
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAnnealing activated nickel–molybdenum oxide as an efficient electrocatalyst toward benzyl alcohol upgrading†
Shunda
Hu
a,
Xiaoning
Sun
b,
Zimeng
Liu
b,
Lingfeng
Gao
*a,
Xiaoli
Li
a,
Chunyu
Yu
a,
Xiangkang
Han
a,
Junfeng
Xie
 *b and
Xu
Sun
*a
*b and
Xu
Sun
*a
aKey Laboratory of Interfacial Reaction & Sensing Analysis in Universities of Shandong, School of Chemistry and Chemical Engineering Institution, University of Jinan, Jinan, Shandong 250022, P. R. China. E-mail: chm_gaolf@ujn.edu.cn; chm_sunx@ujn.edu.cn
bCollege of Chemistry, Chemical Engineering and Materials Science, Key Laboratory of Molecular and Nano Probes (Ministry of Education), Collaborative Innovation Center of Functionalized Probes for Chemical Imaging in Universities of Shandong, Institute of Molecular and Nano Science, Shandong Normal University, Jinan, Shandong 250014, P. R. China. E-mail: xiejf@sdnu.edu.cn
First published on 28th November 2023
Abstract
Efficient coupling of economically favorable electro-oxidation reactions with the hydrogen evolution reaction (HER) has been considered as a promising way to realize synergistic production of hydrogen and value-added chemicals. In this work, a supported nickel molybdenum oxide catalyst was fabricated, which exhibits enhanced activity towards the benzyl alcohol oxidation reaction (BOR) benefited from the enriched active sites. Further investigations indicate that the nearly complete conversion from benzyl alcohol (BA) to the benzoic acid (BC) product can be achieved with simultaneously realized high selectivity and high faradaic efficiency (FE) for long-term operation. The efficient catalyst explored in this work could offer a new material platform for coupled production and value-added benzoic acid and hydrogen.
Introduction
With the concern of the global energy crisis as well as the depletion of fossil fuel reserves, there is an urgent need to explore clean and sustainable alternative energy sources. Hydrogen (H2), as a carbon-free and sustainable energy carrier, holds immense potential to alleviate the dependence on fossil fuels and related environmental concerns.1,2 The electrocatalytic water splitting process that is composed of the hydrogen evolution reaction (HER) and oxygen evolution reaction (OER) is now a viable method for hydrogen production, with significant progress being made to improve the reaction efficiency.3–9 However, the high overpotential and the sluggish kinetics of the OER process at the anode still impede the whole reaction efficiency for hydrogen production. Hence, optimizing the coupled anodic reaction is of great significance to promote the hydrogen production process.10–16As discussed above, replacing the OER process with thermodynamically favorable electro-oxidation reactions of small molecular chemicals, such as methanol, ethanol, hydrazine and urea,17–28 has been regarded as an advanced way of realizing efficient hydrogen production and even simultaneous valorization to produce value-added chemicals.29,30 As a widely used chemical in the pharmaceutical, dye and fragrance industries, benzoic acid (BC) receives substantial demand, and can be produced from the oxidation of benzyl alcohol (BA).31 However, the current synthesis of BC from BA requires harsh conditions such as the use of harmful reagents, high temperature and noble metal catalysts.32 Therefore, developing a green and economic method for BC production is highly attractive and urgently desired.33,34 With the above considerations, developing an advanced catalyst that could catalyze the BA-to-BC conversion will be of great significance to realize simultaneous production of hydrogen and value-added BC.35,36
Despite the optimization of the oxidation reaction, the design and selection of the appropriate electrocatalyst is also an important factor that would influence the whole reaction efficiency. Recently, transition metal-based compounds have emerged as the preferred choice due to their low cost, high activity, excellent stability, and abundant reserves.37–39 In particular, the synergistic effect between different cationic sites in multi-metal oxides has been extensively studied, which could lead to optimized electronic structures and multi-site active centers.40–46 Among the transition metal oxide catalysts, the Ni–Mo based oxides, have received substantial research interest owing to their abundant high-valence active sites with favorable electronic structure, which have been applied as highly efficient electrocatalysts for the OER and the urea oxidation reaction (UOR).47 For example, Solomon and co-workers fabricated nickel–molybdenum oxide microcrystals covered by Co3O4 nanoparticles via atomic layer deposition, which exhibit high activity towards the OER.48 Fang and co-workers proposed an electrochemical tuning strategy to fabricate Ni3+-rich NiMoO4·xH2O nanorod arrays, and realized electrochemical tuning that effectively enriches the content of Ni3+, finally resulting in the remarkable enhancement of the UOR activity.49 However, the application of Ni–Mo oxides for organic molecule oxidation is still rare to date.50,51 Developing the potential application of Ni–Mo based catalysts for the electrocatalytic oxidation of organic molecules is highly desired.52 In this work, a Ni–Mo oxide catalyst supported on nickel foam (NiMoO-Ar/NF) was successfully fabricated, which exhibits advanced catalytic activity for the benzyl alcohol oxidation reaction. The as-explored NiMoO-Ar/NF catalyst exhibits a low potential of 1.39 V vs. RHE to reach a current density of 10 mA cm−2 in 1 M KOH solution with 15 mM BA. Gas chromatography results indicate that nearly complete conversion from BA to BC can be achieved after electrolysis at 1.5 V vs. RHE for 2 h, and the faradaic efficiency (FE) and yield of the product reach 93% and 95%, respectively. Meanwhile, excellent stability can be achieved even for 4-cycle chronoamperometric tests with a total operation time of 8 hours. This research developed a highly efficient electrocatalyst for coupled green synthesis of value-added benzoic acid and hydrogen.
