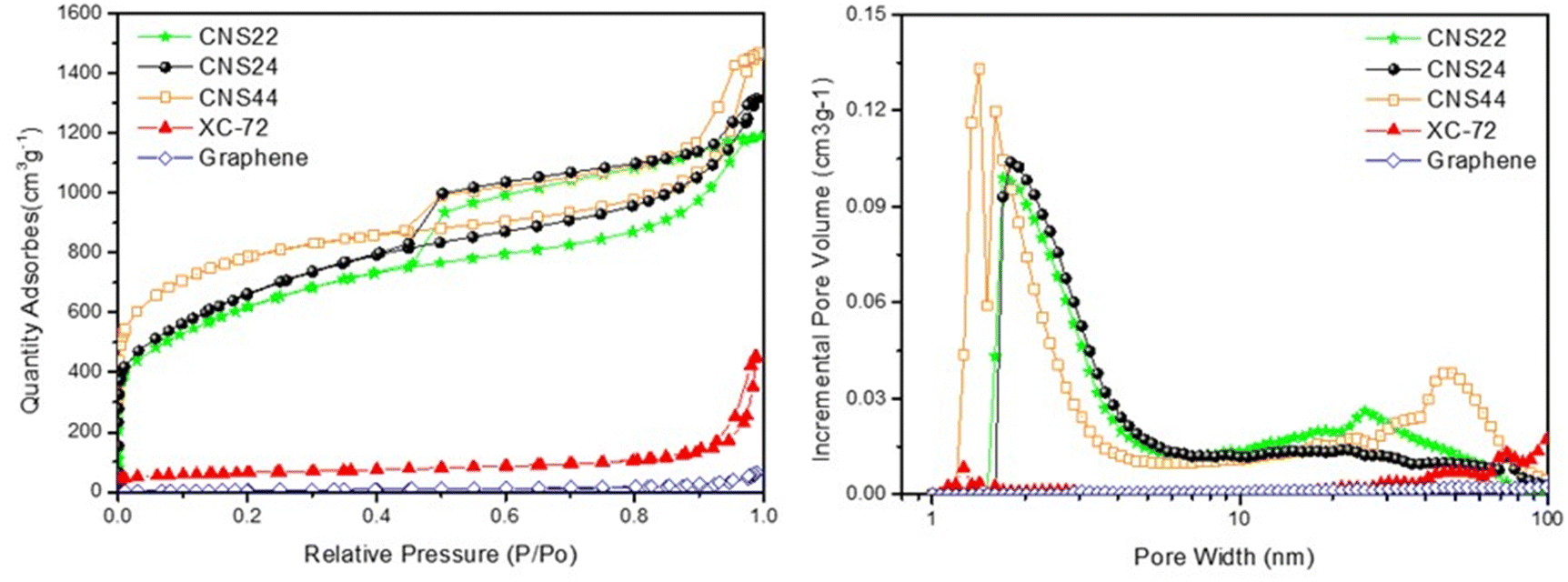 | ||
| Fig. 3 BET analysis of the CNSs, XC-72, and graphene. (left) Absorption/desorption curves and (right) size distributions of pores. | ||
Among the g-ACNS samples, g-ACNS24 exhibits a pore size distribution between 1 and 6 nm, with some larger pores present in the structure. On the other hand, CNS44 shows a high number of micropores, but also some very large pores in the range of 4–8 nm, indicating that these micropores are embedded inside the larger pores. In comparison, CNS22 has poorer pore distribution and volume in the micropore region, resulting in a significantly reduced nitrogen adsorption capacity, which highlights the importance of micropores for hydrogen adsorption.
It has been reported that hydrogen is primarily stored in micropores through physical adsorption at high pressure.2 The micropore size distributions in Fig. 3 (right) supports the higher hydrogen storage capacity of the three g-ACNS structures compared to XC-72 and graphene, due to their higher micropore ratio. In the next section, the hydrogen storage capacity of various carbon materials will be measured and compared using the HPVA (high-pressure volumetric adsorption) method.
