
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhoto-accelerated oxidation of spiro-OMeTAD for efficient carbon-based perovskite solar cells
S. N.
Vijayaraghavan
a,
Kausar
Khawaja
b,
Jacob
Wall
b,
Wenjun
Xiang
b and
Feng
Yan
 *b
*b
aDepartment of Metallurgical and Materials Engineering, The University of Alabama, Tuscaloosa, Alabama 35487, USA
bSchool for Engineering of Matter, Transport and Energy, Arizona State University, Tempe, AZ 85249, USA. E-mail: fengyan@asu.edu
First published on 17th April 2024
Abstract
Metal halide perovskite thin film solar cell (PSC) technology demonstrates promising power conversion efficiency and paves the way to commercialization. So far, the best device performance originates from the n-i-p structure with FTO/SnO2/perovskite/Spiro-OMeTAD/Au device architecture. Traditionally, to achieve high efficiency, several hours of oxidization of a Li-doped Spiro-OMeTAD hole transport layer (HTL) is necessary, which hinders the application of Spiro-OMeTAD for rapid large-scale manufacturing. In particular, the oxidization process significantly relies on ambient conditions, including oxygen and moisture levels. In this work, we report a fast technique to accomplish rapid oxidation in several mins of Spiro-OMeTAD HTL in perovskite solar cells by illuminating in visible light with one Sunlight intensity (100 mW cm−2). Compared to the traditional approach, our photo-accelerated oxidization of Spiro-OMeTAD approach can deliver a high-power conversion efficiency. We employ this photo-accelerated oxidization for the planar carbon-based perovskite solar cell and achieve a champion device efficiency of 17.47%. This strategy overcomes the barriers for a simple and rapid means toward mass manufacturing and industrial applications by utilizing Spiro-OMeTAD as the HTL for the perovskite solar module.
1. Introduction
Metal halide perovskite solar cells (PSCs) have been gaining increasing attention as promising candidates for next-generation photovoltaic (PV) devices.1,2 The PSCs have rapidly progressed, and have now achieved a remarkable power conversion efficiency (PCE) of >26%.3 A traditional PSC contains the active perovskite layer embedded between an electron transport layer (ETL) and a hole transport layer (HTL) in either n-i-p or p-i-n architecture.4,5 Currently, the most efficient PSC is based on the 2,2′,7,7′-tetrakis(N,N-di-p-methoxyphenylamine)-9,9′-spirobifluorene (Spiro-OMeTAD) HTL in an n-i-p structure.3,6,7 Spiro-OMeTAD is one of the most popular HTLs for PSCs due to its amorphous, non-volatile, and non-reactive nature with perovskites, and it has a perfect bandgap matching with perovskites.8–10 However, it suffers from a lack of conductivity, which is a required characteristic that a charge transport layer should possess.11 To address this issue, Spiro-OMeTAD is doped with additives such as bis(trifluoromethane)-sulfonimide lithium salt (LiTFSI), which is a p-type dopant that enhances the conductivity and hole mobility of Spiro-OMeTAD.12–14 The doping process begins when the pristine Spiro-OMeTAD:LiTFSI film is exposed to oxygen.15 The oxygen oxidizes the Spiro-OMeTAD film while LiTFSI acts as a mediator in the reaction between O2 and Spiro-OMeTAD, producing a TFSI-stabilized radical cation along with lithium oxides as proposed by Abate et al. via a two-step mechanism.16,17| Spiro − OMeTAD + O2 ⇔ Spiro − OMeTAD˙+ O2˙− | (1) |
| Spiro − OMeTAD˙+ O2˙− + LiTFSI → Spiro − OMeTAD˙+ TFSI− + LixOy | (2) |
So far, several modified methods to shorten the oxidization of Spiro-OMeTAD have been proposed. The introduction of other dicationic salts in Spiro-OMeTAD (e.g., Co-TFSI, Zn-TFSI, FeCl3),20–22 non-contact electrochemical route,23 pre-fabrication CO2 doping,16 and O3 exposure under UV light24 have been studied to shorten the oxidation/doping period. However, these alternative methods still require either time-intensive oxidation of the doped Spiro-OMeTAD, a complicated setup, or the UV treatment may even degrade the organic HTLs, which could lead to more costs for manufacturing and long-term stability issues of the HTLs. It has been demonstrated that a short period of light soaking the perovskite cells can significantly improve the device during measurement.25 This performance improvement was attributed to the dopants, e.g., Li+ ion migration and Spiro-OMeTAD oxidation, which provides evidence that photons (e.g., partial UV spectrum) in sunlight can directly oxidize Spiro-OMeTAD.26 In response to the elevated expense of noble electrodes such as Au and Ag for perovskite solar cells, economical carbon materials have been utilized.5 The compatibility of carbon electrodes with perovskite solar cells is due to their optimal work function (∼5 eV) for band alignment with Spiro-OMeTAD and perovskite, as well as their potential for stability, scalability and flexibility.4
In this study, we systematically investigate the sunlight photo-accelerated oxidization (PAO) of the Li-doped Spiro-OMeTAD by using a solar simulator for carbon based perovskite solar cells. It is observed that sunlight illumination could significantly accelerate the Spiro-OMeTAD oxidization to 5 mins, which is on par with our traditional time-intensive oxidization. We utilize this newly developed PAO-treated Spiro-OMeTAD and accomplished a champion device performance of 17.49% on a carbon-based perovskite, which is similar to the time-intensive, traditional oxidization approach. This photo-accelerated oxidization approach can be directly integrated into the perovskite manufacturing line by shortening the Spiro-OMeTAD HTL oxidization process.