Results and discussion
The NiMoO-Ar/NF catalyst was synthesized via a two-step reaction (Fig. 1), and the experimental details are provided in the ESI.† Typically, the NiMoO/NF precursor was firstly fabricated via a hydrothermal reaction, followed by annealing in an Ar atmosphere, which finally leads to the formation of NiMoO-Ar/NF. To analyze the crystal structure of the product, the X-ray diffraction (XRD) pattern of the NF-supported NiMoO after calcination in Ar was provided in Fig. 2A, and the XRD pattern of the NiMoO/NF precursor without calcination was provided as well for comparison. As presented by the purple line, all the peaks can be indexed to the NiMoO4·xH2O phase as reported in the literature.48 In contrast, a series of peaks appeared at 14.2°, 25.3°, 28.8° and 32.5° for NiMoO-Ar/NF, which match well with the standard card of NiMoO4 (JCPDS card no. 33-0948), proving the removal of crystal water in the NiMoO4·xH2O precursor. The peaks located at 44.5°, 51.8° and 76.3° correspond to metallic nickel of the NF support (JCPDS card no. 04-0850). As reported in previous literature, the removing of crystal water is beneficial for the enhancement of the catalytic activity since crystal water may occupy the active sites and block the catalytic reaction.53,54 Hence, enhanced catalytic activity can be expected for the NiMoO4-based catalyst without crystal water. | ||
| Fig. 1 Schematic illustration of the synthesis process of the NiMoO-Ar/NF catalyst. | ||
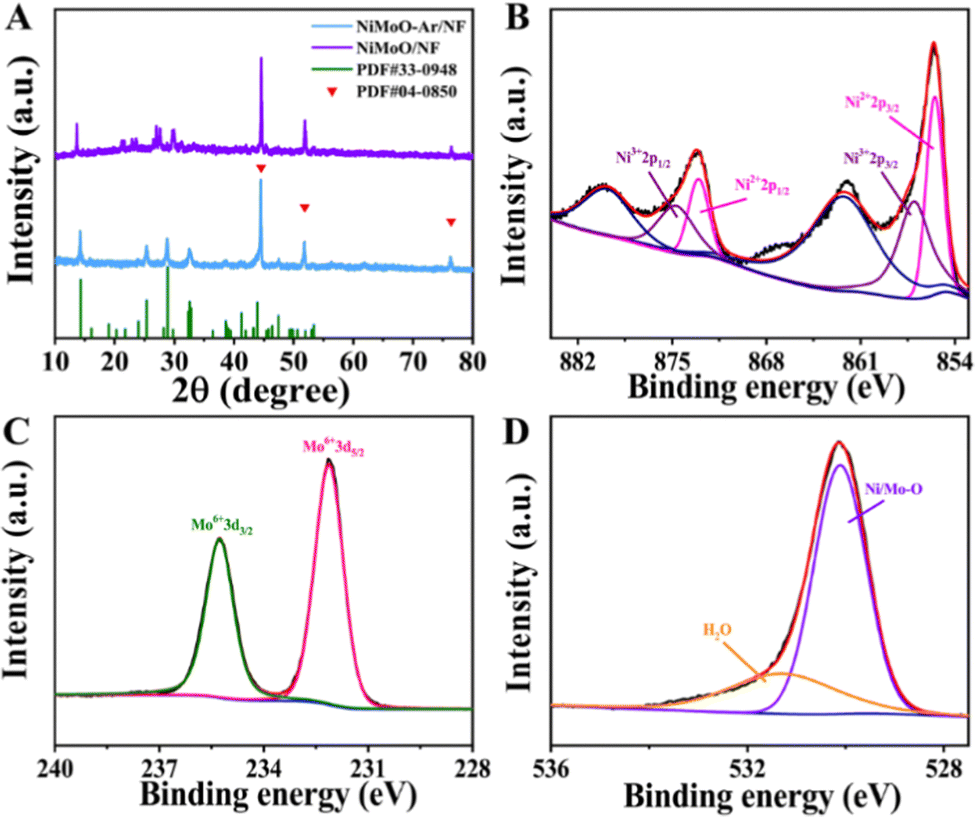 | ||
| Fig. 2 (A) XRD patterns of NiMoO-Ar/NF and NiMoO/NF. (B) Ni 2p spectra. (C) Mo 3d spectra. (D) O 1s spectra. | ||
To further analyze the chemical composition and valence states of the as-synthesized NiMoO-Ar/NF catalyst, X-ray photoelectron spectroscopy (XPS) was conducted. As shown in the survey spectrum, Fig. S1 (ESI†), the characteristic peaks corresponding to Ni, Mo and O elements can be observed, confirming the presence of these elements in the catalyst. As shown in the high-resolution Ni 2p spectrum in Fig. 2B, two intense peaks can be identified at 873.0 and 855.4 eV, which can be indexed to the 2p1/2 and 2p3/2 peaks of Ni2+ ions, respectively. Meanwhile, the peaks corresponding to Ni3+ emerge at 874.8 eV and 856.9 eV, confirming the coexistence of Ni2+ and Ni3+ in the NiMoO-Ar/NF catalyst.45,55 The Mo 3d spectrum was provided as Fig. 2C, from which two peaks corresponding to Mo6+ species can be identified at 232.1 and 235.2 eV, respectively.55,56 As reported in previous literature, the presence of Mo species with high oxidation valence can significantly influence the electronic structure of neighboring metals, which might bring about the variation of the catalytic performance.48,55 Considering the above fact, the electro-oxidation performance can be expected for NiMoO-Ar/NF. In addition, the O 1s spectrum in Fig. 2D exhibits two peaks centered at 530.0 and 531.3 eV, assigned to the metal–oxygen bonds and the adsorbed water molecules, respectively.8,57 Furthermore, considering the existence of [MoO4]2− oxyanion counterparts, which are vibrationally active, both the Raman and Fourier transform infrared (FTIR) spectra of NiMoO-Ar/NF and NiMoO/NF were provided for better structure analysis, as shown in Fig. S3A and B (ESI†). As shown in the FTIR spectral in Fig. S3A (ESI†), it can be seen that an obvious peak appeared at 3400.3 cm−1, which should be attributed to the O–H stretching of water molecules.48 Besides, the peak corresponding to the absorbed water molecules was observed as well at 