HPVA analysis and cycle test for the CNSs, graphene, and XC-72
Fig. 4(a) illustrates the comparison between hydrogen storage capacity measured by HPVA and BET test results for various materials. It shows that the slope of hydrogen adsorption is different at low pressure (0.1100 bar). Notably, graphene exhibits a significantly different slope at low pressure compared to high pressure, indicating different storage patterns at these pressure ranges. It is speculated that the hydrogen adsorption behavior at high pressure is mainly dominated by physical adsorption due to pressure, with hydrogen predominantly adsorbed in the micropores of the carbon materials. On the other hand, adsorption behavior at low pressure is primarily influenced by sp bonds in carbon materials. By comparing the g-ACNSs with XC-72 and graphene, it can be inferred that the g-ACNSs not only store more hydrogen in the micropores under high pressure but also a considerable proportion of hydrogen is adsorbed in sp bonds by defects under low pressure. This suggests that the g-ACNSs exhibit high hydrogen storage capacity through a combination of physical adsorption and chemical adsorption.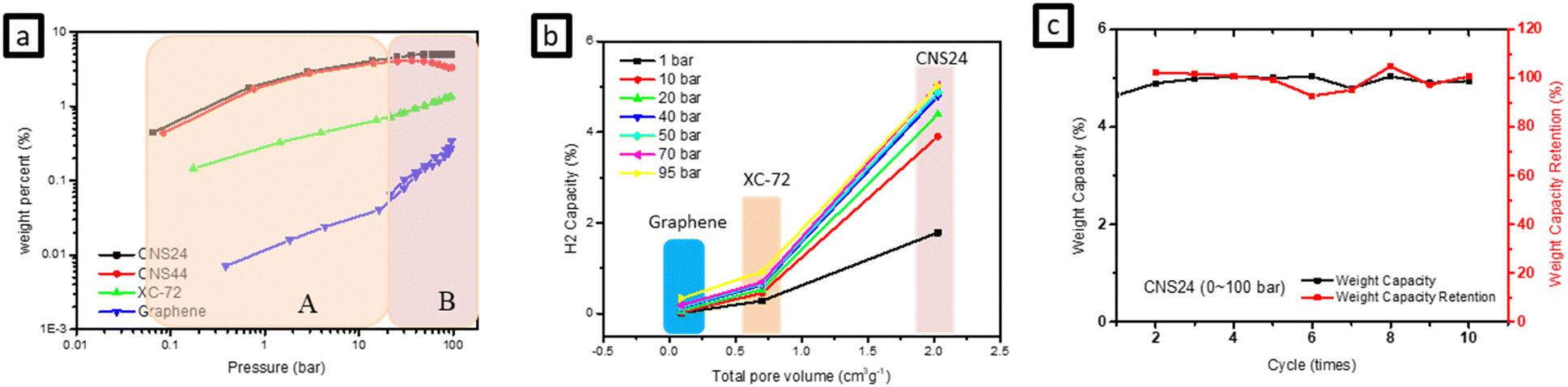 | ||
| Fig. 4 The comparison of the CNSs, XC-72 and graphene in (a) hydrogen adsorption vs. pressure, (b) hydrogen adsorption vs. total pore volume and (c) CNS24 cycling tests (0–100 bar). | ||
Furthermore, the activated carbon nanospheres (ACNSs) themselves possess a high total porosity, and the hydrogen storage capacity is positively correlated with the total pore volume, which is a direct factor affecting the hydrogen storage capacity. Table 3 presents the micropore volume and specific surface area of the g-ACNSs, which are much higher than those of XC-72 and graphene, resulting in significantly improved hydrogen storage capacity. Additionally, Fig. 4(b) shows that a larger pore volume corresponds to better gas storage ability. Moreover, the influences of specific surface area, pore volume, and defect ratio on the hydrogen storage efficiency of the three g-ACNS samples are compared in Tables 2 and 3. It can be observed that the dominant factors affecting the hydrogen storage capacity are the specific surface area and defect ratio. CNS22, due to its lower specific surface area, exhibits a lower hydrogen storage capacity compared to the other two samples. Conversely, although CNS24 has a slightly lower specific surface area than CNS44, its higher defect ratio compensates for this, resulting in a higher hydrogen storage capacity than CNS44. Therefore, the optimal ratio for the mixing of CNSs with KOH in the secondary activation process is 1:2 for the first activation and 1:4 for the second activation, which leads to the best hydrogen storage efficiency for the g-ACNSs.