2. Materials and methods
2.1. Materials
Perovskite precursor compounds lead iodide (PbI2) (Alfa Aesar, 99.999%), lead bromide (PbBr2) (Alfa Aesar, 99.98%), formamidinium iodide (FAI) (GreatCell Solar), methylammonium bromide (MABr) (GreatCell Solar), cesium iodide (CsI) (BeanTown Chemical, 99.9%), materials for charge transporting layers (SnCl2·2H2O (Acros Organics, 97%), thiourea (Alfa Aesar, 99%), Spiro-OMeTAD (Sigma-Aldrich, 99%), LiTFSI (Sigma-Aldrich, 99.95% trace metals basis)), and solvents (N,N-dimethylformamide (DMF) (Sigma-Aldrich, anhydrous, 99.8%), dimethyl sulfoxide (DMSO) (Alfa Aesar, >99%), diethyl ether (DEE) (Sigma-Aldrich, anhydrous, >99.7%), chlorobenzene (CB) (Sigma-Aldrich, anhydrous, 99.8%), acetonitrile (ACN) (Sigma-Aldrich) (anhydrous, 99.8%), 4-tert-butylpyridine (tBP) (Sigma-Aldrich, 98%), ethanol (EtOH) (Sigma-Aldrich)) were purchased from the stated chemical vendors and were used as received without further purification.2.2. Preparation of Spiro-OMeTAD and oxidation
The Spiro-OMeTAD solution was prepared by dissolving 72.6 mg of Spiro-OMeTAD in 1 mL of CB, 28.8 μL of tBP, and 17.5 μL of LiTFSI solution (520 mg of LiTFSI in 1 mL of ACN) were added into the Spiro-OMeTAD solution. The solution was then filtered with PTFE hydrophobic filters (pore size ∼0.22 μm) and used for HTL. For traditional oxidization (TO) of Spiro-OMeTAD films, the films were kept in an oxygen-filled dark chamber with R.H. <5% overnight. For photo-accelerated (PA)-Spiro-OMeTAD films, the solution was kept for light-soaking in sealed glass vials that were exposed to various gas environments in a glovebox before being sealed, including vacuum, N2, dry air, and O2.2.3. Fabrication of planar carbon-based perovskite solar cells
Indium tin oxide (ITO)-coated glass substrates were cleaned sequentially via sonication using detergent, deionized water (DIW), acetone, and isopropanol (IPA). SnO2 ETL was prepared as explained in our previous work.5 In short, a 0.15 M SnCl2·2H2O solution in DIW with thiourea was prepared and stirred overnight until the solution turned yellow. The solution was then filtered with a 0.45 μm PTFE filter before use. The SnO2 ETL was then spin-coated at 3000 rpm for 30 s and then annealed at 180 °C for 1 hour. The ETL-covered ITOs were then transferred into a nitrogen-filled glovebox after 10 minutes of UV-Ozone treatment. A Cs0.04FA0.81MA0.15PbI2.55Br0.45 precursor solution (1.4 M) was prepared in a 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v:v) solvent mixture of DMF:DMSO. A two-step spin-coating procedure (1000 rpm 10 s + 6000 rpm 30 s) was applied for the formation of the perovskite layer. 600 μL of DEE was then dropped 10 s before the end of the spinning procedure. The films were then dried at 100 °C for 1 hour. After cooling the substrates to room temperature, Spiro-OMeTAD HTL was then spin-coated on the perovskite layer at 3000 rpm for 30 s within a dry N2-filled glovebox. The traditional oxidization of Spiro-OMeTAD films was performed for the Spiro-OMeTAD solution without PAO treatment, while the PAO-treated Spiro-OMeTAD solution can be directly applied on the perovskite film without further oxidization. Finally, 75 μm of carbon was then hot-pressed on top of Spiro-OMeTAD to form top contacts with an area of 0.08 cm2.27
:1 (v:v) solvent mixture of DMF:DMSO. A two-step spin-coating procedure (1000 rpm 10 s + 6000 rpm 30 s) was applied for the formation of the perovskite layer. 600 μL of DEE was then dropped 10 s before the end of the spinning procedure. The films were then dried at 100 °C for 1 hour. After cooling the substrates to room temperature, Spiro-OMeTAD HTL was then spin-coated on the perovskite layer at 3000 rpm for 30 s within a dry N2-filled glovebox. The traditional oxidization of Spiro-OMeTAD films was performed for the Spiro-OMeTAD solution without PAO treatment, while the PAO-treated Spiro-OMeTAD solution can be directly applied on the perovskite film without further oxidization. Finally, 75 μm of carbon was then hot-pressed on top of Spiro-OMeTAD to form top contacts with an area of 0.08 cm2.27