1621.8 cm−1, which is well consistent with the previous literature.58,59 This confirms the existence of water in the NiMoO/NF structure. Besides, a serious peak appeared at about 449.8 to 964.7 cm−1, which should be attributed to the symmetric and asymmetric stretching of Mo–O bonds.48,60 As for the NiMoO-Ar/NF, similar peaks were observed as well at 455.6 cm−1 to 960.8 cm−1. Meanwhile, the two peaks that were indexed to the water molecules were decreased in the NiMoO-Ar/NF catalyst, proving the removal of water molecules. From the Raman spectra in Fig. S3B (ESI†), as for the NiMoO/NF sample, an obvious peak appeared at about 942.8 cm−1, which should be attributed to the symmetric stretching of the Mo–O bands. Besides, two peaks that were indexed to the asymmetric stretching of O–Mo–O appeared as well at 891 and 818.7 cm−1, respectively. Furthermore, the band corresponding to the bending mode appeared as well at the low energy area of about 300–400 cm−1. These values were well consistent with the previous literature, confirming the successful fabrication of NiMoO4·xH2O.58–60 As for the NiMoO-Ar/NF obtained after annealing treatment, similar peaks were observed as well at 952.8, 892, 817 and 342 cm−1, confirming the well-kept Mo–O and O–Mo–O structure. Furthermore, a new peak corresponding to NiMoO4 appeared at 706 cm−1, which is in agreement with the annealed NiMoO4 sample as reported in the literature.61 The peak at 952.8 cm−1 corresponds to the symmetric stretching mode of the Mo–O bond, while the peaks at 706 and 342 cm−1 correspond to the asymmetric stretching mode of the Ni–O–Mo bond and the bending mode of Mo–O, respectively.62
Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images are provided in Fig. 3. As shown in the SEM images in Fig. 3A, a thick oxide layer comprised from nanorods can be observed on the surface of the Ni foam. The magnified SEM image was provided in Fig. 3B, from which the square-shaped cross section of the nanorods can be observed. Meanwhile, the TEM images of the powdery sample detached from NiMoO-Ar/NF are shown in Fig. 3C and D, further proving the nanorod morphology with a width of about 500 nm, which is consistent with the SEM result. To figure out the crystal structure, a high-resolution TEM (HRTEM) image is provided in Fig. 3E. As can be seen, uniform lattice fringes with an interplanar spacing of 0.619 nm can be identified, which match well with the (110) plane of the NiMoO4 phase. In addition, the elemental distribution based on the energy-dispersive X-ray spectroscopy (EDS) technique is shown in Fig. 3F. It can be observed that Ni (green), Mo (red), and O (blue) are homogeneously distributed across the nanorods. In addition, Fig. S4 (ESI†) illustrates the SEM of NiMoO/NF.
 | ||
| Fig. 3 (A) SEM images of NiMoO-Ar/NF. (B) Magnified SEM images. (C) and (D) TEM images of NiMoO-Ar/NF. (E) HRTEM images of NiMoO-Ar/NF. (F) Element mapping of Ni, Mo, and O in NiMoO-Ar/NF. | ||
The electrochemical measurements were conducted by using a typical three-electrode system. The linear sweep voltammetry (LSV) curves of NiMoO-Ar/NF were firstly analyzed to unravel the different behavior between the OER and BOR. Typically, the polarization curves for BOR and OER were obtained from 1 M KOH electrolyte with and without 15 mM benzyl alcohol. As can be seen from Fig. S6 (ESI†), the required potential for BOR to drive a 10 mA cm−2 anodic current density is only 1.39 V vs. RHE. In contrast, the required potential for the OER to reach the same current density is 1.49 V vs. RHE.