| Sample | H2ads capacity (wt%) | Average pore size | Specific surface area | Total pore volume | Micro-pore volume | Micro to meso pore volume ratio |
|---|---|---|---|---|---|---|
| 0–100 bar | nm | m2 g−1 | cm3 g−1 | cm3 g−1 | % | |
| CNS22 | 4.61 | 3.44 | 2137.0 | 1.80 | 0.4257 | 32.4 |
| CNS24 | 5.04 | 3.55 | 2291.0 | 2.03 | 0.3977 | 32.4 |
| CNS44 | 4.91 | 3.51 | 2584.0 | 2.09 | 0.8313 | 79.6 |
| XC-72 | 1.39 | 13.03 | 215.2 | 0.41 | 0.0275 | 37.1 |
| Graphene | 0.34 | 19.30 | 20.5 | 0.10 | 0 | 0 |
In addition to the hydrogen storage capacity, the reversibility and repeatability of the CNSs are also crucial. Fig. 4(c) shows the results of 10 cycles of hydrogen adsorption and desorption tests performed on CNS24 samples at 77 K and between 0–100 bar. The experimental data indicate that there is only about a 5% drop in hydrogen adsorption/desorption for CNS24, demonstrating its decent capability for cycling hydrogen storage/release.
Computational analysis of hydrogen adsorption and saturation
We first compare the hydrogen adsorption behaviors on both graphene and amorphous carbon models which mimic experimental g-ACNSs. Fig. 5(a) shows that hydrogen adsorption on the graphene surface with only 3-fold coordinated carbon atoms is mainly dominated by physisorption with adsorption energy per hydrogen atom (Eads/H) of ∼−0.6 eV; its chemisorption is thermodynamically unfavorable with Eads/H of larger than 0.9 eV. In contrast, for amorphous carbon models, Fig. 5(b) shows that spontaneous dissociation of the H2 takes place in the vicinity of 2-fold coordinated carbon atoms; with chemisorption energies ranging from −1.38 to −0.55 eV, and with increasing H–H distances of 1.792 and 1.737 Å. While the H2 molecule is physisorbed on the 3-fold coordinated carbon atoms with the Eads/H of −0.08 and −0.07 eV and with the H–H distances of 0.752 and 0.754 Å, their adsorption energy magnitudes and