2.4. Materials and device characterizations
Photocurrent density and voltage (J–V) were measured with a solar simulator (Newport, Oriel Class AAA 94063A, 1000-Watt Xenon light source) with a Keithley 2420 source meter under simulated AM1.5G (100 mW cm−2, calibrated with a standard Si solar cell) solar irradiation. The masked active area was 0.08 cm2, and the current–voltage scan rate was 100 mV s−1. The external quantum efficiency (EQE) was obtained using an EnliTech QE measurement system. The capacitance–voltage (C–V) measurement was performed in the dark at room temperature using a Keithley 4200 semiconductor parameter analyzer (Tektronix 4200A-SCS, USA). The EIS measurement was conducted by a Solartron Analytical 1260 impedance analyzer at a bias potential of 0.5 V in the dark with the frequency ranging from 1 Hz to 1 MHz. The hall measurement system was used to characterize the hole mobility using an HMS-5000 Hall Effect Measurement System.3. Results and discussion
Spiro-OMeTAD has a large molecular weight as shown in Fig. 1a, and LiTFSI is a reagent that is consumed during the traditional, time-intensive oxidization reaction process, with the existence of oxygen, as shown in eqn (2),17 where the molecular oxygens bond with the Li and generate a TFSI-stabilized Spiro-OMeTAD. In the case of the photo-accelerated oxidization process, when the pristine Li-TFSI mixed Spiro-OMeTAD solution is oxidized under the AM1.5G spectrum, the electrons are excited to the lowest unoccupied molecular orbital (LUMO) of Spiro-OMeTAD, where they are then captured by O2. These electrons are then responsible for reducing the molecular oxygen to form a negatively charged O2 (or radical anion), which can then react with the lithium ions, producing LixOy. While this happens, Spiro-OMeTAD is oxidized, forming a radical cation that is then stabilized by TFSI−. The formation of oxidized Spiro-OMeTAD radical cations in an O2-treated Spiro-OMeTAD solution illuminated with a solar simulator with 1 sun light intensity (100 mW cm−2) was confirmed as shown in Fig. 1(c). The light-yellow Spiro-OMeTAD solution gradually changed into a reddish-brown color with increasing illumination time from 0 to 10 min, indicating that Spiro-OMeTAD is significantly affected by the PAO process with sunlight illumination.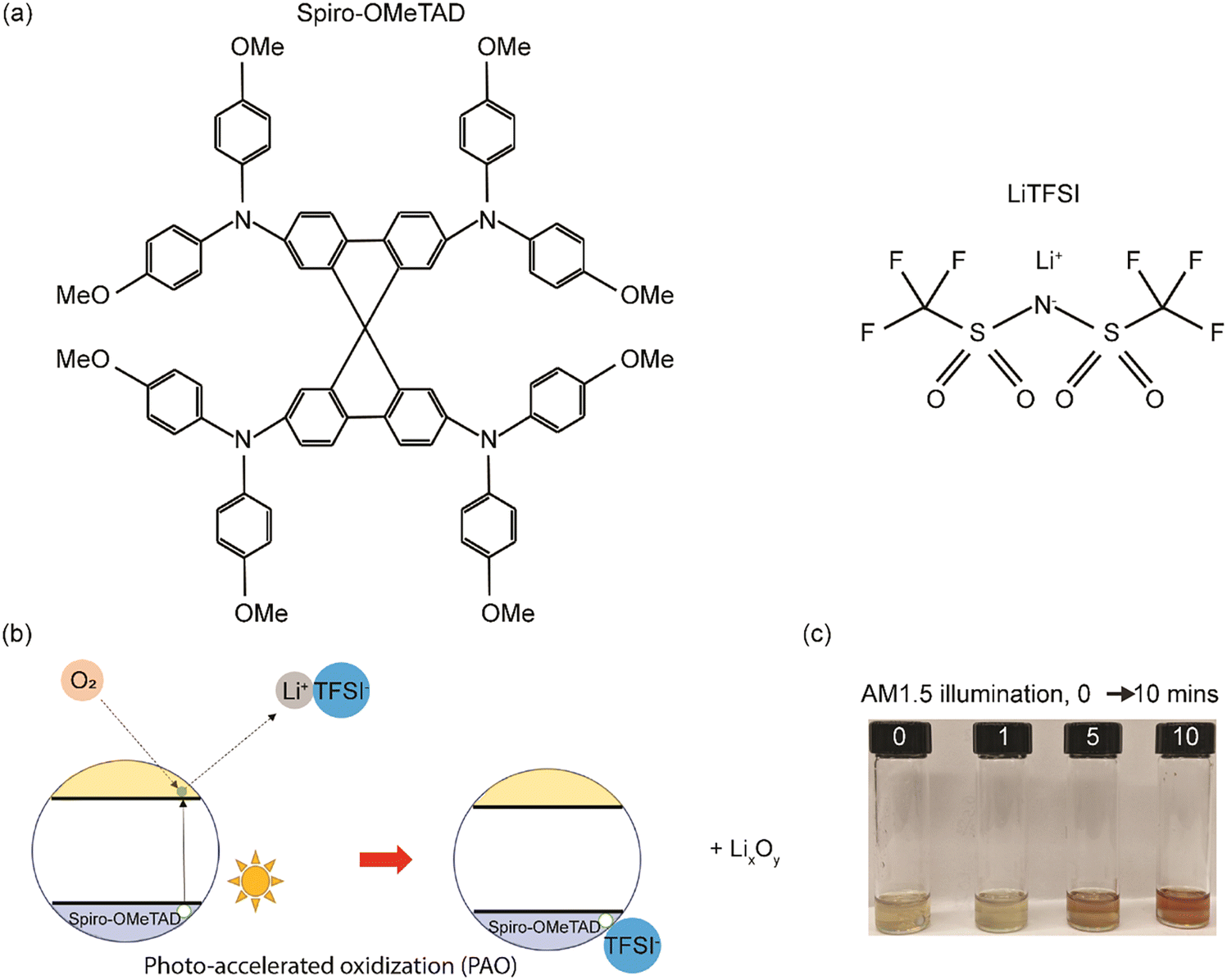 | ||
| Fig. 1 (a) Molecular structures of Spiro-OMeTAD and LiTFSI, (b) schematic of the proposed PAO mechanism of Spiro-OMeTAD, and (c) optical pictures of the Spiro-OMeTAD solution illuminated under 1 sun sunlight after 0, 1, 5, and 10 minutes of PAO treatment in O2. | ||
In addition, ultraviolet-visible (UV-Vis) spectroscopy was utilized to verify the PAO of the Spiro-OMeTAD solution. All the PAO-treated specimens were exposed to AM1.5 illumination for 5 minutes, while the TO sample was kept in an oxygen-filled dark chamber overnight. The oxidized Spiro-OMeTAD has a sharp peak at ∼400 nm and another broader peak around 650 nm.28Fig. 2(a) shows the influence of PAO on Spiro-OMeTAD oxidation at 1 sunlight illumination. The increasing intensity of the absorption peak from 600–800 nm for Spiro-OMeTAD solution treated with O2 (in dry air, O2, and TO) implies the generation of oxidized Spiro-OMeTAD. However, samples that were not exposed to oxygen (in vacuum and N2) showed only very weak absorption