Furthermore, compared with the OER process that only generates oxygen, the electro-oxidation of benzyl alcohol may achieve the production of the value-added benzoic acid, which is considered to be a more promising pathway for coupling with the HER half reaction to reach energy-saving production. To investigate the BOR performance of the catalysts, the LSV curve comparison of NiMoO-Ar/NF, NiMoO/NF and calcined NF for BOR was provided in Fig. 4A. It can be seen that as for the NiMoO-Ar/NF catalyst, the current density reaches 10.72 mA cm−2 at 1.4 V (vs. RHE). Meanwhile, for NiMoO/NF and NF, the current density was only about 0.266 and 1.452 mA cm−2, respectively. Furthermore, the potentials at the fixed current density (20 mA cm−2) were compared as well. This illustrated that the NiMoO-Ar/NF catalyst only needed 399 mV to reach 20 mA cm−2. The potentials for NiMoO/NF and NF to reach the same current density were 563 and 747 mV, respectively, much larger than that of NiMoO-Ar/NF catalyst. Additionally, as shown in Fig. 4B, the Tafel plots also indicate the excellent activity of the NiMoO-Ar/NF catalyst. It can be seen that the Tafel slope of NiMoO-Ar/NF is as small as 42 mV dec−1, significantly lower than that of the NiMoO/NF catalyst (55 mV dec−1) and the NF-Ar catalyst (82 mV dec−1), further demonstrating the enhanced BOR performance of NiMoO-Ar/NF.
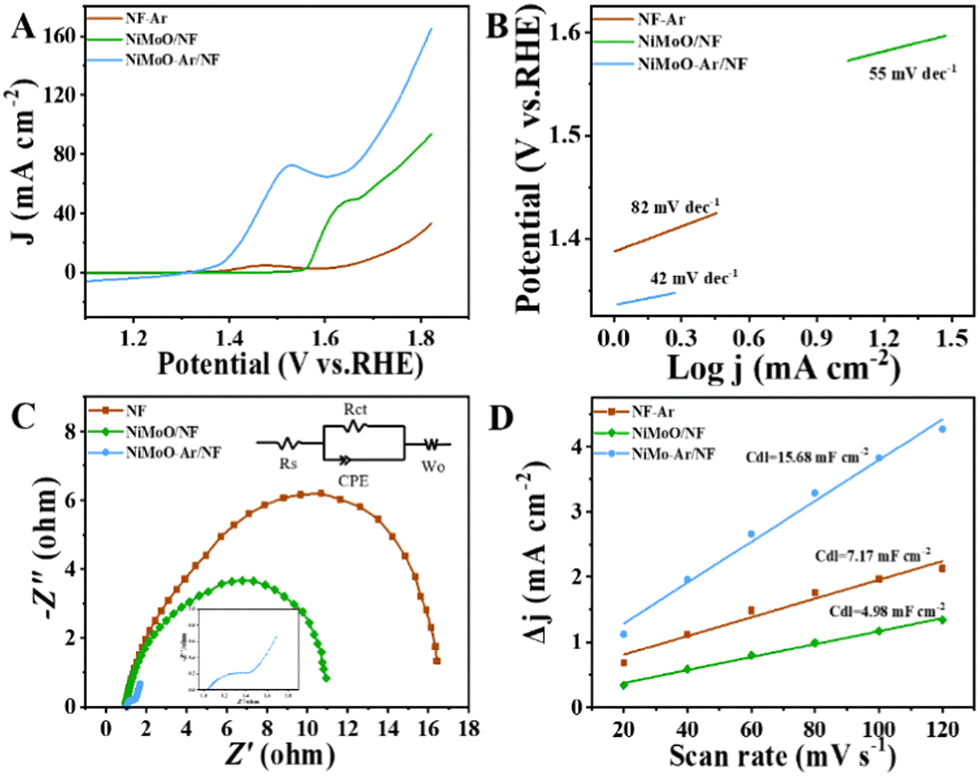 | ||
| Fig. 4 (A) Polarization curve comparison of NF-Ar, NiMoO/NF, and NiMoO-Ar/NF in electrolyte containing 15 mM BA. (B) Tafel curves corresponding to figure A. (C) Comparison of the impedance of NF-Ar, NiMoO/NF and NiMoO-Ar/NF in electrolyte containing 15 mM BA. (D) Cdl curves of NF-Ar, NiMoO/NF and NiMoO-Ar/NF in an electrolyte containing 15 mM BA. | ||
To further analyze the catalytic transfer kinetics of the NiMoO-Ar/NF catalyst, electrochemical impedance spectroscopy (EIS) analysis was performed at 1.5 V vs. RHE, with the corresponding results being shown in Fig. 4C. It reveals that the charge transfer resistance (Rct) of the NiMoO-Ar/NF catalyst is about 1.45 Ω, much smaller than that of NiMoO/NF (10.42 Ω) and NF-Ar (16.98 Ω), indicating the promoted charge transfer process during the BOR catalysis. According to the above analyses, it is concluded that the annealing treatment is beneficial for the enhancement of the catalytic activity. This might be owing to the following two reasons. Firstly, the annealing treatment could lead to the removal of crystal water within the NiMoO4·xH2O lattice, thus increasing the exposure of active sites, which finally results in enhanced catalytic activity. Secondly, the calcination treatment may induce the formation of a porous structure, thereby further increasing the surface area and enriching the catalytic activity. To prove the increment of the surface area, the nitrogen adsorption–desorption isotherms of NiMoO-Ar/NF and NiMoO/NF were measured. As revealed in Fig. S5 (ESI†), NiMoO-Ar/NF displays a larger BET specific surface area compared with that of NiMoO/NF, therefore confirming the enhancement of the surface area owing to the calcination. In addition, the electrochemical active surface area (ECSA) that is linearly proportional