configurations are similar to H2 adsorption on the 3-fold coordinated carbon atom of the graphene surface.41 These results show that the H2 interaction with amorphous carbon can lead to both chemisorption as well as physisorption on the surface, which depends on the nature of the carbon adsorption site. We anticipate that high hydrogen adsorption capacity can be achieved by saturating the amorphous carbon models with multiple layer hydrogen molecules for mimicking the high pressure H2 adsorption conditions.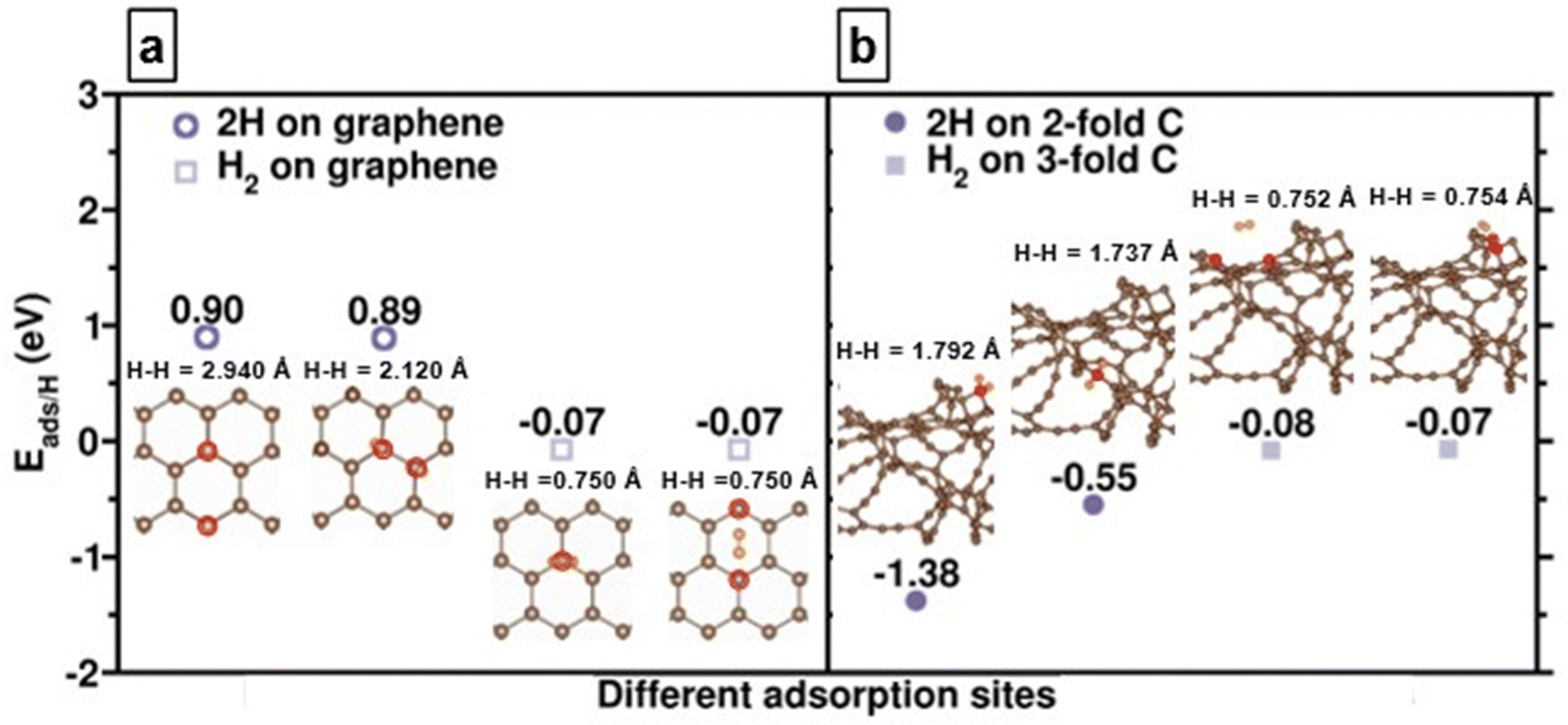 | ||
| Fig. 5 Adsorption energies per hydrogen atom (Eads/H) and the optimized structures of H2 dissociative and molecular adsorption on (a) graphene and (b) amorphous carbon (a-C) surfaces. (brown, C; orange, H) Red circles indicate the nearest C atoms around H atoms. | ||
We further examine the hydrogen saturation in our a-C model by successively adsorbing hydrogen molecules on various carbon sites. In real conditions, the 2-fold coordinated carbon atoms will be saturated by some ligands during the synthesis procedure. Thus, we first saturated all 2-fold coordinated carbon sites and do not count the hydrogen atom numbers as a contribution of hydrogen storage amount since we assume that the hydrogen desorption might be difficult in these strong atomic hydrogen chemisorption sites. The other additional 64 hydrogens molecularly and dissociatively adsorbed on the a-C surface with adsorption energies from −0.69 to −0.60 eV, reaching an unflucturated hydrogen adsorption magnitude (Fig. 6), which implies that the status of hydrogen saturation and the hydrogen adsorption amount corresponds to the theoretical hydrogen gravimetric capacity of 5.10 wt%. These DFT calculation outcomes demonstrate that defects, including various hybridizations of carbon atoms, might assist in the hydrogen adsorption amounts, leading to higher hydrogen storage capacity.