peaks, suggesting that our process does not have an unintentional oxidization process and the oxidization of spiro-OMeTAD was dominated by the ambient control under illumination. The increase in the intensity of the peaks under different oxidation conditions implies higher oxidation degrees of the HTL. Furthermore, FTIR was also used to confirm the oxidation of the Spiro-OMeTAD films. As seen from Fig. 2(b), TO and PAO films in dry air and O2 show a series of conspicuous peaks at 1248 cm−1, 1466 cm−1, and 1498 cm−1 that can be assigned to methoxy phenyl of the Spiro-OMeTAD molecule.29 In addition, the peak at ∼1750 cm−1 shows the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch in the oxidized Spiro-OMeTAD in air and oxygen. Note that films PAO in vacuum and N2 do not show these peaks indicating a lack of oxidized Spiro-OMeTAD.
O stretch in the oxidized Spiro-OMeTAD in air and oxygen. Note that films PAO in vacuum and N2 do not show these peaks indicating a lack of oxidized Spiro-OMeTAD.
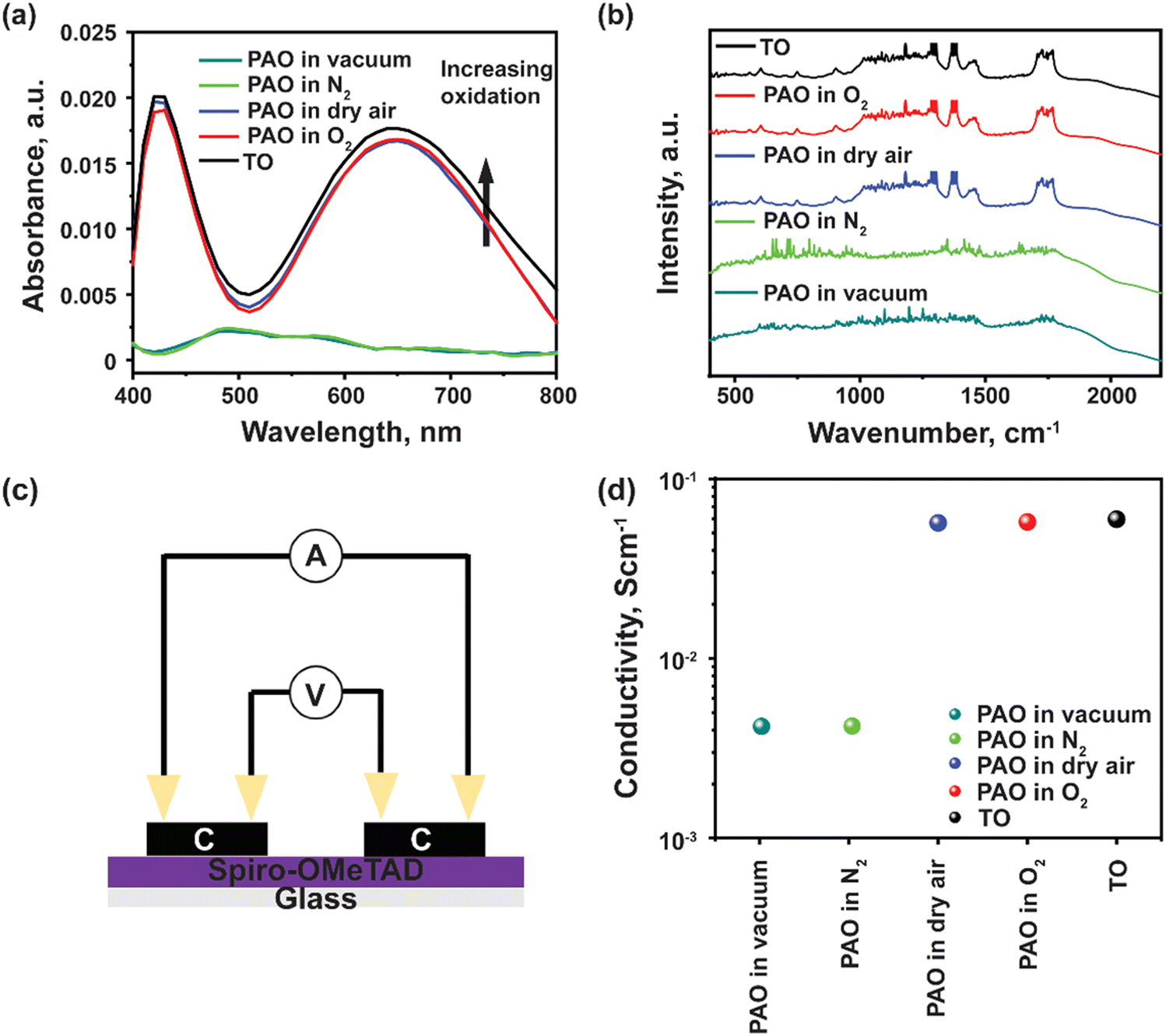 | ||
| Fig. 2 (a) UV-Vis absorption spectra, and (b) FTIR of Spiro-OMeTAD films PAO in different ambient conditions, (c) testing device structure for conductivity measurements, and (d) effective conductivity of Spiro-OMeTAD PAO in different media measured by a four-point probe. | ||
Four-probe conductivity measurements of HTL films deposited in a nitrogen atmosphere under dark conditions, and later oxidized under different conditions were acquired, as shown in Fig. 2(c). Films that were PAO in dry air or pure O2, showed comparable conductivity as that with TO film (Fig. 2(d)). A significant drop in conductivity was observed for films PAO in vacuum and N2. The increased conductivity of PAO films in dry air and O2 can be mainly attributed to increased hole mobility and charge carrier concentration. The carrier mobilities are 5.15 × 10−5, 4.25 × 10−2 and 1.75 × 10−1 cm2 V−1 s−1, respectively for the as-deposited, PAO and TO Spiro-OMeTAD, respectively. The results of the conductivity test are in excellent agreement with the UV-Vis characterization; Spiro-OMeTAD films were oxidized under AM1.5 illumination in the presence of dry air or pure O2.
Next, the devices with ITO/SnO2/Perovskite/Spiro-OMeTAD/C were fabricated, incorporating PAO Spiro-OMeTAD in different conditions (PAO in a vacuum, N2, dry air, and pure O2 ambient, respectively), and compared to devices using TO-Spiro-OMeTAD that had undergone traditional oxidization in dry air for a longer time in the dark. By varying the duration for PAO of Spiro-OMeTAD in different conditions, the optimized duration was observed to be 5 minutes as shown by the J–V characteristics performed under the AM1.5G simulated sunlight illumination in Fig. 3(a). As shown in Fig. 4(a), as the PAO conditions are changed from vacuum to pure O2, the VOC, JSC, and FF improve drastically. The solar cell containing Spiro-OMeTAD that was PAO in either vacuum or N2 showed poor device performance, exhibiting a PCE of 3.63% (open-circuit voltage VOC = 0.68 V, short-circuit current density JSC = 17.02 mA cm−2, and fill factor FF = 31.28%) and 3.68% (VOC = 0.64 V, JSC = 17.81 mA cm−2, and FF = 32.06%), respectively. The solar cell with TO Spiro-OMeTAD film showed the best device performance of 17.47% (VOC = 1.07 V, JSC = 21.48 mA cm−2, and FF = 76.02%). By contrast, the solar cells containing PAO Spiro-OMeTAD in dry air and pure O2 showed comparable device performance of 17.04% (VOC = 1.09 V, JSC = 20.83 mA cm−2, and FF = 74.89%) and 17.17% (VOC = 1.07 V, JSC = 21.01 mA cm−2, and FF = 76.35%), respectively. For devices with TO Spiro-OMeTAD, the films are exposed to air for several hours, or sometimes even overnight depending on the