to the double-layer capacitance (Cdl) is estimated as presented in Fig. 4D and Fig. S7 (ESI†). The results showed that the NiMoO-Ar/NF holds the largest Cdl value of 15.68 mF cm−2. Meanwhile, for the NiMoO/NF catalyst and NF-Ar catalyst, the Cdl values were just 4.98 mF cm−2 and 7.17 mF cm−2, respectively. The much larger Cdl value of the NiMoO-NF catalyst confirmed its larger specific surface area, which is essential for increasing the exposure of active sites and thus improving the catalytic activity.
Based on the above analysis, it is concluded that the NiMoO-Ar/NF is an ideal catalyst for BOR. To further investigate the oxidation products of BOR, chronoamperometry measurement was performed at 1.5 V vs. RHE with the gas chromatography (GC) results, as shown in Fig. S10 (ESI†). As can be seen from Fig. S10A (ESI†), before the reaction, an intensive peak that corresponds to BA was observed at about 10.2 min. After the reaction for half an hour, a new peak located at approximately 12.2 min emerges, corresponding to BC, as shown in Fig. S10B (ESI†). The formation of the BC product indicates the occurrence of the electro-oxidation of benzyl alcohol. Besides, another new peak that was attributed to benzaldehyde (BH) can be observed at 9.0 min, which was brought by the side reaction. It is noteworthy that the peak area of BC is larger than that of BH as presented in Table S2 (ESI†), suggesting that the side reaction is slight. Furthermore, as illustrated in Fig. S10C and D (ESI†), along with the increment of the reaction time to 1 h and 1.5 h, the peak for BC was further increased, indicating that more BA was converted to the product. Then, after the reaction for 2 h (Fig. S10E, ESI†), the peak for BA nearly disappeared, while the BC peak is dominant, indicating the high conversion efficiency from BA to BC. The concentration of BC was calculated according to the GC results (Fig. 5A). It can be seen that along with the increment of the reaction time, the concentration of BC increased, achieving a high value of 14.21 mM after reaction for 2 h. Hence, the high activity of NiMoO-Ar/NF catalyst towards the BOR can be confirmed.
 | ||
| Fig. 5 (A) Evolution of the concentration of BA, BC and BH under various reaction times at 1.5 V vs. RHE. (B) Con., Sel., yield and FE at different potentials. (C) The chronoamperometry tests were repeated four times. (D) Con., Sel., yield and FE of BC in four electrochemical cycles. | ||
To survey the influence of applied potential, chronoamperometry measurements at various potentials were conducted, and the corresponding GC results are provided in Fig. 5B and Fig. S11, Table S3 (ESI†). It can be seen that high selectivity (Sel.) can be achieved at various potentials, proving a wide potential window for high-performance BOR. In contrast, the conversion percentage (Con.), yield and faradaic efficiency (FE) display variation along with the change of potential, and the optimal performance is determined to be 1.5 V vs. RHE. In detail, the Con. of BA reaches 94.27%, and the Sel. to BC and FE are as high as 100% and 91.25%. When the applied potential further increases, the competitive OER process becomes obvious, and the BOR efficiency declines.