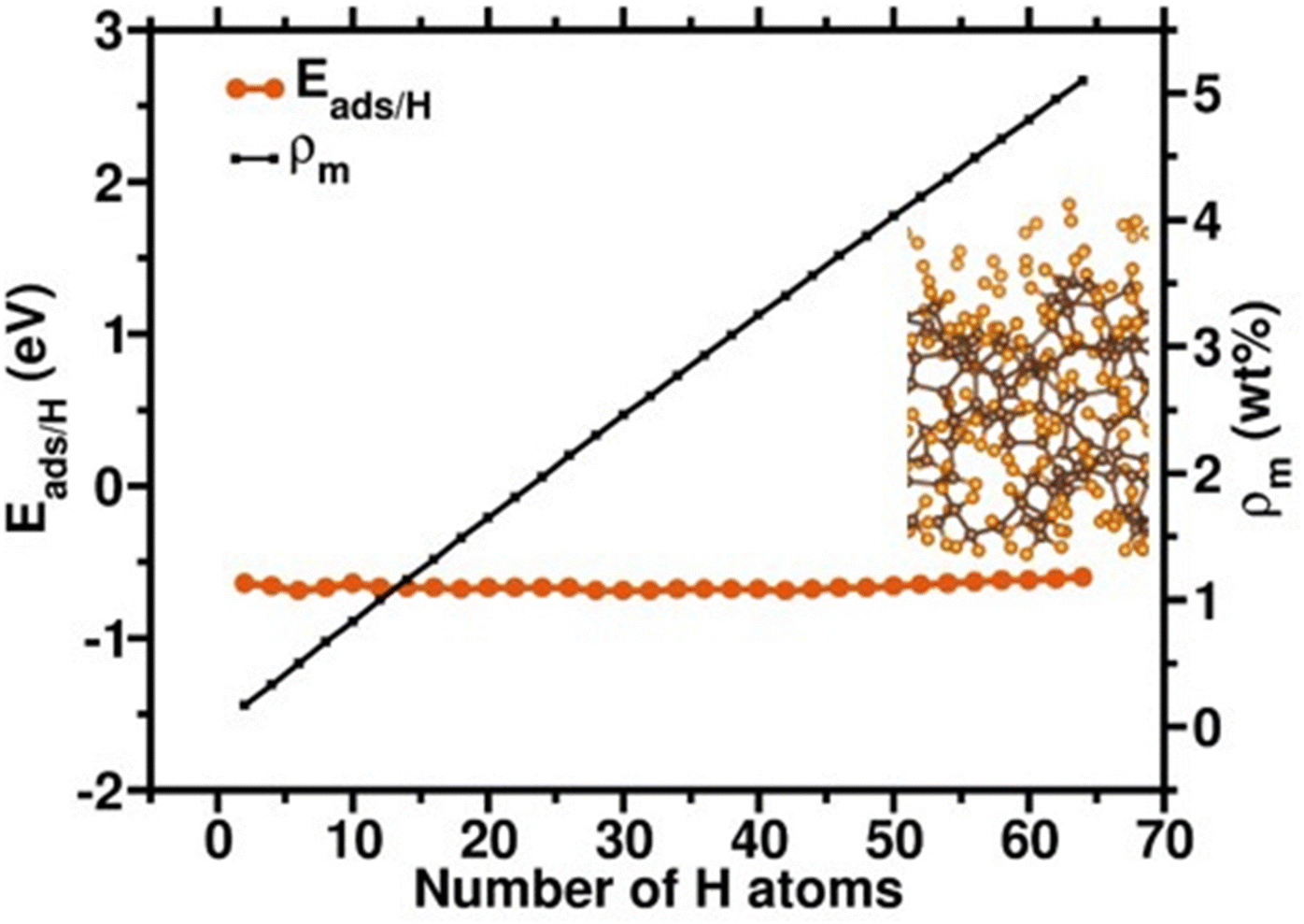 | ||
| Fig. 6 Adsorption energies per hydrogen atom (Eads/H) of an additional 64 hydrogens molecularly and dissociatively adsorbed on the a-C surfaces to the number of hydrogen atoms, after saturating 2-fold coordinated C sites. The insert is the corresponding optimized structure. ρm is the H gravimetric capacity with the unit of weight percent (wt%). | ||
Based on the results of the previous material analysis, it can be observed that g-ACNS (graphene-based activated carbon nanospheres) has a predominantly spherical structure, in contrast to the lamellar structure of general carbon XC-72 and graphene. TEM images also reveal that the atomic structure arrangement of g-ACNS is relatively non-uniform, indicating the presence of defects or void structures on the carbon spheres, which is further supported by Raman analysis showing a higher defect ratio in g-ACNS compared to XC-72 and graphene. BET analysis results show that the specific surface area of g-ACNS is 10 times higher than that of XC-72 and 100 times higher than that of graphene, with significantly higher total pore volume and micropore volume as well. This indicates that g-ACNS possesses the desirable characteristics of a good hydrogen storage material, including high specific surface area, high pore volume, and a significant number of defects.