ambient conditions.30 For devices with PAO Spiro-OMeTAD films in dry air and pure O2, the devices were found to be comparable in terms of performance with that of TO film-based devices, which can be mainly attributed to the increasing amount of oxidized Spiro-OMeTAD.31,32 This results in the improved conductivity and hole mobility of Spiro-OMeTAD films as shown in Fig. 2(d). Thus, by replacing the TO technique with PAO of films in dry air or pure O2, the oxidation duration can be shortened from several hours to a few minutes, which is a huge boost towards the commercialization of this technology. Note that our device performance is slightly lower than the recently reported high PCE of perovskite solar cells. This is mainly because of two reasons: (1) the precursor, Cs0.04FA0.81MA0.15PbI2.55Br0.45, has a bandgap of around 1.6 eV, which delivers a short circuit current of approximately 20 mA cm−2. In comparison, the more popular perovskite precursor composition at FAPbI3 has a bandgap of around 1.5 eV, and the Jsc can be 23–25 mA cm−2. (2) The carbon electrode has a high resistivity of around 50 Ω □−1, contributing to the low fill factor of approximately 76%. In contrast, the Au electrode with 0.5 Ω □−1 possesses a high fill factor of around 80%.33
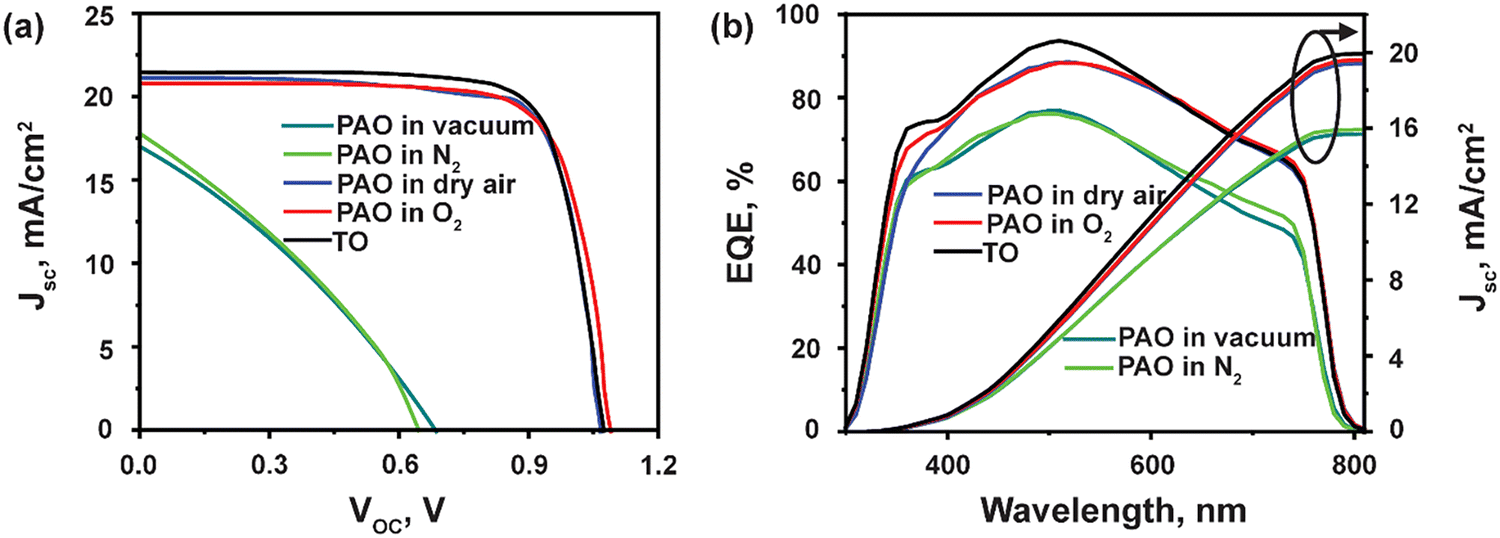 | ||
| Fig. 3 (a) J–V characteristics and, (b) EQE spectra of PSCs using TO and PAO Spiro-OMeTAD in different conditions. | ||
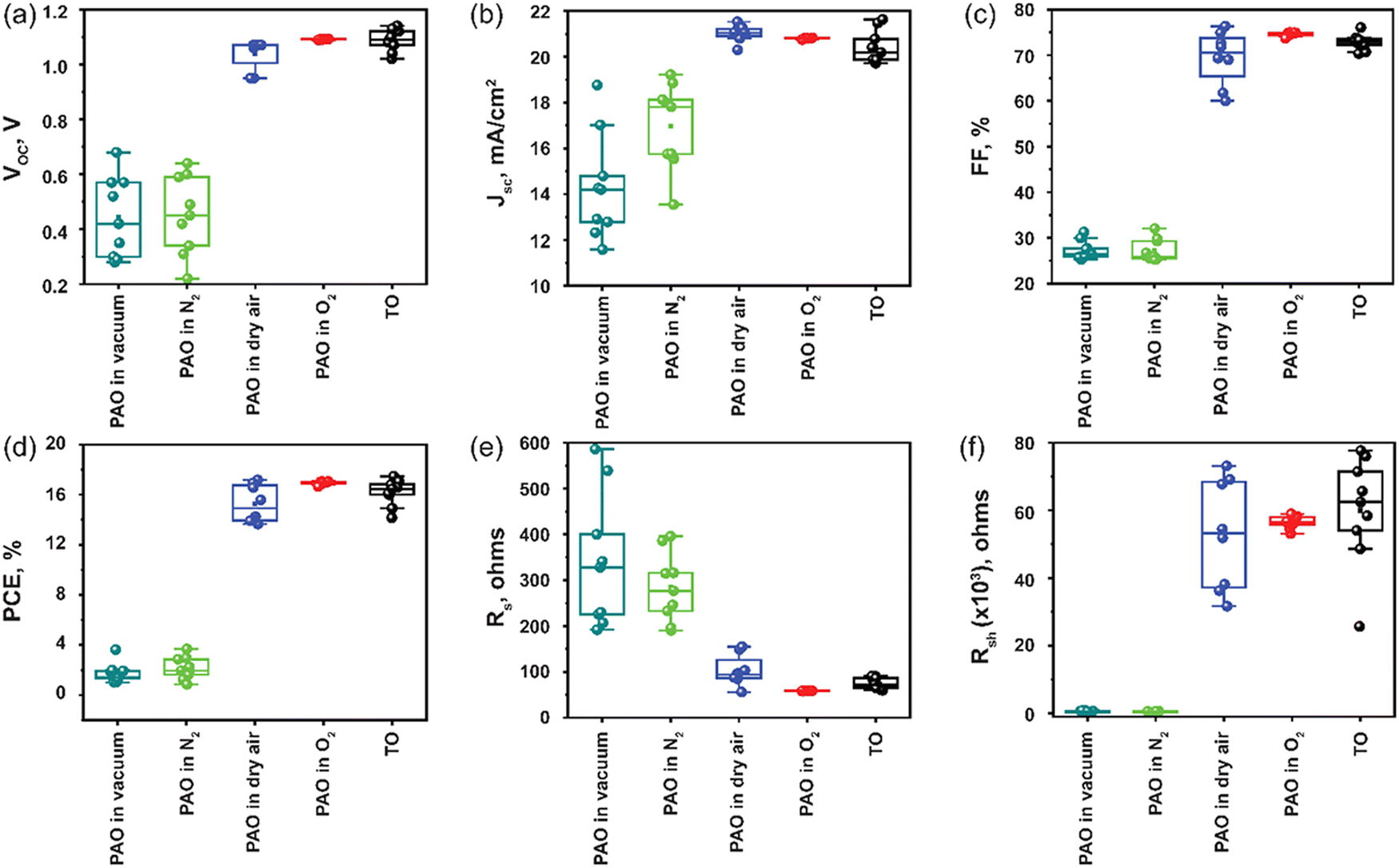 | ||
| Fig. 4 Statistical distribution diagram of the photovoltaic parameters of the PSCs prepared with TO and PAO Spiro-OMeTAD in different conditions. (a) Voc, (b) Jsc, (c) fill factor, FF, (d) PCE, (e) Rs, and (f) Rsh. | ||
The EQE was measured to confirm the accuracy of the J–V measurements, as shown in Fig. 3(b). The integrated current densities are 15.69 mA cm−2, 15.93 mA cm−2, 19.41 mA cm−2, 19.6 mA cm−2, and 19.93 mA cm−2 for PAO films in vacuum, N2, dry air, O2 and TO cells, respectively. The JSC values obtained from the J–V characteristic were well matched (within 5%) with those obtained by integration of the spectral response. Nevertheless, the EQEs for devices with films PAO in vacuum and N2 exhibit lower responses due to the poor conductivity and hole mobility in these films. On the other hand, devices with films that were PAO in dry air, pure O2, and TO showed EQE > 80% from 420–620 nm due to the increased oxidation of Spiro-OMeTAD films. The EQEs for all the devices exhibit lower responses at longer wavelengths mainly due to the carbon contact. Unlike a metal electrode, the carbon electrode absorbs the remanent long-wavelength transmission light directly without reflection for second absorption.34–37
The statistical optoelectronic parameters of the devices with different modes of oxidation for Spiro-OMeTAD are demonstrated in Fig. 4, illustrating the excellent reproducibility of devices with the HTL PAO in dry air and pure O2. Compared to other modes of oxidation, PAO in pure O2 delivers devices with high reproducibility that rivals that of devices with TO films. The standard deviation (SD) for devices with PAO films in O2 was observed to be 0.13, while for devices with TO films it was 0.99. This is a clear indication that the oxidation degree of films via TO can fluctuate in a wider range depending on the ambient conditions, and this can affect the repeatability of the devices. Over-oxidation of films can result in damage to the structure of the HTL, and hence, it can adversely affect the device's performance. On the other hand, devices with PAO films in pure O2 had a very narrow data spreading, and this degree of repeatability is highly desirable when it comes to the commercialization of the technology (Table 1).