Operational stability is another important criterion to judge an advanced catalyst. We further performed cyclic voltammetry (CV) cycling tests and compared the polarization curves before and after 1000 CV cycles. The electrolyte for the CV stability test was 1 M KOH + 15 mM BA. As shown in Fig. S12 (ESI†), the anodic current density shows negligible degradation, confirming the high stability of the NiMoO-Ar/NF catalyst. In addition, a long-term chronoamperometry test was also performed, and the electrolyte was refreshed per 2 h to reduce the influence of the concentration of BA to the activity. As can be seen from Fig. 5C, the current density decreases along with the BOR catalysis, and the current density can be well recovered after refreshing the electrolyte, indicating that the decreasing current density originates from the efficient BOR process that rapidly consumes BA. The conversion percentage, selectivity, yield and FE were also measured after 4 cycles of 2 hour BOR operation. As shown in Fig. 5D, all the parameters exhibit negligible decay, and the FE still reaches ∼93%, further confirming the superior operational stability of NiMoO-Ar/NF towards the BOR. The high activity and stability of NiMoO-Ar/NF for BOR catalysis make the catalyst a promising catalyst for the green production of value-added benzoic acid.
Conclusions
In summary, a NiMoO-Ar/NF nanorod array catalyst was synthesized for an electrocatalytic benzyl alcohol oxidation reaction to produce benzoic acid. By means of annealing treatment, the crystal water within the lattice of the hydrated Ni–Mo oxide precursor can be removed, leading to highly exposed active sites and enhanced catalytic activity. Meanwhile, along with the calcination process, porous structure with increased surface area can be obtained, thereby further facilitating mass transport and enriching the active sites. Benefited from the above structural merits, the NiMoO-Ar/NF catalyst exhibited excellent electrocatalytic activity for BOR catalysis with a low required potential of 1.39 V vs. RHE to achieve a 10 mA cm−2 current density. In addition, nearly complete conversion from BA to BC can be realized after 2-hour electrolysis at 1.5 V vs. RHE with high faradaic efficiency and yield of product. Superior operational stability can also be achieved even for 4-cycle chronoamperometric tests with only slight performance decay. This work presents a new electrocatalyst for green electrochemical synthesis of benzoic acid, which may shed light on the future design of advanced catalysts for the electro-oxidation of organic molecules.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Talent Introduction and Training Program for Youth Innovation teams in Colleges and Universities of Shandong Province, the Natural Science Foundation of Shandong Province (ZR2020MB136) and the National Natural Science Foundation of China (22171167).Notes and references
- Y. Li, X. Wei, L. Chen and J. Shi, Angew. Chem., Int. Ed., 2021, 60, 19550–19571 CrossRef CAS PubMed.
- O. J. Guerra, J. Eichman, J. Kurtz and B.-M. Hodge, Joule, 2019, 3, 2425–2443 CrossRef.
- H. Sun, Z. Yan, F. Liu, W. Xu, F. Cheng and J. Chen, Adv. Mater., 2020, 32, 1806326 CrossRef CAS.
- Z. Chen, X. Duan, W. Wei, S. Wang and B.-J. Ni, J. Mater. Chem. A, 2019, 7, 14971–15005 RSC.
- X. Shang, B. Dong, Y.-M. Chai and C.-G. Liu, Sci. Bull., 2018, 63, 853–876 CrossRef CAS PubMed.
- J. Xie, X. Yang and Y. Xie, Nanoscale, 2020, 12, 4283–4294 RSC.
- J. Xie, J. Qi, F. Lei and Y. Xie, Chem. Commun., 2020, 56, 11910–11930 RSC.
- J. Xie, X. Yang, Y. Wang, L. Kang, J. Li, Z. Wei, P. Hao, F. Lei, Q. Wang and B. Tang, Chem. Commun., 2021, 57, 11517–11520 RSC.
- X. Han, H. Sheng, C. Yu, T. W. Walker, G. W. Huber, J. Qiu and S. Jin, ACS Catal., 2020, 10, 6741–6752 CrossRef CAS.
- M. Yu, E. Budiyanto and H. Tüysüz, Angew. Chem., Int. Ed., 2022, 61, e202103824 CrossRef CAS.
- L. Li, P. Wang, Q. Shao and X. Huang, Chem. Soc. Rev., 2020, 49, 3072–3106 RSC.
- Q. Hu, G. Li, Z. Han, Z. Wang, X. Huang, H. Yang, Q. Zhang, J. Liu and C. He, J. Mater. Chem. A, 2019, 7, 14380–14390 RSC.
- W. Sun, J. Li, W. Gao, L. Kang, F. Lei and J. Xie, Chem. Commun., 2022, 58, 2430–2442 RSC.
- X. Yang, L. Kang, Z. Wei, S. Lou, F. Lei, P. Hao, J. Xie and B. Tang, Chem. Eng. J., 2021, 422, 130139 CrossRef CAS.
- M. Hao, J. Chen, J. Chen, K. Wang, J. Wang, F. Lei, P. Hao, X. Sun, J. Xie and B. Tang, J. Colloid Interface Sci., 2023, 642, 41–52 CrossRef CAS.
- J. Li, C. Dong, M. Guo, W. Gao, L. Kang, F. Lei, P. Hao, J. Xie and B. Tang, Chem. Commun., 2022, 58, 6845–6848 RSC.
- Y. Wang, X. Yang, K. Wang, Z. Liu, X. Sun, J. Chen, S.-S. Liu, X. Sun, J. Xie and B. Tang, Green Chem., 2023, 25, 8216–8225 RSC.