Furthermore, a comparison of g-ACNS samples formed through three different processes reveals differences in pore size distribution, total pore volume, specific surface area, and defects, which in turn affect the hydrogen storage capacity. g-ACNS22, formed with a low KOH ratio, has fewer micropores and defects, resulting in a lower hydrogen storage capacity. Conversely, g-ACNS44, formed with a higher KOH ratio in both KOH reaming steps, exhibits a high ratio of micropores, but also a large number of large pores (40–80 nm) due to excessive reaming, leading to pore collapse and reduced defect rate, thereby affecting hydrogen storage efficiency. In contrast, g-ACNS24, formed with an intermediate KOH ratio, can generate sufficient micropores without excessive pore expansion, thus maintaining a high proportion of micropores and defects, and consequently exhibiting the best hydrogen storage results among the three materials. These findings are also supported by DFT calculations, indicating that an appropriate micropore ratio and high defect structure are beneficial for improved hydrogen storage and release, resulting in efficient hydrogen storage performance.
Conclusions
An activated carbon with super high specific surface area (up to 2584 m2 g−1) and high porosity has been prepared and investigated for hydrogen adsorption at 77 K and up to 100 bar. CNS24 showed a hydrogen absorptivity of 5.04 wt%, which is the highest absorptivity of activated carbon derived from KOH activation to date. The high uptake for CNS24 was attributed to high specific surface area and defect ratio. In addition, the comparison of the hydrogen storage efficiency between the CNS series samples and commercial carbon materials (XC-72) confirms once again that high surface area and porosity play a key role in the adsorption of hydrogen at a low temperature of 77 K, because hydrogen is more likely to condense into the carbon matrix. A highly porous activated carbon material with an exceptionally high specific surface area of up to 2584 m2 g−1 has been successfully prepared and investigated for hydrogen adsorption at 77 K and pressures up to 100 bar. Among the samples tested, CNS24 exhibited the highest hydrogen adsorption capacity of 5.04 wt%, which is the highest reported absorptivity for activated carbon derived from the KOH activation method. This exceptional performance of CNS24 can be attributed to its high specific surface area and defect ratio, indicating that these factors play a crucial role in hydrogen adsorption. Furthermore, a comparison of the hydrogen storage efficiency between the CNS series samples and commercial carbon materials, such as XC-72, reaffirms the importance of high surface area and porosity in hydrogen adsorption at the low temperature of 77 K. The high surface area and porosity of the activated carbon facilitate the condensation of hydrogen into the carbon matrix, leading to higher hydrogen adsorption capacity. DFT calculations show that the chemisorption hydrogen adsorption enhances in the amorphous model with mixed coordinated carbon atoms, compared to the perfect six-membered graphene surface. The increased hydrogen chemisorption is attributed to the enhanced hydrogen adsorption amounts. This suggests that materials with defects, high surface area, and porosity are favorable for efficient hydrogen storage at low temperatures.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Science and Technology Council, Taiwan (NSTC 111-2221-E-007-087-MY3, NSTC 111-2112-M-007-028-MY3, NSTC 112-2112-M-007-026- and NSTC 111-2113-M-007-025) and National Tsing Hua University (111Q2711E1). Tsan-Yao Chen also appreciates the support from the industrial collaboration project (MA-tek 2023-T-004 and Taiwan Space Agency TASA-S-1120691). The computational resources were supported by TAIWANIA at the National Center for High-Performance Computing (NCHC) of the National Applied Research Laboratories (NARLabs) in Taiwan.Notes and references
- M. Mohan, V. K. Sharma, E. A. Kumar and V. Gayathri, Energy Storage, 2019, 1, e35 CrossRef CAS.
- G. Sethia and A. Sayari, Carbon, 2016, 99, 289–294 CrossRef CAS.
- H. Barthelemy, M. Weber and F. Barbier, Int. J. Hydrogen Energy, 2017, 42, 7254–7262 CrossRef CAS.
- D. J. Durbin and C. Malardier-Jugroot, Int. J. Hydrogen Energy, 2013, 38, 14595–14617 CrossRef CAS.
- A. Karkamkar, C. Aardahl and T. Autrey, Mater. Matters, 2007, 2, 6–9 CAS.
- J. Graetz, J. Reilly, G. Sandrock, J. Johnson, W. M. Zhou and J. Wegrzyn, Aluminum hydride, A1H3, as a hydrogen storage compound, Brookhaven National Lab.(BNL), Upton, NY (United States), 2006 Search PubMed.
- J. J. Reilly and G. D. Sandrock, Sci. Am., 1980, 242, 118–131 CrossRef CAS.
- B. Panella, M. Hirscher and S. Roth, Carbon, 2005, 43, 2209–2214 CrossRef CAS.
- F. H. Stephens, V. Pons and R. T. Baker, Dalton Trans., 2007, 2613–2626 RSC.