| Samples | V oc (V) | J sc (mA cm2) | FF (%) | PCE (%) |
|---|---|---|---|---|
| PAO in vacuum | 0.68 | 17.02 | 31.28 | 3.63 ± 0.10 |
| (0.42) | (14.51) | (26.52) | (2.12) | |
| PAO in N2 | 0.64 | 17.81 | 32.06 | 3.68 ± 0.12 |
| (0.46) | (17.72) | (25.6) | (2.45) | |
| PAO in dry air | 1.09 | 20.83 | 74.89 | 17.04 ± 0.15 |
| (1.05) | (20.53) | (71.5) | (15.54) | |
| PAO in O2 | 1.07 | 21.01 | 76.35 | 17.17 ± 0.05 |
| (1.06) | (20.60) | (75.32) | (16.12) | |
| Control | 1.07 | 21.48 | 76.02 | 17.47 ± 0.17 |
| (1.04) | (20.05) | (75.41) | (16.34) |
To understand the charge recombination mechanism, we carried out the JSC and VOCversus light intensity measurements. The power-law dependence of JSC upon the light intensity with a slope approaching 1 is observed for devices with PAO films in dry air and O2 and TO films, as shown in Fig. 5(a), implying negligible bimolecular recombination in the devices.38 The ideality factor (n) was obtained from the fitted curves of VOC at various light intensities (I) (Fig. 5(b)) to elucidate the trap-assisted Shockley-Read-Hall (SRH) molecular recombination within the devices, according to the equation VOC = nkT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) ln(I)/q + constant, where T is the absolute temperature, k is the Boltzmann constant, and q is the elementary charge.39 As shown in Fig. 5(b), the ideality factor of devices with PAO films in dry air and pure O2 is similar to that of the films TO, while those of the devices with films PAO in vacuum and N2 are higher. The lower ideality factor implies suppressed SRH recombination and efficient extraction of charge carriers in the devices with PAO films in dry air and O2.40,41
ln(I)/q + constant, where T is the absolute temperature, k is the Boltzmann constant, and q is the elementary charge.39 As shown in Fig. 5(b), the ideality factor of devices with PAO films in dry air and pure O2 is similar to that of the films TO, while those of the devices with films PAO in vacuum and N2 are higher. The lower ideality factor implies suppressed SRH recombination and efficient extraction of charge carriers in the devices with PAO films in dry air and O2.40,41
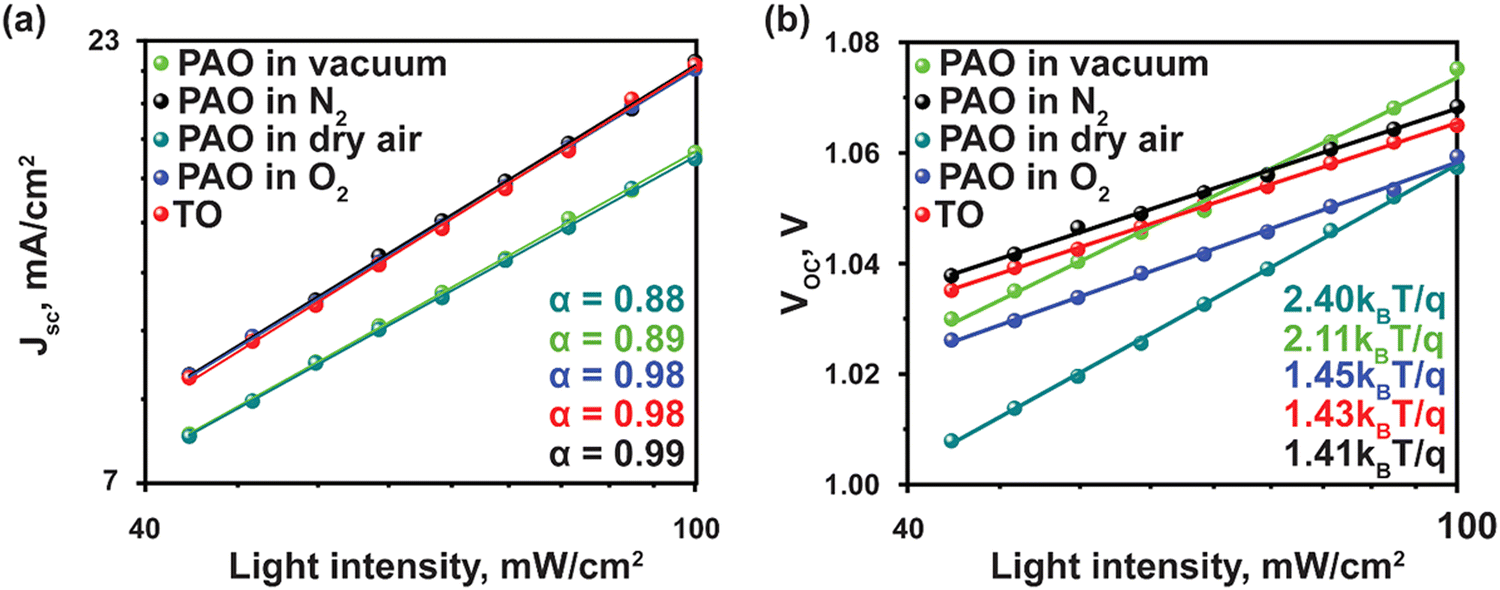 | ||
| Fig. 5 Light-intensity-dependent (a) Jsc, and (b) Voc of PSCs with TO and PAO Spiro-OMeTAD in different conditions. | ||
Fig. 6 shows the electrochemical impedance spectroscopy (EIS) results of the devices with the ITO/SnO2/PVSK/Spiro-OMeTAD/C structure. The most notable feature in Fig. 6 is the significant decrease of the impedance (three orders of magnitude) over the full range of frequency for devices with PAO-treated Spiro-OMeTAD under vacuum and in N2. The impedance of the device could be satisfactorily modeled by an equivalent circuit consisting of series resistance (Rs), a parallel combination of charge recombination resistance (Rrec), and a constant phase angle element (CPE) as shown in the inset of Fig. 6(b).42 In this fitted circuit model, Rs is equivalent to the internal resistance, and Rrec is associated with the charge transport process at the interface.43 The fitting results are shown in Table 2. After simulation, we found that devices with Spiro-OMeTAD PAO in dry air, O2, and TO, had the highest Rrec values, which suggests lower charge recombination at the interface. This is attributed to the higher conductivity and charge extraction capability of the HTL and hence the effective suppression of the carrier recombination at the backside of the device.44 These devices were also found to have lower Rs than that of devices with HTL PAO under vacuum and in N2. After analysis, we think that similar values of Rs and Rsh for PAO films in dry air and O2 and TO films are due to the optimized oxidation conditions. If the oxidation activity of atomic oxygen is too strong, then the structure of the HTL will be destroyed by excessive oxidation resulting in a marked increase in the resistivity of the devices.10 The light soaking for the unencapsulated devices with TO and PAO spiro-OMeTAD was tested under 1 sun light intensity at room temperature. The cells show similar stability behavior within 4 days (96 hours) as shown in Fig. 6c, suggesting that the PAO treated spiro-OMeTAD does not damage the stability of the perovskite cells. However, the overall device stability using the spiro-OMeTAD needs to be further improved.