- Z. Liang, D. Jiang, X. Wang, M. Shakouri, T. Zhang, Z. Li, P. Tang, J. Llorca, L. Liu, Y. Yuan, M. Heggen, R. E. Dunin-Borkowski, J. R. Morante, A. Cabot and J. Arbiol, Adv. Funct. Mater., 2021, 31, 2106349 CrossRef CAS.
- R. Li, P. Kuang, L. Wang, H. Tang and J. Yu, Chem. Eng. J., 2022, 431, 134137 CrossRef CAS.
- G. Liu, W. Zhou, Y. Ji, B. Chen, G. Fu, Q. Yun, S. Chen, Y. Lin, P.-F. Yin, X. Cui, J. Liu, F. Meng, Q. Zhang, L. Song, L. Gu and H. Zhang, J. Am. Chem. Soc., 2021, 143, 11262–11270 CrossRef CAS PubMed.
- M. T. Bender, X. Yuan and K.-S. Choi, Nat. Commun., 2020, 11, 4594 CrossRef CAS.
- J. Xie, W. Liu, X. Zhang, Y. Guo, L. Gao, F. Lei, B. Tang and Y. Xie, ACS Mater. Lett., 2019, 1, 103–110 CrossRef CAS.
- J. Xie, L. Gao, S. Cao, W. Liu, F. Lei, P. Hao, X. Xia and B. Tang, J. Mater. Chem. A, 2019, 7, 13577–13584 RSC.
- J. Xie, H. Qu, F. Lei, X. Peng, W. Liu, L. Gao, P. Hao, G. Cui and B. Tang, J. Mater. Chem. A, 2018, 6, 16121–16129 RSC.
- L. Gao, J. Xie, S. Liu, S. Lou, Z. Wei, X. Zhu and B. Tang, ACS Appl. Mater. Interfaces, 2020, 12, 24701–24709 CrossRef CAS PubMed.
- M. Li, W. Qi, J. Yu, L. Shen, X. Yang, S. Liu and M.-Q. Yang, Chin. J. Struct. Chem., 2022, 41, 15–24 Search PubMed.
- W. Qiao, X. Huang and L. Feng, Chin. J. Struct. Chem., 2022, 41, 16–34 Search PubMed.
- J. Li, J. Xie, X. Wang, Y. Dai, X. Xu, J. Liu, Z. Cai, X. Meng and J. Zou, Chin. J. Struct. Chem., 2022, 41, 59–67 Search PubMed.
- Y. Huang, X. Chong, C. Liu, Y. Liang and B. Zhang, Angew. Chem., Int. Ed., 2018, 57, 13163–13166 CrossRef CAS PubMed.
- M. T. Bender, Y. C. Lam, S. Hammes-Schiffer and K.-S. Choi, J. Am. Chem. Soc., 2020, 142, 21538–21547 CrossRef CAS PubMed.
- E. Tiburcio, R. Greco, M. Mon, J. Ballesteros-Soberanas, J. Ferrando-Soria, M. López-Haro, J. C. Hernández-Garrido, J. Oliver-Meseguer, C. Marini, M. Boronat, D. Armentano, A. Leyva-Pérez and E. Pardo, J. Am. Chem. Soc., 2021, 143, 2581–2592 CrossRef CAS.
- H. Huang, X. Song, C. Yu, Q. Wei, L. Ni, X. Han, H. Huang, Y. Han and J. Qiu, Angew. Chem., Int. Ed., 2023, 62, e202216321 CrossRef CAS PubMed.
- B. You, X. Liu, X. Liu and Y. Sun, ACS Catal., 2017, 7, 4564–4570 CrossRef CAS.
- D. Feng, Y. Dong, L. Zhang, X. Ge, W. Zhang, S. Dai and Z.-A. Qiao, Angew. Chem., Int. Ed., 2020, 59, 19503–19509 CrossRef CAS PubMed.
- J. Zheng, X. Chen, X. Zhong, S. Li, T. Liu, G. Zhuang, X. Li, S. Deng, D. Mei and J.-G. Wang, Adv. Funct. Mater., 2017, 27, 1704169 CrossRef.
- H. Huang, C. Yu, X. Han, H. Huang, Q. Wei, W. Guo, Z. Wang and J. Qiu, Energy Environ. Sci., 2020, 13, 4990–4999 RSC.
- Y. Xu, M. Liu, S. Wang, K. Ren, M. Wang, Z. Wang, X. Li, L. Wang and H. Wang, Appl. Catal., B, 2021, 298, 120493 CrossRef CAS.
- J. Cao, H. Li, R. Zhu, L. Ma, K. Zhou, Q. Wei and F. Luo, J. Alloys Compd., 2020, 844, 155382 CrossRef CAS.
- Y. Wang, Y. Sun, F. Yan, C. Zhu, P. Gao, X. Zhang and Y. Chen, J. Mater. Chem. A, 2018, 6, 8479–8487 RSC.