- B. Schmitz, U. Müller, N. Trukhan, M. Schubert, G. Férey and M. Hirscher, ChemPhysChem, 2008, 9, 2181–2184 CrossRef CAS PubMed.
- H.-L. Jiang and Q. Xu, Catal. Today, 2011, 170, 56–63 CrossRef CAS.
- C.-H. Liu, Y.-C. Wu, C.-C. Chou, B.-H. Chen, C.-L. Hsueh, J.-R. Ku and F. Tsau, Int. J. Hydrogen Energy, 2012, 37, 2950–2959 CrossRef CAS.
- M. Valcárcel, S. Cárdenas, B. M. Simonet, Y. Moliner-Martínez and R. Lucena, TrAC, Trends Anal. Chem., 2008, 27, 34–43 CrossRef.
- P. Mohanty, L. D. Kull and K. Landskron, Nat. Commun., 2011, 2, 401 CrossRef PubMed.
- Y. Gong, Z. Wei, J. Wang, P. Zhang, H. Li and Y. Wang, Sci. Rep., 2014, 4, 6349 CrossRef CAS PubMed.
- A. Ferrari, B. Kleinsorge, N. Morrison, A. Hart, V. Stolojan and J. Robertson, J. Appl. Phys., 1999, 85, 7191–7197 CrossRef CAS.
- J. L. Figueiredo, M. Pereira, M. Freitas and J. Orfao, Carbon, 1999, 37, 1379–1389 CrossRef CAS.
- R. Andrews, D. Jacques, D. Qian and E. Dickey, Carbon, 2001, 39, 1681–1687 CrossRef CAS.
- M. Jordá-Beneyto, F. Suárez-García, D. Lozano-Castello, D. Cazorla-Amorós and A. Linares-Solano, Carbon, 2007, 45, 293–303 CrossRef.
- Q. Wang and J. K. Johnson, J. Chem. Phys., 1999, 110, 577–586 CrossRef CAS.
- H. Wang, Q. Gao and J. Hu, J. Am. Chem. Soc., 2009, 131, 7016–7022 CrossRef CAS PubMed.
- K.-Y. Zhang, Y.-Q. Fu, H.-H. Liu, J.-L. Yang, M.-Y. Su, Y. Wang and X.-L. Wu, Phys. Scr., 2023, 98, 125977 CrossRef.
- C. Sun, Z. Han, X. Wang, B. Liu, Q. Li, H. Li, J. Xu, J.-M. Cao and X.-L. Wu, Adv. Funct. Mater., 2023, 33, 2305606 CrossRef CAS.
- J.-M. Cao, K.-Y. Zhang, J.-L. Yang, Z.-Y. Gu and X.-L. Wu, Chin. Chem. Lett., 2024, 35, 109304 CrossRef CAS.
- F. Caturla, M. Molina-Sabio and F. Rodriguez-Reinoso, Carbon, 1991, 29, 999–1007 CrossRef CAS.
- H. Teng and L.-Y. Hsu, Ind. Eng. Chem. Res., 1999, 38, 2947–2953 CrossRef CAS.
- H. Teng, J.-A. Ho and Y.-F. Hsu, Carbon, 1997, 35, 275–283 CrossRef CAS.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef CAS.
- T. FC and T. SL, Mater. Trans., 2006, 47, 1847–1852 CrossRef.
- P. Hohenberg and W. Kohn, Phys. Rev., 1964, 136, B864 CrossRef.
- J. Hutter and D. Marx, Ab Initio Molecular Dynamics: Basic Theory and Advanced Methods, Cambridge University Press, Cambridge, 2009 Search PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- X. Li, P. Guo, L. Sun, A. Wang and P. Ke, ACS Appl. Mater. Interfaces, 2015, 7, 27878–27884 CrossRef CAS PubMed.
- D. McCulloch, D. McKenzie and C. Goringe, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 2349 CrossRef CAS.
- R. Jana, D. Savio, V. L. Deringer and L. Pastewka, Modell. Simul. Mater. Sci. Eng., 2019, 27, 085009 CrossRef CAS.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 15410 CrossRef PubMed.
- J. Petucci, C. LeBlond, M. Karimi and G. Vidali, J. Chem. Phys., 2013, 139, 044706 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ya00126e |
| This journal is © The Royal Society of Chemistry 2024 |