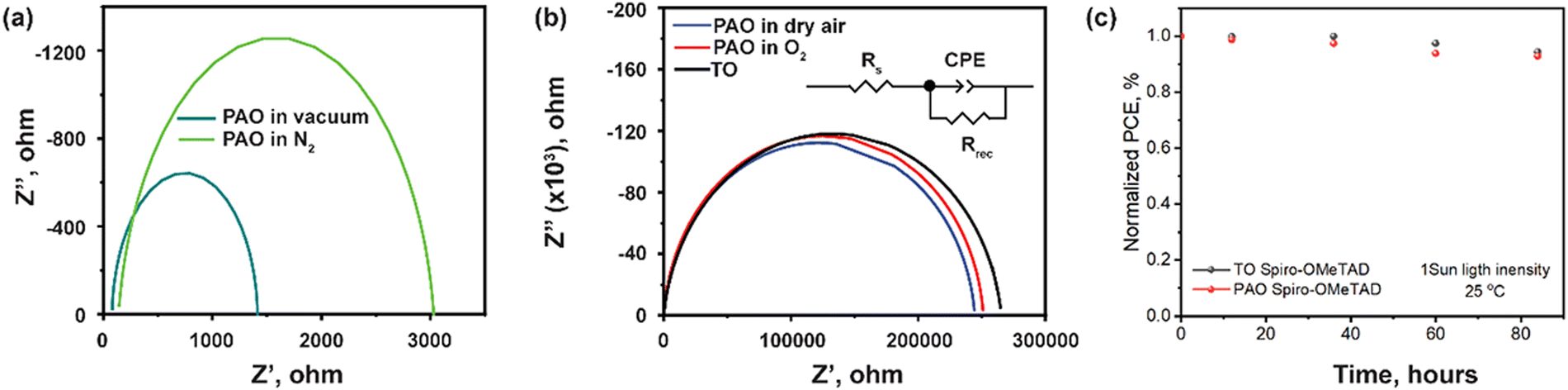 | ||
| Fig. 6 (a) Nyquist plot for the devices with PAO Spiro-OMeTAD in vacuum and N2, and (b) Nyquist plot for the devices with TO and PAO Spiro-OMeTAD in O2 and dry air. (c) Light soaking stability test for the TO and PAO Spiro-OMeTAD based perovskite solar cells with a carbon electrode. | ||
| Samples | R s (Ω cm−2) | R rec (Ω cm−2) | CPE (F cm−2) |
|---|---|---|---|
| PAO in vacuum | 125.1 | 1331 | 9.069 × 10−9 |
| PAO in N2 | 112.6 | 3124 | 4.487 × 10−9 |
| PAO in dry air | 98.24 | 244270 |
1.078 × 10−8 |
| PAO in O2 | 94.84 | 250660 |
1.0125 × 10−8 |
| Control | 80.57 | 263290 |
1.3447 × 10−8 |
4. Conclusion
In summary, we conducted a comprehensive study on the oxidation mechanism of the Spiro-OMeTAD in a planar n-i-p carbon-electrode-based PSC using sunlight illumination. By combining the UV-Vis absorption, FTIR, and conductivity measurements of the Spiro-OMeTAD films, we confirmed that oxidation of the Spiro-OMeTAD layer was achieved via 1 sun illumination in a matter of a few minutes as compared to several hours for traditional oxidation in a dark and ambient environment. The oxidized Spiro-OMeTAD possessed improved conductivity and enhanced charge collection efficiency at the HTL/C interface. Through this PAO Spiro-OMeTAD approach, the device reproducibility and uniformity were significantly improved. The champion device for a carbon-based perovskite was achieved using the PAO-treated Spiro-OMeTAD with a champion device performance of 17.17%. In particular, the shortened oxidization process via a simple sunlight simulator could significantly reduce the cost and time for the large-scale manufacturing of perovskite solar modules.Author contributions
S. N. V. prepared the samples and wrote the manuscript. S. N. V., J. W., W. X., and K. K. carried out the experiments and analyzed the data. F. Y. provided facilities and supervised the research work. All authors discussed the results and reviewed the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This material is based upon work supported by the U.S. Department of Energy's Office of Energy Efficiency and Renewable Energy (EERE) under the Solar Energy Technologies Office Award Number DE-EE0009833. This work is supported by the National Science Foundation under contract No. DMR 2330738, ECCS-2413632, CMMI-2226918, and, TI-2329871.References
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef CAS PubMed.
- G. Ding, Y. Zheng, X. Xiao, H. Cheng, G. Zhang, Y. Shi and Y. Shao, J. Mater. Chem. A, 2022, 10, 8159–8171 RSC.
- H. Min, D. Y. Lee, J. Kim, G. Kim, K. S. Lee, J. Kim, M. J. Paik, Y. K. Kim, K. S. Kim and M. G. Kim, Nature, 2021, 598, 444–450 CrossRef CAS PubMed.
- S. Vijayaraghavan, J. Wall, H. G. Menon, X. Duan, L. Guo, A. Amin, X. Han, L. Kong, Y. Zheng and L. Li, Sol. Energy, 2021, 230, 591–597 CrossRef CAS.
- S. Vijayaraghavan, J. Wall, L. Li, G. Xing, Q. Zhang and F. Yan, Mater. Today Phys., 2020, 13, 100204 CrossRef.
- Y. Rong, M. Jin, Q. Du, Z. Shen, Y. Feng, M. Wang, F. Li, R. Liu, H. Li and C. Chen, J. Mater. Chem. A, 2022, 10, 22592–22604 RSC.
- H. Zhang, K. Song, L. Zhu and Q. Meng, Carbon, 2020, 168, 372–391 CrossRef CAS.
- L. Yang, U. B. Cappel, E. L. Unger, M. Karlsson, K. M. Karlsson, E. Gabrielsson, L. Sun, G. Boschloo, A. Hagfeldt and E. M. Johansson, Phys. Chem. Chem. Phys., 2012, 14, 779–789 RSC.
- T. Leijtens, I.-K. Ding, T. Giovenzana, J. T. Bloking, M. D. McGehee and A. Sellinger, ACS Nano, 2012, 6, 1455–1462 CrossRef CAS PubMed.
- Y. Wang, H. Qu, C. Zhang and Q. Chen, Sci. Rep., 2019, 9, 1–9 CrossRef PubMed.
- Z. Shariatinia and S.-I. Sarmalek, Sci. Rep., 2022, 12, 1–17 CrossRef PubMed.
- Z. Hawash, L. K. Ono, S. R. Raga, M. V. Lee and Y. Qi, Chem. Mater., 2015, 27, 562–569 CrossRef CAS.
- G. Ren, W. Han, Y. Deng, W. Wu, Z. Li, J. Guo, H. Bao, C. Liu and W. Guo, J. Mater. Chem. A, 2021, 9, 4589–4625 RSC.