- D. Li, F. Ruan, Y. Jin, Q. Ke, Y. Cao, H. Wang, T. Wang, Y. Song and P. Cui, Catal. Sci. Technol., 2019, 9, 418–424 RSC.
- J. Zhang, S. Guo, B. Xiao, Z. Lin, L. Yan, D. Du and S. Shen, Chem. Eng. J., 2021, 416, 129127 CrossRef CAS.
- D. Yang, L. Yang, L. Zhong, X. Yu and L. Feng, Electrochim. Acta, 2019, 295, 524–531 CrossRef CAS.
- X. Cui, M. Chen, R. Xiong, J. Sun, X. Liu and B. Geng, J. Mater. Chem. A, 2019, 7, 16501–16507 RSC.
- W. Sun, Y. Wang, S. Liu, F. Lei, J. Xie and B. Tang, Chem. Commun., 2022, 58, 11981–11984 RSC.
- M. Guo, P. Li, A. Wang, J. Wang, J. Chen, F. Lei, P. Hao, X. Sun, J. Xie and B. Tang, Chem. Commun., 2023, 59, 5098–5101 RSC.
- J. Li, M. Guo, X. Yang, J. Wang, K. Wang, A. Wang, F. Lei, P. Hao, J. Xie and B. Tang, Prog. Nat. Sci.: Mater. Int., 2022, 32, 705–714 CrossRef CAS.
- Z. Ma, H. Meng, M. Wang, B. Tang, J. Li and X. Wang, ChemElectroChem, 2018, 5, 335–342 CrossRef CAS.
- G. Solomon, A. Landström, R. Mazzaro, M. Jugovac, P. Moras, E. Cattaruzza, V. Morandi, I. Concina and A. Vomiero, Adv. Energy Mater., 2021, 11, 2101324 CrossRef CAS.
- M. Fang, W.-B. Xu, Y. Shen, P. Cao, S. Han, W. Xu, D. Zhu, Y. Lu and W. Liu, Appl. Catal., A, 2021, 622, 118220 CrossRef CAS.
- D. M. Morales, D. Jambrec, M. A. Kazakova, M. Braun, N. Sikdar, A. Koul, A. C. Brix, S. Seisel, C. Andronescu and W. Schuhmann, ACS Catal., 2022, 12, 982–992 CrossRef CAS.
- L. A. Wolzak, F. J. de Zwart, J.-P. H. Oudsen, S. A. Bartlett, B. de Bruin, J. N. H. Reek, M. Tromp and T. J. Korstanje, ChemCatChem, 2022, 14, e202201033 CrossRef.
- Y. Wang, B. Zhang, W. Pan, H. Ma and J. Zhang, ChemSusChem, 2017, 10, 4170–4177 CrossRef CAS.
- P. Wang, D. Li, H. Chi, Y. Zhao, J. Wang, D. Li, S. Pang, P. Fu, J. Shi and C. Li, Angew. Chem., Int. Ed., 2021, 60, 6691–6698 CrossRef CAS PubMed.
- H. Wu, T. He, M. Dan, L. Du, N. Li and Z.-Q. Liu, Chem. Eng. J., 2022, 435, 134863 CrossRef CAS.
- C. Dong, M. Guo, W. Gao, P. Hao, F. Lei, J. Xie and B. Tang, J. Colloid Interface Sci., 2022, 627, 891–899 CrossRef CAS.
- S. Cao, J. Qi, F. Lei, Z. Wei, S. Lou, X. Yang, Y. Guo, P. Hao, J. Xie and B. Tang, Chem. Eng. J., 2021, 413, 127540 CrossRef CAS.
- J. Qi, J. Xie, Z. Wei, S. Lou, P. Hao, F. Lei and B. Tang, Chem. Commun., 2020, 56, 4575–4578 RSC.
- A. P. de Moura, L. H. de Oliveira, I. L. V. Rosa, C. S. Xavier, P. N. Lisboa-Filho, M. S. Li, F. A. La Porta, E. Longo and J. A. Varela, Sci. World J., 2015, 2015, 315084 Search PubMed.
- K. Eda, Y. Kato, Y. Ohshiro, T. Sugitani and M. S. Whittingham, J. Solid State Chem., 2010, 183, 1334–1339 CrossRef CAS.
- H. Wan, J. Jiang, X. Ji, L. Miao, L. Zhang, K. Xu, H. Chen and Y. Ruan, Mater. Lett., 2013, 108, 164–167 CrossRef CAS.
- O. Popovych, I. Budzulyak, V. Vashchynskyi, M. Khemii, R. Ilnytskyi and L. Yablon, Appl. Nanosci., 2023, 13, 6803–6809 CrossRef CAS.
- P. R. Jothi, K. Shanthi, R. R. Salunkhe, M. Pramanik, V. Malgras, S. M. Alshehri and Y. Yamauchi, Eur. J. Inorg. Chem., 2015, 3694–3699 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ya00447c |
| This journal is © The Royal Society of Chemistry 2024 |