- X. Sallenave, M. Shasti, E. H. Anaraki, D. Volyniuk, J. V. Grazulevicius, S. M. Zakeeruddin, A. Mortezaali, M. Grätzel, A. Hagfeldt and G. Sini, J. Mater. Chem. A, 2020, 8, 8527–8539 RSC.
- Z. Ahmad, A. Mishra, S. M. Abdulrahim, D. Taguchi, P. Sanghyun, F. Aziz, M. Iwamoto, T. Manaka, J. Bhadra and N. J. Al-Thani, Sci. Rep., 2021, 11, 1–11 CrossRef PubMed.
- J. Kong, Y. Shin, J. A. Röhr, H. Wang, J. Meng, Y. Wu, A. Katzenberg, G. Kim, D. Y. Kim and T.-D. Li, Nature, 2021, 594, 51–56 CrossRef CAS PubMed.
- A. Abate, T. Leijtens, S. Pathak, J. Teuscher, R. Avolio, M. E. Errico, J. Kirkpatrik, J. M. Ball, P. Docampo and I. McPherson, Phys. Chem. Chem. Phys., 2013, 15, 2572–2579 RSC.
- B. Tan, S. R. Raga, A. S. Chesman, S. O. Fürer, F. Zheng, D. P. McMeekin, L. Jiang, W. Mao, X. Lin and X. Wen, Adv. Energy Mater., 2019, 9, 1901519 CrossRef.
- C. C. Boyd, R. Cheacharoen, T. Leijtens and M. D. McGehee, Chem. Rev., 2018, 119, 3418–3451 CrossRef PubMed.
- X. Gu, Y. Li, Y. Mu, M. Zhang, T. Lu and P. Wang, RSC Adv., 2018, 8, 9409–9413 RSC.
- Y. Saygili, H.-S. Kim, B. Yang, J. Suo, A. B. Muñoz-Garcia, M. Pavone and A. Hagfeldt, ACS Energy Lett., 2020, 5, 1271–1277 CrossRef CAS.
- J.-Y. Seo, S. Akin, M. Zalibera, M. A. R. Preciado, H.-S. Kim, S. M. Zakeeruddin, J. V. Milić and M. Grätzel, Adv. Funct. Mater., 2021, 31, 2102124 CrossRef CAS.
- C. Ding, R. Huang, C. Ahlaeng, J. Lin, L. Zhang, D. Zhang, Q. Luo, F. Li, R. Österbacka and C.-Q. Ma, J. Mater. Chem. A, 2021, 9, 7575–7585 RSC.
- E.-D. Jia, X. Lou, C.-L. Zhou, W.-C. Hao and W.-J. Wang, Chin. Phys. B, 2017, 26, 068803 CrossRef.
- C. L. Watts, L. Aspitarte, Y.-H. Lin, W. Li, R. Elzein, R. Addou, M. J. Hong, G. S. Herman, H. J. Snaith and J. G. Labram, Commun. Phys., 2020, 3, 1–10 CrossRef.
- L. Yang, B. Xu, D. Bi, H. Tian, G. Boschloo, L. Sun, A. Hagfeldt and E. M. Johansson, J. Am. Chem. Soc., 2013, 135, 7378–7385 CrossRef CAS PubMed.
- H. Wei, J. Xiao, Y. Yang, S. Lv, J. Shi, X. Xu, J. Dong, Y. Luo, D. Li and Q. Meng, Carbon, 2015, 93, 861–868 CrossRef CAS.
- S. Fantacci, F. De Angelis, M. K. Nazeeruddin and M. Grätzel, J. Phys. Chem. C, 2011, 115, 23126–23133 CrossRef CAS.
- J. Wang, J. Li, X. Xu, Z. Bi, G. Xu and H. Shen, RSC Adv., 2016, 6, 42413–42420 RSC.
- G. Liu, X. Xi, R. Chen, L. Chen and G. Chen, J. Renewable Sustainable Energy, 2018, 10, 043702 CrossRef.
- X. Zhao, M. L. Ball, A. Kakekhani, T. Liu, A. M. Rappe and Y.-L. Loo, Nat. Commun., 2022, 13, 1–10 Search PubMed.
- C. Cho, S. Feldmann, K. M. Yeom, Y.-W. Jang, S. Kahmann, J.-Y. Huang, T. C. J. Yang, M. N. T. Khayyat, Y.-R. Wu and M. Choi, Nat. Mater., 2022, 1–8 Search PubMed.
- S. N. Vijayaraghavan, J. Wall, W. Xiang, K. Khawaja, L. Li, K. Zhu, J. J. Berry and F. Yan, ACS Appl. Mater. Interfaces, 2023, 15, 15290–15297 CrossRef CAS PubMed.
- H. Zhang, Y. Li, S. Tan, Z. Chen, K. Song, S. Huang, J. Shi, Y. Luo, D. Li and Q. Meng, J. Colloid Interface Sci., 2022, 608, 3151–3158 CrossRef CAS PubMed.
- K. Lee, J. Kim, H. Yu, J. W. Lee, C.-M. Yoon, S. K. Kim and J. Jang, J. Mater. Chem. A, 2018, 6, 24560–24568 RSC.
- M. Omrani, R. Keshavarzi, M. Abdi-Jalebi and P. Gao, Sci. Rep., 2022, 12, 1–10 CrossRef PubMed.
- P. Jiang, T. W. Jones, N. W. Duffy, K. F. Anderson, R. Bennett, M. Grigore, P. Marvig, Y. Xiong, T. Liu and Y. Sheng, Carbon, 2018, 129, 830–836 CrossRef CAS.
- C. Xu, L. Zuo, P. Hang, X. Guo, Y. Pan, G. Zhou, T. Chen, B. Niu, X. Xu and Z. Hong, J. Mater. Chem. A, 2022, 10, 9971–9980 RSC.
- D. Liu, M. Chen, Z. Wei, C. Zou, X. Liu, J. Xie, Q. Li, S. Yang, Y. Hou and H. G. Yang, J. Mater. Chem. A, 2022, 10, 10865–10871 RSC.
- W. Tress, M. Yavari, K. Domanski, P. Yadav, B. Niesen, J. P. C. Baena, A. Hagfeldt and M. Graetzel, Energy Environ. Sci., 2018, 11, 151–165 RSC.
- Y. S. Kim, H.-J. Jin, H. R. Jung, J. Kim, B. P. Nguyen, J. Kim and W. Jo, Sci. Rep., 2021, 11, 1–10 CrossRef PubMed.
- D. Xu, Z. Gong, Y. Jiang, Y. Feng, Z. Wang, X. Gao, X. Lu, G. Zhou, J.-M. Liu and J. Gao, Nat. Commun., 2022, 13, 1–8 Search PubMed.
- K. Liu, Y. Luo, Y. Jin, T. Liu, Y. Liang, L. Yang, P. Song, Z. Liu, C. Tian and L. Xie, Nat. Commun., 2022, 13, 1–10 CAS.
- P. You, G. Tang, J. Cao, D. Shen, T.-W. Ng, Z. Hawash, N. Wang, C.-K. Liu, W. Lu and Q. Tai, Light: Sci. Appl., 2021, 10, 1–12 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2024 |